

Abstract
Oxidative modifications of protein thiols are important mechanisms for regulating protein functions. The present study aimed to compare the relative effectiveness of two thiol-specific quantitative proteomic techniques, difference gel electrophoresis (DIGE) and isotope coded affinity tag (ICAT), for the discovery of redox-sensitive proteins in heart tissues. We found that these two methods were largely complementary; each could be used to reveal a set of unique redox-sensitive proteins. Some of these proteins are low-abundant signaling proteins and membrane proteins. From DIGE analysis, we found that both NF-κB-repressing protein and epoxide hydrolase were sensitive to H2O2 oxidation. In ICAT analysis, we found that specific cysteines within sacroplasmic endoplamic reticulum calcium ATPase 2 and voltage-dependent anion-selective channel protein 1 were sensitive to H2O2 oxidation. From these analyses, we conclude that both methods should be employed for proteome-wide studies, to maximize the possibility of identifying proteins containing redox-sensitive cysteinyl thiols in complex biological systems.
Keywords: oxidation, DIGE, ICAT, cysteine thiol, redox proteomics
Introduction
Oxidative modifications of protein thiols serve important roles in regulating cellular physiology. Under mild oxidative stress, reversible oxidation of selective protein residues may serve as redox sensors and signal transducers for conveying cellular anti-stress responses. When encountering severe oxidative insults, many proteins undergo irreversible oxidative modifications, causing protein degradation and cell death.1 Among the many amino acids that are susceptible to oxidative stress, cysteine is particularly sensitive to oxidative insults, owing to its favorable nucleophilic property.2 Given the unique chemical nature of protein thiols, they play essential roles in maintaining the integrity of protein structure and function. Redox-sensitive cysteines usually possess acidic pKa’s and are likely to deprotonate under physiological pH, rendering them susceptible to oxidant challenge. Redox-sensitive cysteines have been identified in a wide spectrum of proteins including transcription factors: OxyR,3 AP-1,4 NF-κB,5 Ref-1,6 p53,7 and HIF-1α;8 signal transducers: cAMP-dependent protein kinase,9 mitogen-activated protein kinases,10 protein tyrosine phos-phatase,11 and apoptosis signal-regulating kinase 1;12 and stress response proteins, such as peroxiredoxins,13 superoxide dismutase,14 thioredoxin,15 and heat shock proteins.16,17 Oxidative modifications of these crucial cysteines have significant impacts on both protein structures and functions.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the major driving forces for oxidizing proteins in general and cysteine thiols in particular. ROS and RNS, including hydrogen peroxide (H2O2), superoxide ion (O2•−), nitric oxide (NO•), and hydroxyl radical (OH•), can be produced in vivo from a wide range of cellular processes, such as metabolism, proliferation, inflammation, and senescence. Certain reversible cysteine oxidative modifications, such as disulfide bond formation,18 sulfenation,19,20 nitrosylation,21 andglutathionylation,22,23 are suggested to serve cellular protective functions to preserve crucial cysteine residues from irreversible oxidative damage or to regulate redox signal transduction. On the other hand, refractory and irreversible oxidative modifications of cysteine residues such as sulfonic acid formation and 4-hy-droxynonenal adducts formation often lead to the inhibition of protein function and/or protein degradation.1
The labile, transient, and dynamic nature of reversible oxidative modifications poses enormous technical challenges for both accurate proteomic identification and sensitive quantification of cysteine thiols. Such difficulties also reside in the fact that many proteins of biological interest are present in relatively low abundance. A variety of gel-based proteomics methods have been developed to identify redox-sensitive proteins.24–27 Diagonal electrophoresis18 is designed to identify disulfide-bond interlaced proteins according to distinct gel migration profiles of proteins with either intra- or intermolecular disulfide bond linkages, under sequential nonreducing and reducing electrophoresis conditions. Protein glutathionylation and nitrosylation could be detected by 2DE methods by taking advantage of the availability of specific antibodies.28 In addition, Kim et al.27 devised a novel immunoblotting method based on the covalent attachment of a biotin-linked tag to reactive cysteines for immunoblotting detection. However, these immunoblotting methods suffer from moderate resolution, nonspecific detection, and low throughput. Recently, gel-based methods employing cysteine-specific fluorochromes24–26 or radioactive probes22,29 have been developed to identify redox-sensitive proteins within complex protein mixtures. To minimize gel-to-gel variations, it is essential to be able to carry out multiplexed experiments in a high-throughput fashion. Unlike conventional 2DE methods, the saturation-labeling DIGE method has the potential for robust multiplexed analysis of protein thiols. This approach employs a pair of fluorescent CyDyes to specifically label free thiols in multiple protein samples. Mixtures of differentially labeled proteins can then be resolved simultaneously in the same gel, and quantitative differences can be ascertained from the dual channel fluorescent images. Although DIGE was originally designed to measure the alterations of protein expression, Hurd and co-workers30 have successfully tailored this technique to detect redox-sensitive proteins.
A gel-free mass spectrometry (MS)-based method, ICAT,31 is another thiol-specific proteomic technique with multiplexing capability. Each ICAT reagent consists of three essential groups: a thiol-reactive group, an isotope-coded light or heavy linker, and a biotin segment to facilitate peptide enrichment. In an ICAT experiment, protein samples are first labeled with either light or heavy ICAT reagents on cysteine thiols. The mixtures of labeled proteins are then digested by trypsin and separated through a multistep chromatographic separation procedure. Peptides are identified with tandem MS, and the relative quantifications of peptides are inferred from the integrated LC peak areas of the heavy and light versions of the ICAT-labeled peptides. Sethuraman et al.32 successfully applied the ICAT technique to discover 18 potential H2O2-sensitive membrane proteins in rabbit heart.
Despite reports of the successful applications of both DIGE and ICAT methods for the identification of redox-sensitive proteins, comparative studies of their strengths and limitations for analyzing redox proteomes have not been reported. Herein we used these two quantitative proteomic methods to screen for heart proteins that are sensitive to H2O2 oxidation. To our knowledge, this is the first direct comparison of DIGE and ICAT methods for the identification and quantification of redox-sensitive proteins in complex biological mixtures. We found that these two methods were largely complementary and each was able to reveal a set of unique low-abundant redox-sensitive proteins. From DIGE analysis, we were able to identify transcriptional regulators and signaling molecules that were H2O2-sensitive, whereas from ICAT analysis, we were able to discover several redox-sensitive membrane proteins. Based on this study, we conclude that both methods should be employed for redox proteomics studies in order to comprehensively identify redox-sensitive proteins as well as localize their reactive cysteine thiols within complex biological systems.
Materials and Methods
Chemicals and Reagents
HPLC grade acetonitrile (ACN) and water were purchased from J. T. Baker (Phillipsburg, NJ). Tris, α-cyano-4-hydroxycinnamic acid (CHCA), trifluoroacetic acid (TFA), iodoacetamide, dithiothreitol (DTT), and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO). MS calibration standard peptides, Glu-fibrinopeptide, and human adrenocorticotropic hormone 18–39 were also bought from Sigma-Aldrich. Cleavable ICAT reagent was obtained from Applied Biosystems (ABI, Forster city, CA). Porcine malate dehydrogenase (MDH) was purchased from Roche Applied Science (Indianapolis, IN).
Protein Extraction and Oxidation
Eight to ten week old Swiss Webster mice heart tissues (Pel Freeze Biologicals, Rogers, AR) were weighted (from four different animals, ~100 mg each) and lysed with 500 μL of lysis buffer containing 8 M urea, 2% CHAPS, and 0.1% Triton X-100 in 30 mM tris at pH 8.0. A protease inhibitor cocktail was added prior to homogenization. The homogenates were centrifuged at 14 000g at 4 °C for 30 min. Protein concentrations in supernatants were measured by Bradford assay (Bio-Rad, Hercules, CA). Protein solution from each animal was divided into two aliquots of 500 μg each; one was treated with 500 μM H2O2 for 30 min in the dark, and the other was treated with an equal volume of H2O. Protein samples were then desalted with a ReadyPrep 2D Cleanup Kit (Bio-Rad), and the protein pellets were reconstituted with either ICAT or DIGE labeling buffers in accordance to respective experiments.
DIGE Saturation Dye Labeling and 2DE
DIGE experiments were carried out in an octaplexed fashion, where Cy3 male-imide (Cy3m) was used to label the four control and four H2O2 treated samples. In parallel, Cy5 maleimide (Cy5m) was used to label eight equal aliquots of the internal standards. Each Cy3m-labeled sample was then mixed pairwise with one Cy5m-labeled internal standard and used for subsequent 2DE analysis. The DIGE labeling procedure was adapted from the manufacturer’s protocol, except that the tris(2-carboxyethyl)phosphine (TCEP) reduction step was skipped to preserve oxidized cysteines. Briefly, protein samples were first resuspended in DIGE labeling buffer (8 M urea, 2% CHAPS, 0.1% Triton X-100, and 30 mM tris at pH 8.0). Five micrograms of proteins from either control or H2O2-treated samples was labeled with 4 nmol of Cy3m each. Aliquots of 5 μg of the internal standards (prepared by pooling 20 μg of proteins each from all eight samples) were labeled with 4 nmol of Cy5m separately. The labeling reactions were carried out at 37 °C for 1 h and then quenched by adding equal volumes of sample buffer (8 M urea, 2% CHAPS, 2% DTT, and 2% ampholytes 5–8). Each Cy5m-labeled internal standard was combined with a designated Cy3m-labeled sample (either control or H2O2 treated samples) for subsequent 2DE analysis. To achieve optimal DIGE labeling and prevent spontaneous thiol oxidations, all experiments were performed with minimal light exposure. In addition, each tube was flushed with nitrogen prior to labeling. The final volume of each combined reaction mixture was adjusted to 183 μL with rehydration buffer (8 M urea, 2% CHAPS, 1% DTT, and 1% ampholytes 5–8) plus 2 μL of 1% bromophenol blue. A pick gel sample was prepared by combining 25 μg of proteins from all eight samples and paired with a Cy5m-labeled internal standard to facilitate spot matching. Samples were loaded onto 11 cm IPG strips (pH 5–8, Bio-Rad), rehydrated with the sample buffer for 2 h, and subsequently focused in a Protean IEF cell (Bio-Rad) for at least 80 000 V-hr, at 7000 V maximum and a current limit of 50 μA/gel strip. The IEF-focused IPG strips were then sequentially equilibrated with 50 mM DTT and 50 mM iodoacetamide for 15 min each in equilibration buffer (6 M urea, 2% SDS, and 20% glycerol in 50 mM tris at pH 8.3). Proteins were then separated in the second dimension with 12.5% Criterion Tris-HCl gels using a Criterion Doceca cell (Bio-Rad). After electrophoresis, gels were first rinsed with Milli-Q water and then fixed with 40% ethanol and 10% acetic acid. The gels were then scanned on a Typhoon 9400 imager (GE Healthcare, Piscat-away, NJ) with 100 μm resolution and appropriate photomultiplier tube voltages to ensure no spot saturation. The pick gel was stained overnight with Sypro Ruby (Bio-Rad) and scanned. DeCyder software (v5.0, GE Healthcare) was used to analyze the gel images. Gel images were processed with 1500 expected spots, and spots with areas smaller than 400 and slopes larger than 1.5, mostly dust artifacts, were excluded from further analysis. Changed protein spots with p-values ≤ 0.05 were matched to the pick gel and were excised for identification according to a protocol established in this laboratory.33 Tryptic peptides were desalted with C18 Ziptips (Millipore) and spotted onto a MALDI plate by mixing 1:1 with the matrix solution (6 mg/ml CHCA in 60% ACN and 0.1% TFA). MS and MS/MS spectra were acquired using a 4700 MALDI-TOF/TOF mass spectrometer (ABI). Protein identifications were performed by both peptide mass fingerprinting (with Profound, http://prowl.rockefeller.edu/prowl-cgi/profound.exe) and MS/MS spectra matching (using embedded Mascot search engine v1.9 on a GPS server v3.5, ABI). Database search parameters were as followed: mass tolerance of 50 ppm for MS and 0.3 Da for MS/MS, trypsin digestion with maximum one missed cleavage, NCBI mouse database, variable modifications including methionine oxidation, and carboamidomethylation of cysteines. Proteins identified with sequence coverage above 15% and at least two unique peptides identified with confidence interval (C. I.) values above 95% from the MS/MS search were considered significant.
ICAT Labeling and Multidimensional Chromatography
Protein samples were reconstituted with the labeling buffer supplied with the ICAT Reagent Kit (ABI) and processed according to the manufacturer’s protocol, unless stated otherwise. Two ICAT labeling strategies were employed: (1) A forward strategy in which free cysteines were directly labeled in the control and treated samples with either light or heavy ICAT reagents respectively without the reduction of disulfide bonds. In brief, 100 μg of the control sample was labeled with the light ICAT reagent, while the H2O2-treated sample was labeled with the heavy ICAT reagent. The TCEP reduction step was avoided to preserve the native thiol redox-states. The labeling reactions were carried out at 37 °C for 2 h, and excess ICAT reagents were quenched with additional 10 mM DTT. The newly reduced thiols were alkylated with 10 mM iodoacetamide. (2) A backward approach in which we first alkylated all free cysteine thiols with iodoacetamide and reduced the reversibly oxidized cysteines with TCEP. For easier conceptual comparison between the forward and backward labeling ICAT results, we used the light ICAT reagent to label the newly exposed cysteine thiols from H2O2-treated samples and heavy ICAT reagent for labeling the control samples. After the labeling reactions with either strategy, the light and heavy versions of the samples were combined and subjected to tryptic digestion at 50:1 (protein to enzyme) ratio overnight at 37 °C. Tryptic peptides were separated from the excess ICAT reagents and detergents in the lysis buffer and were fractioned with strong cation exchange chromatography (SCX). SCX was carried out on a Biocad Sprint System (ABI) with a PolySulfoethyl A column (200 mm × 4.6 mm, PolyLC Inc., Columbia, MD). The ICAT-labeled peptides were first acidified with mobile phase A (10 mM KH2PO4, 20% ACN at pH 2.7) to a final volume of 500 μL and injected. The peptides were first separated at 1.0 mL/min with 0–25% mobile phase B (0.6 M KCl, 10 mM KH2PO4, and 20% ACN at pH 2.7) for 30 min, followed by 25–100% B over 20 min, and remained at 100% B for an additional 30 min. Peptide fractions were collected at 2 min intervals and dried. ICAT-labeled peptides were then enriched by avidin cartridges (ABI) and dried. Biotin moieties were cleaved off with TFA at 37 °C for 2 h. After the cleavage, the peptides were dried and reconstituted in 5% ACN and 0.1% TFA (mobile phase A for reversed phase LC, RPLC) and were purified with a LC Packings capillary HPLC system (Dionex, Sunnyvale, CA) on a PepMap C18 column (5 μm, 0.075 × 150 mm) along with an inline trapping column (5 μm, 0.3 × 5 mm). RPLC gradient consisted of first 5–30% mobile phase B (MPB, 95% ACN, and 0.1% TFA) over 75 min, then 30–90% MPB for 15 min, and maintained at 90% MPB for an additional 10 min at a flow rate of 400 nL/min. The RPLC eluent was mixed at a 1:2 ratio with the MALDI matrix (6 mg/mL CHCA, 60% ACN, and 0.1% TFA) through a micro tee and spotted onto the MALDI plates with Probot (Dionex).34
Redox Analysis of Purified Malate Dehydrogenase
One hundred micrograms of MDH was dissolved in 80 μL of the lysis buffer (identical to the buffer used for heart protein extraction). Forty microliters each of MDH solution was treated with either H2O or 500 μM H2O2 for 30 min in the dark. Subsequently both DIGE and ICAT analyses were performed as described for the heart samples.
MS and Database Search
An ICAT-ratio-dependent acquisition strategy was used for the quantification and identification of peptides, where both MS and MS/MS spectra were acquired on a 4700 MALDI-TOF/TOF mass spectrometer (ABI). ICAT ion pairs (heavy and light versions with a mass difference of 9.03 ± 0.03 Da) were quantified with the GPS Explorer software (v3.5, ABI). Relative ICAT ratios were computed with the integrated chromatographic areas of the ICAT ions. Only ICAT pairs with at least 20% intensity changes, plus with signal-to-noise ratios over 80 were submitted for MS/MS analysis and the subsequent MASCOT database search. Only peptides identified with C. I. values at or above 95% and mass errors within 20 ppm were considered as confident identifications. The frequency of false identification in ICAT analysis was evaluated with a target-decoy database search strategy proposed by Peng et al.35 The decoy database was constructed with all database protein entry sequences reversed. The false positive rate (FPR) was calculated with the equation FPR= (2 × FP)/(FP + TP). False positive (FP) hit equals to the total number of peptide assignments in the decoy database search with C. I. ≥ 95%, and true positive (TP) hit is the number of total hits after the subtraction of FP hits. The FPR calculated for this study was 3.9%.
Results and Discussion
Establishment of a Model Redox System for the Comparison of ICAT and DIGE
We chose to use hydrogen peroxide treatment of extracted tissue proteins for this study was to create a robust model system with defined redox states for each protein, to compare the effectiveness of these two methods. This model system is different from in vivo redox systems, in which many diverse and somewhat unpredictable oxidative modifications are likely to exist, rendering head-to-head comparisons of the ICAT and DIGE methods difficult. The H2O2 concentration selected was based on both our own observations and previous published reports.36 We wanted to establish a model system that would demonstrate noticeable decrease of free cysteine thiols following oxidative treatment, yet not too severe to induce protein degradation. To correlate the relationship between the degree of protein thiol oxidation and H2O2 dosage, we conducted a Western blotting analysis to assess the extent of thiol alkylation changes with biotinylated iodoacetamide, following the oxidation of the heart proteins with serial-diluted H2O2. The same samples were also analyzed by 2DE to evaluate the degrees of protein degradation. To our surprise, from 50 to 400 μM, no consistent and substantial protein thiol oxidation was observed (data not shown). Above 1 mM, we started to observe protein degradation (data not shown). It is known that H2O2 is not as potent as other agents that include hydroxyl radicals and peroxinitrite for protein oxidation, both in vivo and in vitro.37 Additional reason why appreciable protein oxidation was observed only at 500 μM of H2O2 might also be due to the fact that, a small tripeptide antioxidant, GSH could be present in the protein extracts, and it would quench the effects of H2O2 at lower oxidant concentrations. Given these observations, we chose 500 μM of H2O2 for this methodology comparison study.
Identification of H2O2-Sensitive Heart Proteins by DIGE
Saturation-labeling DIGE utilizes fluorescent CyDyes linked to a thiol-reactive maleimide group. Such that the Cy3m and Cy5m CyDyes can label free thiols in protein samples, facilitating simultaneous protein thiol measurement. Herein we used DIGE method to identify redox-sensitive heart proteins. Individual control and H2O2-treated samples were paired with a common internal standard for accurate gel-to-gel spot matching and quantification. Because we performed CyDye labeling reactions without protein reduction and alkylation, only free thiols should be labeled and their relative ratios measured. The gel images were analyzed with the DeCyder software and the quantification of free thiol levels in the samples was normalized to the pooled internal standards. Only significantly changed (p ≤ 0.05) protein spots with at least 50% DIGE signal decrease were subjected to MS identification. Overall, 1000 proteins spots were detected and matched among the gels; 55 spots were excised for identification by MS. Forty-two spots containing 26 unique proteins were confidently identified (Supplemental Table 1, Supporting Information). An example of the comparison between either a control sample (green, Figure 1A) or a H2O2-treated sample (green, Figure 1B) and their respective internal standards (red) is shown in Figure 1. Among the proteins having significantly decreased thiol signals upon H2O2 treatment, an example of DIGE analysis of malate dehydrogenase is shown (Figure 1C–G). A treatment/control ratio of 0.26 (p-value < 0.05) was observed. We observed that several protein spots shifted to more acidic forms as a result of H2O2 treatment (Figure 1A and B), indicating the possible occurrence of acidic post-translational modifications such as the oxidation of cysteine thiols to sulfenic, sulfinic, or sulfonic acids. Recently, several studies have reported that key cysteines oxidized into these more acidic forms may serve as redox sensors for modulating protein functions.38,39 Given our experimental design, there may be two possibilities for the reduction of fluorescence intensities observed for H2O2-treated proteins: H2O2-induced protein degradation or cysteine thiol oxidation. To distinguish these two possibilities, we also analyzed the samples by conventional 2DE and stained with Sypro Ruby. No significant changes at the protein expression levels were observed (data not shown). Therefore, it is likely that the selective reduction of thiol signals in H2O2-treated samples is primarily due to the oxidation of redox-sensitive cysteines.
Figure 1.

Identification of H2O2-sensitive heart proteins with saturation-labeling DIGE. Mouse heart proteins were incubated with H2O or 500 μM H2O2 and labeled with Cy3m (green), each sample was mixed with an internal standard labeled with Cy5m (red). A mixture of a control sample and the internal standard was resolved in a 2D gel and the superimposed fluorescent image from the control and the standard proteome is shown in (A). The same procedure was applied to a H2O2-treated sample and the gel image is shown in (B). More green spots were observed in the control sample (A) than the H2O2 treated sample (B), when both are compared to the Cy5m labeled internal standard. This observation indicates that fewer free thiols were available for Cy3m labeling in the H2O2 treated sample (B). The white boxes in panels (A) and (B) highlight a group of succinate dehydrogenase isoforms that were sensitive to H2O2 oxidation (less green spots in B and confirmed with DeCyder quantification analysis). It is also noticeable that the acidic end of this group of protein isoforms appeared more red in A and greener in B, suggesting the appearance of more acidic isoforms under H2O2 challenge. Protein spots were detected and matched among the gels with the assistance of preassigned landmark spots. Consistent protein spot detection and matching is demonstrated between the control (C) and the H2O2-treated gels (D). The highlighted spots in panels (C) and (D) represent a protein (later identified as malate dehydrogenase) with significant decrease of fluorescent intensity following H2O2 treatment. This change is more strikingly illustrated in the 3D comparison of the spot volumes between a control (E) and a H2O2-treated sample (F). Quantitative analysis was carried out after spot volume normalization with the internal standard. Statistical evaluation (p < 0.005) (G) confirmed the significant decrease of ~70% of cysteine thiol in malate dehydrogenase following H2O2 treatment.
We also attempted to locate the specific CyDye-labeled cysteines in these protein spots by searching for Cy3m or Cy5m as well as other possible oxidative modifications of protein thiols in both MS and MS/MS spectra. However, these efforts were not fruitful, probably due to (1) possible inefficient ionization of CyDye-labeled peptides, (2) poor recovery of large peptides from the in-gel digestion steps, and (3) the DTT reduction step during in-gel digestion may have reduced some oxidative modifications of cysteines. Although the exact sites and types of oxidative modifications could not be determined solely by the DIGE method, it did provide a panoramic view of proteins that are sensitive to H2O2 treatment. Many proteins found here have previously been reported as sensitive to redox modifications (Table 1). For example, many metabolic proteins, especially those involved in the glycolysis, pyruvate dehydrogenase complex, and the tricarboxylic acid cycle, were found to be susceptible to H2O2 oxidation. In addition, proteins essential for the mitochondrial electron transport chain also underwent oxidative modifications upon H2O2 treatment. Finally, we found a significant impact of H2O2 on antioxidant proteins, structural and signaling proteins as well. The widespread oxidation of proteins involved in such diverse biological processes suggested that oxidative stress may impact many cellular machineries within the cardiovascular system. Interestingly, a low-abundant nuclear protein, NF-κB repressing factor, was found oxidized upon H2O2 treatment. The finding may suggest a novel mechanism for NF-κB regulation. The known pathway for NF-κB activation by oxidative stress is initiated via the oxidation-induced dissociation of NF-κB and its inhibitor, IκB in the cytoplasm. The activated NF-κB will translocate to the nucleus and activate target gene transcription.5,40 Under normal physiological conditions, NF-κB repressing factor is the guardian in the nucleus that prevents abnormal activation of NF-κB by direct protein–protein interaction. The oxidation of the NF-κB repressing factor may abolish its NF-κB binding capability and pave the way for full activation of NF-κB.
Table 1.
Redox-Sensitive Proteins Found in This Study Using the Forward Labeling Strategya
| Protein | DIGE | ICAT | peptide sequence | Previously reported redox features | Ref | ||
|---|---|---|---|---|---|---|---|
| Aconitase 2 | + | + | 379VGLIGSCTNSSYEDMGR395 | Sulfonation of Cys385 | 18, 20, 48 | TCA CYCLE | Metabolic proteins |
| Isocitrate dehydrogenase subunit alpha | + | + | 351CSDFTEEICR360 | Disulfide bond formation of unspecified cysteines. | 18 | ||
| Malate dehydrogenase, cytosolic | + | + | 126VIVVGNPANTNCLTASK142 | Nitrosylation of cys137 | 21 | ||
| Malate dehydrogenase, mitochondrial | − | + | 79GYLGPEQLPDCLK91 | ||||
| 92GCDVVVIPAGVPR104 | |||||||
| 204TIIPLISQCTPK215 | Redox-sensitive cys93 | 20, 32, 49 | |||||
| 270EGVVECSFVQSK281 | |||||||
| 282ETECTYFSTPLLLGK296 | |||||||
| Oxoglutarate dehydrogenase | + | − | Possible carbonylation, unspecified cysteine | 50 | |||
| Succinyl-CoA ligase subunit alpha | − | + | 154LIGPNC*PGVINPGECK169 | ||||
| Succinate dehydrogenase complex, subunit A | + | + | 76AAFGLSEAGFNTACLTK92 | 51 | |||
| 648TLNEADCATVPPAIR662 | Glutathionylation of cys89 | ||||||
| Dihydrolipoamide S-succinyltransferase | + | − | Intermolecular disulfide bonds | 18 | PDHC | ||
| Pyruvate dehydrogenase complex E2 | + | − | Possible carbonylation | 18,50 | |||
| Pyruvate dehydrogenase E1 component subunit beta | − | + | 259EGIECEVINLR269 | Possible carbonylation | 52 | ||
| Aspartate aminotransferase, mitochondrial | − | + | 95EYLPIGGLAEFCK107 | Sulfenation on unspecified cysteine | 20 | Fatty acid | |
| 186TCGFDFSGALEDISK200 | |||||||
| Acyl-CoA dehydrogenase, short chain | + | − | Carbonylation | 52 | |||
| Very-long-chain acyl-CoA dehydrogenase | + | − | Intermolecular disulfide bonds | 18,30 | |||
| Long-chain specific acyl-CoA dehydrogenase | − | + | 166CIGAIAMTEPGAGSDLQGVR185 | ||||
| 346AFVDSCLQLHETK358 | |||||||
| Alpha enolase | − | + | 344VNQIGSVTESLQACK358 | Glutathionylation | 53 | Glycolysis | |
| Fructose-bisphosphate aldolase C | − | + | 174YASICQQNGIVPIVEPEILPDGDHDLKR201 | Redox-sensitive Cys178 | 54 | ||
| Glyceraldehyde-3-phosphate dehydrogenase | − | + | 144IVSNASC*TTNCLAPLAK160 | Redox-sensitive cys150 and cys154 | 18,21,32, 55,56 | ||
| 233VPTPNVSVVDLTCR246 | HNE modified cys245 | ||||||
| Pyruvate kinase 3 | + | + | 44NTGIICTIGPASR56 | ||||
| Triosephosphate isomerase | − | + | 207IIYGGSVTGATCK219 | S-thiolation | 49 | ||
| Creatine kinase, mitochondria | + | + | 311LGYILTCPSNLGTGLR326 | Redox-sensitive cys317 | 18,21,32, 45 | ||
| Lactate dehydrogenase 2, B chain | + | − | Unspecified cysteine | 20 | |||
| Electron transfer flavoprotein subunit alpha | + | + | 147TIYAGNALCT VK158 | OXPHOS | |||
| Ubiquinol-cytochrome-c reductase complex core protein 1 | + | + | 397NALVSHLDGTTPVCEDIGR415 | ||||
| 256VYEEDAVPGLTPCR269 | Unspecified cysteine | 54 | |||||
| 448YFYDQCPAVAGYGPIEQLPDYNR470 | |||||||
| COX Vib-1 | − | + | 29NCWQNYLDFHR39 | ||||
| 48GGDVSVCEWYRR59 | |||||||
| Electron transferring flavoprotein dehydrogenase | + | + | 578LQINAQNCVHCK589 | Carbonylation | 52 | ||
| NADH dehydrogenase | + | Unspecified cysteine | 18,52,54 | ||||
| Sodium/potassium-transporting ATPase subunit beta-1 | − | + | 205YNPNVLPVQCTGK217 | Unspecified cysteine | 52 | Transport | |
| Serotransferrin precursor | − | + | 526CAPNNKEEYNGYTGAFR542 | ||||
| 655CFVKLPEGTTPEK667 | Unspecified cysteine | 57 | |||||
| SERCA 2 | − | + | 437VGEATETALTCLVEK451 | ||||
| 864VSFYQLSHFLQCK876 | Cys875 redox state regulates calcium pump switch | 42 | |||||
| VDAC1 | − | + | 134EHINLGCDVDFDIAGPSIR152 | ||||
| 238YQVDPDACFSAK249 | Redox-sensitive cys140,245 | 32 | |||||
| Actin, alpha cardiac muscle 1 | − | + | 218LCYVALDFENEMATAASSSSLEK240 | Unspecified cysteine | 19 | Structural | |
| Actinin alpha 2 | + | + | 853ELPPDQAQYCIK864 | ||||
| 339CQLEINFNTLQTK351 | Unspecified cysteine | 19 | |||||
| Myosin-binding protein C, cardiac-type | + | + | 416TLTISQCSLADDAAYQC*VVGGEK438 | Unspecified cysteine | 19 | ||
| Myosin light polypeptide 3 | − | + | 80ITYGQCGDVLR90 | ||||
| 181LMAGQEDSNGCINYEAFVK199 | Unspecified cysteine | 19,20 | |||||
| Tropomyosin-1 alpha chain | − | + | 190CAELEEELKTVTNNLK205 | Unspecified cysteine | 19 | ||
| 60 kDa heat shock protein | − | + | 431AAVEEGIVLGGGCALLR446 | Redox-sensitive Cys442 | 21,58 | Others | |
| Peroxiredoxin-5, mitochondrial precursor | − | + | 83GVLFGWGAFTPGCSK98 | Sulfenic acid or disulfide bond | 43 | ||
| Superoxide dismutase 1 | + | + | 5AVCVLKGDGPVQGTIHFEQK24 | ||||
| Rab GDP dissociation inhibitor beta | + | + | 194TDDYLDQPCC*ETINR208 | ||||
| Transcription factor NRF | + | − |
TCA cycle, Tricarboxylic acid cycle; PDHC, Pyruvate dehydrogenase complex; OXPHOS, Oxidative phosphorylation.
Identification of H2O2-Sensitive Heart Proteins by ICAT
We adopted a forward ICAT labeling strategy previously reported by Sethuraman et al.41 to identify redox-sensitive protein thiols. Modifications (as described in the Materials and Methods) were made to enhance both protein digestion and downstream MS/MS peptide fragmentation efficiency. The identification of the H2O2-sensitive cysteine-containing peptides must satisfy the followed criteria: (i) contain at least one ICAT modified cysteine; (ii) at least 20% decrease in ICAT MS ion intensity following H2O2 treatment; (iii) C. I. ≥ 95%, and (iv) each peptide assigned to only one protein without redundancy.
Over 2000 ICAT pairs were observed and quantified by MS; the histogram of all ICAT ratios showed a normal distribution with a median of 0.98 (data not shown), suggesting no significant bias was introduced during sample preparation and labeling. To assess the degree of random analytical variations and to obtain a cutoff value of the ICAT ratio that represents significantly oxidized peptides, a preliminary ICAT experiment of bovine serum albumin (BSA) standards in three predefined heavy-to-light ICAT ratios was performed. We found that 99% of the sample populations were located within two standard deviations (~7%) from the expected values (Supplemental Figure 1, Supporting Information). Therefore, at a stringent cutoff value of 20% ICAT ratio alteration, it was unlikely that the changes were due to random analytical variations. About 300 ICAT ion pairs with at least 20% changes were subjected to downstream MS/MS identification. In total, 71 ICAT-labeled peptides from 50 H2O2-sensitive proteins were identified, out of which 15 were identified with at least 2 peptides (Supplemental Table 2, Supporting Information). A large number of these proteins are metabolic enzymes, components of the mitochondrial electron transport chain, antioxidants and stress response proteins, as well as structural and signaling proteins. A MS/MS spectrum of a peptide (204TIIPLISQCTPK215) from MDH is presented with a rather complete series of y-ions for confident identification (Figure 2). The observation of a mass difference of 339 Da between y3 and y4 ions matched the mass of a heavy ICAT reagent modified cysteine. Quantification of thiol levels was carried out at MS level (see insert in Figure 2). The hallmark of a pair of the heavy and light ICAT-labeled peptides is a mass difference of 9.03 Da between the two adjacent monoisotopic peaks. The relative ICAT ratio for this peptide was 0.73, indicating ~27% loss of the redox-sensitive cysteine thiols upon H2O2 treatment. This is in contrast with ~74% loss of redox sensitive cysteine thiols found in DIGE analysis (Figure 1C–G), perhaps due to the fact that many other cysteines within MDH may also be oxidized21,32 (see later discussion). Many proteins found in our ICAT analysis have previously been identified as redox-sensitive, either with specified or unspecified redox-reactive cysteines (Table 1). Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) is an important transmembrane calcium pump. The SERCA2 pump machinery involves the formation of a redox-regulated disulfide bond that gates the influx of calcium flow as well as oscillation frequency. A site-directed mutagenesis study42 has identified this pair of crucial cysteines (cys875 and cys887) and the formation of the disulfide bond has been shown to be modulated by endoplasmic reticulum (ER) protein disulfide isomerase. We found that the redox-sensitive cys875 in peptide 864VSFYQLSHFLQCK876 (C: ICAT labeled) with a 50% decline of free thiol levels upon H2O2 treatment. The disulfide bond counterpart Cys887 resides in a tryptic peptide spanning 47 amino acid residues, which might be too large for efficient MS/MS fragmentation. Peroxiredoxins (PRX) are important anti-oxidant proteins that serve to detoxify peroxides. Unlike other 2-cys motif-containing PRXs, PRX5 contains only one conserved cys96 at its redox-sensitive catalytic center,43 which may form an intramolecular disulfide bond with cys200. We identified the redox-sensitive cys96 in the ICAT-labeled peptide 83GVLFGVP-GAFTPGCSK98 with a 40% decline of free thiol levels upon H2O2 treatment. To the best of our knowledge, this study represents the first unambiguous identification of this redox-sensitive cysteine in PRX5 by a proteomic method. Many proteins identified here, contain reactive cysteines concurring with published reports (Table 1). They included aconitase 2 (cys385), malate dehydrogenase (cys137), fructose-bisphosphate aldolase C (cys178), creatine kinase (cys317), and HSP 60 (cys442). Redox modifications of these crucial cysteines lead to the modifications of protein activities. Some redox-sensitive cysteine thiols are possibly reported here for the first time, including cyto-chrome C oxidase VIb-1, sodium/potassium transporting ATPase β-1, tropomyosin-1, and actinin α-2.
Figure 2.

Example of ICAT identification of H2O2-sensitive heart proteins. A MS/MS spectrum for a peptide (204TIIPLISQCTPK215) from MDH isolated from mouse heart is presented with a continuous series of y-ions for confident identification. The observation of a mass difference of 339 Da between y3 and y4 ions matched the mass of a heavy ICAT reagent-modified cysteine. The decrease of cysteine thiol was observed in the MS spectrum (see insert). The hallmark of a pair of the heavy and light ICAT-labeled peptides is a mass difference of 9.03 Da between the two adjacent monoisotopic peaks in the MS spectrum. The relative ICAT ratio for this peptide was 0.73, indicating ~27% loss of the redox-sensitive cysteine thiols upon H2O2 treatment.
Comparing our ICAT study results with the observations made by Sethuraman et al.,32 both studies utilized a similar “forward” labeling strategy and have demonstrated the effectiveness of the ICAT approach for redox studies. Sethuraman et al. focused on membrane proteins. Interestingly, several identical peptides were seen in both studies, including peptides from creatine kinase, malate dehydrogenase, GAPDH, VDAC1, and SERCA2, suggesting that the ICAT approach is robust for the detection of redox-sensitive proteins. Furthermore, the degree of oxidation of these cysteines is comparable despite the fact that we used only one-fourth of the H2O2 concentration used in Sethuraman’s work. For example, creatine kinase was identified with 2 peptides, 173GLSLPPACSR182 (9.8% oxidized) and 311LGYILTCPSNLGTGLR326 (42.3% oxidized) in their study and 8.0% and 37.0% in our report, respectively. Both studies demonstrated that cys317 was more sensitive to H2O2 oxidation than cys180. It is notable that the 173GLSLPPACSR182 peptide is not included in Supplemental Table 2 (Supporting Information) in this study, due to the requirement of at least a 20% decrease in ICAT signals.
In addition to identifying diminished free thiol levels upon H2O2 treatment, we also attempted to identify other reversibly oxidized cysteines by including a DTT reduction step after ICAT labeling, followed by alkylation with iodoacetamide. Although in the present ICAT workflow, the opportunities to locate such cysteines were limited by the requirement of the coexistence of at least one ICAT-labeled cysteine in the same tryptic peptide; otherwise those carboamidomethyl modified cysteines would have been washed off during the avidin affinity enrichment step. We did identify 6 proteins containing carboamidomethyl modifications, including GAPDH, in which cys150 has previously been shown to be essential for its function and the reversible oxidation of cys150 could down-regulate GAPDH activity.44 In our study, we observed a carboamidomethyl modified cys150 together with an adjacent ICAT-labeled cys154 in peptide IVSNASC150TTNC154LAPLAK. It is possible that GAPDH underwent reversible cysteine modification (e.g., sulfenation) to conserve the crucial cys150 under H2O2 challenge. Despite the exact identity of the reversible modification could not be revealed by our current method, a modified method with the inclusion of different reducing reagents, such as arsenite (for sulfenic acid)20 or ascorbic acid (for nitrosylation)21 could be used to identify such modifications. In summary, we have shown that ICAT is a versatile and robust method to identify redox-sensitive proteins with precise specification of sites of oxidation.
Comparison of Forward and Backward Labeling Strategies
Recently, Hurd and co-workers30 reported a “backward” labeling strategy to identify redox-sensitive proteins with the DIGE method. With this strategy, free thiols were initially blocked by alkylating reagents, and those reversibly oxidized thiols were then reduced by DTT and labeled with different CyDyes. In contrast to this “backward” labeling strategy, we used a “forward” DIGE labeling strategy in this study, where oxidized thiols were maintained in their native states, and free thiols were labeled with different saturation CyDyes for the quantification of redox-sensitive protein thiols. Both strategies can identify redox-sensitive proteins, but with their unique pros and cons. For example, the forward strategy could be affected by spontaneous cysteine oxidation during sample extraction and analysis. Such oxidation events could be minimized with low temperature, short processing time, and inert gas protection. The backward strategy could be impeded by tedious reagent cleanup steps prior to the DIGE labeling, which is not desirable for routine quantitative study.
Interestingly, despite using different DIGE labeling approaches to unravel redox-sensitive proteins in mouse heart following H2O2 treatments, a comparable number of redox-sensitive proteins were identified in both our and Hurd et al.’s work. Several proteins were identified by both strategies; for example, oxoglutarate dehydrogenase, succinate dehydrogenase, isocitrate dehydrogenase, electron transfer flavorprotein, acyl-CoA dehydrogenase, and superoxide dismutase (see Supplemental Table 1, Supporting Information). On the other hand, each method also identified a unique set of proteins. This observation may in part be due to the different isoelectric focusing pI ranges used (pI 5–8 in our study and pI 3–10 in Hurd’s study). Curiously, several proteins from the mitochondrial electron transport chain as well as cardiac actin and aconitase 2 were not seen as sensitive to H2O2 treatment by Hurd et al. However, we observed significant reduction of thiol levels in these proteins (either by DIGE or ICAT analysis), which are in agreement with published reports of having redox-sensitive cysteines within these proteins.20,32 The reason that these proteins were not detected by the “backward” DIGE labeling strategy is unclear. It is notable that several basic and/or hydrophobic proteins, for example, creatine kinase and voltage-dependent anion channel 1 (VDAC1), were observed by Hurd et al., but not by our forward DIGE strategy. We did identify these proteins as H2O2-sensitive proteins, however, with the “forward” labeling strategy employed for the ICAT analysis.
One possible advantage of the backward labeling strategy is the likelihood of revealing redox-sensitive cysteines buried inside the proteins that may not be accessible by the bulky DIGE or ICAT reagents using the forward strategy. To verify this possibility, we conducted a comparative ICAT study between the forward and backward labeling strategies. It was apparent that using the backward ICAT labeling strategy, we found 35% more peptides sensitive to H2O2 oxidation than using the forward labeling strategy (Supplemental Tables 2 and 3, Supporting Information). This result may be attributed to the reduction of the disulfide bonds, after which cysteines became more accessible for subsequent ICAT labeling reagents. Twenty-seven peptides (in 22 unique proteins) were found to be H2O2-sensitive by both strategies. The common proteins found with both approaches included peptides within aspartate aminotransferase, creatine kinase, PRX5, and triosephosphate isomerase etc. (Supplemental Tables 2 and 3, Supporting Information). Similar to the comparison between forward and backward DIGE strategies, each ICAT labeling strategy also enabled the discovery of a distinct list of nonoverlapping peptides, implying the complementary nature of these two approaches.
Comparison of DIGE and ICAT Methods for Thiol Oxidation Studies
In this study, we have compared two thiol-specific quantitative proteomic techniques for redox-sensitive protein identification. DIGE is a gel-base method and quantifies overall protein thiol levels whereas ICAT utilizes multidimensional chromatographic methods to selectively enrich and quantify isotope-encoded cysteine-containing peptides. These two methods appear to be complementary for the purpose of identifying redox-sensitive proteins (Figure 3). There were 13 proteins commonly found by both methods (Table 1). They included actinin 2, creatine kinase, aconitase 2, malate dehydrogenase, ubiquinol-cytochrome-c reductase complex core protein 1, and superoxide dismutase. Many of these proteins are known to be susceptible to oxidative stress in cells.30,45–47 There were 37 redox-sensitive proteins identified solely by ICAT method. We found that some of these proteins are hydrophobic membrane proteins, which may be poorly resolved in 2DE. Examples of these proteins included SERCA2 and membrane glycoprotein gp42. Another scenario for proteins detected only with ICAT may attribute to the fact that protein pI values exceed the isoelectric focusing range of 2DE (pI 5–8 used in this study). PRX5 (pI 9.1), succinyl-CoA ligase (pI 9.46), cytochrome c oxidase VIb-1 (pI 8.96), CRP2 (pI 8.96), CRP3 (pI 8.90), and tropomyosin-1 alpha chain (pI 4.69) all fell into this category. Additional possibilities for ICAT-identified proteins that were missed in DIGE analysis might be due to the comigration of low and high abundance proteins to the same gel spots. It is conceivable that redox-sensitive proteins may not be identified in the background of abundant proteins. On the other hand, DIGE method also captured 13 proteins that were absent from ICAT analysis (Figure 3). They included oxoglutarate dehydrogenase, dihydrolipoamide S-acetyltransferase, epoxide hydrolase, NF-κB-repressing factor, etc. (Supplemental Table 1, Supporting Information). We speculate that the lack of detection of these proteins with ICAT could result from incomplete enzymatic digestion, weak retentions on the RPLC trapping column, or poor ionization and/or fragmentation in MS. Furthermore, we compared the quantitative aspects of these two methods with regard to the redox-sensitive proteins found within the TCA cycle (Figure 4). Both methods found that aconitase, isocitrate dehydrogenase, succinate dehydrogenase, and malate dehydrogenase were significantly oxidized upon H2O2 challenge. The ICAT method detected thiol reductions of 20–30% whereas the DIGE method observed more dramatic changes ranging from 70 to 90% of the thiol signals for the same proteins. On the other hand, succinyl-CoA ligase was observed solely in ICAT method with a 35% decrease, whereas oxoglutarate dehydrogenase oxidation appeared only in DIGE analysis with an over 95% decrease of free thiol signal. Overall, the trend of protein/peptide thiol signal decreases followed the same direction but to different extents. Almost in all cases, DIGE analysis reported more significant reduction of free thiol levels whereas ICAT only detected a moderate change. This is probably due to the fact that DIGE signals represented the quantification of a specific protein isoform that may differ from other isoforms of the same protein by having multiple oxidatively modified cysteines. In contrast, ICAT analysis detected the averaged changes of a specific cysteine residue among all protein isoforms.
Figure 3.
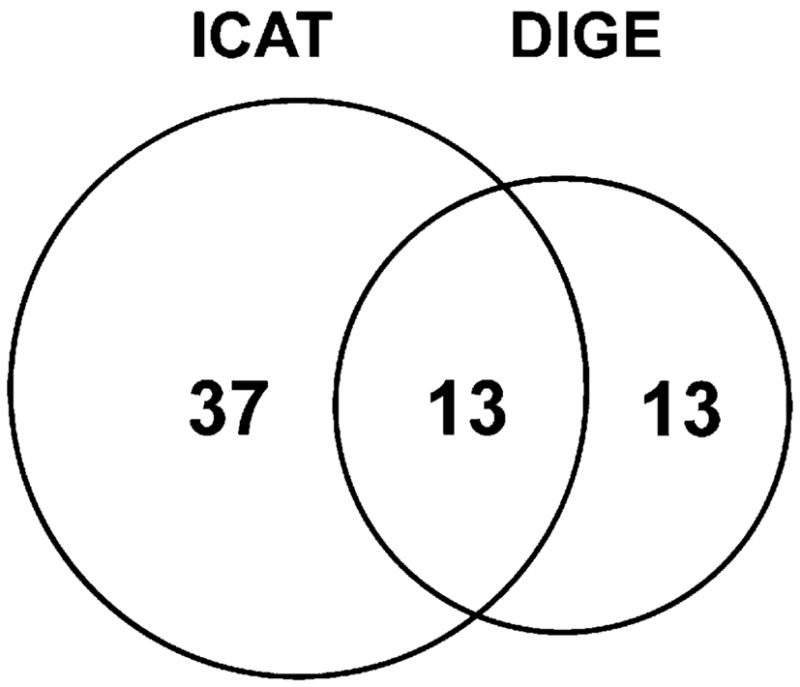
Venn diagram of H2O2-sensitive proteins discovered by DIGE and ICAT methods. We identified 50 proteins as potential targets of H2O2 oxidation by the ICAT method and 26 with the DIGE method. Of these, 13 proteins were identified with both methods. Overall, the ICAT technique enabled us to identify 24 more redox-sensitive proteins than the DIGE method.
Figure 4.

H2O2-sensitive TCA cycle proteins identified in this study. Both ICAT and DIGE methods revealed that many proteins in the TCA cycle were prone to oxidation by H2O2. The quantification of protein thiol level change with the ICAT method is the average of all its ICAT peptides. We detected a reduction of 20–30% of free cysteine thiols in aconitase 2, isocitrate dehydrogenase, succinate dehydrogenase, and malate dehydrogenase with the ICAT method, whereas a more dramatic reduction ranging from 70 to 90% for the same proteins was seen with the DIGE method. Succinyl-CoA ligase was observed solely by the ICAT method with a 35% free thiol level decrease, whereas oxoglutarate dehydrogenase oxidation was observed only with the DIGE method, with an over 95% decrease of free thiols. Student t test was carried out with four independent studies, *, p < 0.05; ***, p < 0.005.
To confirm the hypothesis that the quantitative discrepancies between DIGE and ICAT originated from the formation of a series of acidic protein isoforms following H2O2 oxidation, we have performed additional ICAT and DIGE analyses of a purified malate dehydrogenase (MDH) following identical H2O2 oxidation treatment used for heart proteins. From DIGE analysis, we found clear evidence of the appearance of additional acidic MDH isoforms (Figure 5, red gel spots 1–3) in H2O2-treated MDH. The 7 discrete 2DE spots appeared to represent MDH isoforms with different extent of oxidatively acidified cysteines. The most acidic protein spots were likely to contain many but not all of the 8 MDH cysteines as sulfinic, sulfenic, or sulfonic acids, with the remaining cysteine thiols labeled with CyDyes. The DIGE ratios shown in Figure 5 were indicative of the relative amounts of the remaining cysteine thiol levels, likely barometers of the relative proportion of each MDH isoform in the samples under comparison, assuming overall MDH levels remained the same following H2O2 oxidation. The conclusions that can be drawn from DIGE analysis of MDH included that higher proportion of acidically oxidized MDH isoforms were formed following H2O2 oxidation. It should be mentioned that completely oxidized protein isoform would not be labeled by CyDyes but may be revealed with Sypro Ruby staining of the 2D gels. In ICAT experiments, we successfully identified 5 cysteine-containing MDH peptides, with the remaining peptide too large to be efficiently fragmented by MS/MS in our MS instrument. The quantification of these ICAT-labeled MDH peptides demonstrated varied susceptibility to H2O2 oxidation among the 5 cysteines (Table 2 and Supplemental Figure 2, Supporting Information). As a simplistic interpretation, Cys 69 was the most vulnerable to H2O2 oxidation, with 77% of its free thiol oxidized by H2O2. Cys 65 was also relatively sensitive to H2O2 oxidation, with over 66% of free thiol oxidized. Cys 188, Cys 251, and Cys 261 were only moderately sensitive to H2O2 oxidation (Table 2). Both DIGE and ICAT redox quantification results should be interpreted with the understanding that related yet different biochemical information are obtained with each technique. The quantification results from DIGE method is gel spot-based (or protein isoform-specific) instead of simply protein-specific. From DIGE analysis, we found the increase of MDH oxidative isoforms. It was an effective method for discovering a small fraction of MDH isoforms that were particularly sensitive to redox regulation. On the other hand, with the ICAT method, the quantification is carried out at the peptide level, which is the average of all common peptides derived form various isoforms of the same protein. For the five peptides detected from MDH, the reported ICAT peptide ratios represented the relative amounts of total free thiols of the given peptides derived from the 7 isoforms observed in the DIGE gel. Therefore, potentially pronounced changes of a peptide redox state in one isoform may be “averaged out” by a large proportion of the same peptides present in the less oxidized isoforms, resulting in the overall marginal change reported by the ICAT ratios. Consequently, only the redox trend can be compared between the two methods, indicating overall protein susceptibility to oxidative modifications. Because gel spots representing dramatically oxidized protein isoforms are more easily detected and quantified by DIGE software, it is therefore likely that the degree of oxidation reported from DIGE is more pronounced than ICAT, as reported in this study.
Figure 5.

DIGE analysis of MDH oxidized by H2O2. Cy3m (green)-labeled control sample and Cy5m (red)-labeled H2O2- treated porcine MDH. A series of MDH isoforms were observed with both untreated and H2O2-treated MDH. It was clear that the H2O2-treated MDH contained additional isoforms distributed toward the acidic end of the 2DE gel (red spots 1–3), suggesting the acidification of MDH, likely in the forms of cysteine oxidation to sulfinic, sulfenic, or sulfonic acids. The untreated MDH maintained isoforms clustered around the more basic pI range, as illustrated by the green gel spots 6–7. The relative MDHreduction/MDHoxidation ratios for all MDH isoforms are marked below the corresponding gel spots, with isoform #1 being the most oxidized and isoform #7 being the most reduced. Ratio*: Cy3m/Cy5m ratio represents relative free cysteine thiol levels in control/H2O2-treated MDH isoforms.
Table 2.
Purified Malate Dehydrogenase Peptide Identification and Thiol-Oxidative Quantification by ICAT
| peptide sequencea | C.I. % | observed mass | error (ppm) | ICAT ratio (H/L)b |
|---|---|---|---|---|
| 55GYLGPEQLPDCLK67 | 100 | 1659.8 | −11 | 0.34 |
| 68GCDVVVIPAGVPR80 | 100 | 1508.8 | −13 | 0.23 |
| 180TIIPLISQCTPK191 | 100 | 1540.9 | −16 | 0.49 |
| 246EGVVECSFVK255 | 100 | 1323.5 | 5 | 0.45 |
| 256SQETDCPYFSTPLLLGK272 | 100 | 2126.0 | −4 | 0.47 |
ICAT labeled cysteines are underlined.
H/L: H2O2 oxidized/untreated MDH peptides.
Adaptation of DIGE and ICAT Methods for Redox Proteomics Studies of Biological Systems
We chose denaturing buffer for this study to recover sufficient low-abundant proteins from the tissues, which is important for biological studies. Because in our model system the proteins were oxidized by H2O2 after denaturation, many cysteine thiols might not possess their native protein structures in denaturing lysis buffer; therefore protein sensitivities to oxidant H2O2 reported in this study may not be identical to in vivo conditions. However, because no reduction and alkylation steps were included in the lysis buffer, select cysteine thiols might still contain some structural environments that enabled their interactions with other vicinal amino acids, resulting in varied thiol sensitivity to H2O2 oxidation observed in this study (see example in Figure 5). We can not claim unequivocally whether the isolated heart proteins oxidized by 500 μM of H2O2 are also sensitive to intracellular oxidants under pathophysiological conditions. However, from two subsequent independent studies of heart proteins isolated from animal models with deficiencies of redox modulators, we found many of the same proteins were differentially oxidized (manuscripts in preparation), confirming the effectiveness of both DIGE and ICAT methods for the discovery of distinct groups of redox-sensitive proteins.
Taken together, both methods have demonstrated the effectiveness for the identification of both previously known and unknown redox-sensitive proteins. Meanwhile, each method was useful at discovering a unique set of novel redox-sensitive proteins, and may pave the way for further mechanistic studies. DIGE could be useful for the identification of redox-induced protein isoform formations and ICAT method is more versatile for identifying redox-sensitive peptides with exact localization of the cysteine oxidized. Overall, these two methods are complementary for redox proteomics studies. The selection of the best method is task-dependent (Table 3). If the goal is to quantify the global redox proteomic changes, DIGE is the method of choice. However, if the focus is to identify specific cysteines within key proteins for downstream structural and functional study, ICAT may be more advantageous. If the goal is to identify as many redox-sensitive proteins as possible, both methods may be needed.
Table 3.
Relative Merits of DIGE and ICAT Techniques for Protein Thiol Quantification
| advantages | disadvantages | |
|---|---|---|
| DIGE |
|
|
| ICAT |
|
|
Summary
A systematic comparison between DIGE and ICAT methods was carried out for detecting redox-sensitive proteins following the H2O2 treatment of heart proteins. Each method complemented the other and can be used together to obtain a comprehensive understanding of the changes in heart redox proteome. ICAT is an attractive tool for the precise localization of redox-sensitive cysteine in low-abundant proteins, whereas DIGE is advantageous for easier implementation of multiplexed experiments to discover redox proteomics patterns. Collectively, we have identified 63 unique redox-sensitive proteins, including some previously reported and several novel H2O2 oxidation targets, that may shed light into better understandings of the redox-mediated regulation of cellular processes.
Supplementary Material
Supplemental Tables 1–3 and Supplemental Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research is supported in part by a NIH grant NS046593 to H.L. and by a grant from the foundation of UMDNJ to H.L. and J.S.
References
- 1.Ghezzi P, Bonetto V. Redox proteomics: Identification of oxidatively, modified proteins. Proteomics. 2003;3(7):1145–53. doi: 10.1002/pmic.200300435. [DOI] [PubMed] [Google Scholar]
- 2.Di Simplicio P, Franconi F, Frosali S, Di Giuseppe D. Thiolation and nitrosation of cysteines in biological fluids and cells. Amino Acids. 2003;25(3–4):323–39. doi: 10.1007/s00726-003-0020-1. [DOI] [PubMed] [Google Scholar]
- 3.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279(5357):1718–21. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 4.Abate C, Patel L, Rauscher FJ, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249(4973):1157–61. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 5.Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci USA. 1991;88(10):4328–32. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xanthoudakis S, Miao GG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci USA. 1994;91(1):23–7. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15(7):3892–903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu J, Milligan J, Huang LE. Molecular mechanism of hypoxia-inducible factor 1alpha -p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem. 2001;276(5):3550–4. doi: 10.1074/jbc.M009522200. [DOI] [PubMed] [Google Scholar]
- 9.Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277(45):43505–11. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 10.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 11.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121(5):667–70. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Park HS, Yu JW, Cho JH, Kim MS, Huh SH, Ryoo K, Choi EJ. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem. 2004;279(9):7584–90. doi: 10.1074/jbc.M304183200. [DOI] [PubMed] [Google Scholar]
- 13.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci USA. 2005;102(25):8875–80. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujiwara N, Nakano M, Kato S, Yoshihara D, Ookawara T, Eguchi H, Taniguchi N, Suzuki K. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem. 2007;282(49):35933–44. doi: 10.1074/jbc.M702941200. [DOI] [PubMed] [Google Scholar]
- 15.Sumbayev VV. S-nitrosylation of thioredoxin mediates activation of apoptosis signal-regulating kinase 1. Arch Biochem Biophys. 2003;415(1):133–6. doi: 10.1016/s0003-9861(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 16.Graf PC, Jakob U. Redox-regulated molecular chaperones. Cell Mol Life Sci. 2002;59(10):1624–31. doi: 10.1007/PL00012489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilbert M, Horst J, Ahrens S, Winter J, Graf PC, Lilie H, Jakob U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat Struct Mol Biol. 2007;14(6):556–63. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennan JP, Wait R, Begum S, Bell JR, Dunn MJ, Eaton P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J Biol Chem. 2004;279(40):41352–41360. doi: 10.1074/jbc.M403827200. [DOI] [PubMed] [Google Scholar]
- 19.Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6(9):1473–84. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci USA. 2004;101(52):17982–87. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103(4):1012–17. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA. 2002;99(6):3505–10. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michelet L, Zaffagnini M, Marchand C, Collin V, Decottignies P, Tsan P, Lancelin JM, Trost P, Miginiac-Maslow M, Noctor G, Lemaire SD. Glutathionylation of chloroplast thioredoxin f is a redox signaling mechanism in plants. Proc Natl Acad Sci USA. 2005;102(45):16478–83. doi: 10.1073/pnas.0507498102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yano H, Wong JH, Lee YM, Cho MJ, Buchanan BB. A strategy for the identification of proteins targeted by thioredoxin. Proc Natl Acad Sci USA. 2001;98(8):4794–99. doi: 10.1073/pnas.071041998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Essex DW, Li MR, Miller A, Feinman RD. Protein disulfide isomerase and sulfhydryl-dependent pathways in platelet activation. Biochemistry. 2001;40(20):6070–75. doi: 10.1021/bi002454e. [DOI] [PubMed] [Google Scholar]
- 26.Baty JW, Hampton MB, Winterbourn CC. Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics. 2002;2(9):1261–66. doi: 10.1002/1615-9861(200209)2:9<1261::AID-PROT1261>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal Biochem. 2000;283(2):214–21. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 28.Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J Biol Chem. 2003;278(48):47361–4. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- 29.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2(11):e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of ros-sensitive thiol proteins by redox-difference gel electrophoresis (redox-dige): Implications for mitochondrial redox signalling. J Biol Chem. 2007:22040–51. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 31.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17(10):994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 32.Sethuraman M, McComb ME, Huang H, Huang SQ, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag (ICAT) approach to redox proteomics: Identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J Proteome Res. 2004;3(6):1228–33. doi: 10.1021/pr049887e. [DOI] [PubMed] [Google Scholar]
- 33.Christoffers KH, Li H, Howells RD. Purification and mass spectrometric analysis of the delta opioid receptor. Mol Brain Res. 2005;136(1–2):54–64. doi: 10.1016/j.molbrainres.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Liu T, Donahue KC, Hu J, Kurnellas MP, Grant JE, Li H, Elkabes S. Identification of differentially expressed proteins in experimental autoimmune encephalomyelitis (EAE) by proteomic analysis of the spinal cord. J Proteome Res. 2007;6(7):2565–75. doi: 10.1021/pr070012k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2(1):43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 36.Lee CK, Park HJ, So HH, Kim HJ, Lee KS, Choi WS, Lee HM, Won KJ, Yoon TJ, Park TK, Kim B. Proteomic profiling and identification of cofilin responding to oxidative stress in vascular smooth muscle. Proteomics. 2006;6(24):6455–75. doi: 10.1002/pmic.200600124. [DOI] [PubMed] [Google Scholar]
- 37.Obata T. Nitric oxide and depolarization induce hydroxyl radical generation. Jpn J Pharmacol. 2002;88(1):1–5. doi: 10.1254/jjp.88.1. [DOI] [PubMed] [Google Scholar]
- 38.Yang K-S, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. Inactivation of Human Peroxiredoxin I during Catalysis as the Result of the Oxidation of the Catalytic Site Cysteine to Cysteine-sulfinic Acid. J Biol Chem. 2002;277(41):38029–36. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 39.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA. 2004;101(24):9103–8. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic Biol Med. 2000;28(9):1317–27. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- 41.Sethuraman M, McComb ME, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol Cell Proteomics. 2004;3(3):273–8. doi: 10.1074/mcp.T300011-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Camacho P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol. 2004;164(1):35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Declercq JP, Evrard C, Clippe A, Stricht DV, Bernard A, Knoops B. Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 A resolution. J Mol Biol. 2001;311(4):751–9. doi: 10.1006/jmbi.2001.4853. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima H, Amano W, Fujita A, Fukuhara A, Azuma Y-T, Hata F, Inui T, Takeuchi T. The Active Site Cysteine of the Proapoptotic Protein Glyceraldehyde-3-phosphate Dehydrogenase Is Essential in Oxidative Stress-induced Aggregation and Cell Death. J Biol Chem. 2007;282(36):26562–74. doi: 10.1074/jbc.M704199200. [DOI] [PubMed] [Google Scholar]
- 45.Wolosker H, Panizzutti R, Engelender S. Inhibition of creatine kinase by S-nitrosoglutathione. FEBS Lett. 1996;392(3):274–6. doi: 10.1016/0014-5793(96)00829-0. [DOI] [PubMed] [Google Scholar]
- 46.Sharma AB, Sun J, Howard LL, Williams AG, Jr, Mallet RT. Oxidative stress reversibly inactivates myocardial enzymes during cardiac arrest. Am J Physiol Heart Circ Physiol. 2007;292(1):H198–206. doi: 10.1152/ajpheart.00698.2006. [DOI] [PubMed] [Google Scholar]
- 47.Inarrea P, Moini H, Rettori D, Han D, Martinez J, Garcia I, Fernandez-Vizarra E, Iturralde M, Cadenas E. Redox activation of mitochondrial intermembrane space Cu, Zn-superoxide dismutase. Biochem J. 2005;387(Pt 1):203–9. doi: 10.1042/BJ20041683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44(36):11986–96. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 49.Eaton P, Byers HL, Leeds N, Ward MA, Shattock MJ. Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J Biol Chem. 2002;277(12):9806–11. doi: 10.1074/jbc.M111454200. [DOI] [PubMed] [Google Scholar]
- 50.Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem. 2000;275(35):27393–8. doi: 10.1074/jbc.M003140200. [DOI] [PubMed] [Google Scholar]
- 51.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282(45):32640–54. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 52.Meany DL, Xie H, Thompson LV, Arriaga EA, Griffin TJ. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics. 2007;7(7):1150–63. doi: 10.1002/pmic.200600450. [DOI] [PubMed] [Google Scholar]
- 53.Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J Neurosci Res. 2007;85(7):1506–14. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- 54.Schilling B, Yoo CB, Collins CJ, Gibson BW. Determining cysteine oxidation status using differential alkylation. Int J Mass Spectrom. 2004;236(1–3):117–27. [Google Scholar]
- 55.Mohr S, Stamler JS, Brune B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271(8):4209–14. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 56.Ishii T, Tatsuda E, Kumazawa S, Nakayama T, Uchida K. Molecular basis of enzyme inactivation by an endogenous electrophile 4-hydroxy-2-nonenal: identification of modification sites in glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 2003;42(12):3474–80. doi: 10.1021/bi027172o. [DOI] [PubMed] [Google Scholar]
- 57.Kim BJ, Hood BL, Aragon RA, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6(4):1250–60. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagumo Y, Kakeya H, Shoji M, Hayashi Y, Dohmae N, Osada H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem J. 2005;387(Pt 3):835–40. doi: 10.1042/BJ20041355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables 1–3 and Supplemental Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.