

SUMMARY
Histone lysine residues can be mono-, di-, or trimethylated. These posttranslational modifications regulate the affinity of effector proteins and may also impact chromatin structure independent of their role as adaptors. In order to study histone lysine methylation, particularly in the context of chromatin, we have developed a chemical approach to install analogs of methyl lysine into recombinant proteins. This approach allows for the rapid generation of large quantities of histones in which the site and degree of methylation can be specified. We demonstrate that these methyl-lysine analogs (MLAs) are functionally similar to their natural counterparts. These methylated histones were used to examine the influence of specific lysine methylation on the binding of effecter proteins and the rates of nucleosome remodeling. This simple method of introducing site-specific and degree-specific methylation into recombinant histones provides a powerful tool to investigate the biochemical mechanisms by which lysine methylation influences chromatin structure and function.
INTRODUCTION
Cell-type specificity in multicellular organisms is an epigenetic phenomenon. Studies in model organisms implicate histone lysine methylation as particularly important for defining the epigenetic status of a cell. The ε-amine of lysine is subject to mono-, di-, or trimethylation. Each methylation state may have a distinct regulatory impact through modulating the binding of different effector proteins (Martin and Zhang, 2005; Sims et al., 2003). Consistent with this notion, plant homeodomains (PHD) in the NURF remodeling complex and in the tumor suppressor ING2 bind with specificity for trimethylated over dimethylated Lys4 of histone H3 (Li et al., 2006; Pena et al., 2006; Shi et al., 2006; Wysocka et al., 2006). The functional consequences of lysine methylation, in addition to being degree dependant, are also determined by the site of methylation (Lachner et al., 2003). For example, while trimethylation at Lys4 is associated with euchromatin and transcriptional activation (Santos-Rosa et al., 2002), trimethylation of H3 Lys9 is a well-established marker of heterochromatin and associated with transcriptional repression (Lee et al., 2005; Rea et al., 2000).
Recent years have seen the identification of numerous enzymes responsible for lysine methylation and demethylation, as well as downstream effectors that bind to specific methyl-lysine residues in histones (Grewal and Moazed, 2003; Martin and Zhang, 2005). For instance, the Lys9-specific methyltransferase SUV39H1 (in addition to its orthologs in other organisms) has been implicated in transcriptional silencing (Ivanova et al., 1998; Rea et al., 2000) and interacts genetically and biochemically with the heterochromatin-associated protein HP1α (and its orthologs). Indeed, Lys9 methylation is recognized by the chromodomain of HP1α, which itself recruits SUV39H1 and is believed to oligomerize, causing a repressive chromatin structure by spreading along the chromatin (Grewal and Moazed, 2003).
Methyl-lysine residues in nucleosomal histones are hypothesized to mediate interactions with the macromolecular complexes that regulate transcription, replication, and DNA repair. Investigating how lysine modifications influence the activity of these factors would be facilitated by a biochemical system that allows testing of specific methylation patterns on any histone. In particular, nucleosomes reconstituted fromhomogeneous preparations of recombinant histones, ideally with every possible methylation state at each site of interest, would allowsystematic examination of the events downstream of lysine methylation.
In order to develop such a system, we sought an efficient means to reconstitute nucleosomes with site-specific mono-, di- and trimethylation at positions throughout the entire sequence of each histone. Current methods to introduce methylation into recombinant histones include biosynthetic approaches or semisynthesis. The use of enzymes to methylate lysine residues is limited by the availability of specific methyltransferases. Even in cases where an appropriate methyltransferase is available, these reactions are difficult to drive to completion and can lead to uncontrolled degrees of methylation or heterogeneity with respect to site specificity.
Semisynthetic methods to construct modified histones using native chemical ligation have been reported (He et al., 2003; Shogren-Knaak et al., 2003; Shogren-Knaak and Peterson, 2004). This approach was instrumental in demonstrating a role for H4 Lys16 acetylation in antagonizing chromatin compaction (Shogren-Knaak et al., 2006), underscoring the utility of homogeneously modified histones for investigating the impact of lysine modifications on chromatin function. Nonetheless, the semisynthesis of modified histones is currently limited to modifications at only N-terminal residues (residues 1–30) and requires the synthesis of large quantities of modified peptide thioesters.
As an alternative to existing methods, we hypothesized that it would be possible exploit the latent reactivity of natural amino acid side chains to chemically generate histones with site- and degree-specific methylation. Methods to chemically methylate lysine residues, such as reductive alkylation (Means and Feeney, 1968), are not amenable to controlling which of the numerous histone-lysine residues are modified. Direct site-specific methylation of lysine residues does not appear to be a tractable solution. Cysteine is an attractive target for chemical elaboration due to its distinctive reactivity as a nucleophile. We were inspired by reports dating back half a century regarding the aminoethylation reaction: a cysteine, installed at the desired site of modification, can be alkylated to create aminoethylcysteine, an analog of lysine (see Figure 1A, reviewed in Kenyon and Bruice, 1977). Here, we report the extension of the traditional aminoethylation reaction to introduce N-methylated aminoethylcysteine residues (Figure 1B), thereby allowing the installation of methyl lysine analogs (MLAs). This scheme provides a simple and affordable route to large quantities of specifically methylated histones. The MLA strategy is compatible with the installation of modifications in the nucleosome core including histone H3 Lys79–a modification that, even using state-of-the-art semisynthesis strategies (Muralidharan and Muir, 2006), would be extremely challenging to access. We demonstrate that MLAs function similarly to their natural counterparts and are useful for the study of histone-lysine methylation’s influence on chromatin structure and function.
Figure 1. The Installation of Methyl-Lysine Analogs into Recombinant Proteins by Alkylating Cysteine Residues.

(A) The MLA strategy is an extension of the traditional aminoethylation reaction in which a cysteine is alkylated with an electrophilic ethylamine producing aminoethylcysteine, an analog of lysine.
(B) Cysteine residues can be converted into analogs of mono-, di-, and trimethylated lysine by treatment with alkylating agents 1-3, respectively.
(C) Incubating 2 under basic conditions leads to aziridinium formation. The resulting aziridinium can react with a cysteine residue leading to the desired dimethyl lysine analog.
RESULTS
Design of MLAs
Aminoethylcysteine has proven useful as an analog of lysine for chemical rescue and cysteine mapping applications (Kenyon and Bruice, 1977). For example, a protein that contains a cysteine can be alkylated with an electophillic ethylamine, and the product (aminoethylcysteine, see Figure 1A) can direct protealytic cleavage using a lysine-directed protease such as trypsin (for early examples, see Raftery and Cole, 1966 and references therein). Aminoethylcysteine is structurally and chemically similar to lysine; substituting the lysine γ-methylene with a sulfide causes only a slight lengthening of the side chain (~0.28 Å), and the electron withdrawing effect of the thioether causes only a small increase in the acidity (−1.1 pKa unit) of the ammonium protons (see Gloss and Kirsch, 1995 and references therein).
We reasoned that a related reaction would allow the introduction of analogs of methyl lysine into recombinant proteins. Indeed, the use of (2-bromoethyl)-trimethylammonium bromide (3) was developed as a reagent to block cysteine residues (Itano and Robinson, 1972) but to our knowledge has never been recognized as a useful analog of trimethyl lysine. To develop other reagents that selectively alkylate cysteine residues to generate MLAs, we initially synthesized 2-iodoethyl amines (data not shown). While these reagents convert cysteine to the desired MLAs, we found that the commercially available (2-haloethyl) amines 1–3 (Figure 1B) were similarly able to effect this conversion. While alkyl iodides are generally more reactive than alkyl chlorides and bromides, the high reactivity of (2-cholorethyl)-dimethylammonium chloride (2) can be attributed to an aziridinium intermediate formed under the reaction conditions (Figure 1C). The formation of this reactive intermediate decouples the rate-limiting step (alkylation of the cysteine) from the leaving of the halide leaving group. A similar mechanism has recently been characterized for (2-bromoethyl) ammonium bromide (Hopkins et al., 2005).
Fortuitously, the core nucleosome contains only a single conserved cysteine (H3C110) that can be mutated to alanine without disrupting nucleosome function. Therefore a unique cysteine can be installed at any position (e.g., H4K20C or H3K79C in the background of a C110A mutation) and, upon treatment with an appropriate alkylating agent, the introduced cysteine can be converted into an analog of monomethyl lysine, dimethyl lysine, or trimethyl lysine. Given that N-substituted aminoethylcysteine residues are lysine (K) analogs derived from cysteine (C), we refer to them with the abbreviation KC and otherwise follow standard abbreviations (Turner, 2005) for histone modifications (e.g., KC9me3).
Optimization of Conditions to Install MLAs into Proteins
To determine whether the chemistry to install MLAs can produce homogeneous products for biochemical analysis, we used model cysteine-containing peptides to find conditions that promote specificity for cysteine alkylation over other nucleophilic side chains, minimize variability caused by different sequence contexts, and proceed with high conversion. Controlling the pH of the reaction was critical. At pH < 7.5, cysteine residues are mostly protonated, and the reactions proceeded sluggishly. At pH > 8.5 side reactions were observed. However, reactions performed with reagents 1–3 using optimized buffer conditions, reaction time, temperature, and concentrations of the alkylating agents afford good conversion (>90%) of cysteine to the desired MLAs in the context of a model peptide (Figure 2A).
Figure 2. Cysteine Residues Can Be Efficiently Converted into MLAs in the Context of Peptides and Full-Length Proteins.

(A) A cysteine-containing peptide (Fluroesceine-KACR-OH) is converted to the desired lysine analog under optimized reaction conditions (see Experimental Procedures) as observed by reverse phase HPLC analysis of the crude reaction products. The traces are for the starting peptide (Cys); the unmethylated lysine analog, KC; the monomethylated lysine analog, KC(me1); the dimethylated lysine analog, KC(me2); and the trimethylated lysine analog, KC(me3).
(B) Mass spectra (ESI-oaTOF) demonstrate that MLAs can be cleanly installed at position 9 of full-length histone H3 protein. Starting with a K9C mutation (top left), this histone was treated under conditions to install analogs of monomethyl Lys9 H3 (top right), dimethyl Lys9 H3 (bottom left), or trimethyl Lys9 H3 (bottom right). The asterisk indicates a peak at +42 daltons corresponding to an artifact and is present in both the starting material and the methylated histone products.
(C) Representative ECD spectra of full-length histones acquired from a FT-ICR mass spectrometer are consistent with the installation of desired analogs at histone H3 residue 9.
Extending these conditions to convert cysteine to the desired analogs in the context of full-length histones rather than peptides required increasing the concentration of denaturant (to increase the accessibility of cysteine residues installed at core positions; see Figure S1A) and including free methionine to prevent low levels of oxidation observed in its absence (Figure S1B). With these improvements, the reaction pH, concentration of alkylating agent, time, and temperature were reoptimized to afford maximal conversion of cysteine residues to the desired MLAs (Figure S1). By monitoring the reactions by ESI-oaTOF mass spectrometry, these conditions were found to be robust, leading to high conversion without excessive alkylation (shown for histone H3K9C in Figure 2B). Although a second peak at +42 ± 1 Daltons was routinely observed in the product spectra, this peak was also observed in the starting material and is attributable to a mass spectrometry artifact; examination of the products on other mass spectrometers did not detect this contaminating species.
Comprehensive characterization of the proteins was carried out using electron capture dissociation (ECD) (Zubarev et al., 1998) of the intact H3 Lys9-modified histones on a Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR). Most of the observed peaks in ECD spectra feature the N-terminal (c ions) and the Cterminal (z ions) fragmentation ions. The fragment ion ladder around residue 9 (e.g., C62+, C72+, C82+, C93+, C103+; Figure 2C) indicates complete and homogenous incorporation of the desired modifications on residue 9. When comparing KC9me1, KC9me2, and KC9me3 histones, the mass increment of the MLA-containing ions (colored in magenta) equals the expected 14 amu, indicating they are mono-, di-, and trimethylated, respectively (Figure 2C).
These reaction conditions were found to be robust leading to clean installation of methyl-lysine analogs at every position that we have attempted to modify (see, for example, the modification of histones H3 K4C, H3K36C, and H4K20C shown in Figure S2). Given the routine bacterial expression of large quantities of histone mutants (Luger et al., 1999) and the low cost of alkylating reagents 1–3, the MLA approach allows simple and economic access to large quantities of near-homogeneously (>90%) methylated histones.
Functional Analysis of MLAs
Histone H3 methyl Lys9 analogs were used along with the other core histones to reconstitute histone octamers (Figure 3A, Coomassie). Not only could Lys9 analogs be incorporated into histone ocatmers but also anti-sera raised against natural mono-, di-, and trimethyl Lys9 specifically recognized the appropriate MLAs in western blots (Figure 3A). The ability of antisera to recognize MLAs was not limited to H3 Lys9; similar results were achieved at other positions (H3 Lys4, H3 Lys36), including positions in the nucleosome core (H3 Lys79) and on histone H4 (H4 Lys20) as shown in Figures 3B and 3C. Despite their exquisite sensitivity to the presence or absence of a single methyl group, these antibody preparations react strongly with the appropriate MLAs, indicating that the MLA side chains do not constitute a major perturbation to the natural methyl lysine epitopes, leading us to conclude that MLAs and natural methyl lysine residues are not easily distinguished in binding assays. In direct comparisons between peptides bearing K9me2 and KC9me2, we found that the antibody recognition was still specific, but that KC9me2 lead to a~5-fold weaker signal in a dot blot assay relative to a K9me2 peptide (Figure S3).
Figure 3. MLAs Can Be Incorporated into Histone Octamers and Are Specifically Recognized by Antibodies Raised against the Corresponding Natural Modifications.

(A) Western blot analysis of octamers assembled with MLAs at position 9 of histone H3.
(B) Similar analysis with MLAs incorporated at various core and tail positions.
(C) The positions of modifications in (A) and (B) mapped onto a model of the nucleosome (based on Luger et al., 1997).
To further probe the functional similarity between natural methylated lysine residues and MLAs, we examined HP1 binding to Lys9 modifications. The structural perturbation caused by replacing lysine γ-methylene with a sulfide was addressed by modeling a KC9me2 peptide into the crystal structure (Jacobs and Khorasanizadeh, 2002) of the HP1 chromodomain bound to a K9me2 peptide (Figure 4A). The minimal differences observed in the model of the MLA peptide compared with the natural peptide suggest the analog will bind similarly to HP1. To test this model, synthetic peptides were used as affinity reagents to enrich HP1α from a 293 nuclear extract (Figure 4B). While a K9 peptide did not interact with HP1α, both a K9me2 peptide and a KC9me2 peptide interacted strongly with HP1α. This result with model peptides raised the possibility that MLAs should also be functionally useful in the more complex case of MLA-bearing nucleosomes. Indeed, upon incorporation into biotinylated nucleosomes, KC9me2 enhanced interactions between the modified nucleosome and HP1α relative to an unmodified nucleosome (Figure 4C).
Figure 4. Methyl Lys9 Analogs Behave Similarly to Their Natural Counterparts in Binding Assays.
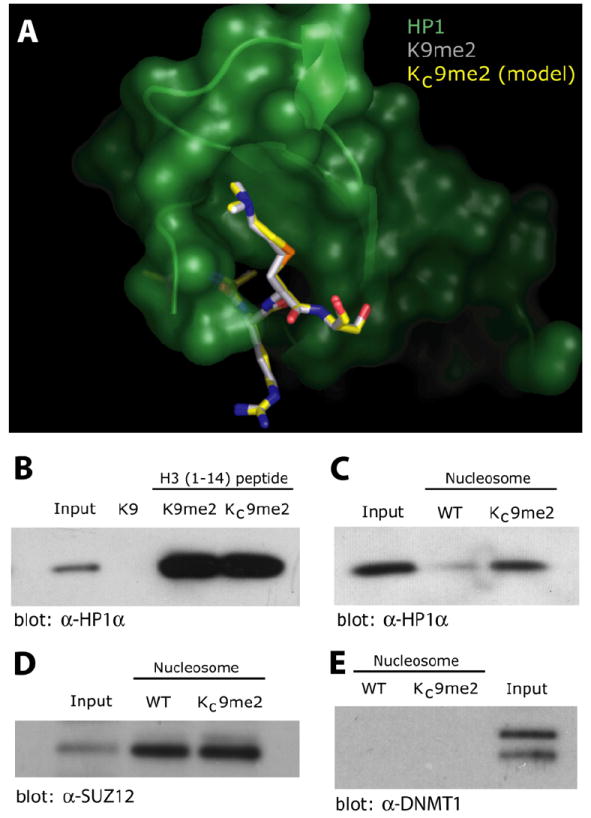
(A) Based on the crystal structure of HP1 (green) bound to a K9me2 peptide (gray backbone, PDB:1KNA, from Jacobs and Khorasanizadeh [2002], a model of a KC9me2 peptide (yellow backbone shown as overlay) bound to HP1 was constructed and minimized using MOLOC.
(B) Analysis of HP1α from 293 nuclear extracts binding to immobilized H3 tail peptides. Peptides (100 μg) with either unmodified lysine at position 9 (K9), with natural dimethylation at Lys9 (K9me2) or containing a dimethyl lysine analog, KC9me2, were used as affinity reagents and bound HP1α was monitored by western blot. In (B)–(E), input represent 5% of the starting nuclear extract.
(C) HP1α from nuclear extracts is specifically enriched upon binding to immobilized nucleosomes (100 pmol) assembled with H3KC9me2 relative to unmodified nucleosomes (WT).
(D) SUZ12 is enriched when using both KC9me2 modified and unmodified (WT) nucleosomes as affinity reagents.
(E) DNMT1 does not pull down using either KC9me2 modified or unmodified (WT) nucleosomes as affinity reagents.
We next used mononucleosomes containing KC9me2 to assay whether incorporation of MLA histones alters the affinity of nucleosomes for proteins in which histone methylation is not implicated in regulation of binding specificity. SUZ12 is important for nucleosome binding of the Polycomb Repressive Complex 2, histone-methyltransferase complex (Nekrasov et al., 2005). In affinity studies, SUZ12 did not distinguish between KC9me2 and wildtype nucleosomes (Figure 4D), indicating that incorporation of MLA histones does not nonspecifically impair nucleosome accessibility and suggesting that methylation at Lys9 does not regulate SUZ12’s nucleosome binding activity.
Histone H3 Lys9 methylation is implicated in regulation of DNA methylation (Tamaru et al., 2003). We examined whetherKC9me2-containing nucleosomes bound theDNA methyltransferase DNMT1. This protein was not enriched when pulling down with either Lys9-modified or -unmodified nucleosomes (Figure 4E).
Although MLAs behave similarly to their natural counterparts in binding assays, the sensitivity of chemical catalysis to small changes in the structure of the reaction’s transition state suggests that analog function in an enzymatic assay is a more stringent criterion for similarity. To test analog function in enzymatic reactions, peptides bearing either lysine or lysine analogs were examined as substrates for the Lys9-specific methyltransferase SUV39H1 (Figure 5A). In this assay, a peptide bearing aminoethylcysteine was nearly as efficient a substrate for methylation as an unmodified lysine peptide. Furthermore, K9me2 and KC9me2 substitutions both resulted in significantly lower SUV39H1 activity as was previously observed for a K9me2 peptide (Rea et al., 2000). Consistent with this trend, a KC9me1 peptide had an intermediate level of reactivity. We conclude that the lysine analogs behave in a manner functionally similar to natural lysine residues even in the geometrically and chemically demanding context of an enzymatic reaction important for epigenetic regulation.
Figure 5. Analysis of MLAs as Substrates in Enzymatic Assays.
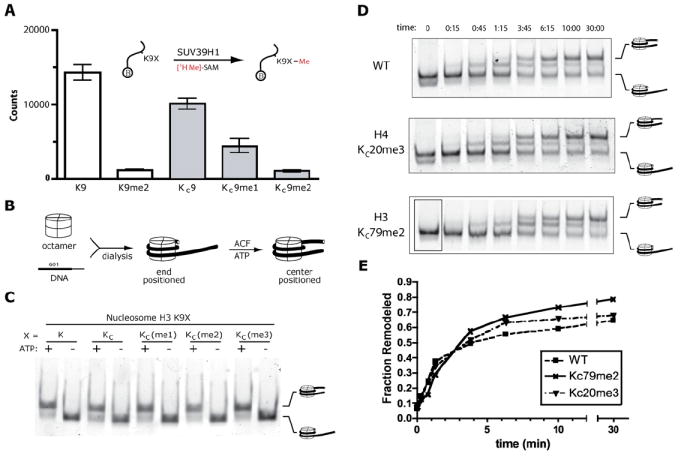
(A) The Lys9 methyltransferase activity SUV39H1 was examined using [3H-Me]-SAM and biotinylated H3 tail peptides (residues 1–14) containing either unmodified lysine at position 9 (K), dimethyl lysine (K9me2), aminoethylcysteine (KC9), the monomethyl lysine analog (KC9me1), or the dimethyl lysine analog (KC9me2). Data are shown as white bars for peptides with natural lysine residues and as gray bars for lysine analogs.
(B) Schematic depicting the assembly and hACF-mediated remodeling of positioned mononucleosomes.
(C) Similar levels of hACF-mediated ATP-dependent remodeling activity are observed using nucleosomes with or without MLAs. Upon native gel electrophoresis, the migration of centrally positioned nucleomsomes is retarded relative to the migration of end-positioned nucleosomes.
(D) The maximal rate of ACF-mediated nucleosome remodeling is not affected by KC20me3 or KC79me2 as demonstrated in a remodeling assay observed at several different time points.
(E) Graphical analysis of the results form (D) demonstrating similar rates of remodeling of wild-type and modified nucleosomes.
To test if nucleosomes prepared from histones with MLAs behave similarly to unmodified nucleosomes, salt dialysis was used to assemble histone octamers into end-positioned nucleosomes with DNA encoding a strong positioning sequence (Lowary and Widom, 1998). The resulting mononucleosomes were tested as substrates for the ATP-dependent remodeling complexh ACF (Figure5B). In the presence of ATP, hACF was able to remodel nucleosomes from their end position to a centered position on the DNA (Figure 5C). It is not surprising that the methyl Lys9 analogs have no apparent effect on the remodeling reaction; even complete removal of the H3 N-terminal tails does not substantially affect ACF activity (Eberharter et al., 2001). Nonetheless, similar remodeling of unmodified and MLA nucleosomes supports the conclusion that the chemical manipulation required to construct MLA histones does not have adverse effects on the histones, consistent with the clean installation of MLAs observed by mass spectrometry. Aside from the site of modification, nucleosomes with MLAs appear similar to unmodified nucleosomes in these assays.
While methylation at H3 K9 was not anticipated to affect the rates of ACF or ISWI-family nucleosome remodelers, H4 K20 is proximal to residues that interact with other members of the ISWI family (Clapier et al., 2001; Clapier et al., 2002; Hamiche et al., 2001). To test if trimethyaltion at histone H4 residue 20 affects the rate of ACF mononucleosome remodeling, we assembled end-positioned nucleosomes with H4 KC20me3 and compared their rate of ACF remodeling with that of wild-type nucleosomes under conditions of excess and saturating ACF. In this assay, no significant differences were observed (data not shown). In efforts to uncover more subtle effects of KC20me3 on remodeling, we repeated the remodeling assay and followed the progress of the reaction using a time course (Figure 5D). No significant differences were observed between the remodeling of H4 KC20me3 and wild-type nucleosomes. These data suggest that, despite the proximity of histone H4 K20 to residues that influence ACF activity, methylation at H4 K20 exerts its regulatory influence by a mechanism other than influencing the maximal rate of ACF nucleosome remodeling. The MLA strategy will be helpful for the investigation of other models connecting K20 methylation to transcriptional repression.
In addition to the potential regulatory roles exerted by posttranslational modifications of histone tails, it has been suggested that modifications of residues in the globular domains in the core of the nucleosome may influence nucleosome stability or ATP-dependent nucleosome remodeling (Cosgrove et al., 2004). The MLA approach allows the generation of nucleosomes specifically methylated at core residues to test these hypotheses. For example, we generated nucleosomes specifically dimethylated at position 79 of histone H3 and tested whether or not this modification affects the rate of remodeling by ACF. End-positioned H3 KC79me2 nucleosomes were remodeled with similar rates as wild-type nucleosomes (Figures 5D and 5E). In addition to demonstrating that KC79me2 does not influence ACF remodeling under these conditions, these results underscore the utility of MLA histones to test nucleosome-level properties of methylated nucleosomes (such as remodeling), especially nucleosomes methylated at residues within the globular core of the nucleosome that are largely inaccessible by other techniques such as native chemical ligation.
DISCUSSION
Understanding the impact of site-specific lysine mono-, di-, and trimethylation on transcriptional regulation, development and tumorigenesis requires biochemical analysis of the effects of these modifications on chromatin structure and function. In some cases, the primary sequence context immediately surrounding the site of methylation appears sufficient to recapitulate a biochemical role for the modification. In such cases, including the binding interactions of H3 Lys9me to HP1α (Nakayama et al., 2001) and Lys27me to Polycomb (Fischle et al., 2003; Min et al., 2003), peptides serve as useful models. In other contexts, however, it is likely that the role of the modification will require recognition elements present in the nucleosome but not necessarily nearby in the primary sequence. This is particularly likely for core modifications. Furthermore, for studies involving lysine modifications that directly affect nucleosome structure, mobility, or stability, peptide models are not applicable. The need for the technology described here is made apparent by recent reports demonstrating H3 Lys4 methylation functions, at least in part, by directly recruiting remodeling factors (Wysocka et al., 2006). Nucleosomes constructed using the MLA approach are useful for testing the regulatory influence of site-specific methylation on nucleosome-level biochemical activities, as we have demonstrated by testing the effects of methylation at positions 9 and 79 of histone H3 and position 20 of histone H4 on nucleosome remodeling (see Figures 5C, 5D, and 5E).
The MLA strategy described here provides an efficient and economical route to large quantities of recombinant methylated histones. The chemistry to install MLAs has been optimized to allow clean conversion of cysteine residues anywhere on the histones. The resulting analogs are stable except that, like methionine, they contain a thioether and are therefore susceptible to oxidation (i.e., sulfoxide formation). Standard precautions to avoid oxidizing conditions are sufficient to prevent these side reactions (Figure S1B).
While MLAs differ from their natural counterparts by replacement of a methylene with a sulfide, we have found that, similar to previous reports using aminoethylcysteine, this perturbation causes minimal impact on function in binding and enzymatic assays. While the extent to which the MLAs mimic their natural counterparts will be context dependent, we found that antibodies generated against K9me1, K9me2, K9me3, K20me1, K4me3, K36me3, and K79me2 all demonstrated specific recognition of the appropriate analogs (Figure 3), albeit in one case (i.e., K9me2) with approximately 5-fold lower activity (Figure S3). Nonetheless, the success with antibodies and the results from other functional assays support the conclusion that MLAs serve as a general means to study lysine methylation. For example, here, we examined the binding of candidate effector proteins and probed the influence of site-specific methylation on ATP-dependent nucleosome remodeling.
As the biochemical functions associated with each lysine methylation are elucidated, it will become increasingly important to study these modifications in combination. While the MLA strategy is not compatible with the installation of different degrees of methylation on the same histone subunit (e.g., H3 KC9me3/KC27me1), it is compatible with the installation of the same modification on different sites (e.g., H3 KC4me3/KC27me3) or different modifications on different subunits (e.g., H3 KC27me3/H4 KC20me1).
The biochemical analysis of the regulatory impact of specific histone lysine methylation marks has been largely limited to studying methylated peptides outside the context of nucleosomes. The MLA strategy facilitates the study of these important epigenetic marks in the context of nucleosomes. Ongoing studies include the use of MLAs to further investigate the role of lysine methylation in nucleosome remodeling studies, enzymatic assays, structural studies, compaction assays, and in vitro transcription, particularly with respect to understanding repressive chromatin structures. Using the MLA strategy, proteins bearing specific methylation are straightforward to generate; their broad use will aid in studying the events downstream of lysine methylation, thereby expanding our understanding of the biochemical mechanisms underlying epigenetic regulation.
EXPERIMENTAL PROCEDURES
Histones, Octamers, and Nucleosomes
Xenopus histones were expressed in E. coli and purified via gel filtration as previously described (Luger et al., 1997, 1999). All histone H3 constructs contain a C110A mutation, including those labeled as wild-type here. Lys-to-Cys mutants constructed using Quikchange mutagenesis (Stratagene) according to the manufacturer’s instructions. Histone octamers and nucleosomes were assembled according to previous reports (Luger et al., 1997, 1999). The DNA used for the assembly of mononucleosomes included the Widom 601 positioning sequence (Lowary and Widom, 1998) with 49 bp of additional DNA and was constructed by PCR using biotinylated and Cy3-labeled primers. Nucleosomes constructed using MLA histones were purified and stored in the presence of 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP).
Peptide Synthesis and Alkylation
Peptides were synthesized using Fmoc-protected amino acids according to standard methods. Fluoresceine-mPEG-GACR-OH, where Fl is Fluoresceine and mPEG is an ethylene glycol linker (building block: Fmoc-mini-PEG, Peptides International) and Biotin-mPEG-ARTKQTARXSTGGK- OH, where X is either cysteine, lysine, or dimethyl lysine as indicated. For peptide alkylations, HPLC-purified peptides were dissolved in guanidinium chloride (10 μl, 8 M) and diluted with HEPES buffer (For KC[me1] and KC[me2]: 85 μl, 1 M, pH 8; for KC[me3] 88 μl, 1M, pH 7.5) and DTT (2 μl of 1 M, dissolved immediately before use). The peptides were reduced for 1 hr at 37°C. Alkylating agents were added as follows. For KC(me1), (2-chloroethyl)-methylammonium chloride (1, Karl Industries, Inc.) was added (10 μl of 3 M, dissolved immediately before use) and the reaction was allowed to proceed for 4h. DTT was added (1 μl of 1 M) and after 30 min at rt, more alkylating agent was added (10 μl of 3 M). The reaction was allowed to proceed for 12 hr. For KC(me2), (2-chloroethyl)-dimethylammonium chloride (2, Aldrich) was added (10 μl of 1 M, dissolved immediately before use), and the reaction was allowed to proceed for 2 hr. DTT was added (1 μl of 1 M) and, after 30 min at room temperature, additional 2 was added (10 μl of 1 M). The reaction was then allowed to proceed for 2 hr. For KC(me3), (2-bromoethyl) trimethylammonium bromide (3, Aldrich) was added as a solid (10 mg), and the reaction was heated to 50°C in the dark. With occasional mixing, the solid dissolved slowly. After 2 hr, DTT was added (1 μl of 1 M), and the reaction was allowed to proceed for another 2 hr at 50°C. In all cases, the reactions were quenched with BME (5 μL, 14.2 M) and then analyzed or purified from the reaction mixture by analytical RP HPLC (A = H2O with 0.1% TFA; B = MeCN with 0.1% TFA). For HPLC of Fluoresceine-labeled peptides, absorbance was monitored at 430 nm. The identities of the products were confirmed by MALDI-TOF MS.
Installation of MLAs into Full-Length Proteins
Lyophilized histones (5–10 mg) were dissolved in alkylation buffer (1 M HEPES pH 7.8, 4 M guanadinium chloride, 10 mM D/L-methionine; for KC[me1], 900 μL; for KC[me2], 930 μL; for KC[me3], 980 μl), and DTT was added (20 μl of 1 M, dissolved immediately before use). The histones were reduced for 1 hr at 37°C. Alkylating agents were added as follows. All reactions were performed in the dark. For KC(me1), 1 was added (100 μl of 1 M, dissolved immediately before use), and the reaction was allowed to proceed for 4 hr at room temperature. DTT was added (10 μl of 1 M), and the reaction was allowed to proceed for at least 10 hr at room temperature. For KC(me2), 2 was added (50 μl of 1 M, dissolved immediately before use), and the reaction was allowed to proceed for 2 hr at room temperature. Then, DTT was added (10 μl of 1 M), the reaction incubated at room temperature for 30 min, treated with additional 2 (50 μL, 1M) and allowed to proceed for an additional 2 hr at room temperature. For KC(me3), 3 was added as a solid (100 mg), and the reaction was heated to 50°C. With occasional mixing, the solid slowly dissolved. After 2.5 hr, DTT was added (10 μl of 1 M), and the reaction was allowed to proceed for another 2.5 hr at 50°C. In all cases, the reactions were quenched with BME (50 μL, 14.2 M) and then purified from the reaction mixture using PD-10 columns pre-equilibrated with BME/H2O (3 mM). The concentration of the eluant was quantified using a protein concentration assay (BioRad), and the histones were aliquoted, lyophilized, and stored as frozen pellets.
Mass Spectrometry
Mass spectrometry was performed on the following instruments. Peptides were analyzed on an ABI Voyager Elite MALDI-TOF using α-cyano-4-hydroxycinnamic acid as matrix. Routine analysis of MLA reactions was preformed using α Waters Micromass LCT premire. For more comprehensive analysis, the intact histones were analyzed on a 7-tesla hybrid linear ion trap (LTQ FT) mass spectrometer (Thermo Electron Corp., Palo Alto, CA) with a nanoelectrospray ion source at a concentration of ~5 pmole/ml. ECD spectra were acquired with a resolution of 100,000 and an isolation window of m/z 20. The activation and delay time in ECD were 5 ms and 8 ms. FT transients were accumulated, and ECD fragment ions were assigned using the Fragment Assignment by Visual Assistance (FAVA) algorithm in the MATLAB environment.
Nucleosome-Remodeling Assays
Analysis of nucleosome remodeling by recombinant hACF was performed essentially as described (He et al., 2006). Briefly, reactions (10 μL) were performed in 12% glycerol, 60 mM KCl, 12 mM HEPES, 4 mM Tris, 3 mM MgCl2, 0.32 mM EDTA, 0.02% NP-40, and 0.4 mg/mL FLAG peptide (Strohner et al., 2005). Mononucleosomes (20 nM) assembled using Cy3-labeled DNA with the Widom 601 positioning sequence (Lowary and Widom, 1998), and 49 bp additional DNA were incubated with recombinant hACF (25 nM). Reactions were started with Mg•ATP (2 mM) and incubated at room temperature for 30 min before addition of stop buffer (2 μL, 0.8 mg/mL plasmid DNA, and 115 mM ADP). The entire 12 μl reaction was analyzed by native PAGE (5% acrylamide, 0.5X Tris borate-EDTA gel, 2 hr, 100 V), and the Cy3 fluorescence of the gel was visualized on an Amersham Typhoon 9400 variable mode imager at 580 nm. The time course experiments were performed similarly except the reactions were performed using subsaturating concentrations of ATP (4 μM Mg•ATP, see Yang et al., 2006), and the reactions were incubated at 30°C.
Affinity Studies
Nuclear extracts were prepared from 293 cells according to (Dignam et al., 1983) and then precleared with strepavidin-coated magnetic beads (1 mg beads per mL extract, Dynal M270). Biotinylated peptides (100 μg) or biotinylated nucleosomes (100 pmol) were diluted to 100 μl with wash buffer (100 mM KCl, 5 mM MgCl2, 20 mM HEPES at pH 7.6, 5% glycerol, 0.1 mg/mL BSA) and incubated with prewashed strepavidin-coated magnetic beads (0.5 mg) for 30 min with mixing. The beads were washed three times with wash buffer. The precleared nuclear extract was added (20 μL), and the bead suspension was mixed for 1 hr at 4°C. The beads were captured, the supernatant was aspirated, and the beads rinsed three times with wash buffer. The bound material was eluted with loading buffer, boiled for 5 min, resolved by denaturing PAGE (5%–20%), transferred to a PVDF membrane, and analyzed by western blot.
Coimmunoprecipitation and HMTase Assays
Human SUV39H1 cDNA was cloned by PCR from HEK293 cDNA, verified by sequencing, and subsequently fused to a GFP tag in the pEGFP-C3 vector (Clontech). The plasmid was transfected into HEK293 cells (5 × 106/10-cm plate) using FuGENE6 (Roche Applied Science) according to manufacturer’s conditions. Forty-eight hours after transfection, HEK293 cells were processed for coimmunoprecipitation as described (Chu et al., 2006). The immunoprecipitation reactions were performed at 4°C overnight. The resulting immunoprecipitates were then subjected to HMTase assays essentially as described (Rea et al., 2000). S-Adenosyl-L-(methyl-3H) Methionine (74.0 Ci/nmol, Amersham) was used as the methyl donor, and 4 μg of biotinylated N-terminal peptides were used as the substrate. After the HMTase assay, peptides were spotted onto SAM2 Biotin Capture Membranes (Promega), washed, and subjected to scintillation counting as described (Lindroth et al., 2004).
Supplementary Material
Acknowledgments
We thank Hiten Madhani for critical reading of this manuscript, Janet Yang for providing ACF, Shenheng Guan for making available his FAVA algorithm, and Zack Knight and other members of the Shokat Lab for helpful comments and suggestions. We thank Sami Mahrus and the Wells Laboratory (UCSF) for assistance and use of their ESI-oaTOF mass spectrometer. Funding was provided by the Volkswagon Foundation and the University of California Office of the President, Dissertation Year Fellowship (to CCC). The UCSF MS Facility is supported by the Biomedical Research Technology Program, NIH NCRR RR001614, and NIH NCRR RR019934.
Footnotes
Supplemental Data include three figures, Supplemental Experimental Procedures, and Supplemental References and can be found with this article online at http://www.cell.com/cgi/content/full/128/5/1003/DC1/.
References
- Clapier CR, Langst G, Corona DF, Becker PB, Nightingale KP. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol Cell Biol. 2001;21:875–883. doi: 10.1128/MCB.21.3.875-883.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Nightingale KP, Becker PB. A critical epitope for substrate recognition by the nucleosome remodeling ATPase ISWI. Nucleic Acids Res. 2002;30:649–655. doi: 10.1093/nar/30.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol Cell Proteomics. 2006;5:194–203. doi: 10.1074/mcp.M500285-MCP200. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Ferrari S, Langst G, Straub T, Imhof A, Varga-Weisz P, Wilm M, Becker PB. Acf1, the largest subunit of CHRAC, regulates ISWI-induced nucleosome remodelling. EMBOJ. 2001;20:3781–3788. doi: 10.1093/emboj/20.14.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloss LM, Kirsch JF. Decreasing the basicity of the active site base, Lys-258, of Escherichia coli aspartate aminotransferase by replacement with gamma-thialysine. Biochemistry. 1995;34:3990–3998. doi: 10.1021/bi00012a017. [DOI] [PubMed] [Google Scholar]
- Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Hamiche A, Kang JG, Dennis C, Xiao H, Wu C. Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc Natl Acad Sci USA. 2001;98:14316–14321. doi: 10.1073/pnas.251421398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Bauman D, Davis JS, Loyola A, Nishioka K, Gronlund JL, Reinberg D, Meng F, Kelleher N, McCafferty DG. Facile synthesis of site-specifically acetylated and methylated histone proteins: reagents for evaluation of the histone code hypothesis. Proc Natl Acad Sci USA. 2003;100:12033–12038. doi: 10.1073/pnas.2035256100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Fan HY, Narlikar GJ, Kingston RE. Human ACF1 alters the remodeling strategy of SNF2h. J Biol Chem. 2006;281:28636–28647. doi: 10.1074/jbc.M603008200. [DOI] [PubMed] [Google Scholar]
- Hopkins CE, Hernandez G, Lee JP, Tolan DR. Aminoethylation in model peptides reveals conditions for maximizing thiol specificity. Arch Biochem Biophys. 2005;443:1–10. doi: 10.1016/j.abb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Itano HA, Robinson EA. 4-Thialaminine, a strongly basic chemical modification of cysteine. J Biol Chem. 1972;247:4819–4824. [PubMed] [Google Scholar]
- Ivanova AV, Bonaduce MJ, Ivanov SV, Klar AJ. The chromo and SET domains of the Clr4 protein are essential for silencing in fission yeast. Nat Genet. 1998;19:192–195. doi: 10.1038/566. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- Kenyon GL, Bruice TW. Novel sulfhydryl reagents. Methods Enzymol. 1977;47:407–430. doi: 10.1016/0076-6879(77)47042-3. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- Lee DY, Teyssier C, Strahl BD, Stallcup MR. Role of protein methylation in regulation of transcription. Endocr Rev. 2005;26:147–170. doi: 10.1210/er.2004-0008. [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroth AM, Shultis D, Jasencakova Z, Fuchs J, Johnson L, Schubert D, Patnaik D, Pradhan S, Goodrich J, Schubert I, et al. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 2004;23:4286–4296. doi: 10.1038/sj.emboj.7600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Means GE, Feeney RE. Reductive alkylation of amino groups in proteins. Biochemistry. 1968;7:2192–2201. doi: 10.1021/bi00846a023. [DOI] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan V, Muir TW. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Nekrasov M, Wild B, Muller J. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 2005;6:348–353. doi: 10.1038/sj.embor.7400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery MA, Cole RD. On aminoethylation of proteins. J Biol Chem. 1966;241:3457–3461. [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407– 411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak MA, Peterson CL. Creating designer histones by native chemical ligation. Methods Enzymol. 2004;375:62–76. doi: 10.1016/s0076-6879(03)75004-6. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak MA, Fry CJ, Peterson CL. A native peptide ligation strategy for deciphering nucleosomal histone modifications. J Biol Chem. 2003;278:15744–15748. doi: 10.1074/jbc.M301445200. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Strohner R, Wachsmuth M, Dachauer K, Mazurkiewicz J, Hochstatter J, Rippe K, Langst G. A ‘loop recapture’ mechanism for ACF-dependent nucleosome remodeling. Nat Struct Mol Biol. 2005;12:683–690. doi: 10.1038/nsmb966. [DOI] [PubMed] [Google Scholar]
- Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng X, Selker EU. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12:110– 112. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;13:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.