

Abstract
Mechanisms utilized by human regulatory T cells (Treg) for elimination of effector cells may vary. We investigated the possibility that the mechanism of Treg suppression depends on Fas/FasL-mediated apoptosis of responder cells (RC). CD4+CD25highFoxp3+Treg and autologous CD4+CD25− and CD8+CD25− subsets of RC were isolated from blood of 25 cancer patients and 15 normal controls and cocultured in the presence of OKT3 and IL-2 (150 or 1000 IU/ml). Suppression of RC proliferation was measured in CFSE assays. RC and Treg apoptosis was monitored by 7-aminoactinomycin D staining in flow-based cytotoxicity assays. Treg from all subjects expressed CD95+, but only Treg from cancer patients expressed CD95L. These Treg, when activated via TCR plus IL-2, up-regulated CD95 and CD95L expression (p < 0.001) and suppressed CD8+ RC proliferation (p < 0.001) by inducing Fas-mediated apoptosis. However, Treg cocultured with CD4+ RC suppressed proliferation independently of Fas/FasL. In co-cultures, Treg were found to be resistant to apoptosis in the presence of 1000 IU/ml IL-2, but at lower IL-2 concentrations (150 IU/ml) they became susceptible to RC-induced death. Thus, Treg and RC can reciprocally regulate Treg survival, depending on IL-2 concentrations present in cocultures. This divergent IL-2-dependent resistance or sensitivity of Treg and RC to apoptosis is amplified in patients with cancer.
Regulatory T cells (Treg)3 maintain peripheral tolerance by suppression of proliferating effector T cells (1–4). Treg are necessary for the control of immune responses and their depletion in autoimmunity, and conversely, their expansion in cancer are associated with human disease. Regulation of developing immune responses requires that the functional potential and ratios of Treg and effector T cells are strictly controlled. Apoptosis or programmed cell death of responder T cells (RC) imposes a degree of control on immune responses that develops following antigenic stimulation, and the integrity of apoptotic pathways is required for the maintenance of peripheral tolerance.
T cell activation involves a highly coordinated sequence of events. Signaling via TCR or signal 1, costimulation (signal 2), and cytokine support (signal 3) are all needed for mounting an effective immune response and for RC proliferation and differentiation (5). However, although proliferation of activated T cells is necessary, it cannot proceed forever, and Treg are expected to play a key role in the contraction of immune responses. Recently, Treg have been shown to suppress immune responses not only to self epitopes but also to foreign Ags, including transplantation and tumor-associated Ags (6, 7). Furthermore, Treg appear to be able to inhibit multiple stages of T cell activity: proliferation, differentiation, as well as effector function (8). Mechanisms employed by Treg to mediate suppression of T cell functions at these various levels are not yet understood and remain a matter of considerable controversy.
To maintain lymphocyte homeostasis, the coordinated engagement of apoptotic events involving activation-induced cell death (AICD) of T cells or spontaneous T cell death is necessary (9–11). T lymphocytes become vulnerable to apoptosis at critical points of their lifespan, at the time when clonal expansion ends and the contraction phase begins (9). However, it remains unclear how the numbers of human CD4+CD25highFoxp3+ T cells are regulated or when the Treg subset expands or how Treg exert suppression in the course of the acute and chronic immune responses. We and others have shown that the frequency and suppressor function of Treg increase in the peripheral circulation of patients with cancer (2, 12–14). Evidence for their depletion in autoimmune diseases is well documented (15–17). These and other observations suggest that the mechanisms involved in control of Treg numbers and function are of critical importance for human health and disease.
Human Treg have been shown to express Fas and FasL (2, 12). Therefore, the use of the Fas/FasL pathway by Treg as one mechanism of suppression is expected based on their phenotypic characteristics (2, 10, 18). We hypothesize that suppression mediated by Treg engages the Fas/FasL pathway, resulting in apoptosis of RC. Also, Treg might become susceptible to Fas-mediated apoptosis during their interaction with RC. However, Treg sensitivity/resistance to this pathway have also been a subject of considerable controversy in the literature (19–21).
In this paper, we use an in vitro system established with circulating CD4+ or CD8+ T lymphocytes and autologous Treg obtained from peripheral blood of patients with cancer and normal controls (NC) to investigate whether the mechanism of Treg suppression depends on Fas/FasL-mediated apoptosis of RC. We demonstrate that in Treg/RC cocultures, these cells can reciprocally regulate each other’s survival. Activated human CD4+CD25high Foxp3+ Treg induce Fas-mediated apoptosis in autologous CD8+ RC, and this mechanism is in part responsible for suppression of RC proliferation. In contrast, CD4+ RC are resistant to Fas-mediated killing by Treg but are able to induce death of Treg in cocultures containing lower concentrations of IL-2. Furthermore, in patients with cancer, these killing mechanisms are amplified, providing a partial explanation for the death of CD8+ effector T cells and a relative resistance of CD4+ T cells to apoptosis observed in cancer (11).
Materials and Methods
Head and neck squamous cell carcinoma (HNSCC) patients and healthy volunteers
Blood samples were obtained from 25 HNSCC patients and 15 age-matched healthy controls (NC). All subjects signed an informed consent approved by the Institutional Review Board of the University of Pittsburgh. All patients were seen at the Outpatient Otolaryngology Clinic at the University of Pittsburgh Cancer Institute between January 2006 and April 2007. The patient cohort included 17 males and 8 females, with a mean age of 60 years (range, 23–82 years). The NC group included 10 males and 5 females, with a mean age of 60 years. Of patients seen, 22 had a primary disease and 3 had a recurrent disease. At the time of the blood draw, 7 patients were untreated and 18 received various forms of oncologic therapy. The therapy, if given, was terminated from 3 wk to 12 mo before the time of phlebotomy for this study. The age, sex, and clinicopathologic characteristics of the patients are listed in Table I.
Table I.
Clinicopathologic characteristics of patients with HNSCC who donated PBMC for this study
| Age (years): mean range | 23–82 |
| Sex | |
| Male | 17 |
| Female | 8 |
| Tumor site | |
| Nasal cavity | 2 |
| Oral cavity | 15 |
| Oropharynx | 2 |
| Hypopharynx | 1 |
| Larynx | 4 |
| Not determined | 1 |
| Tumor differentiation | |
| Poor | 5 |
| Moderate | 15 |
| Well | 3 |
| Not determined | 2 |
| Tumor state | |
| T1 | 2 |
| T2 | 6 |
| T3 | 4 |
| T4 | 11 |
| Unstaged | 2 |
| Nodal status | |
| N0 | 12 |
| N1 | 4 |
| N2 | 7 |
| N3 | 0 |
| Unstaged | 2 |
| Status at blood draw | |
| Primary disease | 22 |
| Recurrent disease | 3 |
| Therapy prior to blood draw | |
| Surgery | 18 |
| Radiotherapy | 11 |
| Radiochemotherapy | 6 |
Collection of PBMC
Peripheral venous blood (20–30 ml) was drawn into heparinized tubes. The samples were hand carried to the laboratory and immediately centrifuged on Ficoll-Hypaque. PBMC were recovered, washed in AIM-V medium (Invitrogen), counted in a trypan blue dye, and immediately used for experiments.
Isolation of T cell subsets from PBMC
CD4+CD25high T cells from PBMC of HNSCC patients and NC were single cell sorted using the gating strategy previously described by us, in which the threshold for CD25high cells was established at the mean fluorescence intensity of 120 (12). A MoFlo high-speed cell sorter (Dako) was used for cell separations. The CD4+CD25− and CD4+CD25high cell fractions were collected and routinely tested for expression of Foxp3 by flow cytometry and viability in a trypan blue dye exclusion assays. The purity of CD4+CD25high cells obtained from the patients was usually ~95–98%, while the CD4+CD25high fractions obtained from NC contained 70–80% CD4+CD25highFoxp3+ Treg. Sorted cells were immediately used for establishing cocultures.
Antibodies
The following anti-human mAbs (mAb) were used for flow cytometry: anti-CD3-ECD, anti-CD4-PC5, anti-CD8-PC5, anti-CD25-FITC, anti-Foxp3-FITC (clone PCH101), anti-Fas-FITC, anti-FasL-PE (NOK-1, 40 and 26 kDa), anti-CD25-PE, and anti-CD152-PE (CTLA-4). Abs and their respective isotypes (purified unlabeled mouse IgG1 and IgG2a), used as negative controls for surface and intracellular staining, were all purchased from Beckman Coulter, except for anti-Foxp3-FITC and the respective rat IgG2a isotype control (eBioscience). Before use, all mAbs were titrated using normal resting or activated PBMC to establish optimal staining dilutions.
Surface and intracellular staining
To determine the frequency of CD4+CD25high, CD8+CD25high, CD4+CD25−, and CD8+CD25− T cells and the expression of Treg markers Foxp3 and CTLA-4, freshly obtained PBMC (at least 2 × 105 cells/tube) were stained with mAbs included in the above-described panel for 15 min at 4°C. Appropriate isotype Ab controls were used in all experiments. Cells were washed and examined by four-color flow cytometry, as previously described (22).
Intracellular staining for Foxp3 or CD152 (CTLA-4) was also performed as previously described (22). Briefly, samples were first incubated with mAbs against surface markers CD4, CD8, CD3, and CD25. After washing, cells were fixed with 4% (v/v) paraformaldehyde in PBS for 20 min at room temperature, washed once with PBS containing 0.5% (v/v) BSA and 2 nM EDTA, permeabilized with PBS containing 0.5% BSA and 0.1% (v/v) saponin, and stained with the pre-titrated anti-Foxp3-FITC for 30 min at room temperature. Cells were further washed twice with PBS containing 0.5% BSA and 0.2% (v/v) saponin, resuspended in FACS flow solution, and analyzed by flow cytometry. Appropriate isotype controls were included for each sample.
Detection of annexin V (AnxV)+ cells
PBMC immediately after their isolation, as well as PBMC ex vivo-activated with OKT3 or IL-2, were analyzed for AnxV binding (% AnxV+ cells) using flow cytometry. Samples were stained with FITC-conjugated AnxV (Molecular Probes) and propidium iodide (Molecular Probes) according to the instructions provided by the manufacturer.
Flow cytometry
Flow cytometry was performed using a FACScan flow cytometer (Beckman Coulter) equipped with Expo32 software (Beckman Coulter). The acquisition and analysis gates were restricted to the lymphocyte gate as determined by their characteristic forward (FSC) and side scatter (SSC) properties. FSC an SSC were set in a linear scale. For analysis of all apoptosis-related proteins, at least 1 × 106 lymphocytes were acquired. Furthermore, analysis gates were restricted to the CD3+CD4+, CD4+CD25+, CD8+CD3+, CD8+CD25high, and CD4+CD25high T cell subsets, as appropriate. Cells expressing Treg marker, apoptosis-related proteins, or death molecules were acquired and analyzed in the FL1 or FL2 logarithmic scale using the set gates.
Flow cytometry-based cytotoxicity assay (FLOCA)
An in vivo FLOCA was a modification of the assay described by Grossman and colleagues (19). Single-cell sorted, fresh CD4+CD25high T cell populations were tested for regulatory function by coculture with at least 0.5 × 105 CFSE-labeled autologous CD4+CD25− or CD8+CD25− RC at the Treg/RC ratios of 1:1 or 1:5. The cocultures were incubated for 5 days. CD4+CD25− or CD8+CD25− T cells separated by SCS were labeled with 1.5 μM CFSE (Molecular Probes/Invitrogen) for 10 min at room temperature. The CFSE label was quenched by the addition of an equal volume of FCS (Invitrogen), and then cells were washed extensively with PBS. Treg/RC in cocultures were stimulated with bead-bound or soluble OKT3 (1 μg/ml) (American Type Culture Collection) and soluble anti-CD28 Ab in the presence of 150 or 1000 IU of IL-2/ml. Immediately before analysis, cell samples were stained for surface CD4 or CD8 with mAb CD4-ECD or CD8-ECD, respectively, for 15 min at 4°C. Cells were then washed and incubated with 20 μg of 7-aminoactinomycin D (7-AAD; Calbiochem) for 20 min at 4°C. After 20 min, samples were resuspended in 500 μl of PBS and immediately analyzed by flow cytometry. In this assay, 7-AAD incorporation was used as a surrogate marker for late cell death/apoptosis, as it intercalates with DNA in cells that have lost membrane integrity. In many instances, diffuse staining of 7-AAD− T cells was observed following coculture. This might be a consequence of down-regulation of CD4 or CD8 molecules in activated T cells.
T cell populations were classified as suppressive if they inhibited proliferation of the CD4+CD25− or CD8+CD25− RC in the coculture assay, and if decreasing the number of CD4+CD25+ T cells relative to the number of CD4+CD25− or CD8+CD25− RC in coculture restored proliferation. CD4+CD25+ T cell populations that satisfied both of these criteria were classified as suppressor T cells. These criteria were applied to all populations so that they could be tested regardless of levels of expansion.
All CFSE data were analyzed using the ModFit software provided by Verity Software House. The percentages of suppression were calculated based on the proliferation index of RC alone compared with the proliferation index of cultures containing RC and Treg. The program determines the percentage of cells within each peak, and the sum of all peaks in the control culture is taken as 100% of proliferation and 0% of suppression.
Transwell assays
To assess whether cell-to-cell contact was necessary for Treg to mediate suppression, polycarbonate 24-well transwell inserts (0.4 μM) (Corning Costar) were used. At least 5 × 104 CFSE-labeled CD4+CD25− or CD8+CD25− T cells were stimulated with soluble or bead-bound OKT3 and soluble anti-CD28 Ab (each at 1 μg/ml) in the presence of 150 or 1000 IU of IL-2/ml in the lower chambers of the plates. Autologous Treg or control cells (CD4+CD25− or CD8+CD25−) were added to the upper chambers at the stimulator/responder ratio of 1:1 or 1:5.
Analysis of mechanisms used by human CD4+CD25high Treg for suppression of CD4+ or CD8+ RC
The FLOCA was modified and used to evaluate the mechanism of suppression by Treg as follows. Expression of FasL on CD4+CD25high Treg was blocked with anti-human FasL-neutralizing Ab before the 5-day coculture with autologous CD4+ or CD8+ RC. Briefly, Treg were incubated with neutralizing anti-human FasL-Ab (NOK-1; BioLegend) at 0.5 μl/100 μl for 2 h at 37°C. Controls were incubated with isotype-matched control Ab (purified unlabeled mouse IgG1). The optimal concentration of neutralizing Ab was predetermined using titration assays with OKT3/CD28 Ab-activated Jurkat cells, which express high levels of FasL and Fas. The percentage inhibition of FasL obtained with various concentrations of NOK-1 Ab on days 1, 3, and 5 of culture was determined by coincubating the NOK-1-treated Jurkat cells with Fas+ microvesicles (250 ng/ml) isolated from supernatants of a HNSCC cell line (23). After 4 h of incubation, the percentage of AnxV+ cells in coculture was determined.
Susceptibility of human CD4+ and CD8+ T cell subsets to Fas-mediated apoptosis
To determine whether Fas-expressing T cells obtained from cancer patients or NC are sensitive to Fas-mediated apoptosis, we incubated freshly isolated PBMC with anti-Fas Ab (CH-11 Ab). PBMC were treated with 250 ng/ml/1 × 106 cells of CH-11 Ab for 4 h. We have shown before that at this concentration 90 × 15% of activated Jurkat cells bind AnxV (22). Additionally, PBMC activated with soluble OKT3 (1 μg/ml) and/or IL-2 (150 or 1000 IU/ml) for 48 h were also incubated with CH-11 Ab.
Statistical analysis
Differences between groups were assessed using Wilcoxon test with p values adjusted by resampling (2000 permutations) and a signed rank test (no adjustment). Values of p ≤0.05 were considered significant.
Results
A phenotypic profile of Treg in the peripheral circulation of NC and patients with HNSCC
Fresh peripheral blood obtained from 25 patients with HNSCC and 15 NC was separated on Ficoll-Hypaque gradients, and PBMC were phenotyped by multiparameter flow cytometry using a previously described panel of markers for Treg (12, 22). The data in Table II show that the mean percentage of CD4+CD25highFoxp3+ T cells within the CD4+CD25high gate was significantly higher for HNSCC patients than for NC in the peripheral circulation, con-firming our earlier results (12). When the patients’ PBMC were single cell sorted based on CD25high expression (i.e., ≥120 mean fluorescence intensity) and checked for purity, the Treg fractions contained from 75% to 95% of CD3+CD25highFoxp3+ T cells. In contrast, the purity of Treg isolated by single cell sorting from PBMC of NC was lower, ranging from 60% to 70%. The purity of single cell-sorted CD8+CD25− or CD4+CD25− T cells ranged from 95% to 98%. All isolated Treg and RC were viable as determined by a trypan blue dye exclusion assay.
Table II.
Percentages of Treg in the peripheral blood of normal controls or patients with HNSCCa
| Marker | % CD4+CD25highFoxp3+ within the CD4+CD25high Gate | ||
|---|---|---|---|
| Normal controls (n = 15) | 65 ± 20 |
|
p < 0.0001 |
| HNSCC patients (n = 25) | 86 ± 12 | ||
The data are mean percentages ± SD of CD4+CD25highFoxp3 cells within the CD4+CD25high gate measured by multicolor flow cytometry in the freshly obtained peripheral blood of NC and patients with HNSCC.
Human CD4+CD25highFoxp3+ Treg suppress proliferation and induce apoptosis in autologous CD8+ RC
We and others have shown that human CD4+CD25high Treg obtained from the peripheral blood suppress proliferation of autologous as well as allogeneic CD4+ or CD8+ T cells via direct cell-to-cell contact (12, 24–26). To determine whether CD4+CD25high Foxp3+ Treg isolated from patients with HNSCC or NC induce apoptosis in CD8+ RC, we used FLOCA. As shown in Fig. 1, FLOCA discriminates between CFSE-labeled, 7-AAD+ (dead) RC (left lower panels) and unlabeled, 7-AAD− (live) Treg (right panels) and simultaneously measures the extent of proliferation inhibition (CFSE dye dilution) in RC populations. In a coculture (RC plus Treg) established with NC cells (Fig. 1A), Treg induced little apoptosis (1.2% of 7-AAD+ RC), and they mediated moderate suppression (32%) of RC proliferation. As indicated in Table III, suppressor function (mean ± SD) for all 15 NC tested was 32 ± 19%, and the percentages of 7-AAD+ cells were 7.2 ± 0.35 and 3.5 ± 2.5 for CD8+ or CD4+ RC, respectively (means ± SD). These results indicate a considerable variability in suppressor function among NC, which is in agreement with our previously reported data (12, 22, 27). Furthermore, a great majority of Treg were 7-AAD− following cocultures (Fig. 1), which were supplemented with 150 IU/ml rIL-2.
FIGURE 1.

FLOCA simultaneously measures induction of apoptosis and suppression of RC proliferation by CD4+CD25highFoxp3+ Treg. FLOCA was used to simultaneously measure proliferation of CD8+ CFSE-labeled autologous RC and their apoptosis. Treg were CFSE−. CD8+ RC alone or RC plus Treg were stimulated with OKT3 and cocultured in the presence of 150 IU/ml IL-2 for 5 days. At harvest, cells were stained with 7-AAD, which stains dead cells and is excluded by live cells, and examined by flow cytometry. FLOCA discriminates CFSE-labeled 7-AAD+ and 7-AAD− RC from unlabeled 7-AAD+ and 7-AAD− Treg after coculture. A, Treg isolated from PBMC of a NC induced low levels of inhibition and little apoptosis in RC. The data are from 1 experiment of 15 performed with T lymphocytes of NC. B, A representative experiment of 10 performed with T cells of HNSCC patients. Treg isolated from PBMC of a HNSCC patient induced strong inhibition of RC proliferation (89%) and death in 80% of RC. In this and all other figures, the Treg/RC ratio was 1:1 in the cocultures. The percentages of proliferation inhibition and of 7-AAD+ cells are indicated in respective panels.
Table III.
Proliferation inhibition and the frequency of 7-AAD+ responder cells or Treg in the coculturesa
| RC | Proliferation Inhibition (%) | Cell death (%)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| RCb | Treg | ||||||||
| CD8+ RC | |||||||||
| NC (n = 15) | 32 ± 19 |
|
p < 0.002 | 7.2 ± 0.35 |
|
p < 0.01 | 12 ± 10 |
|
NS |
| HNSCC patients (n = 25) | 78 ± 8 | 56 ± 12 | 4 ± 2.5 | ||||||
| CD4+ RC | |||||||||
| NC (n = 15) | 12 ± 5.5 |
|
p < 0.0001 | 3.5 ± 2.5 |
|
NS | 14 ± 4.5 |
|
p < 0.02 |
| HNSCC patients (n = 25) | 80 ± 2.6 | 3.2 ± 1.7 | 52 ± 17 | ||||||
Five-day cocultures of Treg with autologous RC were established in the presence of OKT3 and 150 IU/mL rIL-2 as described in Materials and Methods. Treg and RC populations were isolated from the peripheral blood of normal controls or HNSCC patients. Following coculture, the percentage inhibition of RC proliferation and percentages of 7-AAD+ RC and Treg were determined by FLOCA. The data are means ± SD.
RC are either CD8+ or CD4+, as indicated in the first column.
In contrast, CD4+CD25highFoxp3+ Treg obtained from the peripheral blood of patients with HNSCC induced considerably more death in autologous CD8+ RC (up to 80% 7-AAD+ cells) during 5-day cocultures and strongly inhibited RC proliferation (89%) (Fig. 1B and Table III). In control cocultures with autologous CD4+CD25− T cells added in place of Treg, there was no suppression. These observations were highly reproducible, and the data for all patients are shown in Table III. In cocultures established in the presence of OKT3 and 150 IU/ml rIL-2, the difference in suppression between Treg in NC vs patients was highly significant with values of p < 0.002 for CD8+ and p < 0.0001 for CD4+ RC (Table III).
Human CD4+CD25high Treg suppress proliferation but do not mediate apoptosis in autologous CD4+ RC
When CD4+CD25high Treg isolated from PBMC of HNSCC patients were coincubated with autologous CD4+CD25− RC in the presence of OKT3 and 150 IU/ml rIL-2, they suppressed proliferation but did not induce apoptosis in these RC at the Treg/RC ratio of 1:1 (Fig. 2) or 1:5 (data not shown). Treg isolated from NC were only weak suppressors of CD4+ RC proliferation and did not induce apoptosis (data not shown). Surprisingly, CD4+CFSE− Treg cells isolated from HNSCC patients and cocultured with autologous CD4+ RC (at Treg/RC ratios of 1:1 or 1:5) became largely 7-AAD+ (97% in Fig. 2, bottom right panel). For cocultures of all HNSCC patients, 7AAD+ Treg were 52 ± 17% vs 14 ± 4.5% for NC (Table III). These data suggested that Treg in cocultures with CD4+ RC mediate suppression of RC proliferation but then become sensitive to apoptosis and are “killed.” Presumably, activated CD4+ RC in the cocultures containing OKT3 and 150 IU/ml rIL-2 are responsible for Treg death.
FIGURE 2.

Human Treg suppress proliferation but do not mediate apoptosis in CD4+ RC. Proliferation and death (% 7-AAD+) of CD4+ RC alone or CD4+ RC in cocultures with Treg isolated from the peripheral blood of a NC and a HNSCC patient were measured in FLOCA. Note that in the coculture with the patient’s cells nearly all CD4+CSFE− Treg are 7-AAD+ following incubation with CD4+ RC in the presence of OKT3 and 150 IU/ml IL-2. No death is evident in CD4+ RC, although their proliferation is suppressed. In contrast, cocultures set up with NC cells show no Treg apoptosis and little suppression of RC proliferation. Results are representative experiments of 10 performed with cells isolated from PBMC of HNSCC patients and 10 performed with cells isolated from PBMC of NC.
The role of IL-2 in death of CD4+CD25high Treg in autologous Treg/CD4+ RC cocultures
It was unclear why Treg undergo apoptosis following coculture with activated CD4+ RC. However, we have previously observed that OKT3-activated human CD4+CD25high Treg become resistant to apoptosis in the presence of high-dose IL-2 (1000 IU/ml), whereas autologous CD4+CD25− RC are highly sensitive (28). Because it has been reported that human Treg are dependent on exogenous IL-2 for their suppressor activity (29), we suspected that the IL-2 concentration in cocultures played a key role in RC suppression by Treg. To confirm this, we set up cocultures of Treg obtained from patients with HNSCC with OKT3-activated CD4+ RC in the absence of IL-2 as well as in the presence of low (150 IU/ml) and high (1000 IU/ml) doses of IL-2 (Fig. 3). In a representative coculture stimulated with OKT3 in the presence of 1000 IU/ml IL-2, Treg were resistant to apoptosis (7-AAD−), while RC were dying (Fig. 3A, left panels). When cocultures were stimulated with 150 IU/ml IL-2, 7-AAD+ Treg were numerous, while RC were largely 7-AAD− (Fig. 3A, right panels). However, when cultured alone in 150 or 1000 IU/ml IL-2, Treg did not die. Similar results were obtained with cocultures established with RC and Treg obtained from five different HNSCC patients (Fig. 3B). In the absence of IL-2 in the cocultures, variable proportions of Treg and RC were 7-AAD+ on day 5, possibly due to deprivation-induced cell death (Fig. 3B).
FIGURE 3.

The role of IL-2 in death of Treg in autologous Treg/CD4+ RC cocultures. A, Percentages of 7-AAD+ RC as well as 7-AAD+ Treg in a representative coculture performed in the presence of OKT3 and rIL-2 (1000 IU/ml rIL-2 vs 150 IU/ml rIL-2). B, Percentages of 7-AAD+ Treg and RC following cocultures in the presence of different IL-2 concentrations of these cells obtained from PBMC of 10 HNSCC patients. C, Suppression of RC proliferation in response to OKT3 in cocultures of Treg and CD4+ RC obtained from 10 HNSCC patients. The data in B and C are means ± SD. Cell death was measured in FLOCA, and proliferation inhibition in CFSE-based assays is as described in Materials and Methods. Note that IL-2 at high concentrations (1000 IU/ml) protects Treg from RC-mediated death.
As shown in Fig. 3C, Treg cultured in the presence of 1000 IU/ml IL-2 mediated strong suppression of RC proliferation, which was significantly greater than that mediated by Treg activated by 150 IU/ml IL-2 (p < 0.04) or Treg in cocultures with no IL-2 (p < 0.001).
We established comparable cocultures in the presence of the high and low doses of IL-2 with Treg and RC obtained from NC. However, the results were less informative, as percentages of dying cells and proliferation suppression were considerably lower relative to the values obtained with the cells isolated from HNSCC patients (see, e.g., Table III).
Suppression and apoptosis in Treg/RC cocultures is cell contact-dependent
To evaluate whether apoptosis of RC in the presence of IL-2 requires cell-to-cell contact between activated Treg and RC, we established the autologous Treg plus CD4+ RC or Treg plus CD8+ RC cocultures at the Treg/RC ratios of 1:1 and 1:5 using transwell inserts (Tw). In half of the assays, Treg and RC were separated by a membrane eliminating cell-to-cell contact. RC were stimulated with OKT3 in the absence or presence of IL-2 (150 or 1000 IU/ml). With Tw, suppression of CD8+ RC proliferation mediated by CD4+CD25highFoxp3+ Treg isolated from PBMC of three HNSCC patients was nearly completely inhibited at all Treg/RC ratios in cocultures containing IL-2, regardless of its concentration (Fig. 4A, lower panels). Concomitantly, apoptosis of RC in Treg+/CD8+ RC cocultures was significantly, albeit not completely, inhibited in the presence of transwell membranes (Fig. 4A, upper panels). In these cocultures, Treg were not 7-ADD+, except when IL-2 was absent, as noted above.
FIGURE 4.
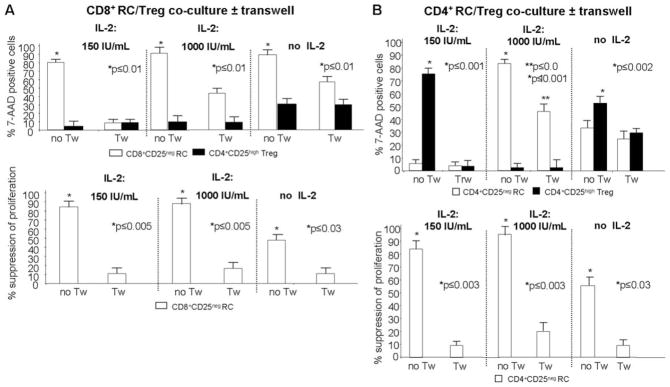
The importance of cell-to-cell contact in Treg-mediated suppression and apoptosis. A, Suppression of CD8+ RC and their death in the presence of IL-2 are cell contact-dependent. In cocultures of Treg with autologous CD8+ RC, suppression of RC proliferation was completely inhibited in the presence of Tw and 1000 IU/ml or 150 IU/ml rIL-2. Death (% 7-AAD+) of CD8+ RC was also inhibited in the presence of Tw, although only partial inhibition was seen in the presence of 1000 IU/ml IL-2. Results are means ± SD from three experiments performed with CD8+ RC/Treg cocultures. B, Suppression of CD4+ RC proliferation is cell contact-dependent in the presence of rIL-2 and so is their apoptosis; however, CD4+ RC apoptosis is only partly inhibited in the Tw system in the presence of 1000 IU/ml IL-2. Results are means ± SD from three experiments performed with CD4+ RC/Treg cocultures. The T cell subsets used in these experiments were isolated from patients with HNSCC.
When Treg/CD4+ RC cocultures were established and activated with lower or higher doses of IL-2, a complete inhibition of CD4+ RC proliferation in the presence of Tw was again observed (Fig. 4B, lower panels). At the lower IL-2 concentration, most Treg became 7-AAD+ in cocultures with no Tw, while in the presence of Tw, death of Treg was abolished (Fig. 4B, upper panels), suggesting that cell-to-cell contact is essential for Treg demise. In cocultures set up in the higher IL-2 concentration, Treg remained 7-AAD− (no death) with or without Tw, while CD4+ RC were 7-AAD+ with or without Tw. This result suggests that death of CD4+ RC is independent on cell-to-cell contact. When cocultures contained no IL-2, death of Treg was significantly greater than for that of CD4+ RC in the absence of Tw, but not in its presence (Fig. 4B). Taken together, these results demonstrate that Treg as well as RC can induce death and deplete their “partner” cells in cocultures activated with OKT3 and IL-2. Proliferation, suppression, and death of the CD8+ partner cell requires direct cell-to-cell contact, but death of the CD4+ partner might depend in part on soluble factors. The fate of Treg and CD8+ or CD4+ RC may be decided by the concentration of IL-2 in the coculture, with low IL-2 concentrations favoring Treg death. Thus, Treg of patients with cancer are highly sensitive to contact-dependent apoptosis at 150 IU/ml IL-2, while at 1000 IU/ml IL2, only CD4+ RC and not Treg undergo apoptosis.
Expression of Fas/FasL on human CD4+CD25high Foxp3+ Treg and autologous CD8+ and CD4+ RC
Our results indicated that apoptosis of RC mediated by human CD4+CD25highFoxp3+ Treg required direct cell-to-cell contact and was regulated by signaling pathways induced by T cell activation with OKT3 and IL-2. It has been well documented that IL-2 mediates AICD via the Fas/FasL pathway (30, 31). We therefore looked for Fas and FasL expression on Treg and autologous CD4+CD25− and CD8+CD25− RC by using flow cytometry immediately after their isolation from blood samples and after their ex vivo activation (for 48 h) with OKT3 and 150 IU/ml or 1000 IU/ml IL-2. Samples from HNSCC patients and NC were analyzed using the anti-FasL Ab NOK-1, which reacts with the membrane-bound 40-, 44-, and 46-kDa isoforms of FasL but also recognizes the cleaved and soluble 26-kDa isoforms of FasL.
As we previously reported (32), CD8+CD25− and CD4+ CD25− RC isolated from peripheral blood in NC or cancer patients expressed low to intermediate levels of Fas, but no FasL (Fig. 5A and Table IV). After in vitro stimulation with OKT3 and IL-2, Fas was up-regulated on CD4+ and CD8+ RC in NC and cancer patients. However, FasL expression was only slightly increased on the cell surface of in vitro-activated human CD4+ and CD8+ RC (Fig. 5A and Table IV).
FIGURE 5.

Expression of Fas and FasL on human Treg and CD4+ or CD8+ RC. A, Freshly harvested or OKT3- and IL-2-activated CD4+ RC or CD8+ RC obtained from PBMC of a representative NC were evaluated for Fas or FasL expression by flow cytometry. B, Fresh and ex vivo-activated CD4+CD25high Treg from a representative patient with HNSCC or a representative NC were similarly tested for Fas and FasL expression. C, Histogram shows mean percentages ± SD of Fas+ and FasL+ CD4+CD25high Treg in peripheral blood of all NC and HNSCC patients included in this study.
Table IV.
Fas and FasL expression on CD4+ or CD8+ RC isolated from NC or patients with HNSCC prior to and after culture with OKT3 and IL-2a
| RC | % Positive Cells
|
p Value | |
|---|---|---|---|
| Prior to culture | After culture | ||
| Fas (CD95) | |||
| CD8+CD25− HNSCC | 38 ± 12 | 68 ± 14 | <0.5 |
| CD8+CD25− NC | 32 ± 10 | 65 ± 13 | <0.5 |
| CD4+CD25− HNSCC | 45 ± 10.5 | 78 ± 6 | <0.5 |
| CD4+CD25− NC | 43 ± 11.2 | 75 ± 6 | <0.5 |
| FasL (CD95L) | |||
| CD8+CD25− HNSCC | 2.1 ± 0.85 | 6.7 ± 3 | <0.5 |
| CD8+CD25− NC | 0 | 0 | |
| CD4+CD25− HNSCC | 0 | 3.2 ± 1 | <0.5 |
| CD4+CD25− NC | 0 | 0 | |
Isolated Treg or RC were obtained from NC (n = 15) or HNSCC patients (n = 25) and studied fresh or after 48 h of culture in the presence of OKT3 and rIL-2 as described in Materials and Methods. The data are mean percentages of positive cells ± SD. The p values refer to differences between fresh vs cultured T cell subsets.
Fas was also highly expressed on fresh as well as OKT3-and IL-2-activated CD4+CD25high Treg obtained from NC and HNSCC patients (Fig. 5B). Importantly, expression of Fas and FasL by the CD4+CD25high Treg in patients with HNSCC was significantly greater (p < 0.03 and p ≤ 0.001, respectively) relative to that in NC (Fig. 5C). FasL was only weakly expressed on a subset of fresh Treg obtained from PBMC of NC or HNSCC patients (Fig. 5C). When we activated Treg of NC with OKT3 and IL-2, FasL expression was not up-regulated (Fig. 5B). Surprisingly, however, in vitro-activated CD4+CD25high Treg cells from HNSCC patients expressed high levels of FasL on the cell surface (Fig. 5, B and C). These results suggest that in patients with cancer either the cleavage or endocytosis of FasL from the cell surface of activated Treg is avoided or delayed, converting these Treg into potent inducers of apoptosis mediated via the Fas/FasL pathway.
CD4+CD25high Treg from HNSCC patients express FasL but are resistant to Fas-mediated apoptosis
Because activated Treg as well as activated RC expressed high levels of Fas (CD95), it was surmised that these cell subsets might be susceptible to Fas/FasL-mediated apoptosis. Therefore, we analyzed the sensitivity of fresh and OKT3- and IL-2 (150 IU/ml)-activated CD4+CD25high Treg and CD4+ or CD8+ RC to Fas-mediated apoptosis. We incubated freshly isolated and activated T cell subsets with CH-11-Ab (or isotype control Ab) for 4 h and measured the percentage of AnxV+ cells (Table V). Fresh CD4+CD25− and CD8+CD25− RC subsets obtained from NC were highly sensitive to CH-11 Ab (45 ± 15% and 85 ± 17% AnxV+ cells, respectively). Susceptibility of CD4+ or CD8+ RC to CH-11 Ab was increased after in vitro stimulation with OKT3 and IL-2 (90 ± 8.5% AnxV+ cells in Table V). Counterintuitively, however, fresh CD4+CD25high Treg isolated from patients with HNSCC and expressing surface FasL (as shown in Fig. 5) were found to be resistant to CH-11 Ab-induced apoptosis (no AnxV+ cells in Fig. 6A). Following activation with OKT3 and 150 IU/ml IL-2, CD4+CD25high Treg obtained from NC or HNSCC patients remained resistant to CH-11 Ab-induced apoptosis (Fig. 6B).
Table V.
Sensitivity of primary CD4+ and CD8+ RC to Fas/FasL-mediated apoptosisa
| Treatment | AnxV binding (%) to Responder Cells
|
|
|---|---|---|
| CD4+CD25− | CD8+CD25− | |
| Plus isotype control Ab | 6 ± 5 | 12 ± 6 |
| Plus CH-11 Ab, fresh PBMC | 45 ± 15 | 85 ± 17 |
| Plus CH-11 Ab, activated PBMC (OKT3/IL-2) | 90 ± 9 | 97 ± 12 |
PBMC were obtained from 10 NC and were treated with CH-11 Ab or isotype control Ab for 4 h at 37°C. Cells were examined for AnxV binding by multicolor flow cytometry, gating on CD4+CD25− and CD8+CD25− RC. The data are means ± SD.
FIGURE 6.

AnxV binding to Treg following ex vivo treatment of PBMC with anti-Fas agonistic CH-11 Ab. A, AnxV+ binding to fresh PBMC treated with CH-11 Ab for 4 h is shown. The gate is set on CD4+CD25high T cells. Note that Treg from a representative HNSCC patient are AnxV−, that is, they are resistant to apoptosis. B, The same experiment with OKT3 and IL-2 (150 IU/ml)-activated PBMC. Note that activated Treg from NC and HNSCC patients are resistant to Fas-mediated apoptosis. Representative data from independent experiments performed with PBMC obtained from 10 patients and 5 NC are shown.
Collectively, these results suggest that 1) while OKT3- and IL-2-activated CD4+CD25high Treg become resistant to Fas-mediated apoptosis, CD4+ RC remain highly sensitive; and 2) freshly isolated CD4+CD25high Treg in patients with HNSCC are highly activated T cells that are protected from, or are resistant to, apoptosis and remain resistant following ex vivo activation.
Treg can “kill” autologous CD8+CD25− RC but not activated CD4+CD25− RC via the Fas/FasL pathway
As FasL and Fas were coexpressed on CH-11-resistant CD4+CD25high Treg, and Fas was present on CH-11 Ab-sensitive CD8+ RC and CD4+ RC, we asked whether Treg could use the Fas/FasL pathway to induce apoptosis in autologous CD8+ or CD4+ RC, and whether Fas/FasL-mediated apoptosis might be in part responsible for suppression of RC proliferation in Treg/RC cocultures. Therefore, Treg obtained from PBMC of HNSCC patients or NC were coincubated with CFSE-labeled autologous CD8+ or CD4+ RC at the Treg/RC ratios of 1:1 or 1:5. As before, RC were stimulated with OKT3 and 150 IU/ml IL-2. Half of the Treg samples were blocked with antagonistic anti-FasL Ab NOK-1 or an isotype control Ab before coculture, as described in Materials and Methods. Suppression of proliferation and apoptosis (7-AAD+ cells) of the CFSE− Treg as well as CFSE-labeled RC population were analyzed by FLOCA. As shown in Fig. 7A, Treg isolated from a HNSCC patient suppressed proliferation of autologous CD8+ RC, and CD8+ RC were sensitive to apoptosis. Treg obtained from patients with HNSCC suppressed proliferation of autologous CD8+ RC, and CD8+ RC were 7-AAD+ (48 ± 18% for all cocultures tested) at the S/RC ratio of 1:1. As expected, Treg from NC were only weakly suppressive and induced little or no apoptosis in autologous CD8+ RC (data not shown). When Treg obtained from HNSCC patients were pretreated with FasL-neutralizing Ab and then coincubated with autologous CD8+ RC, no 7-AAD+ were present in the RC fraction and suppression of RC proliferation was partially relieved (65 ± 12% for all cocultures tested and 62% of suppression in Fig. 7A). Isotype control Ab did not induce these effects (data not shown). These data suggest that suppression of CD8+ RC by Treg is in large part mediated by Fas/FasL-induced apoptosis, although other mechanisms of suppression may also be present.
FIGURE 7.

Blocking of suppression and apoptosis mediated by Treg with FasL-blocking Ab. A, Treg mediate suppression and induce apopto-sis in autologous CD8+ RC, and this apoptosis is inhibited by a FasL-blocking Ab NOK-1. B, Treg mediate suppression but do not induce apoptosis in CD4+ RC. Instead, Treg themselves undergo apoptosis in the cocultures. Death of Treg was not inhibited by NOK-1 Ab, indicating that the Fas/FasL pathway is not involved. Representative experiments of five performed with cocultures of CD8+ RC or CD4+ RC and CD4+CD25high Treg are shown. In the cocultures, cells were incubated in the presence of OKT and rIL-2 (150 IU/ml).
In contrast to Treg/CD8+ RC cocultures, the incubation of Treg with antagonistic FasL-Ab (or isotype control Ab) before coculture with CD4+ RC did not inhibit suppression of their proliferation (Fig. 7B) and had no effect on RC cell death, which remained minimal. Thus, suppression of proliferation of CD4+ RC by Treg was not mediated via the Fas/FasL pathway (Fig. 7B). Surprisingly, however, Treg in Treg/CD4+ RC cocultures became 7-AAD+ after coincubation with CD4+CD25− RC (75 ± 15%) (Fig. 7B). The addition of antagonistic FasL Ab did not substantially reduce the number of 7-AAD+ Treg in these cocultures (Fig. 7B). These results suggest that the Fas/FasL pathway is not involved in suppression of CD4+ RC by Treg and that activated Treg are not depleted via a “suicide” mediated by the Fas/FasL pathway.
Discussion
Peripheral tolerance mediated by CD4+CD25highFoxp3+ nTreg appears to represent a key mechanism for maintaining an immunologic balance and protecting the host from undesirable autoimmune reactivity (33–35). The paucity of nTreg in autoimmune diseases and, conversely, their excess in patients with cancer suggest that Treg imbalance is associated with disease development. Treg abnormalities have been reported in autoimmunity, graft-vs-host disease, chronic infections, parasitic infestation, and cancer (1, 2, 36–39). Given the impact of dysregulated Treg function in many human diseases, it is important to better understand Treg biology to be able to devise therapies able to effectively restore or equilibrate the immune balance. Therefore, the knowledge of molecular pathways that control and drive Treg activity is necessary, and the understanding of mechanisms Treg use for interactions with other subsets of lymphocytes is a key requirement for the development of corrective therapies in the future.
Mechanisms reported to be responsible for suppressor functions of Treg vary. It appears that Treg can engage many different mechanisms, perhaps depending on the environment in which they operate. Aside from the mechanisms requiring direct physical contact between cells such as receptor-ligand interactions involving, for example, CD95 (20, 21, 40), ICOS (41), or CD46 (42), as well as contact-mediated suppression by granzyme B/perforin produced by Treg (19, 43, 44), a variety of soluble factors mediating RC death have been described. Prominent among these factors are immunoinhibitory cytokines IL-10 and TGF-β (2, 12, 45) and adenosine likely derived from an enzymatic cleavage of ATP by Treg-associated ectonucleoside hydrolases (46, 47). Additionally, cytokine deprivation induced by Treg has been recently proposed as a mechanism inhibiting effector T cell responses (48). Most of the above-listed mechanisms have been identified in murine systems, and there is a paucity of information regarding human Treg, especially in patients with cancer.
In this study, we have examined mechanisms of suppression mediated by human Treg during interactions with RC in an ex vivo model system consisting of cocultures of single cell-sorted CD4+CD25highFoxp3+ and CD4+CD25− or CD8+CD25− RC populations. It was possible to differentiate between CFSE-labeled RC and unlabeled Treg in these cocultures by using the FLOCA assay. Pretreatment of these interacting cell populations with mAbs or preventing their cell-to-cell contact by using transwell inserts enabled us to determine, in part, the mechanisms responsible for suppression of RC proliferation and cell survival or death. Sorted CD4+CD25high Treg used in these experiments were derived either from peripheral blood of NC or HNSCC patients and were positive for Foxp3 and CTLA-4 expression. Treg isolated from the blood of patients with cancer were previously shown to mediate high levels of suppression, while Treg isolated from PBMC of NC generally had lower levels of suppressor activity in our cocultures (12, 27). The low levels of suppression mediated by isolated CD4+CD25high Treg of NC we consistently see are likely due to the presence of IL-2 in the cocultures. Other investigators have reported high levels of suppression in cocultures of Treg and RC in the absence of exogenous IL-2 (e.g., see Ref. 49) probably because the competition for cytokines is the primary mode of suppression in this instance. Thus, Treg consume IL-2, depriving RC of cytokines necessary for proliferation, as also suggested by Pandiyan et al. (48). Furthermore, in “traditional” suppressor assays, most investigators do not measure cell death, but only RC proliferation. In such assays, it would not be possible to discriminate inhibition of proliferation due to RC death mediated by IL-2 starvation from that mediated by the Fas/FasL engagement or the granzyme/perforin pathway.
Treg obtained from the peripheral circulation of cancer patients mediate strong suppression of RC proliferation (2, 10, 27). These Treg are activated as evidenced by high levels of Fas and FasL expression on the cell surface. We now show that the “killing” potential of Treg is also higher in cancer patients than in NC, and that human CD4+CD25high Treg arrest proliferation of CD4+ and CD8+ RC by distinct mechanisms involving cell-to-cell contact. While CD8+CD25− RC proliferation is suppressed by Fas/FasL-mediated apoptosis, suppression of CD4+CD25− RC by Treg is Fas-independent and involves cell death mediated by granzyme B/perforin, as recently described by us (L. Strauss, C. Bergmann, H. Rabinowich, and T. L. Whiteside, submitted for publication) and others (19, 20, 50). In cancer patients, both the frequency and function of Treg are elevated relative to those in NC (12–14). The level of Treg activation is amplified in patients with cancer, whether due to an Ag excess and/or up-regulated cytokine networks. The state of “chronic inflammation” accompanying tumor progression in patients with active disease or that due to tissue distinction after oncologic therapies in patients with no evident disease probably favors rapid and effective Treg expansion as well as a high level of suppressor activity.
The most striking observation we made is that both Treg and RC can reciprocally regulate T cell survival in cocultures, and that Treg can be induced to die in cocultures with activated RC. Furthermore, Treg sensitivity or resistance to RC-derived death signals appears to be dependent on the IL-2 concentration in cocultures. Our results extend to the purified human Treg and RC subsets somewhat similar results reported by Banz et al. in mice (40). As it has been documented that human nTreg express CD25 and that Tr1 express CD132 (IL-2Rγ) (45), it is not surprising that exogenous IL-2 plays a key role in their functions (29) and that low vs high IL-2 concentrations might exert differential effects on Treg activities. It is possible that the concentration of 1000 IU/ml IL-2 we used ex vivo is not physiological. However, we and others (e.g., see Ref. 48) have selected this concentration to determine the threshold for Treg survival in Treg-RC cocultures. As Treg are known to avidly consume IL-2, we considered it necessary to use high IL-2 concentrations for ex vivo assays. It is, however, plausible that in the tissue or pericellular microenvironment at the peak of immune responses, levels of IL-2 might be quite substantial. Also, when IL-2 is administered therapeutically in patients with cancer, its levels are high, with a reported concomitant increase in Treg frequency and suppressor activity (51). Thus, our in vitro data could be of biologic if not “physiologic” importance.
In addition to IL-2R, Treg as well as RC express Fas and up-regulate Fas and FasL expression on the cell surface upon activation by OKT3 and IL-2. Thus, activated Treg and RC can potentially eliminate one another by utilizing the Fas/FasL pathway. However, in the presence of high IL-2 concentrations, Treg acquire resistance to Fas-mediated apoptosis and remain viable despite expression of Fas and FasL on their cell surface. In contrast, CD8+ RC remain sensitive to Fas-mediated apoptosis and are readily eliminated by activated FasL+ Treg. Interestingly, CD4+ RC are also resistant to Fas-mediated apoptosis, and suppression of their proliferation by Treg is attributable to different cell contact-dependent mechanisms and/or cytokines, as previously reported by us (2, 45). In the presence of low IL-2 concentrations, Treg are sensitive to death signals induced by CD4+ RC. In aggregate, these cellular interactions in the coculture system suggest that both Treg and RC might contribute to the control of immune responses but perhaps at different points in their evolution. Thus, at the peak of the response, when T cell proliferation and IL-2 production are high, Treg are resistant to apoptosis and exercise their suppressive function, inducing inhibition of RC proliferation and RC death. As the immune response enters a contraction phase, due in part to Treg-mediated suppression, IL-2 production is decreased, and Treg become sensitive to apoptosis mediated by surviving, activated, and FasL+ RC. The ability of T cells to reciprocally induce killing of the partner that expresses the correct receptor profile to down-regulate immune response is well documented in the literature (30). Also, the involvement of the Fas/FasL system in this process has been described (18, 20, 21). The use of this mechanism by interacting activated Treg and RC, as documented in our ex vivo experiments, is an excellent example of how immune inhibitory and stimulatory signals are regulated in the specific micro-environment. It is possible that the reciprocal killing contributes to regulating of Treg homeostasis in vivo in health and disease. An excess or paucity of Treg in a given microenvironment might be regulated by T cells themselves, utilizing Fas-dependent or Fas-independent mechanisms to reduce suppression.
Footnotes
Supported in part by National Institutes of Health Grants P01-CA109688, P01-DE12321, and R01-DE13918 to T.L.W. C.B. was supported by a grant from Philip Morris USA and Philip Morris International.
Abbreviations used in this paper: Treg, regulatory T cells; RC, responder cells; AICD, activation-induced cell death; NC, normal controls; HNSCC, head and neck squamous cell carcinoma; AnxV, annexin V; FLOCA, flow-cytometry cytotoxicity assay; 7-AAD, 7 aminoactinomycin D; Tw, transwell insert
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 2.Strauss L, Bergmann C, Szczepanski MJ, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting IL-10 and TGF-β1 mediates suppression in the tumor microenvironment. Clin Cancer Res. 2007;13:4345–4354. doi: 10.1158/1078-0432.CCR-07-0472. [DOI] [PubMed] [Google Scholar]
- 3.Niedbala W, Cai B, Liu H, Pitman N, Chang L, Liew FY. Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL-2, and OX40. Proc Natl Acad Sci USA. 2007;104:15478–15483. doi: 10.1073/pnas.0703725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adriani M, Aoki J, Horai R, Thornton AM, Konno A, Kirby M, Anderson SM, Siegel RM, Candotti F, Schwartzberg PL. Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin Immunol. 2007;124:41–48. doi: 10.1016/j.clim.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hildeman DA, Zhu Y, Mitchell TC, Kappler J, Marrack P. Molecular mechanisms of activated T cell death in vivo. Curr Opin Immunol. 2002;14:354–359. doi: 10.1016/s0952-7915(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 6.Vence L, Palucka AK, Fay JW, Ito T, Liu YJ, Banchereau J, Veno H. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2007;104:20884–20889. doi: 10.1073/pnas.0710557105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang S, Lechler RI, He XS, Huang JF. Regulatory T cells and transplantation tolerance. Hum Immunol. 2006;67:765–776. doi: 10.1016/j.humimm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression: a diverse arsenal for a moving target. Immunology. 2008;124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold R, Brenner D, Becker M, Frey CR, Krammer PH. How T lymphocytes switch between life and death. Eur J Immunol. 2006;36:1654–1658. doi: 10.1002/eji.200636197. [DOI] [PubMed] [Google Scholar]
- 10.Roberts AI, Devadas S, Zhang X, Zhang L, Keegan A, Greeneltch K, Solomon J, Wei L, Das J, Sun E, et al. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol Res. 2003;28:285–293. doi: 10.1385/IR:28:3:285. [DOI] [PubMed] [Google Scholar]
- 11.Whiteside TL. Lymphocyte homeostasis and the antitumor immune response. Exp Rev Clin Immunol. 2006;1:369–378. doi: 10.1586/1744666X.1.3.369. [DOI] [PubMed] [Google Scholar]
- 12.Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the peripheral circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–6311. doi: 10.1158/1078-0432.CCR-07-1403. [DOI] [PubMed] [Google Scholar]
- 13.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–811. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 14.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 15.Lyssuk EY, Torgashina AV, Soloviev SK, Nassonov EL, Bykovskaia SN. Reduced number and function of CD4+CD25highFoxP3+ regulatory T cells in patients with systemic lupus erythematosus. Adv Exp Med Biol. 2007;601:113–119. [PubMed] [Google Scholar]
- 16.Glisic-Milosavljevic S, Wang T, Koppen M, Kramer J, Ehlenbach S, Waukau J, Jailwala P, Jana S, Alemzadeh R, Ghosh S. Dynamic changes in CD4+CD25+ high T cell apoptosis after the diagnosis of type 1 diabetes. Clin Exp Immunol. 2007;150:75–82. doi: 10.1111/j.1365-2249.2007.03475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114:979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, Powell WS, Monneret G. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol. 2006;177:6540–6547. doi: 10.4049/jimmunol.177.9.6540. [DOI] [PubMed] [Google Scholar]
- 19.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Janssens W, Carlier V, Wu B, VanderElst L, Jacquemin MG, Saint-Remy JM. CD4+CD25+ T cells lyse antigen-presenting B cells by Fas-Fas ligand interaction in an epitope-specific manner. J Immunol. 2003;171:4604–4612. doi: 10.4049/jimmunol.171.9.4604. [DOI] [PubMed] [Google Scholar]
- 21.Fritzsching B, Oberle N, Eberhardt N, Quick S, Haas J, Wildemann B, Krammer PH, Suri-Payer E. Cutting edge: in contrast to effector T cells, CD4+CD25+Foxp3+ regulatory T cells are highly susceptible to CD95 ligand-but not to TCR-mediated cell death. J Immunol. 2005;175:32–36. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- 22.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. FasL+ membraneous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11:1010–1020. [PubMed] [Google Scholar]
- 24.Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4+CD25+ T cells with regulatory properties isolated from peripheral blood. J Exp Med. 2001;193:1285–1294. doi: 10.1084/jem.193.11.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levings MK, Sangregorio R, Roncarolo MG. Human CD25+CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strauss L, Bergmann C, Whiteside TL. Function and phenotype of human CD4+CD25+Foxp3+ T cell clones in patients with head and neck cancer (HNSCC) and normal controls. Int J Cancer. 2007;121:2473–2483. doi: 10.1002/ijc.23001. [DOI] [PubMed] [Google Scholar]
- 28.Strauss L, Bergmann C, Johnson JT, Whiteside TL. Expression of Fas ligand (FasL) isoforms on peripheral T-cell subsets in patients with head and neck squamous cell carcinoma (HNSCC) J Immunother. 2006;29:670. (Abstr.) [Google Scholar]
- 29.Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 30.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis: immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Brunner T, Carter L, Dutton RW, Rogers P, Bradley L, Sato T, Reed JC, Green D, Swain SL. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J Exp Med. 1997;185:1837–1849. doi: 10.1084/jem.185.10.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, Whiteside TL. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. 2002;8:2553–2562. [PubMed] [Google Scholar]
- 33.Shevach EM. CD4+CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 34.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset: I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of regulatory T cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft versus host disease. Blood. 2007;110:3804–3813. doi: 10.1182/blood-2007-05-091074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor MD, Harris A, Babayan SA, Bain O, Culshaw A, Allen JE, Maizels RM. CTLA-4 and CD4+CD25+ regulatory T cells inhibit protective immunity to filarial parasites in vivo. J Immunol. 2007;179:4626–4634. doi: 10.4049/jimmunol.179.7.4626. [DOI] [PubMed] [Google Scholar]
- 38.Belkaid Y, Blank RB, Suffia I. Natural regulatory T cells and parasites: a common quest for host homeostasis. Immunol Rev. 2006;212:287–300. doi: 10.1111/j.0105-2896.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- 39.Peng G, Li S, Wu W, Sun Z, Chen Y, Chen Z. Circulating CD4+CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology. 2008;123:57–65. doi: 10.1111/j.1365-2567.2007.02691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banz A, Pontoux C, Papiernik M. Modulation of Fas-dependent apoptosis: a dynamic process controlling both the persistence and death of CD4 regulatory T cells and effector T cells. J Immunol. 2002;169:750–757. doi: 10.4049/jimmunol.169.2.750. [DOI] [PubMed] [Google Scholar]
- 41.Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of inducible co-stimulatory molecules (ICOS) on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180:2967–2980. doi: 10.4049/jimmunol.180.5.2967. [DOI] [PubMed] [Google Scholar]
- 42.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 43.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 44.Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Bergmann C, Strauss L, Wang Y, Szczepanski MJ, Lang S, Johnson JT, Whiteside TL. T regulatory type 1 cells (Tr1) in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res. 2008;14:3706–3715. doi: 10.1158/1078-0432.CCR-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borsellino G, Kleinewietfeld M, DiMitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 47.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 49.Beacher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 50.Baatar D, Olkhanud P, Sumitomo K, Taub D, Gress R, Biragyn A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4− Tregs, regulate effector T cells using FasL. J Immunol. 2007;178:4891–4900. doi: 10.4049/jimmunol.178.8.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]