

Abstract
The phosphate, uracil, and ribose moieties of uracil nucleotides were varied structurally for evaluation of agonist activity at the human P2Y2, P2Y4, and P2Y6 receptors. The 2-thio modification, found previously to enhance P2Y2 receptor potency, could be combined with other favorable modifications to produce novel molecules that exhibit high potencies and receptor selectivities. Phosphonomethylene bridges introduced for stability in analogues of UDP, UTP and uracil dinucleotides markedly reduced potency. Truncation of dinucleotide agonists of the P2Y2 receptor, in the form of Up4-sugars, indicated that a terminal uracil ring is not essential for moderate potency at this receptor and that specific SAR patterns are observed at this distal end of the molecule. Key compounds reported in this study include: 9, α,β-methylene-UDP, a P2Y6 receptor agonist; 30, Up4-phenyl ester and 34, Up4-[1]glucose, selective P2Y2 receptor agonists; 43, the 2-thio analogue of INS37217 (P1-(uridine 5′)-P4- (2′-deoxycytidine 5′) tetraphosphate), a potent and selective P2Y2 receptor agonist.
Introduction
The P2Y receptor family consists of at least eight human subtypes that are activated by either or both adenine and uracil nucleotides.1,2 P2Y2 and P2Y4 nucleotide receptors respond to uridine 5′-triphosphate (UTP, 1) and its analogues, and the P2Y6 receptor responds to uridine 5′-diphosphate (UDP, 2) and analogues.3 However, this delineation of agonist selectivities is not absolute. For example, Müller and coworkers and Besada et al. reported that certain 5′-triphosphate derivatives potently activate the P2Y6 receptor.4,5 The conformational preference of the ribose moiety in binding to uridine nucleotide-activated P2Y receptors has been explored through substitution with the sterically constrained methanocarba (bicyclo[3.1.0]hexane) ring system. P2Y2 and P2Y4 receptors display a North conformational preference, while the P2Y6 receptor prefers the South.6–8
Several recent studies have explored structure activity relationships (SARs) at the P2Y2, P2Y4, and P2Y6 receptors.4–6,9,10,23 Key pharmacological probes introduced include the nonselective P2Y2 receptor agonists 3 and 4, which have progressed to clinical studies for dry eye syndrome and pulmonary diseases, the potent and selective P2Y2 receptor agonist 5,7 and the selective P2Y6 receptor agonist 6.11,12 Modification of the base moiety of UTP to form C-linked nucleotides is possible in P2Y2 receptor agonists and results in enhanced stability.10 Various dinucleotides tend to activate P2Y2 and P2Y4 receptors (diuridine tetraphosphates) or P2Y6 receptors (diuridine triphosphates) with moderate potency and with greater stability than analogues of UTP and UDP.9 Also, the pharmacological activity of diadenosine polyphosphates at both P2Y and P2X receptor subtypes has been characterized.1,2 Diadenosine tetraphosphate is only 3-fold less potent than ATP at the human P2Y2 receptor.
In the present study, we further investigated structure activity relationships at P2Y receptors through synthesis of molecules with additional substitutions of the uracil, ribose, and phosphates moieties and combinations thereof. These analogues of UDP, UTP and dinucleotides were assayed for capacity to promote P2Y2, P2Y4, and P2Y6 receptor-mediated activation of phospholipase C (PLC).3 These novel derivatives incorporated groups such as 2-thio, found previously to enhance receptor potency.4,7 Phosphonomethylene bridges and nonphosphate linkages were introduced to enhance stability of molecules against the action of ectonucleotidases.13 We also probed the effects of truncation of dinucleotide agonists of the P2Y2 receptor by synthesizing and quantifying the activities of a series of Up4-sugars.7
Results and Discussion
Chemical Synthesis
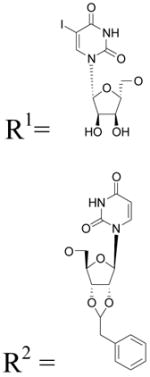
The synthetic routes to the novel nucleotide derivatives (Tables 1 – 3) are shown in Schemes 1 – 6. The potencies of five known reference compounds are listed in Table 1 (P2Y6 agonist UDP 2), Table 2 (P2Y2 agonists UTP 1 and MRS2698 5), and Table 3 (P2Y2 agonists Up4U 3 and INS37217 4). Types of modifications include: UDP analogues containing a methylene-bridged substitute for the diphosphate group (Scheme 1); UTP analogues containing a β,γ-dihalomethylene-bridge in the triphosphate group4 (Scheme 2); UTP analogues with modified uracil and ribose moieties7 (Scheme 3); a 5-iodo analogue of INS488235,12 (Scheme 4); analogues of uridine 5′-tetraphosphate7 (Scheme 5) in which the terminal phosphate moiety was condensed with various alcohols, including sugars. Among the derivatives in Scheme 1, compounds 7 and 10 are new compounds, but 8 and 9 were previously reported.32,33
Table 1.
Relative potencies of UDP, 2, and UDP analogues for activation of the human P2Y6 receptor. Unless noted: R1 = OH;
 and R3 = H.
and R3 = H.
 | |||
|---|---|---|---|
| Compound | Modification | Structure | EC50, µM |
| hP2Y6 receptora | |||
| NUCLEOSIDES | |||
| 7 | Uridine-5′-malonate |
|
NE |
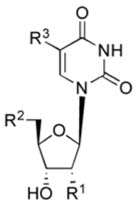
| 8 | Uridine-5′-phosphonoacetate |
|
NE |
| DIPHOSPHATES | |||
| 2 | UDP | 0.30±0.06 | |
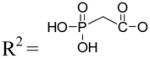
| 9b | Up-CH2-p (α,β-methylene UDP) |
|
0.66±0.11 |
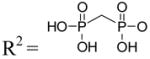
| 10 | Up-CF2-p (α,β-difluoromethylene UDP) |
|
NE |
| 11 | 5-amino-UDP | R3 = NH2 | 0.61±0.17 |
| 12 | 2′-deoxy-2′-ureido-UDP | R1 = NHCONH2 | 4.70±0.44 |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y6 receptor. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
9, MRS2782.
NE - no effect at 10 µM.
Table 3.
Relative potencies of dinucleotide derivatives for activation of the human P2Y2, P2Y4 and P2Y6 receptors. Unless noted: R = Uridine; and X = O
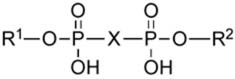 | |||||
|---|---|---|---|---|---|
| Compound | Modification | Structure | EC50, μMa | ||
| hP2Y2 | hP2Y4 | hP2Y6 | |||
| DINUCLEOSIDE TRIPHOSPHATES (n = 1) | |||||
| 38b | Up3U | 1.31±0.21 | 0.87±0.11 | 0.27±0.07 | |
| 39c | 5-I-Up3-(2′,3′-phenylethyl acetal)U |
|
9.97±0.95 | NE | 5.49±0.68 |
| DINUCLEOSIDE TETRAPHOSPHATES (n = 2) | |||||
| 3 | (= Up4U) | 0.21±0.03 | 0.13±0.01 | 1.16±0.42 | |
| 40 | 4-S-Up4(4-S-U) | R1 = R2 = 4-thio-uridine | 0.030±0.010 | 0.08±0.01 | 2.03±0.18 |
| 41 | Up2-CF2-p2-U (β,γ-difluoromethylene Up4U) | X = CF2 | 2.27±1.39 | >10d | NE |
| 42 | Up2-CCl2-p2-U (β,γ-dichloromethylene Up4U) | X = CCl2 | 7.77±1.39 | NE | NE |
| 4 | Up4-2′-dC (INS37217) | R2 = 2′-deoxy-cytidine | 0.14±0.04 | 0.14±0.04 | 0.95±0.06 |
| 43 | 2-thio-Up4-2′-dC | R1 = 2-thio uridine R2 = 2′-deoxy-cytidine | 0.08±0.03 | 0.71±0.15 | 1.05±0.07 |
| 44 | Up4-2′-dG | R2 = 2′-deoxy-guanosine | 0.14±0.04 | 0.54±0.15 | 1.03±0.08 |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y2, P2Y4, or P2Y6 receptor. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
Reported in reference 9.
39, MRS2752; 43, MRS2657.
≤50% effect at 10 μM. Value of >10 μM was determined by extrapolation.
NE - no effect at 10 μM.
Scheme 1.
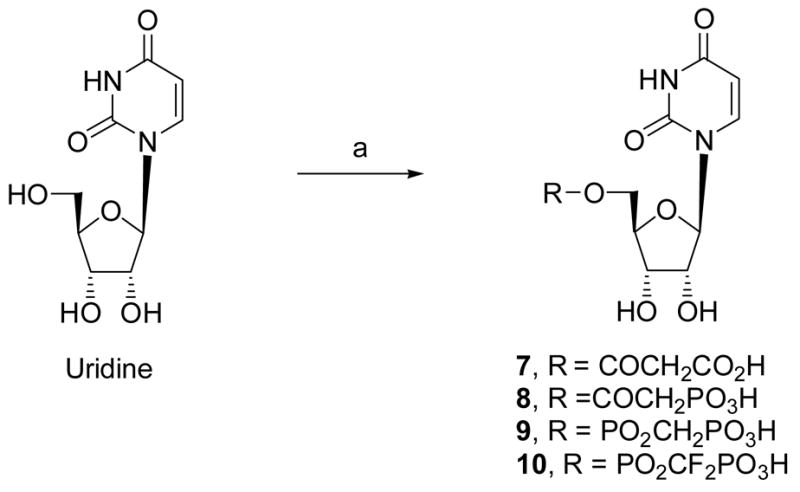
Synthesis of UDP analogues containing a methylene-bridged substitute for the diphosphate group. Reagents and conditions: (a) DCC, ROH, DMF, rt.
Scheme 6.
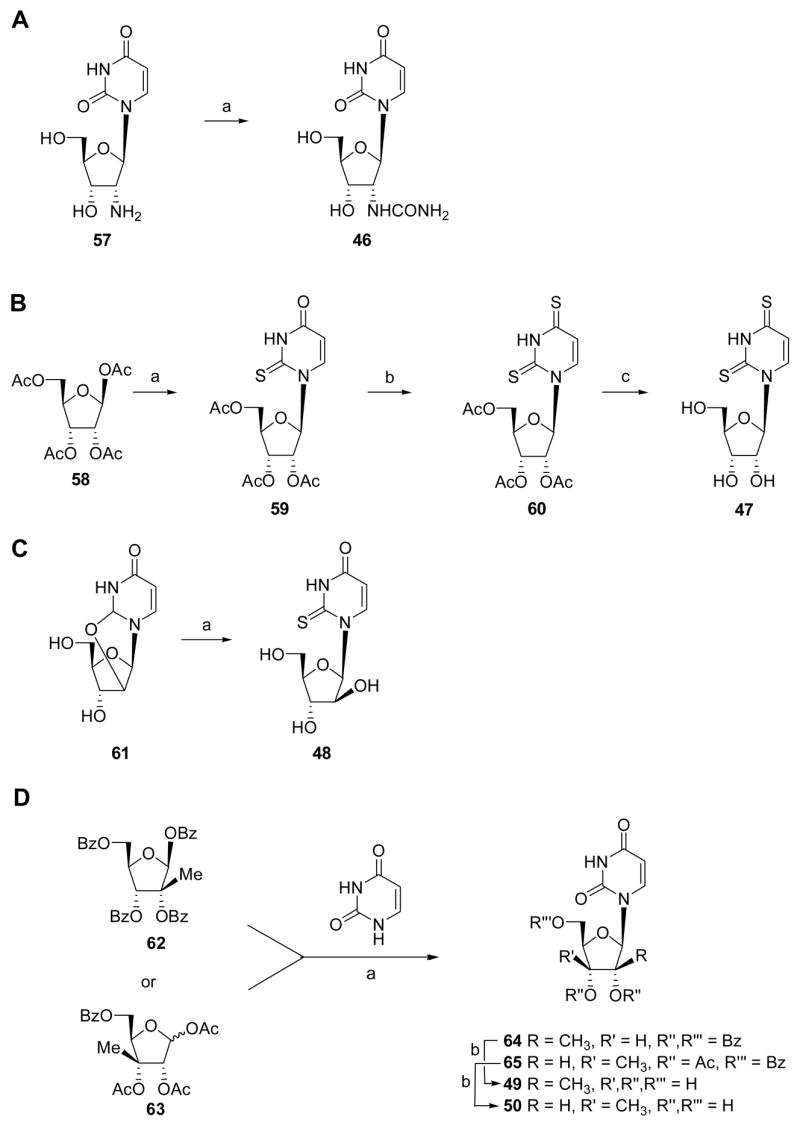
Synthesis of nucleoside intermediates. Reagents and conditions: A, benzotriazole-1-carboxamide, DMF; B, (a) silylated 2-thiouracil, SnCl4, C2H4Cl2; (b) Lawesson’s reagent, toluene, 80°C, overnight; (c) NaOMe, MeOH, reflux, 4 h; C, (a) H2S, Et3N, DMF, 200 psi, 2 days; D, (a) (i) 1,2-dichloroethane, hexamethyldisilazane, trimethylsilylchloride, 80°C, 4 h; (ii) SnCl4, rt, 4 h; (b) NH3/CH3OH, rt, overnight.
Table 2.
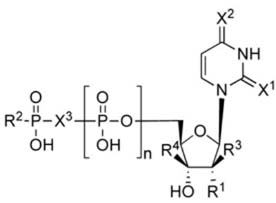
Relative potencies of UTP, 1, and UTP analogues for activation of the human P2Y2, P2Y4 and P2Y6 receptors. Unless noted: R1, R2 = OH; X = O; and R3, R4 = H.
 | |||||
|---|---|---|---|---|---|
| Compound | Modification | Structure | EC50, μMa | ||
| hP2Y2 | hP2Y4 | hP2Y6 | |||
| TRIPHOSPHATES (n = 2) | |||||
| 1b | (= UTP) | 0.060±0.00 | 0.090±0.01 | >10d | |
| 13b | 2′-deoxy-2′-amino-UTP | R1 = NH2 | 0.062±0.008 | 1.2±0.3 | NE |
| 14b | 2-thio-UTP | X1 = S | 0.035±0.004 | 0.35±0.10 | ~1.5f |
| 5b, c | 2-thio-2′-deoxy-2′-amino-UTP | X1 = S, R1 = NH2 | 0.008±0.002 | 2.4±0.8 | NE |
| 15 | 2-thio-β,γ-difluoromethylene-UTP | X1 = S, X3= CF2 | 1.63±0.36 | 8.11±0.69 | 5.15±0.69 |
| 16c | 2-thio-β,γ-dichloromethylene-UTP | X1 = S, X3=CCl2 | 2.51±0.65 | NE | NE |
| 17 | 2-thio-4-methylthio-UTP | X1 = S, X2= SCH3 | 0.91±0.06 | 5.35±1.20 | >10d |
| 18 | 2′-deoxy-2′-ureido-UTP | R1 = NHCONH2 | 1.74±0.25 | 4.64±2.05 | >10d |
| 19 | 2-thio-arabino-UTP | R1 = H, R3 = OH | 0.14±0.01 | 7.93±0.81 | NE |
| 20 | 2′-methyl-UTP | R3 = CH3 | 1.45±0.26 | 1.26± 0.14 | NE |
| 21 | 3′-methyl-UTP | R4 = CH3 | NE | NE | NE |
| TETRA- (n = 3) AND PENTA- (n = 4) PHOSPHATES | |||||
| 22 | Up4 | 2.61±1.39 | 4.64±2.05 | 7.56±1.07 | |
| 23 | 2-thio-Up4 | X1 = S | 0.60±0.20 | 5.52±1.75 | 6.83±1.74 |
| 24 | 4-thio-Up4 | X2 = S | 0.070± 0.01 | 0.28±0.06 | 6.46±0.41 |
| 25 | 2-thio-Up5 | X1 = S, n = 4 | 0.57±0.16 | 5.27±1.24 | 7.33±0.66 |
| 26 | Up4-OMe | R2= OCH3 | 3.95±0.51 | 2.70±0.43 | >10d |
| 27 | Up4-δ-Me- phosphonate | R2 = CH3 | 4.18±0.43 | 2.53±0.57 | 8.16±0.74 |
| 28 | Up4-O(CH2)2CN | R2 = (CH2)2CN | 1.70±0.22 | 1.96±0.53 | >10d |
| 29 | Up4-OCH2CHOHCH2OH | R 2 = OCH2 CHOH-CH2OH | 1.87±0.15 | 1.12±0.04 | 8.19±0.41 |
| 30c | Up4-OC6H5 |
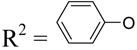
|
1.89±1.07 | NE | NE |
| 31 | Up4-OC6H11 |
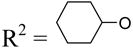
|
5.86±0.33 | >10d | >10d |
| TETRAPHOSPHATE SUGARS (N=3) | |||||
| 32 | Up4-[5]ribose |
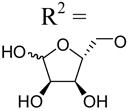
|
1.88±0.03 | 4.78±0.40 | >10d |
| 33 | Up4-[6]fructose |
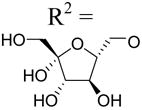
|
3.33±0.42 | 6.30±0.75 | >10d |
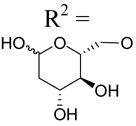
| 34c | Up4-[1]glucose | R2 =
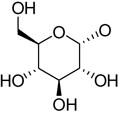
|
0.30±0.13 | 2.06±0.18 | 7.83±0.17 |
| 35 | Up4-[1]galactose |
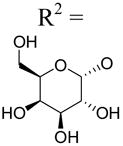
|
4.85±2.07 | 1.77±0.41 | 8.19±0.73 |
| 36 | Up4-[6]mannose |
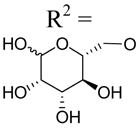
|
>10d | >10d | >10d |
| 37 | Up4-[6]2′-deoxyglucose |
|
3.54±0.96 | 4.32±1.50 | >10d |
Agonist potencies reflect stimulation of phospholipase C in 1321N1 human astrocytoma cells stably expressing the human P2Y2, P2Y4, or P2Y6 receptor. Potencies are presented in the form of EC50 values, which represent the concentration of agonist at which 50% of the maximal effect is achieved. These values were determined using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). The results are presented as mean ± standard error and are the average of three to six different experiments with each molecule.
Agonist potencies from reference 7.
5, MRS2698; 16, MRS2725; 30, MRS2768; 34, MRS2732.
≤50% effect at 10 µM. Values of >10 µM were determined by extrapolation.
NE - no effect at 10 µM.
Scheme 2.
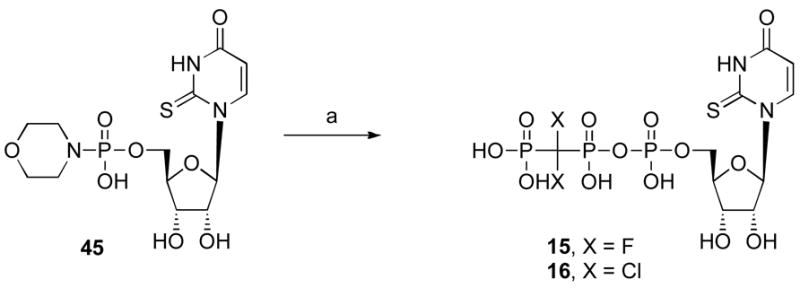
Synthesis of UTP analogues containing a β,γ-dihalomethylene-bridge in the triphosphate group. Reagents and conditions: (a) PO(OH)2CX2PO(OH)2, DMF, rt.
Scheme 3.
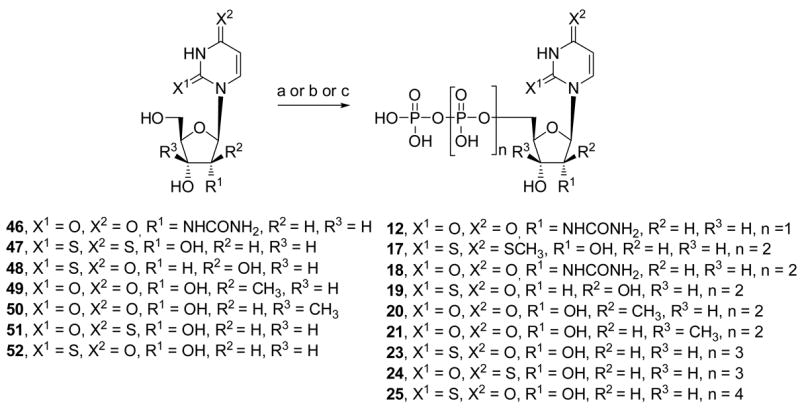
Synthesis of UTP analogues with modified uracil and ribose moieties. Reagents and conditions: (a) (i) POCl3, proton sponge, PO(OMe)3, 0°C; (ii) (Bu3NH)2H2P2O7, Bu3N, DMF, 0°C; (iii) TEAB 0.2M rt.; (b) (i) POCl3, PO(OMe)3, 4 h, 0°C; (ii) conc. NH4OH; (iii) Bu3N, CDI, DMF, 48; (iv) (Bu3NH)2H2P2O7; TEAB 1M for 19; (c) (i) POCl3, PO(OMe)3, 49 or 50, 0°C; (ii) conc. NH4OH; (iii) CDI, DMF, rt; (iv) (Bu3NH)2H2P2O7, DMF, rt for 20 or 21. Note that the UDP analogue 6 (not shown in scheme) was prepared by a similar method to (a), except for use of a phosphoric acid salt in the second step: (ii) (Bu3NH)2H2PO4, Bu3N, DMF, 0°C.
Scheme 4.
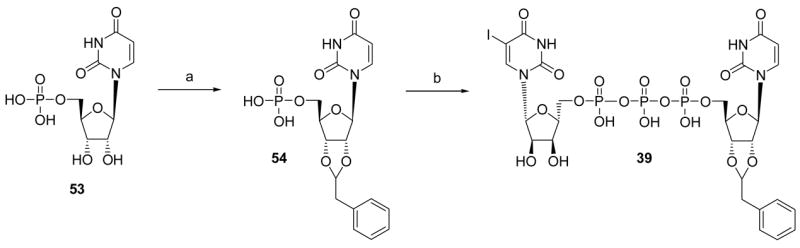
Synthesis of a 5-iodo analogue of INS48823. Reagents and conditions: (a) phenylacetaldehyde dimethylacetal, TFA, rt; (b) 5-iodo-uridine-5′-diphosphoimidazolidate, DMF, rt.
Scheme 5.
Synthesis of 5′-tetraphosphate analogues, including Up4-sugars and dinucleotides. The 2′-deoxyguanosine derivative 44 was prepared in the same manner as 43. Reagents and conditions: (a) (i) DCC, DMF, rt; (ii) RPO(OH)2, DMF, rt.
The synthesis of UTP analogues 12 and 17 – 25 from the corresponding nucleosides was by standard methods of di-, triphosphate formation.6 In each case the unprotected nucleoside was first treated with phosphorous oxychloride (Scheme 3). The reaction mixture was either treated immediately with bis(tri-n-butylammonium) pyrophosphate (phosphoric acid for 12) or the isolated 5′-monophosphate was activated with 1,1′-carbonyldiimidazole (CDI) followed by the pyrophosphate salt. An attempt to synthesize 2,4-dithio-UTP led to isolation only of the 4-methylthio analogue 17. Identification of nucleotide compounds was confirmed by NMR (1H and 13P) and by high-resolution mass spectrometry (HRMS), and purity was demonstrated with high-performance liquid chromatography (HPLC) in two different solvent systems.
The preferred method of synthesis of 2′-MeUTP (20) and 3′-MeUTP (21) was through isolation of the monophosphate. 2′-C-Methyl-uridine-5′-mophosphate14 and 3′-C-methyl-uridine-5′-monophosphate15 were obtained as ammonium salts following the Yoshikawa procedure19 starting from nucleosides 2′-C-methyl-uridine (49) and 3′-C-methyl-uridine (50). The nucleotides also were prepared by the one-pot method using a sequential reaction of 49 and 50 with phosphorus oxychloride and pyrophosphoric acid tributylammonium salt, but the yields were lower.
Most of the required nucleoside precursors were readily available, with several exceptions. 2′-Ureido-2′-deoxyuridine 46 was prepared by a one step method from 2′-amino-2′-deoxyuridine 57 (Scheme 6A).17 The synthesis of 2,4-dithiouridine 47 is depicted in Scheme 6. The commercially available β-D-ribofuranose 1,2,3,5-tetraacetate 58 was coupled with silylated 2-thiouracil under SnCl4-catalyzed Vorbrüggen conditions.18 4-Thionation of the resulting 2′,3′,5′-tri-O-acetyl-2-thiouridine 59 was performed using Lawesson’s reagent.19 Subsequent sugar deprotection of 60 afforded 2,4-dithiouridine 47 in 59% overall yield. The nucleoside 1-(β-D-arabinofuranosyl)-2-thio(1H) pyrimidin-4-one 48 was obtained via opening of 2,2′-O-anhydrouridine 61 with H2S and triethylamine in anhydrous DMF (Scheme 6).20 The synthetic routes to the nucleosides 2′-C-methyl-uridine 49 and 3′-C-methyl-uridine 50, synthesized using the strategy reported by Wolfe & Harry-O’kuru21 and Mikhailov et al.22a with some pmodifications, are outlined in Scheme 6D.
Pharmacological Activity
Activation of PLC by a range of concentrations of each nucleotide derivative (7 – 44) was studied in [3H]inositol-labeled 1321N1 human astrocytoma cells stably expressing the human P2Y2, P2Y4, or P2Y6 receptors (Tables 1 – 3) by methodology (see Experimental Section) we have described previously in detail.1,3,6,24
Table 1 illustrates UDP analogues that were designed for possible interaction with the P2Y6 receptor. Compounds 7 and 8 are derivatives substituted with an anionic carboxylic acid or phosphonate acetyl ester moiety with the goal of approximating the charge and electronic characteristics of the diphosphate moiety for interaction with cationic residues in the ligand binding pocket8. These molecules were inactive at the P2Y6 receptor. Introduction of an α,β-methylene 9 substitution only slightly reduced potency at the P2Y6 receptor (Figure 1), while the α,β-difluoromethylene analogue 10 was strikingly inactive. Nucleobase substitution was also examined in the UDP series. We previously reported that the higher homologue of 11, 5-amino-UTP, displays increased potency relative to UTP as a P2Y6 receptor agonist7. In contrast, the corresponding diphosphate 11 was ~50-fold less potent than UDP. The introduction of a polar ureido group on the ribose moiety of adenosine derivatives provided tailored analogues for selective recognition by adenosine neoceptors.27 Therefore, we replaced the 2′-hydroxy group of UDP with a highly H bonding ureido group. This molecule exhibited a 360-fold reduction of potency at the P2Y6 receptor compared to UDP.
Figure 1.
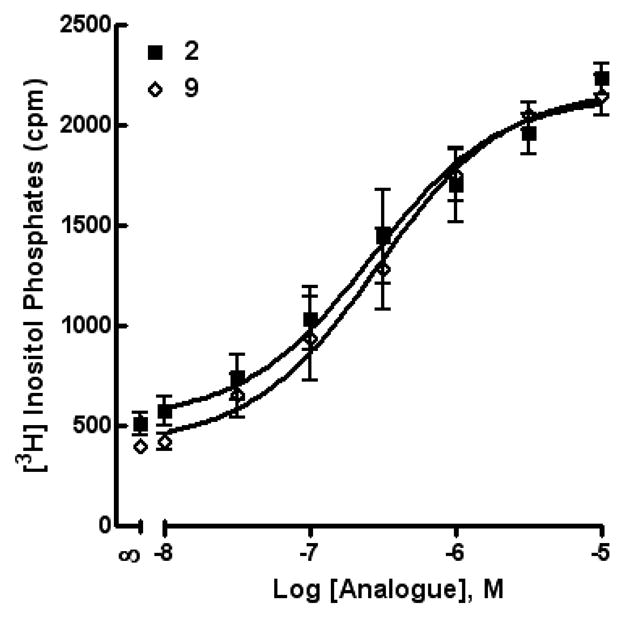
Activity of compounds 2 (the native agonist, UDP) and 9 (α,β-methylene-UDP) at the P2Y6 receptor as indicated by activation of PLC in stably transfected astrocytoma cells.
Table 2 illustrates composite data for UTP analogues designed to activate the P2Y2 and P2Y4 receptors. We previously illustrated that substitution of a 2-thio group enhances potency and/or selectivity of UTP analogues (e.g. 5 and 14) at the P2Y2 receptor.7 The combination of the 2-thio modification with a β,γ-difluoromethylene 15 or β,γ-dichloromethylene 16 substitution of the triphosphate moiety or 4-methylthio 17 group resulted in analogues that were 50 – 100-fold weaker than 14. A report of another UTP analogue,4 5-bromo-β,γ-dichloromethylene-UTP, that displayed submicromolar P2Y2 receptor potency had suggested the possibility of greater potency in 15 and 16 than we observed experimentally in the current study. Nevertheless, the dichloromethylene group of 16 provides moderate selectivity for the P2Y2 receptor, while the equipotent difluoromethylene derivative 15 was only marginally selective. Replacement of the 2′-hydroxy group of UTP with a ureido group 18 resulted in a 36-fold reduction of potency at the P2Y2 receptor and >100-fold reduction at the P2Y4 receptor.
Methyl groups were placed on the ribose ring of UTP at the 2′ and 3′ positions in 20 and 21, respectively. Like the methanocarba modification of ribose,6,28 this approach is a means of conformational control of the ribose ring that has proven effective for achieving selectivity in A1 adenosine receptor agonists.29,30 Compound 20 maintains a North conformation of the ribose ring, and as is the case with UTP, activated the P2Y2 and P2Y4 receptors nearly equipotently. However, a 20–30-fold decrease in potency relative to UTP was observed. In contrast, 21, which maintains a South conformation of the ribose ring, was inactive at both P2Y2 and P2Y4 receptors. These results are consistent with previously reported conformational preferences of these receptors deduced from our studies with methanocarba-derivatives of UTP.6
No previous studies have reported the potency of nucleoside 5′-tetraphosphates at the P2Y2 receptor, although adenosine 5′-tetraphosphate (Ap4) was reported to activate a presumed P2X receptor.31 Therefore, we synthesized and evaluated the activities of 5′-tetraphosphate and pentaphosphate derivatives 22–25. The potency of uridine 5′-tetraphosphate 22 (Up4) was greatly reduced in comparison to UTP 1. The high P2Y2 receptor potency of 2-thio-UTP 14 was remarkably preserved in the corresponding 4-thio-5′-tetraphosphate analogue 24; in contrast, the potency of the 2-thio analogue 23 was reduced. Nevertheless, these results illustrate that 2- or 4-thio substitution of Up4 results in marked increases of potency in tetraphosphate molecules since both 23 and 24 were more potent at the P2Y2 receptor than Up4 22. Homologation of 23 to the pentaphosphate 25 had no effect on potency at the P2Y2 receptor.
Although dinucleoside tetraphosphates, such as 3 and 4, are known to activate P2Y2 and P2Y4 receptors with moderate potency,9 the SAR of the terminal nucleoside moiety mainly has been explored for uridine and other nucleoside units. Therefore, we investigated the effects of substitution of the terminal nucleoside moiety with small organic moieties 26 – 31 or to sugars alone 32 – 37. Most of these modifications led to P2Y2 receptor potencies in the micromolar range. A terminal methyl phosphoester 26 and the corresponding phosphonate 27 were identical in potency as relatively weak P2Y2 receptor agonists, with EC50 values of 4 µM. A terminal cyclohexyl ester 31 was significantly less potent than the corresponding terminal phenyl ester 30, suggesting that aromatic or hydrophilic groups are more favored than simple hydrophobic groups in this region. Curiously, substitution of an acyclic alkyl phosphate or phosphonate at the terminal position (26–29) did not favor selective interaction with the P2Y2 versus P2Y4 receptors, while an aryl phosphate ester (30) substitution resulted in high selectivity for the P2Y2 receptor (Figure 2).
Figure 2.
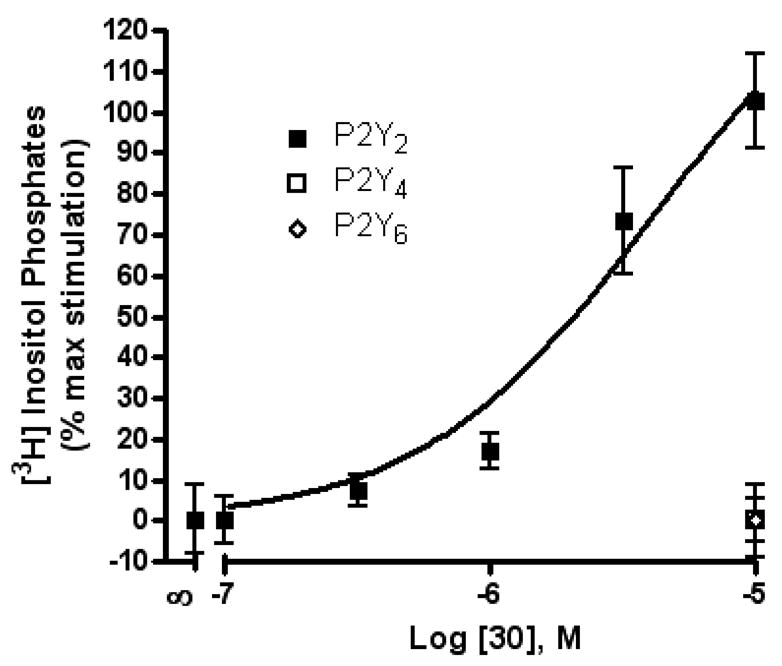
Activity of compound 30 (uridine 5′-tetraphosphate δ-phenyl ester) at P2Y2, P2Y4, and P2Y6 receptors as indicated by activation of PLC in stably transfected astrocytoma cells. The effect of UTP corresponds to 100%.
Among tetraphosphate sugar derivatives, Up4-[1]glucose 34 was the most potent with an EC50 of 0.3 µM (Figure 3). The P2Y2 receptor selectivity of 34 in comparison to P2Y4 and P2Y6 receptors was 7- and 26-fold, respectively. Inversion of the chirality of the 4′-hydroxyl group in the sugar, i.e. galactose rather than glucose, in 35 reduced potency at the P2Y2 receptor (16-fold), but not at the P2Y4 receptor. The [6]mannose adduct 36 of Up4 was essentially inactive at the P2Y2 receptor. Removal of a single hydroxyl group of 36 to form 37 increased the potency at both P2Y2 and P2Y4 receptors. These results are consistent with our recent P2Y2 receptor modeling study, which proposes specific interactions in the region of the distally-binding uridine moiety of Up4U.7
Figure 3.
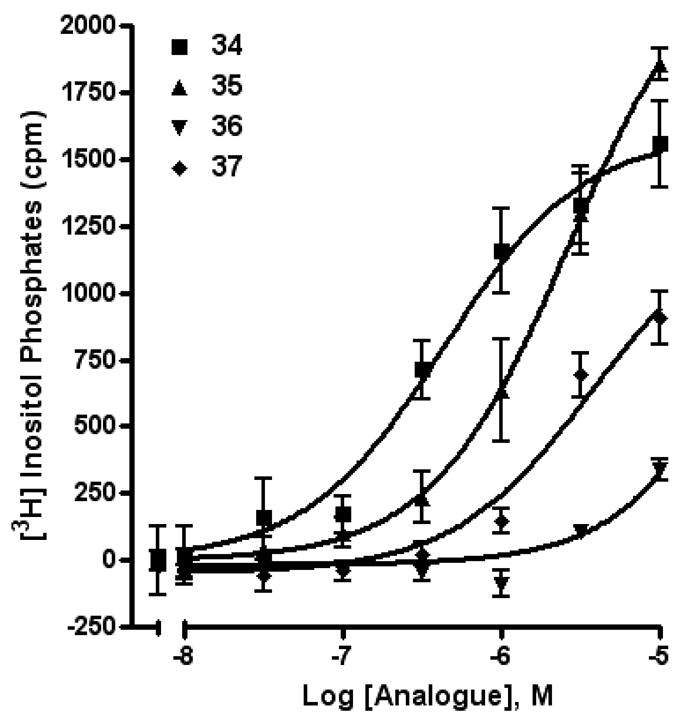
Activity of compounds 34 – 37 (uridine 5′-tetraphosphate δ-sugars) at the P2Y2 receptor as indicated by activation of PLC in stably transfected astrocytoma cells.
The SAR of dinucleotides at the P2Y2 and P2Y6 receptors also was further explored (Table 3). An analogue of a known P2Y6 receptor agonist9, Up3U 38, was designed to combine multiple reported favorable modifications of uracil nucleotides directed toward activation of the P2Y6 receptor based on the agonist INS48823 6.12 The resulting hybrid compound 39, containing 5-iodo substitution and a 2′,3′-phenylmethylacetyl group on opposite uridine moieties of Up3U, was a nonselective agonist of the P2Y6 receptor.
Several new dinucleoside tetraphosphate derivatives 40 – 44 were synthesized, and their activities were compared to previously studied compounds 3, 4.9,11 The inclusion of a β,γ-difluoromethylene 41 or dichloromethylene 42 bridge in the tetraphosphate moiety resulted in analogues that were at least an order of magnitude weaker at the P2Y2 receptor than Up4U 3. The difluoro analogue 41, which exhibited an EC50 of ~2 µM at the P2Y2 receptor, apparently provides a better mimic of the phosphate ester group than does the dichloro substitution in 42. These results are consistent with electronic effects of the difluoromethylene group noted for other phosphonates.25 The P2Y2 receptor agonist INS37217 4 also was prepared for comparison.11 Substitution of a 2-thio group in 43 resulted in enhanced potency (EC50 ~80 nM) and selectivity for the P2Y2 receptor (9-fold in comparison to P2Y4). A 2′-deoxyguanosine analogue 44 also exhibited moderate potency at the P2Y2 receptor, as reported.11
Many of the analogues synthesized and studied including Up4-sugars retained moderate potency at the P2Y4 receptor. However, our work to date has not revealed molecules that exhibit notable selectivity for the P2Y4 receptor subtype over the other uridine nucleotide activated receptors.
In conclusion, we have synthesized novel uracil nucleotide derivatives that are directed toward activation of the P2Y2 or P2Y6 receptor. Key compounds reported in this study include: 34, Up4-[1]glucose, which displayed submicromolar potency at the P2Y2 receptor; 43, the 2-thio analogue of INS37217, which was a potent and selective P2Y2 receptor agonist. Thus, the 2-thio modification, found previously to enhance P2Y2 receptor potency, could be combined with other favorable modifications to produce novel molecules that exhibit high potencies and receptor selectivities. Phosphonomethylene bridges introduced for stability in analogues of UDP, UTP and uracil dinucleotides markedly reduced potency. Truncation of dinucleotide agonists of the P2Y2 receptor, in the form of Up4-sugars, indicated that a terminal uracil ring is not essential for moderate potency at this receptor and that specific SAR patterns are observed at this distal end of the molecule.
Experimental Section
Chemical Synthesis
1H NMR spectra were obtained with a Varian Gemini 300 or Varian Mercury 400 spectrometers using D2O, CDCl3 or DMSO-d6 as a solvent. The chemical shifts are expressed as relative ppm from HOD (4.80 ppm). 13P NMR spectra were recorded at rt (rt) by use of Varian XL 300 (121.42 MHz) or Varian Mercury 400 (162.10 MHz) spectrometers; orthophosphoric acid (85%) was used as an external standard. In several cases the signal of the terminal phosphate moiety was not visible due to high dilution.
Purity of compounds was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax Eclipse 5 μm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 20 min; the flow rate was 1 mL/min. Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm. All derivatives tested for biological activity showed >99% purity by HPLC analysis (detection at 254 nm).
High-resolution mass measurements were performed on Micromass/Waters LCT Premier Electrospray Time of Flight (TOF) mass spectrometer coupled with a Waters HPLC system, unless noted. Purification of the nucleotide analogues for biological testing was carried out on (diethylamino)ethyl (DEAE)-A25 Sephadex columns with a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase. Compounds 16, 20, 21, 39, and 54 were purified by Sephadex alone (and isolated in the ammonium salt form), and all other compounds were additionally purified by HPLC with a Luna 5 μ RP-C18(2) semipreparative column (250 × 10.0 mm; Phenomenex, Torrance, CA) and using the following conditions: flow rate of 2 mL/min; 10 mM triethylammonium acetate (TEAA)-CH3CN from 100:0 to 95:5 (or up to 99:1 to 92:8) in 30 min (and isolated in the triethylammonium salt form).
All reagents were of analytical grade. 2′-Amino-2′-deoxyuridine was from Metkinen Chemistry (Kuusisto, Finland). 2,2′-O-Anhydrouridine 61 was obtained from Wako Chemicals. 5-Amino-UDP was purchased from ALT, Inc. (Lexington, KY). Other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO).
General Procedure for the Preparation of Nucleoside Triphosphates (e.g. 14 and 17), Tetraphosphates (22 – 24), Pentaphosphate (25). Procedure A
To a solution of nucleoside (0.054 mmol) and Proton Sponge (23 mg, 0.11 mmol) in trimethyl phosphate (1 mL) was added phosphorous oxychloride (0.01 mL, 0.11 mmol) at 0°C. The reaction mixture was stirred for 2 h at 0°C and tributylamine (0.03 mL, 0.12 mmol) was added. Tributylammonium pyrophosphate (1.6 moles C12H27N per mole H4P2O7, 110 mg, 0.23 mmol) in DMF (0.3 mL) was added at once to the reaction mixture. After 10 min, 0.2 M triethylammonium bicarbonate solution (2 mL) was added, and the clear solution was stirred at rt for 1 h. The latter was lyophilized overnight, and the resulting residue was purified by ion-exchange column chromatography using a Sephadex-DEAE A-25 resin with a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase to obtain the corresponding nucleotides as the ammonium salts. The collected portions were purified by HPLC again as described above.
General Procedure for the Preparation of Nucleosides Tetraphosphates (26 – 31), Nucleoside Tetraphosphate Sugars (32 – 37), and Dinucleoside Tetraphosphates (40 – 44). Procedure B
Uridine triphosphate trisodium salt (15 mg, 0.027 mmol or UTP analogues for compounds 40 – 44) and the corresponding monophosphate (0.109 mmol) were converted to the tributylammonium salts by treatment with ion-exchange resin (DOWEX 50WX2-200 (H)) and tributylamine. After removal of the water, the obtained tributylammonium salts were dried under high-vacuum overnight. To a solution of uridine triphosphate tributylammonium salt (0.027 mmol) in DMF (2 mL) was added N,N′-dicyclohexylcarbodiimide (DCC, 14 mg, 0.07 mmol). After stirring the reaction mixture at rt for 1 h, a solution of the corresponding monophosphate tributylammonium salt (0.109 mmol) in DMF (1 mL) was added. The reaction mixture was stirred at rt for 48 h. After removal of the solvent, the residue was purified by ion-exchange column chromatography with a Sephadex-DEAE A-25 resin, followed by a semi preparative HPLC purification as described above. Free nucleoside 5′-tetra- and pentaphosphates were not stable upon prolonged storage at 4°C, but were stable at −20°C for at least 3 weeks. Dinucleoside tetraphosphates were stable for several months at −20°C and later showed signs of gradual decomposition.
General Procedure for the Preparation of Compounds (7 – 10). Procedure C
To a solution of uridine (25 mg, 0.1 mmol) and DCC (62 mg, 0.3 mmol) in DMF (1.5 mL) was added the appropriate carboxylic acid or phosphonic acid (0.15 mmol): malonic acid for 7, phosphonoacetic acid for 8, methylene diphosphonic acid for 9, difluoromethylene diphosphonic acid for 10. After stirring the reaction mixture at rt for 24 h to 48 h, the solvent was removed. The residue was purified by ion-exchange column chromatography with a Sephadex-DEAE A-25 resin, followed by a semi preparative HPLC purification as described above.
Uridine-5′-phosphonoacetate triethylammonium salt (8). Procedure C
Compound 8 (13.4 mg, 36%) was obtained as a white solid from uridine (25 mg, 0.1 mmol). 1H NMR (D2O) δ 7.81 (d, J = 8.1 Hz, 1H), 5.94 (d, J = 8.1 Hz, 1H), 5.91 (d, J = 4.5 Hz, 1H), 4.41 (m, 3H), 4.33 (m, 2H), 2.89 (d, J = 20.4 Hz, 2H); 13P NMR (D2O) δ 12.23; 13C NMR (D2O) δ (169.27, 169.19), 164.92, 150.34, 140.53, 101.18, 88.21, 80.14, 72.06, 68.06, 62.87, (36.28, 34.73); HRMS-EI found 367.0658 (M−H)−. C11H16N2O10P requires 367.0543; purity > 99% by HPLC (retention time: 5.9 min).
Uridine-5′-α,β-methylene-diphosphate triethylammonium salt (9). Procedure C
Compound 9 (14 mg, 33%) was obtained as a white solid from uridine (25 mg, 0.1 mmol). 1H NMR (D2O) δ 8.03 (d, J = 8.1 Hz, 1H), 5.98 (m, 2H), 4.41 (m, 2H), 4.28 (m, 1H), 4.19 (m, 2H), 2.18 (t, J = 19.5 Hz, 2H); 13P NMR (D2O) δ 19.37 (m), 14.44 (m); HRMS-EI found 403.0245 (M+H)−. C10H17N2O11P2 requires 403.0308; purity > 99% by HPLC (retention time: 12.6 min).
Uridine-5′-α,β-difluoromethylenediphosphate triethylammonium salt (10). Procedure C
Compound 10 (14 mg, 33%) was obtained as a white solid from uridine (25 mg, 0.1 mmol). 1H NMR (D2O) δ 8.02 (d, J = 8.1 Hz, 1H), 5.98 (m, 2H), 4.40 (m, 2H), 4.26 (m, 1H), 4.19 (m, 2H); 13P NMR (D2O) δ 4.69 (m), 3.48 (m); HRMS-EI found 436.9977 (M−H)−. C10H13N2O11F2P2 requires 436.9963; purity > 99% by HPLC (retention time: 14.1 min).
2′-Deoxy-2′-ureido-uridine-5′-diphosphate triethylammonium salt (12)
A solution of the compound 46 (38 mg, 0.13 mmol) and Proton Sponge (43 mg, 0.20 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0°C. Then phosphorous oxychloride (25 μL, 0.27 mmol) was added dropwise, and the reaction mixture was stirred for 2 h at 0°C. A mixture of tributylamine (0.25 mL, 1.05 mmol) and a solution 0.35 M of bis(tributylammonium) salt of phosphoric acid in DMF (2.28 mL) was added at once. This salt was prepared by mixing tributylamine (0.4 mL, 1.65 mmol) and phosphoric acid (85 mg, 0.87 mmol) in DMF (2.5 mL). After 10 min, 0.2 M triethylammonium bicarbonate (TEAB) solution (3 mL) was added, and the clear solution was stirred at rt for 30 min. The latter was lyophilized overnight. The residue was purified by Sephadex-DEAE A-25 resin ion-exchange column chromatography, followed by semipreparative HPLC as described above to obtain 12 (8 mg, 8%) as a white solid. 1H NMR (D2O) δ 7.98 (d, J = 8.1 Hz, 1H), 6.02 (m, 2H), 4.51 (m, 1H), 4.34 (m, 2H), 4.20 (m, 2H); 13P NMR (D2O) δ −7.24, −10.14 (d, J = 22.0 Hz); HRMS-EI found 445.0172 (M−H)−. C10H15N4O12P2 requires 445.0162; purity > 99% by HPLC (retention time: 12.7 min).
2-Thio-uridine-5′-β,γ-difluoromethylene-triphosphate triethylammonium salt (15)
To a solution of 2-thio-uridine 5′-monophosphate morpholidate 4-morpholine-N,N dicyclohexylcarboxamidine salt, 4526 (10 mg, 0.014 mmol) in DMF (2 mL), difluoromethylene diphosphonate tributylammonium salt (20 mg, 0.021 mmol) was added. After being stirred 3 days at rt, the reaction mixture was evaporated to remove the solvent and purified by Sephadex-DEAE A-25 resin followed by HPLC purification to give 15 (4 mg, 31%). 1H NMR (D2O) δ 8.18 (d, J = 8.1 Hz, 1H), 6.69 (d, J = 3.0 Hz, 1H), 6.28 (d, J = 8.1 Hz, 1H), 4.31–4.47 (m, 5H); 13P NMR (D2O) δ 3.47, −3.80 (m), −10.69 (d, J = 31.2 Hz); HRMS-EI found 532.9375 (M−H)−. C10H14N2O13F2P3S requires 532.9398; purity > 99% by HPLC (retention time: 18.1 min).
2-Thio-uridine-5′-β, γ-dichloromethylene-triphosphate ammonium salt (16)
To a solution of 2-thio-uridine 5′-monophosphate morpholidate 4-morpholine-N,N dicyclohexylcarboxamidine salt, 4526 (7 mg, 0.01 mmol) in DMF (2 mL), dichloromethylene diphosphonate tributylammonium salt (25 mg, 0.025 mmol) was added. After being stirred 3 days at rt, the reaction mixture was evaporated to remove the solvent and purified by Sephadex-DEAE A-25 resin to give 16 (5 mg, 80%). 1H NMR (D2O) δ 8.20 (d, J = 8.1 Hz, 1H), 6.69 (d, J = 2.7 Hz, 1H), 6.29 (d, J = 8.4 Hz, 1H), 4.45 (m, 2H), 4.39 (m, 2H), 4.34 (m, 1H); 13P NMR (D2O) δ 8.08 (d, J = 17.7 Hz), 1.17 (dd, J =17.7, 31.2 Hz), −10.60 (d, J = 31.2 Hz); HRMS-EI found 564.8817 (M−H)−. C10H14N2O13Cl2P3S requires 564.8807; purity > 99% by HPLC (retention time: 19.5 min).
2-Thio-4-methylthio-uridine-5′-triphosphate triethylammonium salt (17). Procedure A
Compound 17 (3.2 mg, 6.4%) was obtained as a white solid from 2,4-dithio-uridine, 47 (15 mg, 0.054 mmol). 1H NMR (D2O) δ 8.51 (d, J = 7.5 Hz, 1H), 7.15 (d, J = 7.2 Hz, 1H), 6.53 (br s, 1H), 4.45 (m, 2H), 4.38 (m, 3H), 2.60 (s, 3H); 13P NMR (D2O) δ −11.00 (d, J = 19.2 Hz), −21.02 (m); HRMS-EI found 528.9315 (M−H)−. C10H16N2O13P3S2 requires 528.9307; purity > 99% by HPLC (retention time: 17.1 min).
2′-Deoxy-2′-ureido uridine-5′-triphosphate triethylammonium salt (18). Procedure A
A solution of the compound 46 (38 mg, 0.13 mmol) and Proton Sponge (43 mg, 0.20 mmol) in trimethyl phosphate (1 mL) was stirred for 10 min at 0°C. Phosphorous oxychloride (25 μL, 0.27 mmol) was then added dropwise, and the reaction mixture was stirred for 2 h at 0°C. A solution of tributylammonium pyrophosphate (377 mg, 0.80 mmol) and tributylamine (0.13 mL, 0.53 mmol) in DMF (1 mL) was added and stirring was continued at 0°C for additional 10 min. Triethylammonium bicarbonate solution (TEAB, 3 mL of 0.2 M) was added, and the reaction mixture was stirred at rt for 30 min. The latter was lyophilized overnight. The residue was purified by Sephadex-DEAE A-25 resin ion-exchange column chromatography, followed by semipreparative HPLC as described above to obtain 18 (9 mg, 7%) as a white solid. 1H NMR (D2O) δ 7.95 (d, J = 8.4 Hz, 1H), 6.05 (d, J = 7.8 Hz, 1H), 6.01 (d, J = 8.4 Hz, 1H), 4.51 (m, 1H), 4.35 (m, 2H), 4.25 (m, 2H); 13P NMR (D2O) δ −10.15, −12.15 (d, J = 19.5 Hz), −23.6; HRMS-EI found 524.9812 (M−H)−. C10H16N4O15P3 requires 524.9825; purity > 99% by HPLC (retention time: 16.6 min).
1-(β-D-Arabinofuranosyl)-2-thio(1H)pyrimidin-4-one 5′-triphosphate triethyl ammonium salt (19)
Solution of 48 (150 mg, 0.58 mmol) in trimethyl phosphate (5.8 mL) was cooled to 0°C, POCl3 (342 μL, 3.8 mmol) was added dropwise and the mixture was stirred for 4 h at 0°C and for 30 min at rt. The mixture was poured into ice-water (10 mL), neutralized with concentrated NH4OH and evaporated to dryness. The resulting residue was purified by column chromatography (i-PrOH:NH4OH:H2O 60:30:5). After lyophilization of the collected pure fractions, the 5′-monophosphate of 48 was obtained as a white solid (124 mg, 60%). 1H NMR (D2O) δ 7.97 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 5.0 Hz, 1H), 6.07 (d, J = 8.2 Hz, 1H), 4.48 (app t, J = 4.8 Hz, 1H), 4.12 (app t, J = 4.8 Hz, 1H), 3.90–4.01 (m, 3H); 13P NMR (D2O) δ 3.42; HRMS-EI found 363.0271 [M+Na]+. C9H12N2O8P1S1Na requires 363.0281. To a solution of 5′-monophosphate of 48 (32 mg, 0.088 mmol) and tributylamine (21 μL, 0.088 mmol) in DMF (3.2 mL) was added CDI (71 mg, 0.44 mmol). After stirring for 3 h at rt, the reaction was quenched by addition of methanol (14 μL). Bis(tri-n-butylammonium)pyrophosphate (228 mg, 0.51 mmol) was added, and the mixture was stirred and subsequently concentrated under reduced pressure. The resulting residue was stirred in 1M triethylammonium bicarbonate (TEAB) buffer (6 mL, pH = 7.4) for 30 min, lyophilized and purified on a preparative HPLC apparatus equipped with a source 15 Q column (100% water → 100% 1 M TEAB/water in 45 min) to yield 10 μmol (11%) of compound 19 after lyophilizing the appropriate fractions. 1H NMR (D2O) δ 7.90 (d, J = 8.1 Hz, 1H), 6.80 (d, J = 5.2 Hz, 1H), 6.08 (d, J = 8.2 Hz, 1H), 4.48 (app t, J = 5.0 Hz, 1H), 4.16 (m, 3H), 4.03 (m, 1H); 13P NMR (D2O) δ −9.54 (d, J = 19.6 Hz), −10.35 (d, J = 19.6 Hz), −22.19 (t, J = 19.6 Hz), HRMS-EI found 498.9099 [M−H] −. C9H14N2O14P3S requires 498.9384. Structural assignment was confirmed with COSY. All signals assigned to hydroxyl groups were exchangeable with D2O. Exact mass measurements were performed on a quadrupole/orthogonal-acceleration time-of-flight (Q/oaTOF) tandem mass spectrometer (qToF 2, Micromass, Manchester, UK) equipped with a standard electrospray ionization (ESI) interface. Samples were infused in a i-PrOH/water (1:1) mixture at 3 μL/min.
General Procedure for the Preparation of the Nucleoside 5′-triphosphates 2′-MeUTP (20) and 3′-MeUTP (21)
To a solution of 2′-14 or 3′-C-methyl-UMP15 (0.15 mmol) dissolved in dry DMF (1.5 mL) was added tri-n-butylamine (0.15 mmol) and the solution was stirred for 20 min at rt. After evaporation under anhydrous condition, the residue was suspended in 1.4 mL of dry DMF and CDI (122 mg, 0.75 mmol) was added and the mixture was stirred for 6 h at rt. Methanol (49 μl, 1.2 mmol) was added and the mixture was stirred for 30 min. Then 6 mL (3 mmol) of a 0.5 M solution of bis(tri-n-butylammonium) pyrophosphate in dry DMF were added. The mixture was stirred for 24 h at rt, and the solvent was removed under high vacuum at rt. The mixture dissolved in water was purified by Sephadex DEAE A-25 resin ion exchange column chromatography with a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate. Compounds 20 and 21 were isolated as ammonium salts (yield 32 and 34%, respectively). Mass spectroscopy was carried out on an HP 1100 series instrument in the negative ion mode using atmospheric pressure electrospray ionization (API-ESI).
2′-C-Methyl-uridine-5′-triphosphate ammonium salt (20)
1H NMR (D2O) δ 7.76 (d, J = 8.1 Hz, 1H), 5.86 (s, 1H), 5.75 (d, J = 8.1 Hz, 1H), 3.85–3.90 (m, 2H), 3.75 (d, J = 9.0 Hz, 1H), 3.68 (dd, J = 4.3, 13.7 Hz, 1H), 1.05 (s, 3H); 13P NMR (D2O) δ ′4.75 (br s), −19.34 (br s), −20.59 (m); MS m/z 497.10 [M+H]−.
3′-C-Methyl-uridine-5′-triphosphate ammonium salt (21)
1H NMR (D2O) δ 7.78 (d, J = 8.1 Hz, 1H), 5.84 (d, J = 7.7 Hz, 1H), 5.75 (d, J = 8.1 Hz, 1H), 4.03 (d, J = 7.7 Hz, 1H), 3.94 (dd, J = 3.4, 5.1 Hz, 1H), 3.65 (dd, J = 3.4, 12.8 Hz, 1H), 3.58 (dd, J = 4.9, 12.6, 1H),1.20 (s, 3H); 13P NMR (D2O) δ 4.83 (d, J = 15.9 Hz), −19.25 (t, J = 15.3 Hz), −20.66 (t, J = 15.9 Hz); MS m/z 497.10 [M+H]−.
2-Thio-uridine-5′-tetraphosphate triethylammonium salt, 2-thio-UP4 (23). Procedure A
Compound 23 (1.5 mg, 4.5%) was obtained as a white solid from 2-thio-uridine (10 mg, 0.038 mmol). 1H NMR (D2O) δ 8.17 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 3.0 Hz, 1H), 6.27 (d, J = 8.1 Hz, 1H), 4.46 (m, 2H), 4.33 (m, 3H); 13P NMR (D2O) δ −11.18 (d, J = 18.9 Hz), −22.55 (m); HRMS-EI found 578.9042 (M−H)−. C9H15N2O17P4S requires 578.9042; purity > 99% by HPLC (retention time: 19.8 min).
4-Thio-uridine-5′-tetraphosphate triethylammonium salt (24). Procedure A
Compound 24 (5.2 mg, 7.6%) was obtained as a white solid from 4-thio-uridine (20 mg, 0.077 mmol). 1H NMR (D2O) δ 7.88 (d, J = 7.8 Hz, 1H), 6.66 (d, J = 7.8 Hz, 1H), 5.95 (d, J = 4.5 Hz, 1H), 4.47 (m, 1H), 4.40 (m, 1H), 4.28 (m, 3H); 13P NMR (D2O) δ −10.91 (d, J = 20.2 Hz), −21.97 (m); HRMS-EI found 578.8890 (M−H)−. C9H15N2O17P4S requires 578.9042; purity > 99% by HPLC (retention time: 19.2 min).
2-Thio-uridine-5′-pentaphosphate triethylammonium salt (25). Procedure A
Compound 25 (1.1 mg, 3%) was obtained as a white solid from 2-thio-uridine 52 (10 mg, 0.038 mmol). 1H NMR (D2O) δ 8.15 (d, J = 7.8 Hz, 1H), 6.70 (d, J = 3.6 Hz, 1H), 6.26 (d, J = 8.1 Hz, 1H), 4.43 (m, 2H), 4.32 (m, 3H); 13P NMR (D2O) δ −11.22 (d, J = 18.3 Hz), −22.78 (m); HRMS-EI found 658.8782 (M−H)−. C9H16N2O20P5S requires 658.8705; purity > 99% by HPLC (retention time: 19.9 min).
Uridine-5′-methyl-tetraphosphate triethylammonium salt (26). Procedure B
Compound 26 (7.8 mg, 33%) was obtained as a white solid using uridine triphosphate (20 mg, 0.036 mmol) and methylphosphate (45 mg, 0.15 mmol). 1H NMR (D2O) δ 8.00 (d, J = 8.1 Hz, 1H), 6.03 (d, J = 5.7 Hz, 1H), 6.00 (d, J = 8.1 Hz, 1H), 4.44 (m, 2H), 4.31 (m, 1H), 4.26 (m, 2H), 3.69 (d, J = 11.7 Hz, 3H); 13P NMR (D2O) δ −8.70 (d, J = 17.7 Hz), −10.59 (d, J = 18.9 Hz), −22.46; HRMS-EI found 576.9375 (M−H)−. C10H17N2O18P4 requires 576.9427; purity > 99% by HPLC (retention time: 18.8 min).
Uridine-5′-methyl(C-P)-tetraphosphate triethylammonium salt (27). Procedure B
Compound 27 (8.6 mg, 34%) was obtained as a white solid using uridine triphosphate (22 mg, 0.04 mmol) and methylphosphoric acid (22 mg, 0.23 mmol). 1H NMR (D2O) δ 7.98 (d, J = 8.4 Hz, 1H), 6.02 (d, J = 5.4 Hz, 1H), 5.99 (d, J = 8.4 Hz, 1H), 4.43 (m, 2H), 4.28 (m, 1H), 4.25 (m, 2H), 1.49 (d, J = 17.1 Hz, 3H); 13P NMR (D2O) δ 19.05, −10.57 (d, J = 18.3 Hz), −22.34 (d, J = 18.9 Hz); HRMS-EI found 560.9476 (M−H)−. C10H17N2O17P4 requires 560.9478; purity > 99% by HPLC (retention time: 19.2 min).
Uridine-5′-(2-cyanoethyl)-tetraphosphate triethylammonium salt (28). Procedure B
Compound 28 (2.5 mg, 13%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and 2-cyanoethyl phosphate (35 mg, 0.11 mmol). 1H NMR (D2O) δ 7.98 (d, J = 8.1 Hz, 1H), 5.99 (m, 2H), 4.41 (m, 2H), 4.29 (m, 1H), 4.24 (m, 2H), 4.18 (t, J = 6.2 Hz, 2H), 2.88 (t, J = 6.2 Hz, 2H); 13P NMR (D2O) δ −10.54 (d, J = 17.7 Hz), −10.83 (d, J = 18.2 Hz), −22.39; HRMS-EI found 639.9489 (M+Na−H)−. C12H19N3O18P4Na requires 639.9512; purity > 99% by HPLC (retention time: 19.4 min).
Uridine-5′-α β-glycerol-tetraphosphate triethylammonium salt (29). Procedure B
Compound 29 (2.1 mg, 11%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and D-L-α-glycerol phosphate (35 mg, 0.11 mmol). 1H NMR (D2O) δ 7.99 (d, J = 7.8 Hz, 1H), 6.01 (m, 2H), 4.44 (m, 2H), 4.30 (m, 1H), 4.26 (m, 2H), 4.01 (m, 3H), 3.67 (m, 2H); 13P NMR (D2O) δ −9.83 (d, J = 17.7 Hz), −10.55 (d, J = 18.3 Hz), −22.40; HRMS-EI found 636.9638 (M−H)−. C12H21N2O20P4 requires 636.9638; purity > 99% by HPLC (retention time: 18.9 min).
Uridine-5′-phenyl-tetraphosphate triethylammonium salt (30). Procedure B
Compound 30 (1.1 mg, 6%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and phenyl phosphate (28 mg, 0.11 mmol). 1H NMR (D2O) δ 7.92 (d, J = 7.8 Hz, 1H), 7.37 (t, J = 7.2 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.17 (t, J = 7.5 Hz, 1H), 5.93 (m, 2H), 4.37 (m, 1H), 4.31 (m, 1H), 4.21 (m, 3H); 13P NMR (D2O) δ −10.51 (d, J = 17.7 Hz), −14.80 (d, J = 17.1 Hz), −22.43; HRMS-EI found 638.9577 (M−H)−. C15H19N2O18P4 requires 638.9583; purity > 99% by HPLC (retention time: 18.6 min).
Uridine-5′-cyclohexane-tetraphosphate triethylammonium salt (31). Procedure B
Compound 31 (6.7 mg, 20%) was obtained as a white solid using uridine triphosphate (20 mg, 0.036 mmol) and cyclohexene monophosphate tributylammonium salt (20 mg, 0.037 mmol). 1H NMR (D2O) δ 8.02 (d, J = 8.1 Hz, 1H), 6.03 (m, 2H), 4.42–4.50 (m, 2H), 4.22–4.34 (m, 4H), 2.01 (m, 2H), 1.74 (m, 2H), 1.33 (m, 6H); 13P NMR (D2O) δ −11.26 (m), −23.10 (m); HRMS-EI found 645.0037 (M−H)−. C15H25N2O18P4 requires 645.0053; purity > 99% by HPLC (retention time: 19.9 min).
Uridine-5′-fructose-6′-tetraphosphate triethylammonium salt (33). Procedure B
Compound 33 (4.2 mg, 19%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and D-fructose-6-phosphate (51 mg, 0.12 mmol). 1H NMR (D2O) δ 7.98 (d, J = 8.3 Hz, 1H), 6.02 (d, J = 5.4 Hz, 1H), 5.99 (d, J = 8.3 Hz, 1H), 4.43 (m, 2H), 4.32–4.06 (m, 7H), 3.95 (m, 1H), 3.63 (m, 1H), 3.56 (m, 1H); 13P NMR (D2O) δ −10.08 (d, J = 15.3 Hz), −10.54 (d, J = 18.3 Hz), −22.27; HRMS-EI found 724.9796 (M−H)−. C15H25N2O23P4 requires 724.9799; purity > 99% by HPLC (retention time: 19.5 min).
Uridine-5′-glucose-1′-tetraphosphate triethylammonium salt (34). Procedure B
Compound 34 (10 mg, 28%) was obtained as a white solid using uridine triphosphate (25 mg, 0.045 mmol) and D-glucose-1-phosphate (68 mg, 0.18 mmol). 1H NMR (D2O) δ 8.01 (d, J = 8.4 Hz, 1H), 6.02 (m, 2H), 5.66 (m, 1H), 4.45 (m, 2H), 4.32 (m, 1H), 4.28 (m, 2H), 4.01–3.77 (m, 4H), 3.55 (m, 1H), 3.46 (m, 1H); 13P NMR (D2O) δ −10.53 (d, J = 15.9 Hz), −11.90 (d, J = 16.5 Hz), −22.27; HRMS-EI found 724.9800 (M−H)−. C15H25N2O23P4 requires 724.9799; purity > 99% by HPLC (retention time: 18.9 min).
Uridine-5′-galactose-1′-tetraphosphate triethylammonium salt (35). Procedure B
Compound 35 (4.5 mg, 16%) was obtained as a white solid using uridine triphosphate (20 mg, 0.036 mmol) and D-galactose-1-phosphate (51 mg, 0.12 mmol). 1H NMR (D2O) δ 7.99 (d, J = 8.3 Hz, 1H), 6.03 (d, J = 5.7 Hz, 1H), 6.00 (d, J = 8.3 Hz, 1H), 5.67 (m, 1H), 4.44 (m, 2H), 4.26 (m, 4H), 4.05 (m, 1H), 3.98 (m, 1H), 3.77 (m, 3H); 13P NMR (D2O) δ −10.54 (d, J = 18.3 Hz), −11.74 (d, J = 18.2 Hz), −22.19; HRMS-EI found 724.9781 (M−H)−. C15H25N2O23P4 requires 724.9799; purity > 99% by HPLC (retention time: 19.0 min).
Uridine-5′-mannose-6′-tetraphosphate triethylammonium salt (36). Procedure B
Compound 36 (2.4 mg, 11%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and D-mannose-6-phosphate (23 mg, 0.08 mmol). 1H NMR (D2O) δ 7.97 (d, J = 8.4 Hz, 1H), 5.99 (m, 2H), 5.18 (m, 3/5H), 4.92 (m, 2/5H), 4.43 (m, 2H), 4.24 (m, 5H), 3.95–3.74 (m, 3H), 3.68 (m, 3/5H), 3.52 (m, 2/5H); 13P NMR (D2O) δ −9.92 (m), −10.46 (m), −22.20 (m); HRMS-EI found 724.9814 (M−H)−. C15H25N2O23P4 requires 724.9799; purity > 99% by HPLC (retention time: 18.6 min).
Uridine-5′-(2′-deoxy-glucose)-6′-tetraphosphate triethylammonium salt (37). Procedure B
Compound 37 (4.2 mg, 20%) was obtained as a white solid using uridine triphosphate (15 mg, 0.027 mmol) and 2′-deoxy-D-glucose-6-phosphate (20 mg, 0.08 mmol). 1H NMR (D2O) δ 7.99 (d, J = 7.8 Hz, 1H), 6.01 (m, 2H), 5.38 (m, 1/2H), 4.96 (m, 1/2H), 4.44 (m, 2H), 4.33–4.09 (m, 6H), 3.93 (m, 1/2H), 3.73 (m, 1/2H), 3.51 (m, 1H), 2.23 (m, 1/2H), 2.13 (m, 1/2H), 1.74 (m, 1/2H), 1.53 (m, 1/2H); 13P NMR (D2O) δ −11.60 (d, J = 14.7 Hz), −12.24 (d, J = 18.9 Hz), −23.58, −24.11 (t, J = 12.2 Hz); HRMS-EI found 708.9827 (M−H)−. C15H25N2O22P4 requires 708.9849; purity > 99% by HPLC (retention time: 18.5 min).
(2-Benzyl-1,3-dioxolo-4-yl)uridine 5′-monophosphate ammonium salt (54)
To a solution of uridine 5′-monophosphate (100 mg, 0.27 mmol) in TFA (1 mL) was added phenylacetaldehyde dimethylacetal (0.3 mL, 1.81 mmol). The reaction mixture was stirred at rt 4 h. After removal of the solvent, the residue was treated with 1 M NaHCO3 (4 mL) and AcOEt (2 mL). Aqueous phase was separated, evaporated and purified by Sephadex-DEAE A-25 resin as described above to obtain 54 (100 mg, 86%) as a white solid. 1H NMR (D2O) δ 7.92 (d, J = 8.3 Hz, 1H), 7.36 (m, 5H), 5.90 (d, J = 8.3 Hz, 1H), 5.63 (m, 1H), 5.47 (m, 1H), 4.95 (m, 1H), 4.90 (m, 1H), 4.42 (m, 1H), 3.89 (m, 2H), 3.16 (m, 2H); 13P NMR (D2O) δ 2.71; HRMS-EI found 425.0748 (M−H)−. C17H18N2O9P requires 425.0750; purity > 99% by HPLC (retention time: 12.9 min).
P1-((2-Benzyl-1,3-dioxolo-4-yl)uridine 5′) P3-(5-iodouridine 5′) triphosphate ammonium salt (39)
To a solution of 5-iodouridine 5′-diphosphate5 (10 mg, 0.017 mmol) in DMF (1 mL) was added CDI (7 mg, 0.04 mmol). The reaction mixture was stirred at rt for 6 h. Methanol (1 mL) was then added, and stirring was continued at rt for an additional 1 h. After removal of the solvent, the residue was dried in high vacuum overnight, was dissolved in DMF (2 mL), and was added compound 54 (11 mg, 0.025 mmol). The reaction mixture was stirred at rt overnight. After removal of the solvent, the residue was purified by Sephadex-DEAE A-25 resin as described above to obtain 39 (2.5 mg, 15%) as a white solid. 1H NMR (D2O) δ 8.17 (s, 1H), 7.77 (d, J=8.1 Hz, 1H), 7.35 (m, 5H), 5.88 (m, 2H), 5.53 (m, 1H), 5.44 (m, 1H), 4.92 (m, 2H), 4.45 (m, 1H), 4.31 (m, 2H), 4.19 (m, 5H), 3.13 (m, 2H); 13P NMR (D2O) δ −10.68 (d, J = 17.7 Hz), −10.97 (d, J = 20.2 Hz), −22.2; HRMS-EI found 936.9611 (M−H)−. C26H29N4O20P3I requires 936.9633; purity > 99% by HPLC (retention time: 18.5 min).
P1,P4-Di(uridine 5′-)β, γ-difluoromethylenetetraphosphate triethylammonium salt (41). Procedure B
Compound 41 (1.4 mg, 21%) was obtained as a white solid from uridine-5′-β, γ-difluoromethylenetriphosphate (3.2 mg, 0.0055 mmol) and uridine-5′-monophosphate (3.2 mg, 0.01 mmol). 1H NMR (D2O) δ 7.97 (br d, J = 6.1 Hz, 2H), 6.01 (m, 4H), 4.41 (m, 4H), 4.28 (m, 6H); 13P NMR (D 2O) δ −6.28 (m), −10.96 (m); HRMS-EI found 822.9910 (M−H)−. C19H25N4O22F2P4 requires 822.9879; purity > 99% by HPLC (retention time: 19.8 min).
P1,P4-Di(uridine 5′-)β, γ-dichloromethlyenetetraphosphate triethylammonium salt (42). Procedure B
Compound 42 (2.2 mg, 19%) was obtained as a white solid from uridine-5′-β, γ-dichloromethylenetriphosphate (5 mg, 0.009 mmol) and uridine-5′-monophosphate (6.4 mg, 0.019 mmol). 1H NMR (D2O) δ 7.99 (d, J = 8.1 Hz, 2H), 5.98 (m, 4H), 4.45 (m, 2H), 4.40 (m, 2H), 4.29 (m, 6H); 13P NMR (D2O) δ −1.61 (m), −10.94 (m); HRMS-EI found 854.9203 (M−H)−. C19H25N4O25Cl2P4 requires 854.9288; purity > 99% by HPLC (retention time: 20.0 min).
P1-(2-Thiouridine 5′-)-P4-(2′-deoxycytidine 5′-)tetraphosphate triethylammonium salt (43). Procedure B
Compound 43 (1.1 mg, 7.1%) was obtained as a white solid from 2-thio-uridine triphosphate tributylammonium salt, (14 mg, 0.013 mmol) and 2′-deoxycytidine mono phosphate tributylammonium salt (27 mg, 0.055 mmol). 1H NMR (D2O) δ 8.16 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 6.63 (d, J = 3.0 Hz, 1H), 6.31 (t, J = 6.6 Hz, 1H), 6.26 (d, J = 8.4 Hz, 1H), 6.15 (d, J = 7.2 Hz, 1H), 4.62 (m, 1H), 4.36 (m, 5H), 4.20 (m, 3H), 2.42 (m, 1H), 2.28 (m, 1H); 13P NMR (D2O) δ −11.10 (m), −22.94 (m); HRMS-EI found 787.9837 (M−H)−. C18H26N5O20P4S requires 787.9842; purity > 99% by HPLC (retention time: 19.5 min).
2′-Deoxy-2′-ureidouridine (46)
Benzotriazole-1-carboxamide17 (20 mg, 0.12 mmol) was added to a solution of 2′-amino-2′-deoxyuridine 57 (20 mg, 0.08 mmol) in DMF (4 mL). The reaction mixture was stirred at rt 6 h. After removal of the solvent, the residue was purified by preparative thin-layer chromatography (CH2Cl2-MeOH, 8:2) to obtain 46 (21 mg, 89%) as a white solid. 1H NMR (CD3OD) δ 7.99 (d, J = 8.3 Hz, 1H), 5.98 (d, J = 8.1 Hz, 1H), 5.71 (d, J = 8.3 Hz, 1H), 4.41 (m, 1H), 4.20 (m, 1H), 4.03 (m, 1H), 3.75 (m, 2H); HRMS-EI found 285.0859 (M−H)+. C10H13N4O6 requires 285.0835.
2′,3′,5′-Tri-O-acetyl-2-thiouridine (59)
A suspension of 2-thiouracil (2 g, 15.6 mmol) and trimethylsilyl chloride (1.8 mL) in hexamethyldisilazane (80 mL) was treated with a few crystals of ammonium sulphate and refluxed overnight. The clear greenish solution was evaporated and a solution of β-D-ribofuranose 1,2,3,5-tetraacetate (5.5 g, 17.3 mmol) in 20 mL dry dichloroethane was added. After a few minutes, stannic chloride (2.4 mL, 20.8 mmol) was added and after 1h, the mixture was poured into a saturated aq. NaHCO3 solution under vigorous stirring and then allowed to stand for 1 h. The suspension was filtered over a silica gel pad, which was washed with CH2Cl2. The organic layer was separated, dried over MgSO4 and evaporated to dryness. Silica gel chromatography (CH2Cl2:MeOH 99:1) yielded 4.75 g (79%) of compound 59. 1H NMR (DMSO-d6) δ10.64 (br s, 1H), 7.49 (d, J = 7.9 Hz, 1H), 6.68 (d, J = 3.8 Hz, 1H), 6.62 (dd, J = 2.3, 7.9 Hz, 1H), 5.45 (dd, J = 3.8, 5.6 Hz, 1H), 5.20 (app t, J = 5.7 Hz, 1H), 4.41–4.47 (m, 2H), 4.31–4.37 (dd, J = 3.2, 13.5 Hz, 1H), 2.09 (s, 6H), 2.06 (s, 3H); HRMS-EI found 387.08604 (M+H)+. C15H19N2O8S1 requires 387.08620.
2′,3′,5′-Tri-O-acetyl-2,4-dithiouridine (60)
To a solution of 59 (290 mg, 0.75 mmol) in dry toluene (10 mL), Lawesson’s reagent (304 mg, 0.75 mmol) was added. After heating the reaction mixture at 80°C overnight, insoluble materials were filtered off and the filtrate was purified by silica gel chromatography (Hex:EtOAc 65:35) yielding compound 60 as a yellow foam (250 mg, 83%). 1H NMR (DMSO-d6) δ 9.97, (brs, 1H), 7.65 (d, J = 8.2 Hz, 1H), 6.76 (d, J = 4.1 Hz, 1H), 5.98 (d, J = 8.2 Hz, 1H), 5.37 (dd, J = 4.1, 5.6 Hz, 1H), 5.16 (app t, J = 5.7 Hz, 1H), 4.34–4.40 (m, 2H), 4.25–4.30 (dd, J = 3.5,13.5 Hz, 1H), 2.09 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H); HRMS-EI found 403.06338 (M+H)+ C15H19N2O7S2. requires 403.06335.
2,4-Dithiouridine (47)
Compound 60 (240 mg, 0.60 mmol) was dissolved in dry methanol (15 mL) and the warmed solution was treated with 130 μL of a solution of NaOMe (30% w/w) in methanol. The reaction was refluxed for 4 h and then treated with diluted acetic acid to pH 5, evaporated to dryness and purified on a silica gel column (CH2Cl2:MeOH 93:7) to obtain compound 47 as a yellow foam (150 mg, 91%).1H NMR (DMSO-d6) δ 13.80 (br s, 1H), 8.07 (d, J = 7.7 Hz, 1H), 6.63 (d, J = 7.7 Hz, 1H), 6.40 (d, J = 2.7 Hz, 1H), 5.53 (d, J = 5.3 Hz, 1H), 5.29 (t, J = 5.0 Hz, 1H), 5.11 (d, J = 5.9 Hz, 1H), 4.08 (m, 1H), 3.93 (m, 2H), 3.72–3.78 (m, 1H), 3.58–3.64 (m, 1H); HRMS-EI found 275.01637 [M−H] −. C9H11N2O4S2 requires 275.01602.
1-(β-D-Arabinofuranosyl)-2-thio(1H)pyrimidin-4-one (48)
In a parr apparatus 2,2′-O-anhydrouridine (61, 2 g, 8.8 mmol) was dissolved in dry DMF (40 mL) and triethylamine (6 mL). The solution was saturated with H2S at −40°C and allowed to warm to rt resulting in a pressure of 200 psi. After stirring for two days, the remaining H2S was released and the solvent evaporated to dryness. The brown residue was purified on a silica gel column (CH2Cl2:MeOH 96:4), yielding compound 48 (1.6 g, 70%). 1H NMR (DMSO-d6) δ 12.59 (br s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 3.9 Hz, 1H), 5.94 (d, J = 8.4 Hz, 1H), 5.59 (d, J = 5.4 Hz, 1H), 5.48 (d, J = 3.9 Hz, 1H), 5.04 (t, J = 5.4 Hz, 1H), 4.18 (m, 1H,), 3.91 (m, 1H), 3.84 (m, 1H), 3.62 (app t, J = 5.1 Hz, 2H); HRMS-EI found 261.0540 (M+H)+. C9H13N2O5S1 requires 261.0545.
General Procedure for Synthesis of 2′-C-Methyl-uridine (49) and 3′-C-Methyl-uridine (50)
To dry uracil (0.45 g, 4 mmol) in dry 1,2-dichloroethane (20 mL) were added HMDS (0.68 ml, 0.8 eq.) and trimethylsilyl chloride (TMSCl, 0.3 mL, 0.8 eq.). The reaction mixture was heated at 80°C for 4 h in the absence of moisture. After cooling to rt, 1,2,3,5-tetra-O-benzoyl-2-C-methyl-β-D-ribofuranose (62)21 or 1,2,3-tri-O-acetyl-5-O-benzoyl-3-C-methyl-β-D-ribofuranose (63)22b (1 eq) in 1,2-dichloroethane (20 mL) was added followed by SnCl4 (0.93 mL, 2 eq.) dropwise. The mixture was stirred at rt for 4 h and quenched by NaHCO3 saturated water solution and extracted with CHCl3 (3 × 10 mL). The organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to give compounds 64 or 65 which were purified by chromatography on a silica gel column eluting with CHCl3. Compounds 64 and 65 (1 mmol) were treated with methanol (20 mL) saturated with ammonia at 0°C stirring at rt overnight. Evaporation of the solvent gave the desired compounds 49 and 50, which were purified by chromatography on a silica gel column.
1-(2,3,5-Tri-O-benzoyl-2-C-methyl-β-D-ribofuranosyl)uracil (64)
The title compound was obtained as a white foam (67% yield). 1H NMR (CDCl3) δ 9.35 (br s, 1H), 8.06 (d, J = 7.7 Hz, 4H.), 7.86 (d, J = 7.7 Hz, 2H), 7.4–7.6 (m, 10H), 6.48 (s, 1H), 5.76 (d, J = 5.7 Hz, 1H), 5.66 (d, J = 7.7 Hz, 1H), 4.82 (dd, J = 4.7, 6.2 Hz, 2H), 4.62 (m, 1H), 1.72 (s, 3H).
1-(2-C-Methyl-β-D-ribofuranosyl)uracil (49)
The title compound was obtained as a white foam after chromatography on a silica gel column eluting with CHCl3-MeOH (86:14) (80% yield). 1H NMR (DMSO-d6) δ 11.35 (br s, 1H), 8.05 (d, J = 8.1 Hz, 1H), 5.78 (s, 1H), 5.58 (d, J = 8.1 Hz, 1H), 5.15 (br s, 2H), 5.10 (s, 1H), 3.40–3.80 (m, 4H), 1.0 (s, 3H). 1-(2,3-Di-O-Acetyl-5-O-benzoyl-3-C-methyl-β-D-ribofuranosyl)uracil (65). The title compound was obtained as a white foam after chromatography on a silica gel column eluting with CHCl3-MeOH (86:14) (80% yield). 1H NMR (DMSO-d6) δ 11.35 (br s, 1H), 8.05 (d, J = 8.1 Hz, 1H), 5.78 (s, 1H), 5.58 (d, J = 8.1 Hz, 1H), 5.15 (br s, 2H), 5.10 (s, 1H), 3.40–3.80 (m, 4H), 1.0 (s, 3H).
1-(3-C-Methyl-β-D-ribofuranosyl)uracil (50)
The title compound was obtained as a white foam after chromatography eluting with CHCl3-MeOH (88:12) (82% yield). 1H NMR (DMSO-d6) δ 11.3 (br s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 5.85 (d, J = 8.1 Hz, 1H), 5.65 (d, J = 7.7 Hz, 1H), 5.33 (d, J = 6.6 Hz, 1H), 5.10 (t, J = 4.9 Hz, 1H), 4.72 (s, 1H), 3.85 (dd, J = 6.6, 7.7 Hz, 1H), 3.75 (pseudo t, 1H), 3.55 (m, 2H), 1.20 (s, 3H).
Assay of PLC activity stimulated by P2Y2, P2Y4, and P2Y6 receptors
Stable cell lines expressing the human P2Y2, P2Y4, or P2Y6 receptor in 1321N1 human astrocytoma cells were generated as described.3 Agonist-induced [3H]inositol phosphate production was measured in 1321N1 cells plated to 20,000 cells/well on 96-well plates two days prior to assay. Sixteen h before the assay, the inositol lipid pool of the cells was radiolabeled by incubation in 100 μL of serum-free inositol-free Dulbecco’s modified Eagle’s medium, containing 1.0 μCi of myo-[3H]inositol. No changes of medium were made subsequent to the addition of [3H]inositol. On the day of the assay, cells were challenged with 25 μL of the five-fold concentrated solution of receptor agonists in 200 mM Hepes (N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid), pH 7.3 in HBSS, containing 50 mM LiCl for 30 min at 37°C. Incubations were terminated by aspiration of the drug-containing medium and addition of 450 μL of ice-cold 50 mM formic acid. [3H]Inositol phosphate accumulation was quantified using scintillation proximity assay methodology as previously described in detail.24
Data Analysis
Agonist potencies (EC50 values) were determined from concentration-response curves by non-linear regression analysis using the GraphPad software package Prism (GraphPad, San Diego, CA). All experiments examining the activity of newly synthesized molecules also included full concentration effect curves for the cognate agonist of the target receptor: UTP for the P2Y2 receptor, UTP for the P2Y4 receptor, and UDP for the P2Y6 receptor. Each concentration of drug was tested in triplicate assays, and concentration effect curves for each test drug were repeated in at least three separate experiments with freshly diluted molecule. The results are presented as mean ± SEM from multiple experiments or in the case of concentration effect curves from a single experiment carried out with triplicate assays that were representative of results from multiple experiments.
Supplementary Material
NMR spectra and HPLC traces for selected derivatives are available.
Chart 1.

Agonists of the P2Y2 and P2Y6 receptors.
Acknowledgments
Mass spectral measurements were performed by Dr. Noel Whittaker and NMR by Wesley White (NIDDK). We thank Dr. Andrei A. Ivanov (NIDDK) for helpful discussions. This research was supported in part by research grant GM38213, by the Intramural Research Programs of NIDDK, National Institutes of Health, and by MIUR (Italian Ministry for the Research and University) (PRIN 2006).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagalli M, King BF, Gachet C, Jacobson KA, Weisman GA. Pharmacol Rev. 2006;58:281. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunschweiger A, Müller CE. P2 Receptors Activated by Uracil Nucleotides - An Update. Current Med Chem. 2006;13:289. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 3.Nicholas RA, Lazarowski ER, Watt WC, Li Q, Harden TK. Mol Pharmacol. 1996;50:224. [PubMed] [Google Scholar]
- 4.El-Tayeb A, Qi A, Müller CE. J Med Chem. 2006;49:7076. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 5.Besada P, Shin DH, Costanzi S, Ko H, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. J Med Chem. 2006;49:5532. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg A-K, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 2002;45:208. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanov AA, Ko H, Cosyn L, Maddileti S, Besada P, Fricks I, Costanzi S, Harden TK, Van Calenbergh S, Jacobson KA. J Med Chem. 2007;50:1166. doi: 10.1021/jm060903o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa M, Marquez VE, Harden TK, Jacobson KA. J Med Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR. Purinergic Signalling. 2005;1:183. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport RJ, Diaz P, Galvin FC, Lloyd S, Mack SR, Owens R, Sabin V, Wynn J Bioorg Med Chem Lett. 2007;17:558. doi: 10.1016/j.bmcl.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 11.Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC. J Pharmacol Exp Ther. 2002;302:871. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- 12.Korcok J, Raimundo LN, Du X, Sims SM, Dixon SJ. J Biol Chem. 2005;280:16909. doi: 10.1074/jbc.M410764200. [DOI] [PubMed] [Google Scholar]
- 13.Atkinson B, Dwyer K, Enjyoji K, Robson SC. Blood Cells Mol Dis. 2006;36:217. doi: 10.1016/j.bcmd.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 14.Ong SP, McFarlan SC, Hogenkamp HPC. Biochemistry. 1993;32:11397. doi: 10.1021/bi00093a017. [DOI] [PubMed] [Google Scholar]
- 15.Ong SP, Nelson LS, Hogenkamp HPC. Biochemistry. 1992;31:11210. doi: 10.1021/bi00160a035. [DOI] [PubMed] [Google Scholar]
- 16.Yoshikawa M, Kato T, Takenishi T. Tetrahedron Lett. 1967;50:5065. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 17.Katritzky AR, Kirichenko N, Rogovoy BV. ARKIVOC. 2003;viii:8. [Google Scholar]
- 18.Dunkel M, Cook DP, Acevedo OL. J Heterocyclic Chem. 1993;30:1421. [Google Scholar]
- 19.(a) Cherkasov RA, Kutyrev GA, Pudovik AN. Tetrahedron. 1985;41:2567. [Google Scholar]; (b) Cava MP, Levinson MI. Tetrahedron. 1985;41:5061. [Google Scholar]
- 20.Sekiya T, Ukita T. Chem Pharm Bull. 1967;15:1498. doi: 10.1248/cpb.15.1498. [DOI] [PubMed] [Google Scholar]
- 21.Wolfe MS, Harry-O’kuru RE. Tetrahedron Lett. 1995;36:7611. [Google Scholar]
- 22.(a) Mikhailov SN, Beigelman LN, Gurskaya GV, Padyukova NSh, Yakovlev GI, Karpeisky MYa. Carbohydr Res. 1983;124:75. [Google Scholar]; (b) Franchetti P, Cappellacci L, Pasqualini M, Petrelli R, Vita P, Jayaram HN, Horvath Z, Szekeres T, Grifantini M. J Med Chem. 2005;48:4983. doi: 10.1021/jm048944c. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson KA, Costanzi S, Ivanov AA, Tchilibon S, Besada P, Gao ZG, Maddileti S, Harden TK. Biochem Pharmacol. 2006;71:540. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Methods in Enzymology. 2006;406:489. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- 25.Spelta V, Mekhalfia A, Rejman D, Thompson M, Blackburn GM, North RA. Br J Pharmacol. 2003;140:1027. doi: 10.1038/sj.bjp.0705531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko H, Fricks I, Ivanov AAT, Harden TK, Jacobson KA. J Med Chem. 2007;50:2030. doi: 10.1021/jm061222w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palaniappan KK, Gao ZG, Ivanov AA, Greaves R, Adachi H, Besada P, Kim H, Kim A, Choe S, Jeong L, Jacobson KA. Biochemistry. 2007;46:7437. doi: 10.1021/bi7001828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 2002;45:2090. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cappellacci L, Franchetti P, Pasqualini M, Petrelli R, Vita P, Lavecchia A, Novellino E, Costa B, Martini C, Klotz KN, Grifantini M. J Med Chem. 2005;48:1550. doi: 10.1021/jm049408n. [DOI] [PubMed] [Google Scholar]
- 30.Maione S, de Novellis V, Cappellacci L, Palazzo E, Vita D, Luongo L, Stella L, Franchetti P, Marabese I, Rossi F, Grifantini M. Pain. 2007;131:281. doi: 10.1016/j.pain.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Gómez-Villafuertes R, Javier Gualix J, Miras-Portugal MT, Pintor J. Neuropharmacology. 2000;39:2381. doi: 10.1016/s0028-3908(00)00070-8. [DOI] [PubMed] [Google Scholar]
- 32.Manfredini S, Solaroli N, Angusti A, Nalin F, Durini E, Vertuani S, Pricl S, Ferrone M, Spadari S, Focher F, Verri A, De Clercq E, Balzarini J. Antiviral Chem Chemother. 2003;14:183. doi: 10.1177/095632020301400403. [DOI] [PubMed] [Google Scholar]
- 33.Kalek M, Jemielity J, Stepinski J, Stolarski R, Darzynkiewicz E. Tetrahedron Lett. 2005;46:2417. doi: 10.1081/ncn-200060103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NMR spectra and HPLC traces for selected derivatives are available.