

Abstract
Endothelial cells and mural cells (smooth muscle cells, pericytes, or fibroblasts) are known to communicate with one another. Their interactions not only serve to support fully functional blood vessels, but also can regulate vessel assembly and differentiation or maturation. In an effort to better understand the molecular components of this heterotypic interaction, we utilized a 3-dimensional model of angiogenesis and screened for genes, which were modulated by coculturing of these two different cell types. In doing so, we discovered that NOTCH3 is one gene whose expression is robustly induced in mural cells by coculturing with endothelial cells. Knockdown by siRNA revealed that NOTCH3 is necessary for endothelial-dependent mural cell differentiation, whereas overexpression of NOTCH3 is sufficient to promote smooth muscle gene expression. Moreover, NOTCH3 contributes to the proangiogenic abilities of mural cells cocultured with endothelial cells. Interestingly, we found that the expression of NOTCH3 is dependent upon Notch signaling, as the γ-secretase inhibitor DAPT blocked its upregulation. Furthermore, in mural cells, a dominant-negative Mastermind-like1 construct inhibited NOTCH3 expression, and endothelial-expressed JAGGED1 was required for its induction. Additionally, we demonstrated that NOTCH3 could promote its own expression and that of JAGGED1 in mural cells. Taken together, these data provide a mechanism by which endothelial cells induce the differentiation of mural cells through activation and induction of NOTCH3. These findings also suggest that NOTCH3 has the capacity to maintain a differentiated phenotype through a positive feedback loop that includes both autoregulation and JAGGED1 expression.
Keywords: Endothelial cells, mural cells, fibroblasts, smooth muscle cells, NOTCH3, JAGGED1
Introduction
Within the vasculature, endothelial cells and mural cells (defined here as vascular support cells that include smooth muscle cells, pericytes, and fibroblasts) are closely associated, and can regulate each other's activity throughout development and into adulthood.1-3 Several groups have shown that mural cells influence blood vessel assembly by controlling such events as endothelial cell proliferation, migration, sprouting and regression.4-10 Later, in intact vessels, these cells influence how endothelial cells respond to humoral and hemodynamic cues.11 Likewise, endothelial cells are known to modulate mural cell phenotype and function. In addition to proliferation and migration, endothelial cells can promote smooth muscle differentiation, and influence contractile activity. 12, 13 Despite their intimate association and obvious abilities to respond to one another in intact vessels, there is still much to be learned about the nature of their interactions, particularly during blood vessel formation.
A handful of signaling mediators form the basis of our understanding about how these two cell types communicate during vasculogenesis and angiogenesis. Growth factor/receptor families including PDGF-B/PDGFR-β, TGF-β, and Angiopoietin-Tie2, have established roles in their interactions.1, 2 For these, secreted factors uniquely expressed from one cell type bind to a receptor on the other cell type to trigger downstream events that influence behavior. Additionally, cell-specific extracellular matrix deposition, matrix metalloproteinases (MMPs), and their inhibitors (TIMPs) can directly affect the activities of these cellular neighbors. 2, 4, 10, 14 These modes of communication act independently of cell contact, however direct cell-cell contact has also been shown to play a role in endothelial/mural cell crosstalk. 15 Recently, the Notch signaling pathway has emerged as an interesting candidate that may have a vital function in modulating the interactions of endothelial cells and mural cells. 16, 17
The Notch family of membrane-bound receptors have a prominent place in vascular development.18-20 Notch receptors (NOTCH1-4), their ligands (Delta-like, and Jagged), and downstream mediators (HES/HEY) are highly expressed in both endothelial cells and mural cells (smooth muscle cells) in distinct combinations. A host of studies have helped to define their specific activities within the vasculature that include regulation of angiogenic remodeling, arterial/venous specification, and tip cell differentiation.18, 19 For example, loss of Notch1 in mice, which is abundantly present in endothelial cells, results in an array of vessel abnormalities such as disorganized intersomitic vessels, an absence of remodeling, and defects in arteriogenesis. 21-23 In vascular smooth muscle cells, Notch3 is the predominant Notch receptor, and is the causal gene for the neurovascular disorder CADASIL. 24, 25 Targeted inactivation of the mouse Notch3 gene revealed defects in smooth muscle maturation, 26 indicating a potential role in differentiation, among other things. In support of this, neural crest ablation of Notch activity similarly prevents smooth muscle differentiation.27 Several independent studies examining the role of Notch on smooth muscle gene expression have shown that an overexpressed Notch intracellular domain can promote the expression of smooth muscle genes. 28-31 Yet, these reports contrast others, which have demonstrated robust inhibition of smooth muscle gene expression by Notch. 32-34 A more recent report has hinted that these discrepancies might be explained by context-dependent activity that is controlled by the ratio of the Notch activator relative to its downstream repressor proteins (HES/HEY). 31
Given that Notch signaling is initiated by cell-cell contact of membrane bound receptor-ligand combinations, it is reasonable to assume that these proteins regulate the communication between endothelial and mural cells within the vasculature. Data supporting this hypothesis comes from an endothelial-specific knockout of Jagged1 that gives rise to an embryonic lethal phenotype with an absence of smooth muscle gene expression in the vasculature. 16 These findings strongly support a role for Notch signaling in the association of these two cell types, but the extent and the precise mechanisms underlying Notch-dependent interactions remain to be elucidated.
Materials and Methods
Cell culture methods, including angiogenesis assays, transwell culture experiments, transfections and lentiviral infections were performed with primary cultures of human endothelial cells, dermal fibroblasts and smooth muscle cells. Plasmid constructs for transfections and lentiviral infections were generated using standard cloning strategies. Expression analysis of protein by Western blotting and immunohistochemistry, and RNA by quantitative reverse transcriptase-polymerase chain reaction (qPCR) were carried out with common established methods. A detailed version of the materials and methods used can be found in an online supplement.
Results
Endothelial cells and mural cells interact in a 3-dimensional model of angiogenesis
Endothelial cells and mural cells (pericytes, smooth muscle cells and fibroblasts) are known to communicate with one another. Several groups, including ours, have demonstrated that angiogenesis is enhanced in the presence of mural cells, indicating they are important mediators of endothelial cell function during blood vessel assembly. 4-10 Shown in Figure 1A-D, a 3-dimensional angiogenesis assay using human umbilical vein endothelial cells (HUVECs) cultured alone or with human dermal neonatal fibroblasts (HDFNs), umbilical artery (HUASMCs) or coronary artery (HCASMCs) smooth muscle cells revealed robust enhancement of endothelial-derived blood vessel formation by cocultured mural cells. In this model, communication is bidirectional, as mural cells also responded to endothelial cells and were seen surrounding nascent vessels in an apparent mimic of the in vivo circumstance (Figure 1E). The inference from these observations is that these cells are programmed to respond to one another in defined ways, of which we still have a limited understanding.
Figure 1. Endothelial cells and mural cells interact.
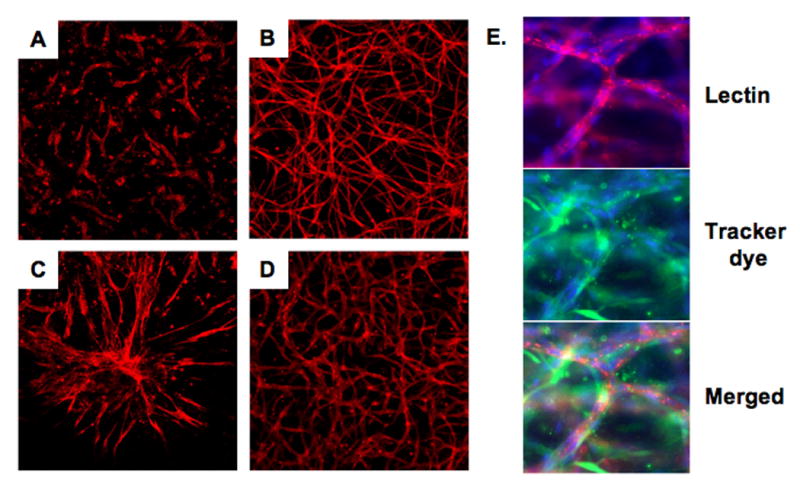
Images of blood vessels formed by endothelial cells (HUVECs) cultured alone (A), or cocultured with human dermal neonatal fibroblasts (HDFNs) (B), human umbilical artery smooth muscle cells (HUASMCs) (C) and human coronary artery smooth muscle cells (HCASMCs) (D) in a 3-dimensional angiogenesis assay. Cells were grown in a collagen matrix for five days, fixed, and stained with TRITC labeled endothelial-specific lectin (red), 100× magnification. (E) Triple labeling with lectin (red), a preloaded CellTracker dye (green) to visualize HDFNs surrounding vessels, and DAPI (blue), 400× magnification.
Endothelial cells increase NOTCH3 expression in mural cells
The obvious physical and functional interactions of these cell types in this in vitro model of angiogenesis lead us to ask what genes may be regulated by their unique association. To address this, we performed a microarray to identify genes that were specifically modulated in this 3-dimensional coculture assay using HUVECs and HDFNs. Fibroblasts were used to emulate naïve mural cells, with the intent of identifying endothelial-regulated specification or differentiation genes. In doing so, we identified NOTCH3 as one gene that was robustly increased in fibroblasts when cocultured with endothelial cells. Western blot analysis revealed low levels of NOTCH3 protein in HDFNs cultured by themselves, while much higher levels of NOTCH3 were observed in fibroblasts cocultured with endothelial cells (Figure 2A). Endothelial cells and fibroblasts were separated after coculture using endothelial-specific anti-PECAM1-conjugated beads. The expression and induction of NOTCH3 were specific to HDFNs, as no protein could be detected in endothelial cells cultured alone. In cocultured endothelial cells, a very small amount of NOTCH3 was detected after separation, and likely reflects contaminating fibroblasts due to incomplete separation. 35
Figure 2. Endothelial cells induce NOTCH3 expression in mural cells.
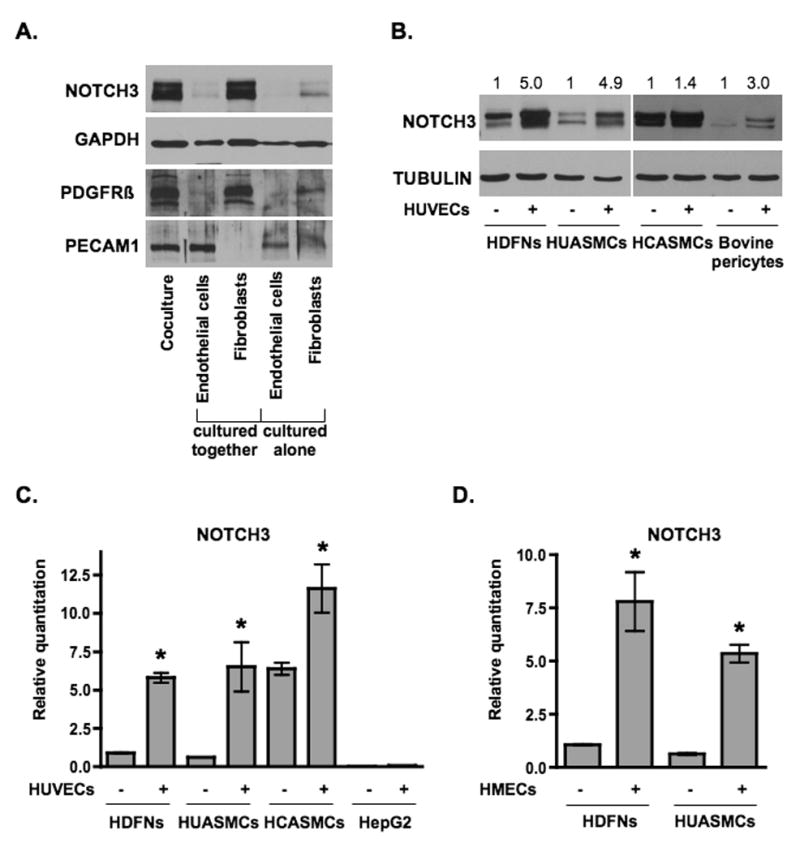
(A) Endothelial cells (HUVECs) and fibroblasts (HDFNs) were cultured alone or cocultured in 3-dimensional angiogenesis assays. After five days, cells were separated by anti-PECAM1-conjugated Dynabeads for Western blot analysis. The purity of the fibroblast and endothelial fractions were evaluated by probing for PDGFRβ as a marker for fibroblasts and PECAM1 as a marker for endothelial cells. (B) HDFNs, HUASMCs, HCASMCs, and bovine pericytes were cultured in 2-dimensions in the presence or absence of HUVECs for 48 hours, separated and subjected to Western analysis. Numbers reflect relative protein expression determined by average pixel intensity from 3 experiments normalized to respective control, P < 0.05. (C) qPCR analysis of NOTCH3 mRNA in HDFNs, HUASMCs, HCASMCs, and HepG2, cultured with or without HUVECs. (D) qPCR analysis of NOTCH3 mRNA in HDFNs and HUASMCs which were cultured alone or cocultured with HMECs. * P < 0.05 compared to control.
Though our initial observation was in a 3-dimensional assay, NOTCH3 protein expression was also increased in 2-dimensional coculture conditions, with peak levels occurring at 48 hours (Figure 2B) and remaining for up to 96 hours, the longest time tested (not shown). Western blot and quantitative RT-PCR (qPCR) indicated that NOTCH3 could be induced in fibroblasts, smooth muscle cells, and bovine retinal pericytes. The increase in NOTCH3 transcript expression ranged from 6 to 10-fold in dermal fibroblasts and umbilical artery smooth muscle cells (Figure 2C). The increase in coronary artery smooth muscle cells was the least dramatic at both RNA and protein levels, however as can be seen in Figure 2, the basal level of NOTCH3 was considerably higher in these cells. In contrast, no induction of NOTCH3 by endothelial cells was observed in HepG2 liver cells, suggesting that this activation is specific to mural cells. We further tested if other endothelial cells could affect NOTCH3 expression. Using human microvascular endothelial cells (HMECs) we demonstrated that NOTCH3 was increased in fibroblasts and umbilical artery smooth muscle cells (Figure 2D), however HepG2 cells were not able to produce a similar enhancement (data not shown). Taken together, these results indicate that endothelial cells can specifically increase the expression of NOTCH3 in mural cells, which may explain how endothelial cells modulate their function.
Endothelial cell activation of Notch signaling in mural cells is dependent upon NOTCH3
The induction of NOTCH3 implies that endothelial cells are activating Notch signaling through an increase in receptor expression. To examine Notch activity in mural cells, we transfected luciferase reporter constructs with or without five Notch-sensing CBF1 binding sites upstream of an SV40 promoter into fibroblasts. In the absence of endothelial cells, the CBF1 reporter had no activity above the basal level of the SV40 promoter, however in the presence of endothelial cells, the CBF1 reporter exhibited a ∼10-fold increase in activity (Figure 3A). Thus, these results indicate that in the absence of endothelial cells, there is little or no CBF-dependent Notch signaling in fibroblasts, and HUVECs can strongly activate this, which is consistent with a previous report. 31 To examine if Notch activity was augmented as a result of an increase in NOTCH3 expression, we examined downstream targets of Notch. As described for previous experiments, cells were cocultured for 48 hours and separated using anti-PECAM1-conjugated beads to measure expression exclusively in mural cells. Indeed, HES1 and HEYL/HRT3 RNA levels were increased in fibroblasts and smooth muscle cells upon coculture with endothelial cells (Figure 3B, C). Similar increases were seen for HRT1, HRT2, and HES4 (data not shown). Furthermore, the expression of HEYL/HRT3 was reduced when NOTCH3 levels were blocked by siRNA, indicating a direct effect of NOTCH3 in the upregulation of this factor (Figure 3D-F).
Figure 3. Notch signaling is activated in mural cells upon coculture with endothelial cells.
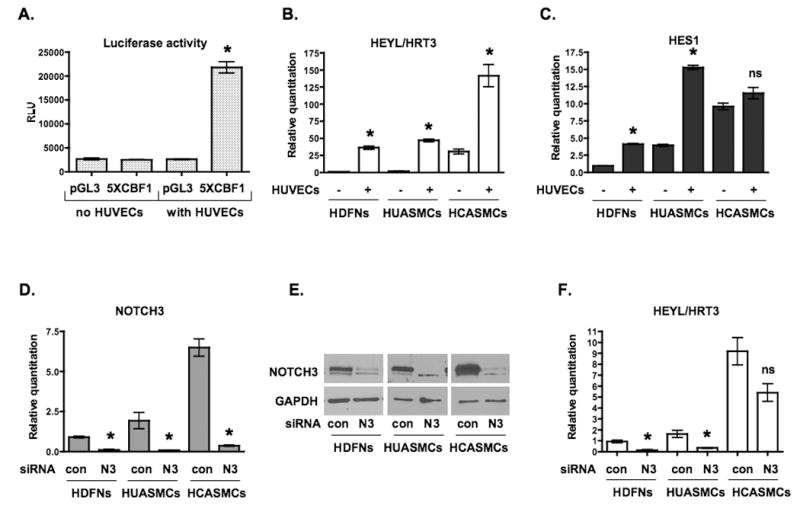
(A) Luciferase reporter assay to assess Notch activity. HDFNs were transfected with pGL3-promoter-luciferase (pGL3) plasmid as control, or plasmid with 5 CBF1 binding elements upstream of the promoter (5XCBF1), cultured in the presence or absence of HUVECs, and luciferase activity was measured. RLU, relative light units. (B, C) qPCR analysis of Notch target genes, HEYL/HRT3 and HES1 in mural cells cultured with or without HUVECs. (D) Knockdown of NOTCH3 by siRNA in HDFNs, HUASMCs and HCASMCs was confirmed by qPCR and (E) Western blot analysis. (F) qPCR analysis of HEYL/HRT3 mRNA expression after NOTCH3 knockdown. Mural cells were transiently transfected with control siRNA (con) or NOTCH3 siRNA (N3). HUVECs were added, and cocultured for an additional 48 hours. * P < 0.05 relative to relevant control; ns, not significant.
NOTCH3 is essential for endothelial-induced smooth muscle gene expression
Given that Notch signaling has been shown to regulate the expression of smooth muscle genes, 28-31 we next examined if stimulation of NOTCH3 by endothelial cells facilitated endothelial-induced smooth muscle gene expression. Coculture of endothelial cells with fibroblasts and smooth muscle cells resulted in a significant increase in smooth muscle marker gene expression in all but two instances as determined by qPCR of transcript levels (Figure 4A). Significant increases were observed for smooth muscle α-ACTIN, and CALPONIN in HDFNs, HCASMCs and HUASMCs. SM22α expression was increased in HDFNs and HCASMCs, but the increase in HUASMCs was not statistically different. Smooth muscle myosin heavy chain (SM-MHC) expression showed increases in both smooth muscle cell types, but was not significantly increased in fibroblasts, which is indicative of a myofibroblast transition rather than smooth muscle differentiation per se. Protein expression of these genes was confirmed by Western analysis and exhibited similar increases relative to RNA levels (Online Figure I). Under coculture conditions, siRNA knockdown revealed that the expression of these genes was largely contingent upon NOTCH3 (Figure 4B), with two cell-specific exceptions that imply the presence of additional regulators. Overall, these data show for the first time that endothelial-induced smooth muscle differentiation is dependent upon NOTCH3 expression.
Figure 4. Endothelial-induced smooth muscle gene expression is dependent on NOTCH3.
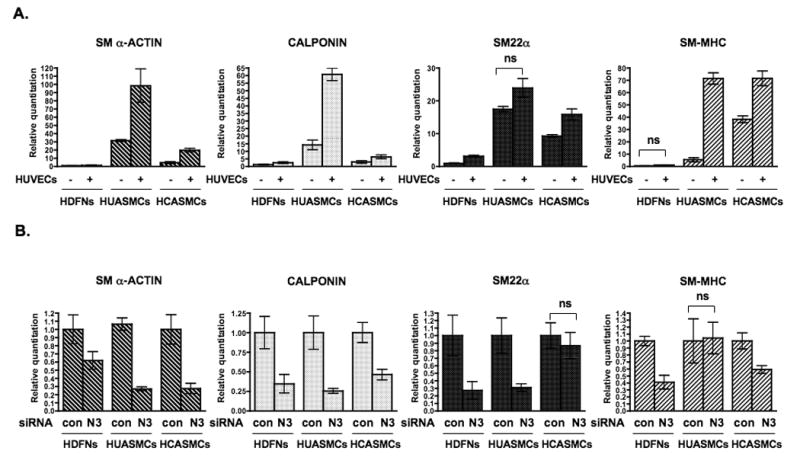
(A) Fibroblasts and smooth muscle cells were cultured with or without HUVECs, separated by anti-PECAM1-conjugated Dynabeads and RNA was collected for qPCR to detect SM α–ACTIN, CALPONIN, SM22α, and SM-MHC transcripts. (B) HDFNs, HUASMCs and HCASMCs were transfected with control siRNA (con) or NOTCH3 siRNA (N3), and cocultured with HUVECs as described. RNA was extracted for qPCR. Significant differences (P < 0.05) between control and respective experimental values were observed in all but those noted as not significant (ns).
NOTCH3 expression depends on Notch transcriptional activity
As a means to define the signal from endothelial cells that was responsible for causing NOTCH3 expression, we cultured cells together or prevented their contact using a 0.4 micron transwell filter. Shown in Figure 5A, separation of the endothelial cells and fibroblasts by a porous filter blocked the upregulation of NOTCH3, suggesting that direct cell-cell contact is critical for the inductive abilities of endothelial cells. Accordingly, endothelial cell-conditioned media did not induce expression of NOTCH3 (data not shown). In testing various pathway inhibitors that could abolish NOTCH3 induction, we observed that the γ-secretase inhibitor, DAPT, robustly abrogated the ability of HUVECs to increase NOTCH3 in HDFNs (Figure 5B, C). Similar results were also observed in HUASMCs and HCASMCs (data not shown). γ-secretase facilitates Notch signaling by a cleavage event that releases the Notch intracellular domain. 36 Based on this, Notch signaling could be responsible for the upregulation of NOTCH3, and further supports the notion that it could regulate itself. To address this hypothesis, we performed experiments to determine if Notch receptor signaling via its transcriptional activity was necessary for NOTCH3 expression in fibroblasts.
Figure 5. Endothelial-induced NOTCH3 expression requires Notch signaling.
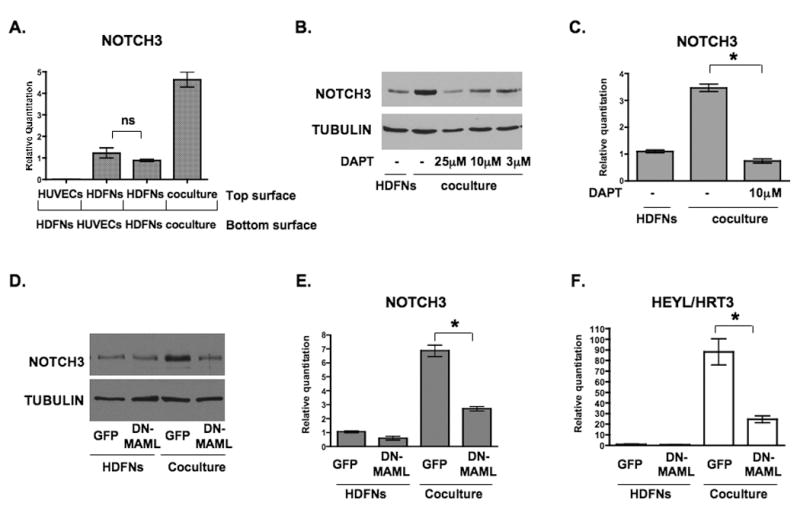
(A) HUVECs and HDFNs were plated on either side of the transwell insert as indicated, cultured for 48 hours, and the cells from the top surface were harvested for qPCR to examine NOTCH3 mRNA expression. (B) Western blot to detect NOTCH3 expression in HDFNs cocultured with HUVECs with varied amounts of the γ-secretase inhibitor (DAPT). Fibroblasts cultured alone were used as a control. (C) qPCR to measure NOTCH3 mRNA expression in HDFNs cocultured with HUVECs in the presence or absence of DAPT. (D-F) HDFNs were lentivirally transduced with GFP or DN-MAML, then cultured alone or cocultured with HUVECs. Fibroblasts were separated from HUVECs for Western analysis to detect NOTCH3 protein (D), or for qPCR to measure NOTCH3 (E) and HEYL/HRT3 (F) mRNA. * P < 0.05; ns, not significant.
Using a dominant-negative Mastermind-like1 (DN-MAML) construct, 27, 37 we tested if inhibition of this Notch coactivator would affect NOTCH3 induction by endothelial cells. The DN-MAML was introduced into fibroblasts by viral transduction, and 48 hours later the cells were cocultured with endothelial cells or cultured by themselves as a control. While the control virus harboring GFP did not block an increase in NOTCH3, DN-MAML significantly blunted this response, exhibiting a reduced level of endothelial-induced NOTCH3 protein and RNA (Figure 5D, E). HEYL/HRT3 expression showed a similar profile, with attenuated expression in the presence of DN-MAML (Figure 5F). Thus, inhibition of Notch signaling at two independent steps within the pathway show that Notch activity is an important component in the upregulation of NOTCH3 by endothelial cells.
NOTCH3 induction requires JAGGED1 on endothelial cells
Because Notch receptor signaling and transcriptional activity were required in mural cells for the expression of NOTCH3, we reasoned that Notch ligands on endothelial cells were responsible for NOTCH3 induction by these cells. The Notch ligand JAGGED1 is strongly expressed by endothelial cells, and therefore we targeted this ligand by siRNA to determine its role in endothelial-induced NOTCH3 expression. Endothelial cells were transiently transfected with JAGGED1 siRNA and 24 hours later cocultured with fibroblasts for 48 hours. Examination of NOTCH3 expression by Western blot and qPCR revealed that knockdown of JAGGED1 in endothelial cells significantly abrogated the induction of NOTCH3 in fibroblasts (Figure 6). Consistent with this, HEYL/HRT3 RNA expression was also blocked by the loss of endothelial-expressed JAGGED1. Although endothelial-expressed JAGGED1 has previously been shown to transduce Notch signaling, 16, 28 our data have extended these findings by demonstrating that an increase in NOTCH3 expression is a JAGGED1-dependent consequence of vascular cell heterotypic interactions.
Figure 6. JAGGED1 on endothelial cells is necessary for NOTCH3 induction.
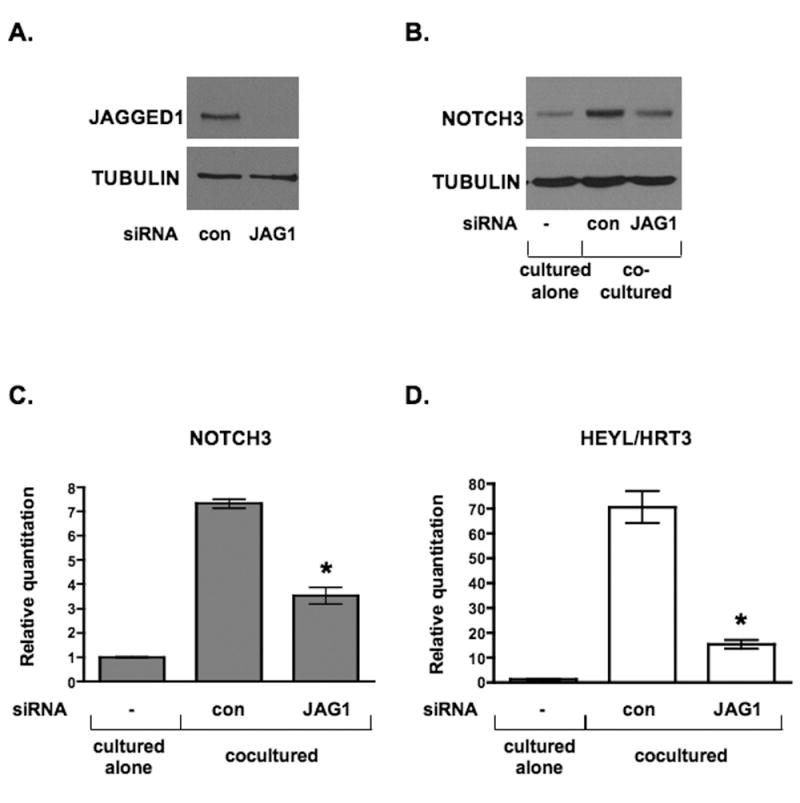
HUVECs were transiently transfected with control siRNA (con) or JAGGED1-specific siRNA (JAG1). HDFNs were added, cocultured for 48 hours, and the cells separated by Dynabeads for analysis. (A) Western blot to confirm knockdown of JAGGED1 protein in HUVECs. (B) Western blot for NOTCH3 protein expression. (C, D) qPCR to detect NOTCH3 and HEYL/HRT3 mRNA. * P < 0.05 relative to respective control.
NOTCH3 promotes its own expression and that of JAGGED1
Given that inhibition of Notch signaling prevents NOTCH3 induction, we next tested if an activated form of NOTCH3 could promote its own transcription. A human NOTCH3 intracellular domain (NICD3) was introduced into fibroblasts and smooth muscle cells by viral transduction, and NOTCH3 transcript expression was examined by qPCR using primers that recognized the extracellular region of NOTCH3 to distinguish endogenous expression from the virally produced intracellular domain. Interestingly, NICD3 strongly induced NOTCH3 transcripts in fibroblasts and smooth muscle cells (Online Figure II). It is important to note that the infection efficiency of fibroblasts was 80-90%, while the infection percentage in both smooth muscle cell types was considerably lower (between 40-60%). Hence, we do not know if the reduced induction in smooth muscle cells is a cell-specific difference or a reflection of reduced amounts of NICD3 in these cells. As expected, the expression of HEYL/HRT3 was also robustly increased by NICD3. Furthermore, consistent with previous studies, 28-31 NICD3 was able to increase transcript expression of smooth muscle genes in fibroblasts and smooth muscle cells (Online Figure II).
Because NOTCH3 was able to autoregulate its expression, we wondered if it also could regulate other Notch signaling mediators, particularly Notch ligands. Examination of JAGGED1 expression in cells expressing NICD3 revealed that JAGGED1 transcript and protein levels were increased in each cell type (Online Figure III). Moreover, like NOTCH3, JAGGED1 was also increased in mural cells cocultured with endothelial cells. We believe these results are the first to demonstrate that NOTCH3 can control its own expression, and corroborates findings showing its ability to regulate one of its ligands. 38, 39 To determine if NOTCH3 regulates JAGGED1 expression in vivo, we examined Jagged1 protein levels in blood vessels of Notch3 null and heterozygous mice. 26 In the mouse retina, Jagged1 is robustly expressed in pericytes and smooth muscle cells surrounding mature arteries. 40 We measured Jagged1 expression by immunostaining of retinas isolated from mice at postnatal day 15. The level of Jagged1 in the retinal arteries of Notch3 null mice was significantly less compared to heterozygous animals (Figure 7); thus indicating that the absence of Notch3 in vivo results in reduced expression of the Jagged1 ligand.
Figure 7. Jagged1 expression is reduced in the absence of Notch3 in vivo.
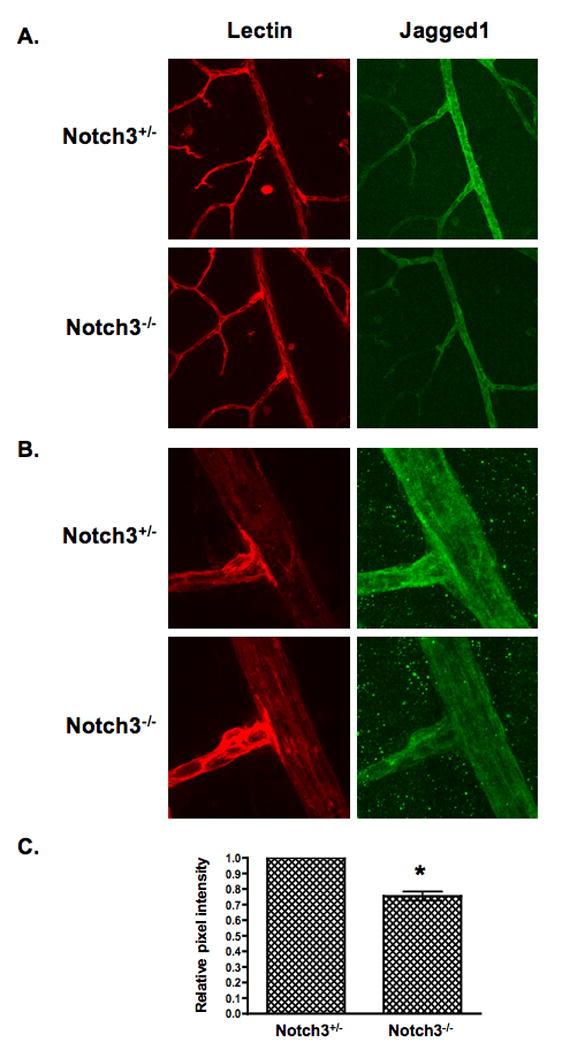
Retinas isolated from Notch3 null (Notch-/-) and heterozygous (Notch+/-) mice at postnatal day 15 were immunostained to detect Jagged1 protein (green). An endothelial-specific lectin (red) was used to highlight the vasculature. Confocal images were taken at (A) 400× and (B) 630× (zoom2) magnification and pixel intensity of Jagged1 staining was quantified (C). Graph represents average Jagged1 expression from 8 null and 8 heterozygous retinas. * P = 0.0004.
Taken together, these data strongly support the role of NOTCH3 as a conduit of smooth muscle differentiation promoted by endothelial cells. Endothelial cells, through the JAGGED1 ligand induce NOTCH3 expression, which autoregulates itself by a positive feedback loop that in turn activates smooth muscle-specific gene expression and maintains vascular support cells in a differentiated phenotype.
NOTCH3 modulates angiogenesis
The ability of mural cells to enhance angiogenesis likely depends on multiple factors that cooperate to facilitate blood vessel formation. Because NOTCH3 expression was strongly induced in mural cells by endothelial cells, we asked if NOTCH3 might facilitate fibroblast-enhanced angiogenesis. To address this, we knocked down NOTCH3 expression by lentivirally-transduced shRNA in fibroblasts. Angiogenesis assays were performed by coculturing endothelial cells with NOTCH3-deficient or control fibroblasts to examine potential differences in blood vessel formation (Online Figure IV). Interestingly, we observed a significant decrease in vessel structures in the presence of NOTCH3-deficient fibroblasts compared to control by measuring total vessel area. These results convincingly demonstrate that NOTCH3 has a role in fibroblast-mediated angiogenesis.
Discussion
Endothelial/mural cell interactions are an important component of blood vessel formation and function. Their functional and physical crosstalk is evident under 3-dimensional coculture conditions in which mural cells can enhance vessel assembly, while being recruited to encase the newly formed vessels. This striking in vitro interaction prompted us to ask what genes might be regulated by their close association. In doing so, we discovered that endothelial cells can promote the expression of NOTCH3 in mural cells and this requires direct cell-cell contact between the two cell types. This was an intriguing result considering what is already known about NOTCH3 in vascular smooth muscle cells. In a carotid artery balloon injury model, Notch3 was shown to be acutely downregulated in denuded smooth muscle, with expression reappearing seven-days post injury. 41-43 The reduction in expression was attributed to release of PDGF-BB, which was shown to directly decrease Notch3 levels. However, in the context of our results, loss of endothelial cell contact may be the main reason for the reduction in Notch3 expression within these injured arteries. In accordance with this, Lindner et. al., reported that Notch3 expression was only observed where endothelial cells were still present in balloon injured vessels.42
Solid evidence has linked Notch3 to the control of smooth muscle maturation. The smooth muscle cells of the small arteries and arterioles in Notch3 null mice exhibit reduced expression of smooth muscle differentiation genes, show defects in elongation and orientation, and display impairments in arteriolization, while large elastic and major muscular arteries are unaffected. 26 Given that endothelial cells are known to promote differentiation, our initial result hinted that endothelial-induced differentiation might be dependent upon NOTCH3. Indeed, knockdown of NOTCH3 inhibited endothelial-induced smooth muscle gene expression, providing a novel mechanism by which endothelial cells regulate this process in neighboring mural cells. These data help to bridge a gap in our understanding of endothelial-induced differentiation and Notch signaling. High et al., nicely showed the importance of endothelial-expressed JAGGED1 on smooth muscle differentiation, 16 while other groups have used overexpressed Notch intracellular domains to activate smooth muscle gene expression. 28-31 Here, we show that JAGGED1 specifically activates NOTCH3 in mural cells, and this activity is both necessary and sufficient for the expression of smooth muscle genes in three distinct mural cell populations. We did observe some differences in the ability of HUVECs and NOTCH3 to regulate certain smooth muscle genes in the different cells (Figure 4). In particular, HCASMCs, which showed robust basal expression of NOTCH3, appeared less responsive to endothelial-induced Notch signaling, possibly because the Notch pathway was already highly activated. Although the reasons for this are unknown, it points to additional complexities in the role of Notch and the transcriptional control of these key smooth muscle markers.
Our data show that the expression of NOTCH3 requires the activity of the Notch signaling mediators, γ-secretase and Mastermind-like1, in mural cells. In addition, we demonstrate that JAGGED1 is necessary for NOTCH3 expression. Although Notch receptors and their ligands have been shown to utilize various positive and negative feedback mechanisms to control availability and ultimately signaling, 20, 44, 45 there are limited reports examining their effect on each other's expression. Most described mechanisms utilize reciprocal regulation of a receptor/ligand pair that acts to contain and amplify the signal.21 Our results not only show that mural cell expression of NOTCH3 is dependent upon endothelial-expressed JAGGED1, but also demonstrate that activated NOTCH3 can promote its own expression as well as that of its ligand JAGGED1 in the same mural cell. Consistent with this, using Notch3 knockout mice we show that Jagged1 protein is decreased in the retinal vasculature in vivo. To our knowledge, these results represent the first direct evidence of a positive autoregulatory loop that involves the upregulation of a Notch receptor/ligand combination in the same cell. While NOTCH3 expression exists in an autoregulatory loop, our data also show that endothelial cells may utilize mural cell-expressed NOTCH3 to modulate their own function. Ablation of NOTCH3 in fibroblasts results in a decrease in their pro-angiogenic abilities, which suggests that endothelial cells promote NOTCH3 expression as a means to facilitate their vessel-forming potential.
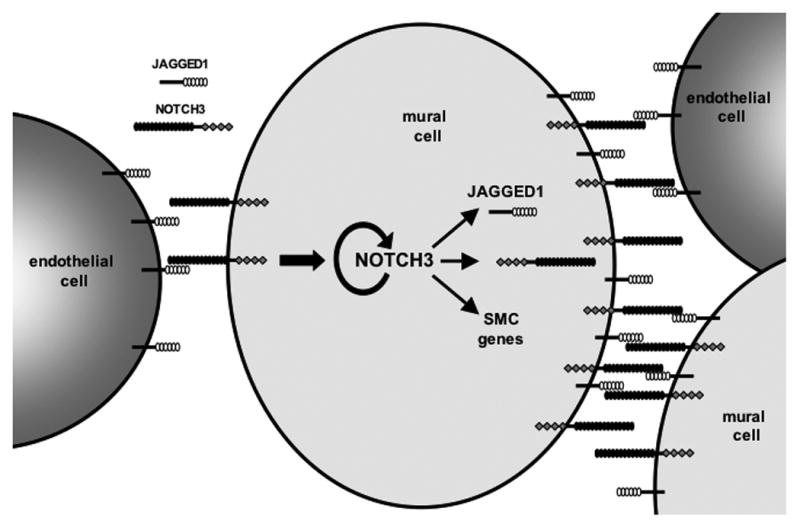
The ability of NOTCH3 to regulate its own expression and that of JAGGED1 offers evidence for a model in which endothelial cells initiate differentiation of mural cells that can maintain or amplify this signal through a positive feedback loop. As shown in Figure 8, the physical association of endothelial cells and mural cells allows for Notch signaling to be initiated by a JAGGED1/NOTCH3 interaction. In mural cells, this augments smooth muscle gene transcription, and causes an increase in NOTCH3 and JAGGED1 expression, leading to more NOTCH3 receptors on the cell surface, as well as JAGGED1 ligands. With an increase in both the receptor and ligand, mural cells can interact more efficiently with endothelial cells, or can activate Notch signaling through homotypic interactions of neighboring mural cells presenting NOTCH3 and JAGGED1. This model suggests that once endothelial cells initiate the cascade, mural cells are then able to maintain the signal and distribute it to other mural cells through continued activation and expression of NOTCH3. Whether the continued presence of endothelial cells is required and if “activated” mural cells are able to pass on this Notch signal remains to be determined. Irrespective of these possibilities, our data conclusively show the importance of Notch signaling in the interactions of these two cell types, and demonstrate one mechanism through which these cells likely communicate with one another within the vasculature.
Figure 8. Model of sustained Notch signaling in mural cells.
Acknowledgments
We are grateful to Wenbo Zhang and Irfan Ali for critical reading of the manuscript. The support of the Metabolic Vascular Disease Group of the Vascular Biology Center is greatly appreciated.
Source of Funding: This work was supported by NIH grant R01 HL076428 to BL.
Footnotes
Disclosures: None
References
- 1.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 2.Hughes CC. Endothelial-stromal interactions in angiogenesis. Curr Opin Hematol. 2008;15:204–209. doi: 10.1097/MOH.0b013e3282f97dbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 4.Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol. 2006;175:179–191. doi: 10.1083/jcb.200603176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montesano R, Pepper MS, Orci L. Paracrine induction of angiogenesis in vitro by Swiss 3T3 fibroblasts. J Cell Sci. 1993;105:1013–1024. doi: 10.1242/jcs.105.4.1013. [DOI] [PubMed] [Google Scholar]
- 6.Villaschi S, Nicosia RF. Paracrine interactions between fibroblasts and endothelial cells in a serum-free coculture model. Modulation of angiogenesis and collagen gel contraction. Lab Invest. 1994;71:291–299. [PubMed] [Google Scholar]
- 7.Kunz-Schughart LA, Schroeder JA, Wondrak M, van Rey F, Lehle K, Hofstaedter F, Wheatley DN. Potential of fibroblasts to regulate the formation of three-dimensional vessel-like structures from endothelial cells in vitro. Am J Physiol Cell Physiol. 2006;290:C1385–1398. doi: 10.1152/ajpcell.00248.2005. [DOI] [PubMed] [Google Scholar]
- 8.Ozerdem U, Stallcup WB. Early contribution of pericytes to angiogenic sprouting and tube formation. Angiogenesis. 2003;6:241–249. doi: 10.1023/B:AGEN.0000021401.58039.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darland DC, Massingham LJ, Smith SR, Piek E, Saint-Geniez M, D'Amore PA. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264:275–288. doi: 10.1016/j.ydbio.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Chen B, Lilly B. Fibroblasts potentiate blood vessel formation partially through secreted factor TIMP-1. Angiogenesis. 2008;11:223–234. doi: 10.1007/s10456-008-9102-8. [DOI] [PubMed] [Google Scholar]
- 11.Mombouli JV, Vanhoutte PM. Endothelial dysfunction: from physiology to therapy. J Mol Cell Cardiol. 1999;31:61–74. doi: 10.1006/jmcc.1998.0844. [DOI] [PubMed] [Google Scholar]
- 12.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 13.Segal SS. Regulation of blood flow in the microcirculation. Microcirculation. 2005;12:33–45. doi: 10.1080/10739680590895028. [DOI] [PubMed] [Google Scholar]
- 14.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 15.Hirschi KK, Burt JM, Hirschi KD, Dai C. Gap junction communication mediates transforming growth factor-beta activation and endothelial-induced mural cell differentiation. Circ Res. 2003;93:429–437. doi: 10.1161/01.RES.0000091259.84556.D5. [DOI] [PubMed] [Google Scholar]
- 16.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sainson RC, Harris AL. Regulation of angiogenesis by homotypic and heterotypic notch signalling in endothelial cells and pericytes: from basic research to potential therapies. Angiogenesis. 2008;11:41–51. doi: 10.1007/s10456-008-9098-0. [DOI] [PubMed] [Google Scholar]
- 18.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 19.Shawber CJ, Kitajewski J. Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays. 2004;26:225–234. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- 20.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 21.Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 23.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 25.Wang T, Baron M, Trump D. An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol. 2008;96:499–509. doi: 10.1016/j.pbiomolbio.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Manabe I, Arai M, Nagai R, Kurabayashi M. Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J Biol Chem. 2006;281:28555–28564. doi: 10.1074/jbc.M602749200. [DOI] [PubMed] [Google Scholar]
- 29.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth Muscle alpha-actin is a direct target of Notch/CSL. Circ Res. 2006;98:1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- 30.Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, Shahidi R, Dorovini-Zis K, Li L, Beckstead B, Durand RE, Hoodless PA, Karsan A. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. 2004;94:910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 31.Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–668. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennard S, Liu H, Lilly B. Transforming growth factor-beta (TGF- 1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. J Biol Chem. 2008;283:1324–1333. doi: 10.1074/jbc.M706651200. [DOI] [PubMed] [Google Scholar]
- 33.Morrow D, Scheller A, Birney YA, Sweeney C, Guha S, Cummins PM, Murphy R, Walls D, Redmond EM, Cahill PA. Notch-mediated CBF-1/RBP-J{kappa}-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am J Physiol Cell Physiol. 2005;289:C1188–1196. doi: 10.1152/ajpcell.00198.2005. [DOI] [PubMed] [Google Scholar]
- 34.Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem. 2005;280:8994–9004. doi: 10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- 35.Lilly B, Kennard S. Differential gene expression in a coculture model of angiogenesis reveals modulation of select pathways and a role for Notch signaling. Physiol Genomics. 2008 doi: 10.1152/physiolgenomics.90318.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxena MT, Schroeter EH, Mumm JS, Kopan R. Murine notch homologs (N1-4) undergo presenilin-dependent proteolysis. J Biol Chem. 2001;276:40268–40273. doi: 10.1074/jbc.M107234200. [DOI] [PubMed] [Google Scholar]
- 37.Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC, Aster JC. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–664. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross DA, Kadesch T. Consequences of Notch-mediated induction of Jagged1. Exp Cell Res. 2004;296:173–182. doi: 10.1016/j.yexcr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P, Bonafe M. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofmann JJ, Luisa Iruela-Arispe M. Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr Patterns. 2007;7:461–470. doi: 10.1016/j.modgep.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ Res. 2002;91:999–1006. doi: 10.1161/01.res.0000044944.99984.25. [DOI] [PubMed] [Google Scholar]
- 42.Lindner V, Booth C, Prudovsky I, Small D, Maciag T, Liaw L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol. 2001;159:875–883. doi: 10.1016/S0002-9440(10)61763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Campos AH, Prince CZ, Mou Y, Pollman MJ. Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. J Biol Chem. 2002;277:23165–23171. doi: 10.1074/jbc.M201409200. [DOI] [PubMed] [Google Scholar]
- 44.Carmena A, Buff E, Halfon MS, Gisselbrecht S, Jimenez F, Baylies MK, Michelson AM. Reciprocal regulatory interactions between the Notch and Ras signaling pathways in the Drosophila embryonic mesoderm. Dev Biol. 2002;244:226–242. doi: 10.1006/dbio.2002.0606. [DOI] [PubMed] [Google Scholar]
- 45.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]