

Abstract
Biochemical and genetic data suggest that synaptotagmin-2 functions as a Ca2+ sensor for fast neurotransmitter release in caudal brain regions, but animals and/or synapses lacking synaptotagmin-2 have not been examined. We have now generated mice in which the 5′ end of the synaptotagmin-2 gene was replaced by lacZ. Using β-galactosidase as a marker, we show that, consistent with previous studies, synaptotagmin-2 is widely expressed in spinal cord, brainstem, and cerebellum, but is additionally present in selected forebrain neurons, including most striatal neurons and some hypothalamic, cortical, and hippocampal neurons. Synaptotagmin-2-deficient mice were indistinguishable from wild-type littermates at birth, but subsequently developed severe motor dysfunction, and perished at ∼3 weeks of age. Electrophysiological studies in cultured striatal neurons revealed that the synaptotagmin-2 deletion slowed the kinetics of evoked neurotransmitter release without altering the total amount of release. In contrast, synaptotagmin-2-deficient neuromuscular junctions (NMJs) suffered from a large reduction in evoked release and changes in short-term synaptic plasticity. Furthermore, in mutant NMJs, the frequency of spontaneous miniature release events was increased both at rest and during stimulus trains. Viewed together, our results demonstrate that the synaptotagmin-2 deficiency causes a lethal impairment in synaptic transmission in selected synapses. This impairment, however, is less severe than that produced in forebrain neurons by deletion of synaptotagmin-1, presumably because at least in NMJs, synaptotagmin-1 is coexpressed with synaptotagmin-2, and both together mediate fast Ca2+-triggered release. Thus, synaptotagmin-2 is an essential synaptotagmin isoform that functions in concert with other synaptotagmins in the Ca2+ triggering of neurotransmitter release.
Keywords: asynchronous release, endplate, neuromuscular junction, striatum, synapse, synaptotagmin
Introduction
Neurotransmitter release from presynaptic nerve terminals is triggered when an action potential gates Ca2+ influx into the terminal, and Ca2+ induces exocytosis of synaptic vesicles (Katz and Miledi, 1967). Neurotransmitter release occurs in two modes: fast synchronous release that is induced by brief transients of high Ca2+ concentrations, and slow asynchronous release that is induced at a slower rate by lower Ca2+ concentrations (Barrett and Stevens, 1972; Goda and Stevens, 1994; Cummings et al., 1996; Atluri and Regehr, 1998; Lu and Trussell, 2000; Hagler and Goda, 2001; Otsu et al., 2004). In the forebrain, synaptotagmin-1 functions as the Ca2+ sensor for fast synchronous release (Geppert et al., 1994; Fernandez-Chacon et al., 2001). Synaptotagmin-1 belongs to a large family of proteins (15 members in mouse) that contain similar domain structures, with an N-terminal transmembrane region, a linker sequence, and two C-terminal C2 domains that bind Ca2+ in most but not all synaptotagmins (for review, see Südhof, 2002). Among synaptotagmins, synaptotagmin-2 shares the highest homology with synaptotagmin-1, has similar biochemical characteristics, and can functionally replace synaptotagmin-1 in neurons and chromaffin cells that lack synaptotagmin-1 (Stevens and Sullivan, 2003; Nagy et al., 2006). These experiments suggested that synaptotagmin-1 and -2 have similar functions. However, synaptotagmin-1 and -2 are not entirely functionally redundant because synaptotagmin-1 knock-out (KO) mice express normal levels of synaptotagmin-2 but nevertheless exhibit a severe phenotype that in hippocampal and cortical neurons manifests as a loss of fast synchronous release and perinatal lethality (Geppert et al., 1994; Nishiki and Augustine, 2004; Maximov and Südhof, 2005). A possible explanation for this finding is based on the differential expression of synaptotagmin-1 and -2, with the former being present primarily in forebrain and the latter in caudal brain regions (Geppert et al., 1994; Ullrich et al., 1994; Marqueze et al., 1995). These expression patterns suggested that synaptotagmin-1 and -2 may have similar functions in different types of neurons, thereby making each essential in the type of neuron in which they are expressed.
Recent studies on a mutant mouse carrying a point mutation in synaptotagmin-2 confirmed the importance of synaptotagmin-2 in neurotransmitter release in brainstem synapses and in neuromuscular junctions (NMJs) (Pang et al., 2006). However, in these mice, the mutant synaptotagmin-2 was functional, and the mice survived, raising the question whether synaptotagmin-2 normally functions as an essential Ca2+ sensor similar to synaptotagmin-1, or performs a more ancillary, possibly regulatory role. To address this question, we have generated and analyzed synaptotagmin-2 KO mice that also allowed us to characterize synaptotagmin-2 expression by means of a knocked-in lacZ sequence. We find that synaptotagmin-2 is abundantly expressed in caudal brain neurons and in restricted populations of forebrain neurons. Using electrophysiological studies, we demonstrate that Ca2+-triggered neurotransmitter release is impaired in synaptotagmin-2-deficient striatal neurons and NMJs. However, this impairment was less severe than that of cortical and hippocampal neurons lacking synaptotagmin-1. Consistent with this unexpectedly limited release phenotype, we observed coexpression of synaptotagmin-1 with synaptotagmin-2 in NMJs, suggesting that, at least in NMJs, Ca2+ triggering of release is driven by both synaptotagmin-1 and -2.
Materials and Methods
Generation and maintenance of synaptotagmin-2 knock-out mice.
The targeting vector (see Fig. 1A) was constructed using routine techniques. The homology arms were obtained from a genomic clone containing the entire synaptotagmin-2 gene (GenBank accession number AF257303) isolated from a 129 SVJ genomic library (Lambda Fix II; Stratagene, La Jolla, CA) using the cDNA for synaptotagmin-2 as template for the probe (Baram et al., 1999). The 5′ arm is a 3.8 kb BglII–NcoI fragment that contains exon 1 and part of exon 2, and the 3′ arm is a 3.1 kb BamHI–SacII fragment that includes exon 8. The poliovirus internal ribosomal entry site (IRES) was cloned from pSBC-1 (Dirks et al., 1993), lacZ with a nuclear localization signal from pPD 46.21 (Goldhamer et al., 1992), and PGK-Neo and MC1-TK from NTKV 1907 (pKO Scrambler; Lexicon, The Woodlands, TX). The construct was electroporated into R1 embryonic stem (ES) cells. Homologous recombination events were enriched under Geneticin (Invitrogen, San Diego, CA) and FIAU selection and confirmed by Southern blot of SalI-digested genomic DNA using as template for the probe a fragment upstream of the 5′ arm obtained from a BglII digest (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Chimeric males were generated by injection of selected ES cell clones into C57BL/6 blastocysts, and then used to found mutant lines. PCR with primers (P1, AGAAGACATGTTCGTCCAAGC; P2, TCATGTTCATGGCGTTCTTC; and P3, ACGGACACCCAAAGTAGTCG) was used to detect the wild-type (WT) (P1 plus P2 product, ∼1 kb) and mutant (P1 plus P3 product, ∼1.5 kb) synaptotagmin-2 allele and to genotype wild-type, heterozygous, and homozygous mice (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Mutant animals were propagated as heterozygous colonies in a barrier facility while being backcrossed for nine generations into a C57BL/6 background before generating homozygous animals by heterozygous crossings.
Figure 1.

Generation of synaptotagmin-2 KO mice by homologous recombination. A, Strategy for mutating synaptotagmin-2 in the mouse. The diagrams depict the structures of the wild-type synaptotagmin-2 gene, which contains all eight exons for synaptotagmin-2 (top), the targeting vector constructed for the mutation of the synaptotagmin-2 gene (middle), and the mutant gene resulting from homologous recombination (bottom). In the final product, part of exon 2 through exon 7 were replaced by the poliovirus IRES (IR), the cDNA for lacZ with a nuclear localization signal (lacZ) and PGK-Neo (Neo). This last gene was used for positive selection, whereas the MC1 thymidine kinase gene (TK) at the 3′ end of the construct was used for negative selection. The positions of the oligonucleotide primers used to identify the wild-type and mutant alleles are indicated by arrows (P1, P2, and P3). The SalI recognition sites (S) and template for the probe used for Southern blotting are also indicated. B, Western blots indicate the lack of synaptotagmin-2 in mutant mouse brain and spinal cord. Other synaptic proteins that were quantified (Table 1) in homogenate of brain, cerebellum/brainstem, and spinal cord homogenates are also showed in the figure. C, X-gal staining of whole-mount mouse CNS in WT and synaptotagmin-2 KO. Note that dorsal root ganglia attached to the spinal cord also show positive staining. Syt 2, Synaptotagmin-2; Syt 1, synaptotagmin-1; Syb 2, synaptobrevin-2; Syp 1, synaptophysin-1.
X-gal (β-galactosidase) staining.
Wild-type and synaptotagmin-2 KO mice at ages of postnatal day 14 (P14) to P16 were deeply anesthetized with halothane and transcardially perfused with 5 ml of PBS followed by 15 ml of 2% paraformaldehyde and 0.25% glutaraldehyde. Brains, spinal cords, and eyeballs were dissected out and postfixed in the same fixative for 30 min at 4°C. For experiments with brain sections, tissues were submerged in 30% sucrose overnight at 4°C for cryoprotection, and then embedded in Tissue-Tek OCT (Ted Pella, Redding, CA) on dry ice. Brain sections at 20 μm were obtained using a Leica cryostat (CM3050S; Leica, Nussloch, Germany) and were mounted onto slides. Slides with sections were rinsed in buffer A (100 mm PBS, pH 7.4, 2 mm MgCl2, 5 mm EGTA) and buffer B (100 mm PBS pH 7.4, 2 mm MgCl2, 0.01% Na-deoxycholate, 0.02% NP-40) twice, each for 5 min. Sections were then developed in buffer C (buffer B containing 5 mm K-ferricyanide, 5 mm K-ferrocyanide; and 0.5 mg/ml X-gal) at 37°C for 6 h in the dark. Samples were twice rinsed with distilled water, counterstained with neutral red (1% neutral red, in pH 4.8 acetate buffer) for 1 min, and washed in distilled water. Finally, sections were air-dried, dehydrated with gradients of ethanol, cleared with xylene, and mounted with coverslips using Permount (SP15-100; Fisher Scientific, Houston, TX). Microscopy was performed after 24 h. In the case of whole-mount tissues (brain and spinal cord), tissues were rinsed twice for 15 min each in buffer A and buffer B, and developed in buffer C overnight at 37°C in the dark.
Electrophysiological analyses of cultured striatal neurons.
Primary striatal neurons were cultured as described previously (Mao and Wang, 2001) with modifications. Briefly, dorsal striatum was isolated from 1-d-old pups of wild-type or synaptotagmin-2 KO mice, dissociated by trypsin digestion, and plated on matrigel-coated glass coverslips. Neurons were cultured in Modified Eagle Medium (Invitrogen) supplemented with B27 (Invitrogen), glucose, transferrin, fetal bovine serum, and Ara-C (Sigma, St. Louis, MO). To monitor synaptic responses, whole-cell patch-clamp recordings were made with neurons at 14–16 d in vitro. Synaptic responses were triggered by a 1 ms current pulse (900 μA) through a local extracellular electrode (FHC, Bowdoinham, ME), and recorded in whole-cell voltage-clamp mode using a Multiclamp 700A amplifier (Molecular Devices, Union City, CA). Data were digitized at 10 kHz with a 2 kHz low-pass filter. The pipette solution contained the following (in mm): 135 CsCl, 10 HEPES, 1 EGTA, 4 Mg-ATP, 0.4 Na-GTP, and 10 QX-314 (lidocaine N-ethyl bromide), pH 7.4. The bath solution contained the following (in mm): 140 NaCl, 5 KCl, 2 or 10 CaCl2, 0.8 MgCl2, 10 HEPES, and 10 glucose, pH 7.4. IPSCs were isolated pharmacologically by including 50 μm d-AP-5 and 20 μm CNQX in the bath solution. Series resistances were compensated 60–70%, and recordings with series resistances >15 MΩ were excluded. Data were analyzed using Clampfit 9.02 (Molecular Devices) or Igor 4.0 (Wavemetrics, Lake Oswego, OR).
Electrophysiological analysis of the neuromuscular junction.
NMJ recordings were performed at room temperature using intracellular recordings on acutely isolated phrenic nerve/diaphragm preparations at P14–P16. Muscles were dissected in oxygenated normal Ringer's solution containing the following (in mm): 136.8 NaCl, 5 KCl, 12 NaHCO3, 1 NaH2PO4, 1 MgCl2, 2 CaCl2, and 11 glucose, pH 7.4 (Liley, 1956), pinned onto Sylgard-coated dishes, and continuously superfused with oxygenated Ringer's solution. Sharp glass microelectrodes were filled with 3 m KCl. Spontaneous synaptic responses were recorded in normal Ringer's solution or with Ringer's solution containing 10 μm EGTA-AM. For experiments with EGTA-AM, phrenic nerve–muscle preparations were treated with EGTA-AM for at least 20 min before recordings. Evoked endplate potentials were elicited by suprathreshold stimulation (2–6 V; 1 ms) of the phrenic nerve via a suction electrode. To prevent muscle contractions, which would destabilize the recordings, 2 μm ω-conotoxin were added to the bath solution to block the muscle specific sodium channel (Cruz et al., 1985). The number of acetylcholine vesicles released after one single nerve impulse (i.e., the quantal content) was calculated as the mean evoked endplate potential (EPP) amplitude divided by the mean amplitude of miniature endplate potential (mEPP). Data were collected with an AxonClamp 2B amplifier, digitized at 10 kHz, and analyzed using Clampfit 9.02 (Molecular Devices) and MiniAnalysis (Synaptosoft, Decatur, GA).
Immunohistochemistry.
Whole mounts of diaphragmatic muscle were dissected and fixed with 2% paraformaldehyde overnight at 4°C. Samples were rinsed thoroughly in PBS, and incubated in 0.1 m glycine/PBS, pH 7.3, at room temperature for 30 min. Coronal 20 μm sections of diaphragmatic muscle were made after overnight immersion of tissues in PBS containing 30% sucrose. Sections were directly mounted onto slides. Slides with muscle sections were incubated with 0.1 mg/L Alexa 594-conjugated α-bungarotoxin (Invitrogen) in antibody dilution buffer (0.5 m NaCl, 10 mm phosphate buffer, pH 7.3, 3% bovine serum albumin, 0.1% sodium azide, 0.3% Triton X-100) for 1 h at room temperature. Sections were permeabilized in 100% methanol at −20°C for 7 min, and washed with 0.5% Triton X-100 in PBS. Samples were incubated with primary antibodies (for synaptotagmin-2, A320, 1:500; synaptotagmin-1, Cl41.1, 1:3000 dilution) overnight at 4°C. Sections were washed in 0.5% Triton X-100 three times, and incubated with secondary antibodies (Alexa Fluor 488-conjugated anti-rabbit or -mouse IgG) at 4°C overnight. Samples were then washed in PBS, mounted with VectaShield mounting medium (H-1000; Vector, Burlingame, CA), and examined by confocal microscopy.
Miscellaneous procedures.
For immunoblotting analyses, forebrains, cerebellums, and spinal cords of P15 wild-type and mutant mice were homogenized in PBS. Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL). Equivalent amounts of brain proteins from wild-type and synaptotagmin-2 KO mice were analyzed by SDS-PAGE and immunoblotting using antibodies as follows: synaptotagmin-2 (A320), synaptotagmin-1 (Cl41.1), syntaxin 1 (I6251), synaptosome-associated protein of 25 kDa (SNAP-25) (P913), synaptobrevin 2 (Cl69.1), Rab3A (42.2), secretory carrier membrane proteins (SCAMP) (R806), synaptophysin (7.2), synapsin (E028), postsynaptic density-95 (PSD-95) (L667), complexins (L668), Munc18 (J371), calmodulin-associated serine/threonine kinase (CASK) (N3927), synuclein (U1126), cysteine-string protein (CSP) (R807), GDP-dissociation inhibitor (GDI) (81.2), and vasolin-containing protein (VCP) (K330; used as an internal control). For quantitations, 125I-labeled secondary antibodies and PhosphorImager detection (Molecular Dynamics) were used. GDI and VCP were used as internal standards.
Statistical analysis.
Data are presented as means ± SEMs. Unpaired Student's t tests were used. Differences were considered significant at p < 0.05.
Results
Generation of synaptotagmin-2 KO mice
We generated synaptotagmin-2 KO mice by homologous recombination in ES cells using the strategy depicted in Figure 1A. In the mutant allele, the synaptotagmin-2 gene sequence containing exons 2–7 was replaced by three termination codons, the poliovirus IRES element followed by the cDNA for LacZ with a nuclear localization signal, and by PGK-Neo. Southern blots with a probe outside the targeting construct and PCR were used to demonstrate successful homologous recombination (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). We maintained two independent lines (S2KO-D, S2KO-K) originating from two different ES cell electroporations, and backcrossed them into a C57BL/6 background. The phenotype of both lines is identical, confirming that it is the result of the synaptotagmin-2 deletion and not of another mutation introduced by ES cell manipulation. For this study, we used mice from the S2KO-D line. To confirm that the homologous recombination created a null allele for synaptotagmin-2, we performed immunoblotting analyses on forebrain, cerebellum, and spinal cord samples from littermate wild-type and homozygous mutant mice (Fig. 1B). As expected, in wild-type mice, synaptotagmin-2 was almost undetectable in forebrain, but abundantly present in cerebellum and spinal cord. In homozygous mutant mice, no synaptotagmin-2 was detectable in any brain region (Fig. 1B). Although the coding region for the first 69 aa of synaptotagmin-2 was not deleted, we did not detect expression of the predicted 7.6 kDa band in our immunoblots using an antibody raised against the N terminus the protein (data not shown). We also analyzed the levels of 14 other synaptic proteins in the same brain samples, using quantitative immunoblotting with 125I-labeled secondary antibodies (Table 1). We detected no significant changes in any synaptic protein tested except for synaptotagmin-1, which exhibited a moderate increase in spinal cord but not in forebrain (Fig. 1B, Table 1). These data suggest that absence of synaptotagmin-2 does not induce a massive increase of synaptotagmin-1 expression or a restructuring of the protein composition of rostral or caudal brain regions.
Table 1.
Levels of synaptic proteins in synaptotagmin-2 knock-out mice
| Protein | Cortex |
Cerebellum |
Spinal cord |
||||
|---|---|---|---|---|---|---|---|
| Wild type | Knock-out | Wild type | Knock-out | Wild type | Knock-out | ||
| Syt 1 | (n = 4) | 100 ± 2.1 | 94.2 ± 3.8 | 100 ± 4.8 | 112.8 ± 7.4 | 100 ± 7.2 | 132.3 ± 2.9* |
| Syntaxin | (n = 4) | 100 ± 3.2 | 102.6 ± 4.5 | 100 ± 10.6 | 100.5 ± 10.7 | 100 ± 6.7 | 83.6 ± 11.5 |
| SNAP-25 | (n = 4) | 100 ± 2.2 | 100.3 ± 2.3 | 100 ± 9.6 | 90.0 ± 8.5 | 100 ± 9.1 | 83.4 ± 10.4 |
| Syb | (n = 4) | 100 ± 5.0 | 111.3 ± 3.9 | 100 ± 8.2 | 92.8 ± 3.8 | 100 ± 10.9 | 106.9 ± 6.5 |
| Rab 3 | (n = 4) | 100 ± 6.2 | 101.2 ± 9.9 | 100 ± 12.2 | 80.7 ± 7.1 | 100 ± 7.7 | 86.0 ± 8.7 |
| Syp 1 | (n = 4) | 100 ± 2.7 | 105.6 ± 3.4 | 100 ± 8.5 | 85.7 ± 3.1 | 100 ± 5.1 | 91.1 ± 8.1 |
| Synapsin | (n = 4) | 100 ± 1.8 | 87.9 ± 1.0 | 100 ± 5.9 | 91.3 ± 4.4 | 100 ± 6.0 | 86.2 ± 6.3 |
| PSD-95 | (n = 3) | 100 ± 2.4 | 103.8 ± 4.2 | 100 ± 3.1 | 97.9 ± 6.0 | 100 ± 2.3 | 98.8 ± 0.5 |
| Cpx | (n = 3) | 100 ± 3.7 | 95.5 ± 10.2 | 100 ± 4.4 | 89.3 ± 0.8 | 100 ± 4.2 | 103.0 ± 6.2 |
| Munc18 | (n = 3) | 100 ± 6.2 | 98.3 ± 2.9 | 100 ± 2.0 | 102.4 ± 1.3 | 100 ± 3.7 | 109.3 ± 3.7 |
| CASK | (n = 3) | 100 ± 4.3 | 89.8 ± 0.3 | 100 ± 0.6 | 101.2 ± 3.7 | 100 ± 8.5 | 93.3 ± 5.6 |
| SCAMP | (n = 3) | 100 ± 1.6 | 100.8 ± 3.0 | 100 ± 2.4 | 98.0 ± 7.5 | 100 ± 2.1 | 98.3 ± 0.8 |
| Synuclein | (n = 3) | 100 ± 6.6 | 95.7 ± 5.6 | 100 ± 2.7 | 95.5 ± 8.5 | 100 ± 3.6 | 99.5 ± 14.2 |
| CSP | (n = 3) | 100 ± 4.6 | 104.5 ± 0.8 | 100 ± 1.3 | 112.3 ± 7.8 | 100 ± 5.1 | 93.2 ± 7.4 |
Total proteins in brain homogenates were analyzed by SDS-PAGE and Western blots. 125I-conjugated secondary antibodies were used and analyzed by a PhosphorImager. Cpx, Complexin; Syb, synaptobrevin 2; Syp, synaptophysin; Syt 1, synaptotagmin-1. The numbers of samples are listed in parentheses.
*p < 0.05.
Synaptotagmin-2 KO mice are postnatally lethal and have motor defects
To test the effect of the synaptotagmin-2 deletion on mouse development and survival, we systematically examined the offspring from heterozygous matings. At birth, synaptotagmin-2 homozygous and heterozygous mutant and wild-type control mice were present at close to Mendelian ratio [wild-type/heterozygous/homozygous ratio, 1.10:2.00:1.05 (n = 447)]. However, homozygous synaptotagmin-2 KO mice began to display severe motor dysfunction in the second postnatal week, and were almost unable to move at P15 (see supplemental movie 1, available at www.jneurosci.org as supplemental material). Whereas wild-type and heterozygous mice grew vigorously in the first 4 weeks of life, KO mice stopped growing in the second postnatal week (Fig. 2A,B). At P19, synaptotagmin-2 KO mice started to die, and all mice perished by P24 (Fig. 2C). However, we observed no seizures, different for example from SV2 KO mice, which also die at a similar age (Janz et al., 1999). These data indicate that synaptotagmin-2 is essential to the well-being and survival of mice.
Figure 2.
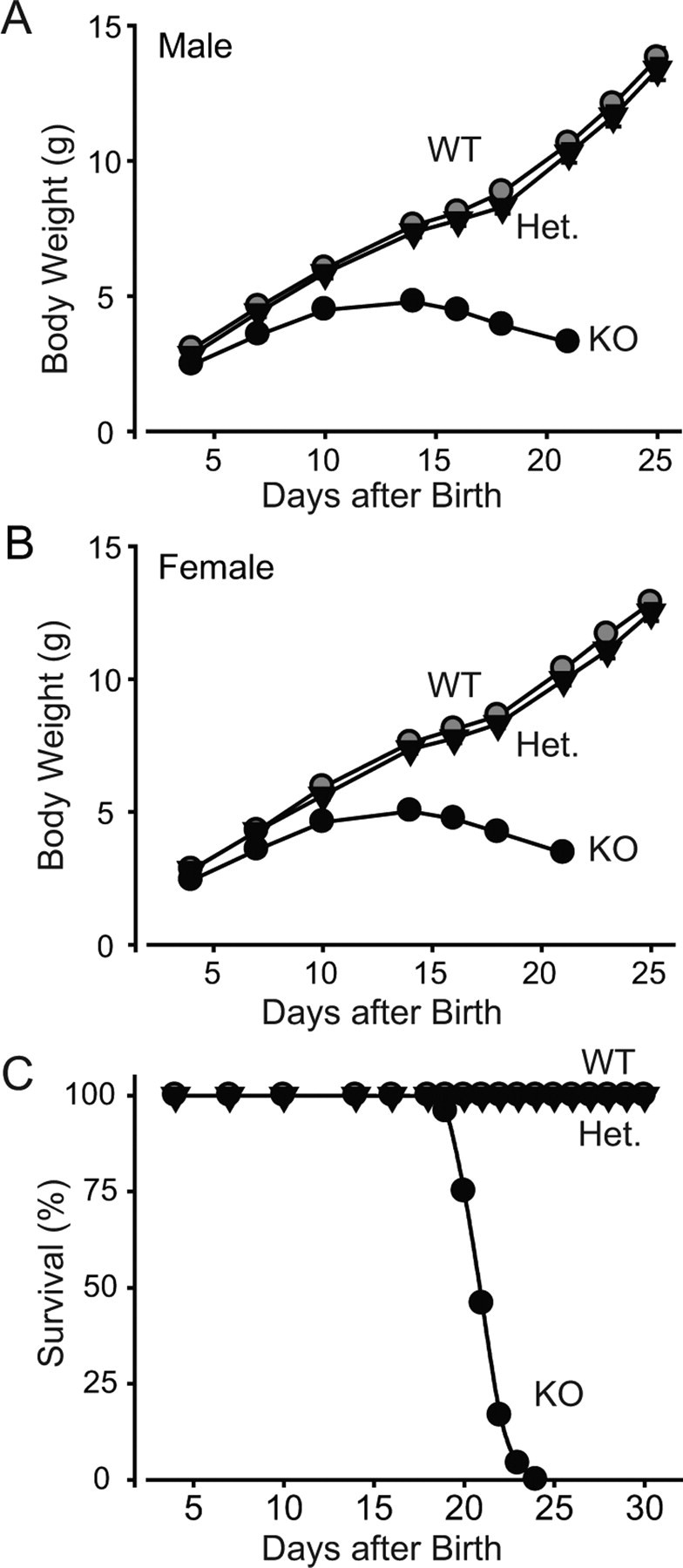
Weight and survival of synaptotagmin-2 KO mice. A, B, Body weight of littermate male (A) and female (B) wild-type and mutant mice as a function of age. Homozygous wild-type (WT), heterozygous mutant (Het), and homozygous mutant (KO) mice were examined (n = 10–21 for male and 10–41 for female animals in each genotype group; p < 0.05 at all ages for the KO mice compared with wild-type or heterozygous mice, which are not significantly different from each other). C, Survival of wild-type, synaptotagmin-2 heterozygous, and synaptotagmin-2 KO mice. All synaptotagmin-2 KO mice die between the ages of P19 and P24 (n = 35, 60, 24 for wild-type, heterozygous, and KO, respectively).
Expression pattern of synaptotagmin-2 in mice
The insertion of lacZ after the IRES element into the synaptotagmin-2 gene placed the expression of β-galactosidase encoded by the lacZ sequences under the transcriptional control of the synaptotagmin-2 gene regulatory elements (Fig. 1A). We used the β-galactosidase activity to follow the precise expression pattern of synaptotagmin-2 in the mutant mice (Figs. 1C, 3, 4). Consistent with previous studies (Geppert et al., 1991; Ullrich et al. 1994; Marqueze et al., 1995), examination of the entire nervous system from mutant mice revealed that synaptotagmin-2 was primarily expressed in the hindbrain, cerebellum, and spinal cord (Fig. 1C). Although on a macroscopic level, synaptotagmin-2 was absent from forebrain, coronal sections revealed considerable expression of synaptotagmin-2 in selected forebrain neurons (Fig. 3A,F). Unexpectedly, the majority of neurons in the striatum and the zona incerta appear to express synaptotagmin-2 (Fig. 3C,F). Moreover, considerable synaptotagmin-2 expression was observed in the reticular nucleus of the thalamus (Fig. 3I) and the ventromedial nucleus of the hypothalamus (Fig. 3J). In contrast, the major thalamic nuclei, the cortex (Fig. 3B), and the hippocampal formation (Fig. 3G,H) expressed little synaptotagmin-2. In the latter structures, a few isolated synaptotagmin-2-positive neurons were observed as exemplified in the image of the CA3 region of the hippocampus in Figure 3H, but the vast majority of neurons did not express synaptotagmin-2. Although it is not possible to determine the nature of the synaptotagmin-2-expressing neurons from these analyses, most of these neurons appear to be inhibitory based on their location. This is suggested by the fact that the majority of neostriatal neurons are inhibitory and express synaptotagmin-2, that the reticular nucleus of the thalamus is mostly composed of inhibitory neurons, and that the location of the scattered synaptotagmin-2-positive cells in the cortex (Fig. 3B) hippocampus corresponds to those of inhibitory interneurons, especially in the hippocampal hilus region (Fig. 3G,H). Some of the synaptotagmin-2-positive neurons found here may correspond to recently discovered hippocampal parvalbumin-expressing basket cells that are still capable of mediating fast synchronous inhibitory synaptic responses even in the absence of synaptotagmin-1 (Kerr et al. 2006).
Figure 3.
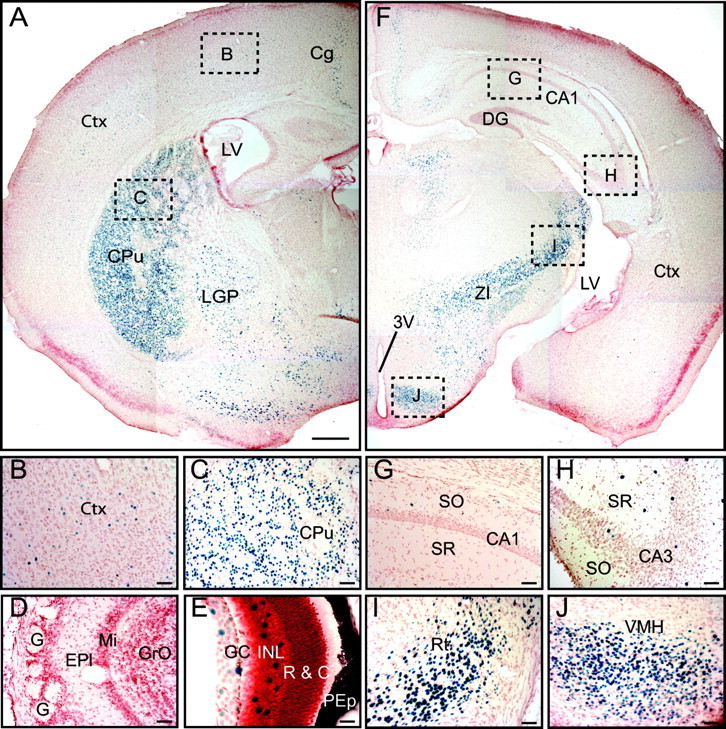
Expression pattern of synaptotagmin-2 in forebrain. β-Galactosidase activity is revealed by X-gal staining in synaptotagmin-2 KO homozygous mice. A, The majority of striatal neurons show β-galactosidase activity. B, C, Higher magnifications of brain areas indicated by dashed squares in A. D, No synaptotagmin-2 expression is found in olfactory bulb. E, Scattered neurons in retina showing β-galactosidase positivity. F, Very few neurons in cortex and hippocampus are β-galactosidase positive. G–J, Higher magnifications of brain areas indicated by dashed squares in F. 3V, Third ventricle; Cg, cingulate cortex; Ctx, cortex; CPu, caudate–putamen (striatum); DG, dentate gyrus; EPl, external plexiform layer of olfactory bulb; G, glomeruli; GC, ganglion cell layer; GrO, granular cell layer of olfactory bulb; INL, inner nuclear layer; LGP, lateral globus pallidus; LV, lateral ventricle; Mi, mitral cell layer of olfactory bulb; PEp, pigment epithelium; SO, stratum oriens; SR, stratum radiatum; R & C, layer of rods and cones; Rt, reticular thalamic nucleus; VMH, ventromedial hypothalamic nucleus; ZI, zona incerta. Scale bars: A, F, 50 μm; B–D, G–J, 5 μm; E, 2.5 μm.
Figure 4.
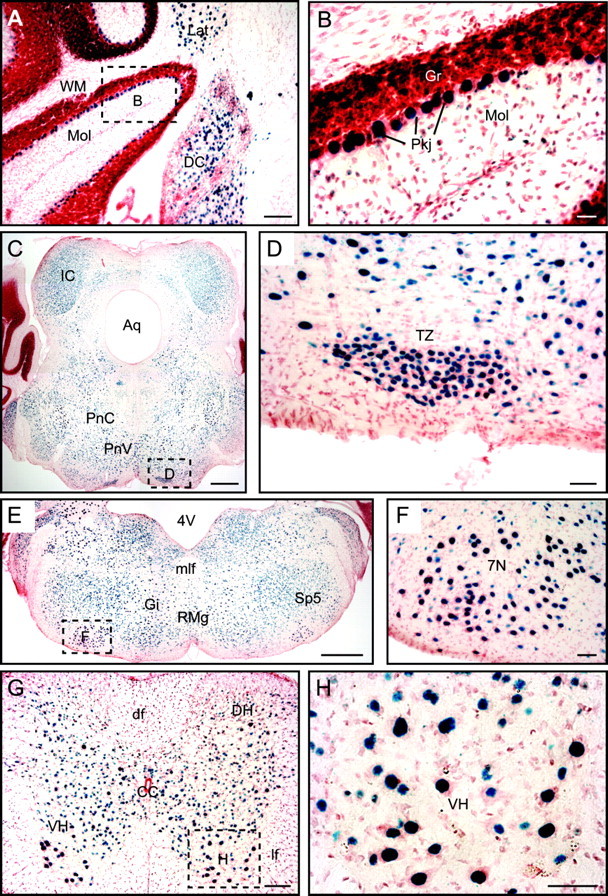
Expression pattern of synaptotagmin-2 in caudal brain. X-gal staining in synaptotagmin-2 KO homozygotes is shown. A, Cerebellum; C, E, brainstem; G, spinal cord. B, D, F, H, Higher magnifications of brain areas as indicated by dashed squares in A, C, E, and G. 4V, Fourth ventricle; 7N, facial nucleus; Aq, aqueduct; CC, central canal; DC, dorsal cochlear nucleus; df, dorsal fasciculus; DH, dorsal horn; Gi, gigantocellular reticular nucleus; Gr, granular layer; IC, inferior colliculus; Lat, lateral cerebellar nucleus; lf, lateral fasciculus; mlf, medial longitudinal fasciculus; Mol, molecular layer; PnC, caudal pontine reticular nucleus; PnV, ventral pontine reticular nucleus; Pkj, Purkinje cells; RMg, raphe magnus nucleus; Sp5, interpolar subnucleus of the spinal trigeminal nucleus; Tz, nucleus of trapezoid body; VH, ventral horn; WM, white matter. Scale bars: A, G, 10 μm; B, 2.5 μm; C, E, 50 μm; D, F, H, 5 μm.
The pattern of inhibitory neurons expressing synaptotagmin-2 is also present in the cerebellum where all Purkinje neurons and the majority of the neurons of deep cerebellar nuclei express synaptotagmin-2 (Fig. 4A,B). In contrast, few neurons in the granule cell layer (which primarily contains excitatory granule cells) contain synaptotagmin-2, and scattered synaptotagmin-2-positive cells are observed in the molecular layer (which contains scattered inhibitory interneurons).
A different expression pattern for synaptotagmin-2 emerges in the brainstem and spinal cord where a majority of neurons appears to express synaptotagmin-2. In the brainstem, abundant expression of synaptotagmin-2 is observed for example in the inferior colliculus, the pontine reticular nuclei, the nuclei of the fifth and seventh cranial nerves, and the nucleus of the trapezoid body (Fig. 4C–F). Similarly, in the spinal cord, most neurons appear to express synaptotagmin-2, including all motoneurons (Fig. 4G,H). Most of the neurons that express synaptotagmin-2 in the brainstem and spinal cord correspond by location to excitatory neurons, either glutamatergic or cholinergic, suggesting that different from the forebrain where synaptotagmin-2 appears to be concentrated in inhibitory neurons, synaptotagmin-2 is more widely expressed in all classes of neurons in the brainstem and spinal cord.
Synaptotagmin-2 expression in the NMJ
The X-gal staining showed that synaptotagmin-2 is expressed in motoneurons, consistent with the NMJ phenotype in hypomorphic synaptotagmin-2I377N mutant mice (Pang et al., 2006). To determine whether synaptotagmin-2 is indeed present in NMJs and whether it is coexpressed with synaptotagmin-1, we performed an immunofluorescence localization study of synaptotagmin-1 and -2 in NMJs in the diaphragmatic muscle from wild-type and KO mice at P14–P16 (Fig. 5). Conjugated fluorescent α-bungarotoxin was used to label postsynaptic acetylcholine receptors. The diaphragmatic muscle is innervated by phrenic nerves that branch on reaching the muscle to form discrete NMJ synapses that are distributed in a characteristic branching pattern in the middle of the diaphragm; this pattern is controlled by synaptic activity, and becomes abnormal when synaptic activity is altered (Buffelli et al., 2003). However, the distribution of NMJs, as revealed by staining with fluorescent α-bungarotoxin (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), was not significantly altered in synaptotagmin-2 KO mice.
Figure 5.
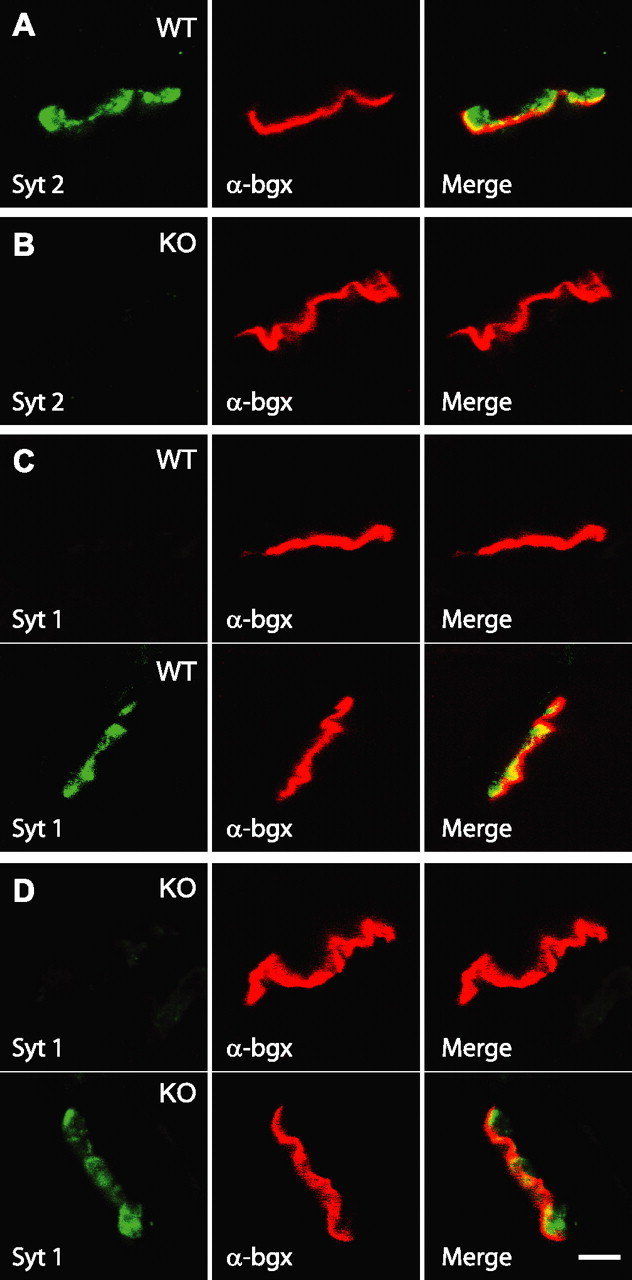
Immunostaining of synaptotagmin-1 and -2 in NMJ. A, All endplates express synaptotagmin-2 in wild-type (WT) NMJ. B, No synaptotagmin-2 was detected in synaptotagmin-2 KO NMJ. C, D, Synaptotagmin-1 was detected in some (bottom panel) but not other endplates (top panel) in both wild type and synaptotagmin-2 KO. Scale bar, 10 μm (applies to all subpanels). Abbreviations: Syt 1, Synaptotagmin-1; Syt 2, synaptotagmin-2; α-bgx, α-bungarotoxin.
We next examined individual NMJs for the presence of synaptotagmin-1 and -2. Synaptotagmin-2 was present in every endplate in wild-type diaphragmatic muscle but absent from KO NMJs (Fig. 5A,B). Three-dimensional reconstructions by confocal microscopy revealed that individual NMJ endplates in synaptotagmin-2-deficient mice were structurally similar to those from wild-type mice (supplemental movies 2, 3, available at www.jneurosci.org as supplemental material). A subpopulation of wild-type NMJs expressed synaptotagmin-1 (∼40%), whereas an appreciably higher percentage of synaptotagmin-2-deficient NMJs (∼90%) contained synaptotagmin-1 (Fig. 5C,D). This finding agrees well with the modest but significant increase in the overall levels of synaptotagmin-1 in spinal cord (Table 1).
Evoked synaptic release in cultured neurons from the neostriatum
To examine the functional consequences of the deletion of synaptotagmin-2 on synaptic transmission, we performed electrophysiological recordings in cultured neurons from the dorsal neostriatum of newborn mice (Fig. 6), and in NMJs in the phrenic nerve/diaphragm preparation from adolescent mice (P14–P16) (Figs. 7–11). In the neostriatal neurons, we analyzed IPSCs as an example of inhibitory synaptic transmission effected by forebrain neurons that express synaptotagmin-2 (Fig. 3C), and NMJs as an example of excitatory synaptic transmission in a caudal synapse that expresses synaptotagmin-2 (Fig. 5). As with all other experiments performed in the present study, all recordings were made with samples from littermate wild-type and mutant mice, with the genotype “blinded” to the experimentalist.
Figure 6.
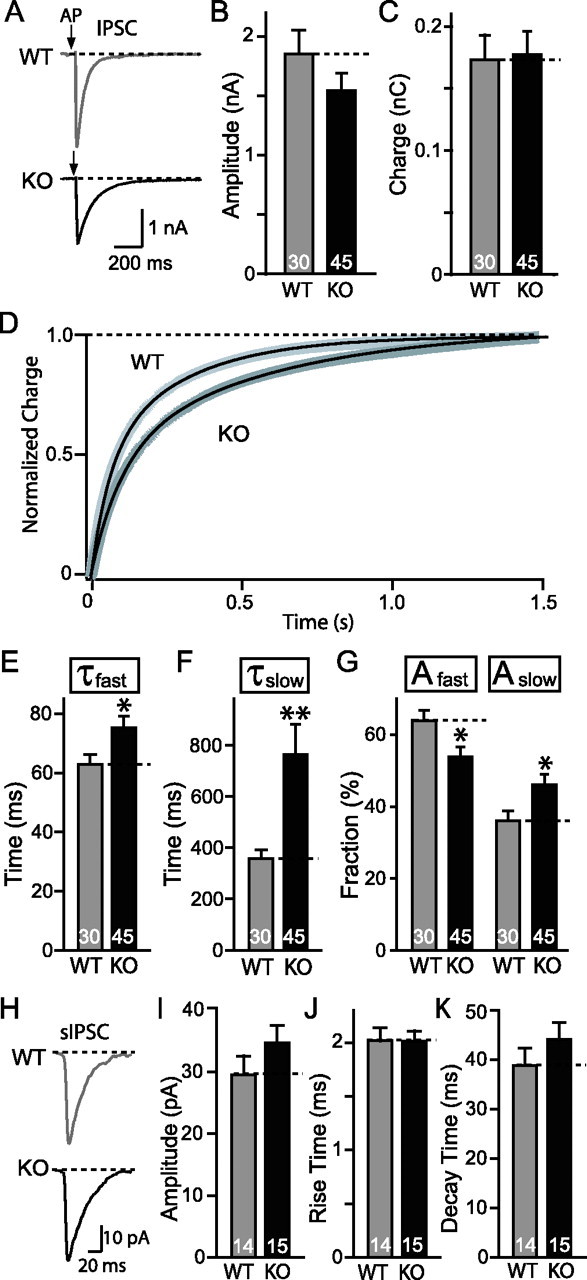
Synaptic release in striatal neuronal cultures. A, Representative traces of evoked IPSC in both WT and synaptotagmin-2 KO. AP, Action potential. Pooled data for both amplitude (B) and charge transfer over a time period of 1.5 s (C). D, Normalized average charge integration as a function of time over 1.5 s. WT, n = 16; KO, n = 17. The integrated charge transfer can be well fitted by a double exponential function (black solid line superimposed on to the integrated lines). The fitting gave out a fast decay (τfast) and slow decay (τslow). E, F, Pooled data indicate both τfast and τslow increased in synaptotagmin-2 KO striatal synapses. G, The fraction of the slow constituent (Aslow) increased in KO synapses, and fast constituent (Afast) decreased accordingly. Numbers of neurons recorded are indicated by numbers within each bar. H, Representative traces of sIPSCs in both WT and synaptotagmin-2 KO mice. There are no significant differences in average sIPSC amplitudes (I), 20–80% rise time (J), and 80–20% decay time (K). Events with amplitudes >150 pA, possibly attributable to spontaneous firing of presynaptic neurons, are excluded from analyses. Error bars indicate SEM. *p < 0.05; **p < 0.01.
Figure 7.

Spontaneous miniature synaptic responses in NMJ. A, Representative traces of spontaneous mEPPs of WT and synaptotagmin-2 KO. B, Pooled data indicate a significant increase in the frequency of miniature synaptic release, whereas amplitude (C), rise slope (D), and rise time (E) show no differences in both WT and synaptotagmin-2 KO. F, In the presence of EGTA-AM (10 μm), a significant reduction of mEPP frequency is found in synaptotagmin-2 KO but not in WT NMJ. Frequency of mEPPs is still significantly higher compared with wild-type control in the presence of EGTA. The numbers of neurons recorded are indicated by the numbers above or within each bar. Error bars indicate SEM. **p < 0.01; ***p < 0.001.
Figure 8.

Evoked synaptic responses in NMJ. A, Representative traces of phrenic nerve evoked EPP by a suction electrode in WT and synaptotagmin-2 KO. Maximum stimulus (2–6 V) intensities were used to evoke synaptic responses. B–D, Pooled data of the amplitude (B), quantal content (C), and speed of the EPP increase (D) in wild-type and synaptotagmin-2-deficient NMJs. The gray and black circles indicate individual evoked responses of each recording. The numbers of recordings are indicated in each bar. Error bars indicate SEM. **p < 0.01; ***p < 0.001.
Figure 9.

Paired-pulse facilitation. A, Representative traces of paired-pulse stimuli induced synaptic responses in WT and synaptotagmin-2 KO NMJ. B, Paired-pulse ratio of evoked EPP when given two stimuli at different interstimulus intervals ranging from 20 to 100 ms. The numbers of recordings are indicated in parentheses. Error bars indicate SEM. **p < 0.01; ***p < 0.001.
Figure 10.

Short-term plasticity of evoked EPPs in NMJ. A, Representative traces of EPPs evoked by a 20 Hz stimulus train in WT and synaptotagmin-2 KO NMJs. B, C, Pooled data showing the absolute (B) and normalized amplitudes (C) of EPPs evoked at 20 Hz. Wild-type EPPs are larger (see Fig. 8B) and exhibit a moderate initial facilitation followed by depression. KO EPPs show facilitation. D, Normalized amplitudes of the steady-state EPPs as a function of stimulation frequency. Steady-state EPP amplitudes are the average of the last five responses during 1 s of train stimulation at 10, 20, and 50 Hz. Steady-state EPP values are the averages of the last five responses during the train of stimulation. The numbers of recordings are indicated in parentheses. Error bars indicate SEM. **p < 0.01.
Figure 11.
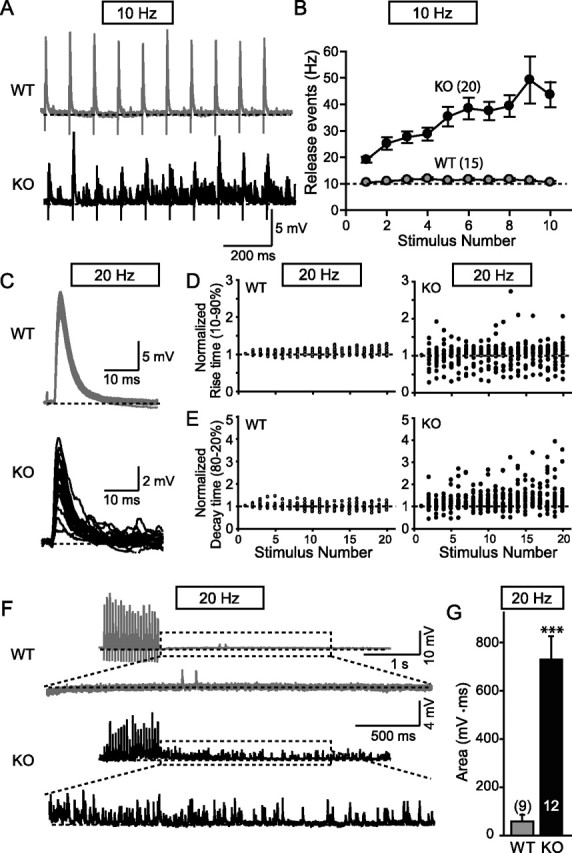
Desynchronization of EPPs during high-frequency stimulus trains in synaptotagmin-2-deficient NMJs. A, Representative traces (superimposed 4 consecutive traces at 7 s interval) of WT and synaptotagmin-2 KO when given 10 Hz stimulation to the phrenic nerve. B, Frequency of individual release events during 10 Hz stimulus trains plotted as a function of the stimulus number. Synaptic events were counted manually. C, Superimposed individual responses after phrenic nerve stimulation at 20 Hz for 1 s. D, E, Distributions of rise time (10–90%) and decay time (80–20%) normalized to the first response plotted as a function of stimulus number (WT, n = 15; KO, n = 19). F, Representative traces of repetitive stimulation at 20 Hz for wild-type and synaptotagmin-2-deficient NMJs. The bottom traces represent enlargements of the traces for 3 s after 100 ms of the last stimulus. G, Integrated area under the peaks for 3 s after 50 ms of the last stimulus after a 1 s 20 Hz stimulus train. Integrated areas were plotted instead of event frequencies because the synaptotagmin-2-deficient NMJs have such high frequencies that they are impossible to accurately measure. The numbers of recordings are indicated above or within each bar. Error bars indicate SEM. ***p < 0.001.
IPSCs could be efficiently induced by focal stimulation in cultured neostriatal neurons from both wild-type and KO mice. We observed no significant decrease in the amplitude or total synaptic charge transfer per stimulus as integrated over 1.5 s (Fig. 6A,B). However, we found that the synaptic charge transfer exhibited a significantly slower time course in synaptotagmin-2-deficient striatal neurons than in wild-type control neurons (Fig. 6D). The integrated charge transfer can be fitted by a two-exponential function. The mean decay time constants for both the fast and slow constituent [called “constituent” to avoid confusion of these different kinetic components with the slow and fast component of release that are differentially affected in synaptoagmin-1 KO neurons (Geppert et al., 1994)] were significantly increased in synaptotagmin-2-deficient neurons. This effect is relatively small for the fast decay constant (∼20%), but very large for the slow time constant (>100%) (Fig. 6E,F). Moreover, consistent with a shift to a slower time course of release, the relative contributions of the fast and slow processes to the overall release are significantly shifted in favor of the slow process (Fig. 6G). Please note that, in these calculations, the two time constants of the integrated IPSCs are purely descriptive tools to characterize the changes in release time course observed in synaptotagmin-2-deficient striatal neurons. We noticed in a subpopulation of neurons (7 of 45) that release was mostly desynchronized similar to synaptotagmin-1-deficient cortical neurons (Maximov and Südhof, 2005), but the majority of neurons did not exhibit this phenotype, presumably because other synaptotagmins are also expressed in these neurons and functionally compensate (Ullrich et al., 1994; Marqueze et al., 1995) (J. Xu, T. Mashimo, T. C. Südhof, unpublished observations). To eliminate the possibility that the change in IPSC kinetics induced by the synaptotagmin-2 deletion is attributable to an alteration in the properties of postsynaptic GABA receptors, we analyzed the kinetics of spontaneous miniature IPSCs (sIPSCs) in cultured striatal neurons. Spontaneous events with amplitude that exceeded 150 pA were excluded from this analysis because of the possibility that bigger responses resulted from spontaneous firing of presynaptic neurons. We found no changes in the average amplitude, rise time, or decay time of sIPSCs (Fig. 6H–K). Thus, the changes in the delayed release time course are most likely caused by a presynaptic defect attributable to the elimination of synaptotagmin-2.
Spontaneous neurotransmitter release in NMJs
To examine synaptic transmission in the NMJ, we performed sharp-electrode intracellular recordings from diaphragmatic muscle. We first studied spontaneous release events (mEPPs for miniature endplate potentials) in normal Ringer's solution. The frequency of mEPPs was almost 10-fold higher in synaptotagmin-2 KO mice than in littermate wild-type control mice at an age of P14–P16 (Fig. 7B), whereas the average amplitude, the rise slope, and rise time of the mEPPs were the same in wild-type and KO mice (Fig. 7C–E). To determine whether the increased mEPP frequency is attributable to residual Ca2+ in nerve terminals, we applied 10 μm EGTA-AM for >20 min in the bath solution before recording mEPPs (Fig. 7F). The EGTA slightly decreased the mEPP frequency in both wild-type and synaptotagmin-2 mutant NMJs, but the difference between the two synapses remained the same (Fig. 6F), demonstrating that the increase in mEPPs frequency in the mutant NMJs is Ca2+ independent.
Evoked synaptic responses in synaptotagmin-2-deficient NMJs
We stimulated the phrenic nerve with a suction electrode using a maximal stimulus intensity (2–6 V), and recorded evoked EPPs in the muscle (Fig. 8A). In these recordings, we applied 2 μm ω-conotoxin to prevent muscle contractions that interfere with stable recordings (Cruz et al., 1985).
The amplitude and the quantal content of the EPPs were decreased approximately twofold in synaptotagmin-2-deficient NMJs when compared with wild-type littermate controls (Fig. 8B,C) [amplitude, KO, 8.6 ± 1.5 mV, n = 25; WT, 23.0 ± 1.4, n = 28; p < 0.001; quantal content (evoked EPP amplitude/mEPP amplitude), WT, 15.3 ± 0.9, n = 28; KO, 7.0 ± 1.2, n = 25; p < 0.001]. Thus, less acetylcholine is released per action potential in NMJs from mutant mice than from control mice. At the same time, the speed with which the EPPs increases when an action potential is triggered was decreased approximately threefold in synaptotagmin-2-deficient NMJs compared with wild-type control NMJs (Fig. 8C) (KO, 6.2 ± 1.0 mV/ms, n = 25; WT, 20.4 ± 1.4 mV/ms, n = 28; p < 0.01). Because the amplitudes and the rise kinetics of mEPPs were not altered in the synaptotagmin-2 KO mice (Fig. 7), these data demonstrate that the Ca2+-triggered stimulation of vesicle release but not the filling or fusion of individual vesicles are impaired in the synaptotagmin-2-deficient NMJs.
A plausible explanation for the changes in evoked EPPs in synaptotagmin-2-deficient NMJs is that the release probability is decreased. To test this hypothesis with an independent method, we made use of the fact that a decreased release probability induces increased facilitation in response to either two closely spaced action potentials (paired-pulse facilitation) or action potential trains (Zucker and Regehr, 2002). When two or more action potentials invade a nerve terminal in rapid succession, residual Ca2+ in the vicinity of presynaptic Ca2+ channels builds up and enhances the probability of vesicle release within this local Ca2+ domain for each subsequent action potential (Katz and Miledi, 1968). The lower the initial release probability, the more facilitation can be achieved.
We first examined paired-pulse facilitation with interstimulus intervals ranging from 20 to 200 ms (Fig. 9A). Under the conditions used, we observed little facilitation in wild-type control NMJs, but a large degree of paired-pulse facilitation in synaptotagmin-2-deficient NMJs than in control NMJs at all intervals tested except for the 200 ms interval (Fig. 9B).
We next monitored EPPs that were evoked by 10–50 Hz stimulus trains (Fig. 10). At 20 Hz, wild-type NMJs exhibited a small amount of initial facilitation followed by moderate depression that leads to a steady-state response. Both the facilitation and depression are evident in plots of either the absolute or the normalized EPP amplitudes (Fig. 10A–C). At 10 and 20 Hz stimulation frequencies, synaptotagmin-2-deficient NMJs different from control NMJs exhibited continued facilitation without depression. The facilitation in the mutant NMJs was much larger than the initial facilitation observed in wild-type NMJs (Fig. 10D). The initial EPP amplitude in synaptotagmin-2-deficient NMJs was approximately twofold lower than in wild-type control NMJs (see also Fig. 8B), but the facilitation increased the steady-state EPP amplitudes in the mutant NMJs to a level that was almost the same as that in the wild-type NMJs (Fig. 10B). These results strongly support the notion of a decreased release probability, a conclusion that is further supported by the finding that synaptotagmin-2-deficient NMJs exhibited a failure rate at 10 and 20 Hz of 6.0 ± 1.6 and 1.5 ± 0.7%, respectively, whereas no failures were detected in wild-type control synapses. The relative decrease in the failure rate at 20 Hz was probably attributable to the facilitation during repetitive stimulations. Note that during 50 Hz stimulus trains, however, we observed no facilitation in the mutant NMJs and could not determine the failure rate, probably because release becomes mostly desynchronized at 50 Hz (see below).
Desynchronization of evoked vesicle release during repetitive stimulation
During studies of EPPs in response to high-frequency stimulus trains, we observed that release became highly desynchronized in mutant but not wild-type NMJs (Fig. 11A). Desynchronization became evident already at 10 Hz stimulation frequencies, resulting in an increasing number of submaximal release events during the stimulus train (Fig. 11B). Similar increases of synaptic release were seen at other stimulus frequencies (20, 50, 100, and 200 Hz) (data not shown). Moreover, the evoked responses in wild-type NMJs remained completely synchronized throughout the stimulus trains (Fig. 11C) as revealed by plotting the 10–90% rise times and the 80–20% decay times as a function of stimulus numbers (Fig. 11D,E). In contrast, in synaptotagmin-2-deficient NMJs evoked responses became desynchronized as uncovered by the same plots, which demonstrate that the rise and decay times become highly variable with increasing number of stimuli applied at 20 Hz (Fig. 11D,E), indicating the release is mostly asynchronous. A similar desynchronization was seen in synaptotagmin-2-deficient NMJs under other frequencies stimulation (data not shown). These results suggest that, during high stimulus trains, synchronous release is increasingly ineffective in synaptotagmin-2-deficient NMJs, and the accumulating Ca2+ during the stimulus train as a result begins to trigger more and more asynchronous release.
Our findings for the EPPs at the NMJ differ from those obtained with synaptotagmin-1-deficient cultured forebrain neurons in which we observed no increase in asynchronous release during the stimulus trains, but increased “delayed” release after the stimulus trains (Maximov and Südhof, 2005). However, even in wild-type forebrain neurons, asynchronous release becomes the dominant form of release during the stimulus trains (Hagler and Goda, 2001; Ohtsuka et al., 2002), suggesting that asynchronous release is already saturated during the stimulus train in wild-type neurons, and thus cannot be further increased in mutant neurons. Because delayed release observed after the stimulus trains is significantly increased in synaptotagmin-1-deficient forebrain neurons (Maximov and Südhof, 2005), we tested whether delayed release is also increased in NMJs lacking synaptotagmin-2 (Fig. 11F). We observed a dramatic (∼10-fold) increase in delayed release in the NMJs (Fig. 11G). In these measurements, we integrated the area under the events for 3 s after a 1 s stimulus train applied at 20 Hz, because individual release events could not be resolved as a result of their high frequency in synaptotagmin-2-deficient NMJs.
Discussion
Synaptotagmin-1 and -2 are closely related synaptotagmin isoforms with similar, although slightly different Ca2+-binding properties (Sugita et al., 2002; Nagy et al., 2006), but distinct expression patterns (Perin et al., 1990; Geppert et al., 1991; Ullrich et al., 1994; Marqueze et al., 1995). Rescue experiments of synaptotagmin-1-deficient neurons and chromaffin cells revealed that synaptotagmin-2 can rescue the synaptotagmin-1 deficiency phenotype (Stevens and Sullivan, 2003; Nagy et al., 2006). Moreover, a mutant mouse in which synaptotagmin-2 carries a point mutation exhibits a defect in synaptic transmission in selected synapses (Pang et al., 2006). These experiments suggested that synaptotagmin-2 functions as a Ca2+ sensor analogous to synaptotagmin-1 in release, but left open the question of the biological significance of synaptotagmin-2. Specifically, because no null mutant of synaptotagmin-2 was available, these previous experiments raised the question whether synaptotagmin-2 is an essential synaptotagmin isoform, and if so, what its essential functions are.
In the present study, we generated KO mice in which lacZ was knocked into exon 2 of the synaptotagmin-2 gene, creating a synaptotagmin-2 null allele that places expression of β-galactosidase under the control of the synaptotagmin-2 promoter. These mice allowed us to address four questions: (1) Where precisely is synaptotagmin-2 expressed in brain? (2) What is the consequence of deleting synaptotagmin-2 for mouse development, survival, and behavior? (3) What role does synaptotagmin-2 have in synapses formed by forebrain neurons that express synaptotagmin-2? (4) What is the role of synaptotagmin-2 in NMJ synapses formed by spinal cord neurons? Overall, our results support the previous hypothesis that synaptotagmin-2 functions as a Ca2+ sensor in release similar to synaptotagmin-1, but reveal a vital biological difference between these two synaptotagmins: whereas synaptotagmin-1 is absolutely essential for fast Ca2+-triggered release in excitatory and inhibitory cortical and hippocampal neurons, in those synapses that we studied here (synapses formed by neostriatal neurons and NMJ synapses formed by spinal cord neurons), synaptotagmin-2 contributes to fast Ca2+-triggered release, but is not solely responsible for it. At least in the case of the NMJs, synaptotagmin-2 is complemented by coexpressed synaptotagmin-1. Thus, although synaptotagmin-1 and -2 likely perform similar functions, they perform these functions in a distinct biological context.
Expression pattern of synaptotagmin-2
Our data, in extension of previous studies that mapped the expression of synaptotagmin-2 by immunoblotting and in situ hybridization (Geppert et al., 1991; Ullrich et al., 1994; Marqueze et al., 1995), demonstrate that synaptotagmin-2 is expressed in a restricted subset of forebrain neurons and in the majority of brainstem and spinal cord neurons. The forebrain and cerebellar expression pattern is interesting in that it appears to selectively involve inhibitory neurons (e.g., striatal neurons, neurons in the reticular nucleus of the thalamus, cerebellar Purkinje cells, and hypothalamic neurons). In some areas, most inhibitory neurons appear to express synaptotagmin-2 (e.g., striatum and cerebellar Purkinje cells), whereas in others, only a tiny percentage of inhibitory neurons seem to contain synaptotagmin-2 (e.g., cerebral cortex and hippocampus). In contrast to the forebrain, the majority of both excitatory and inhibitory neurons and cholinergic motoneurons in the brainstem and spinal cord express synaptotagmin-2. This leads to a strong expression of synaptotagmin-2 in NMJs. Somewhat surprisingly, we found that, at least in the case of NMJs, synaptotagmin-2 and synaptotagmin-1 are coexpressed with an expression pattern that appears to be stochastic (Fig. 5) in that NMJs formed on the same muscle sometimes contain both synaptotagmin-1 and -2, and other times contain only synaptotagmin-2.
Synaptotagmin-2 is essential for survival of adolescent mice
Deletion of synaptotagmin-2 does not have a major effect on mouse survival or growth in the first postnatal week, but the mutant mice stop growing at the end of the second postnatal week, and die without exception in the third postnatal week. Thus, synaptotagmin-2 is clearly not essential for fundamental nervous system activities such as breathing or feeding, a surprising result considering its prominent expression in motoneurons and the brainstem. The mice do not appear to have a developmental abnormality based on studies of brain morphology by light microscopy, and on the fact that the protein composition of the brain and spinal cord is not significantly altered apart from a moderate increase in synaptotagmin-1 (Table 1). Immunofluorescence experiments indicated that there is no major developmental switch in synaptotagmin-2 expression in the second or third postnatal week that would explain the period of lethality of the synaptotagmin-2 deletion. Although it is at present unclear why synaptotagmin-2 KO mice initially appear normal, but then suffer from increasing weakness and die after 3 weeks, a plausible explanation based on the electrophysiological recordings and immunocytochemistry is that initially the coexpression of synaptotagmin-1 and -2 at least in the NMJ enables NMJs to compensate for the loss of synaptotagmin-2, but that in adolescent mice this compensation increasingly fails because the NMJs have to function more stringently as the mice become older.
Effect of the synaptotagmin-2 deficiency on inhibitory synaptic transmission in neostriatal neurons
We analyzed IPSCs elicited by action potentials in interneuronal synapses formed by cultured neostriatal neurons (Fig. 6). Deletion of synaptotagmin-2 caused a discrete release phenotype in these neurons: although the amplitude and charge transfer of the IPSCs was not significantly altered, the time course of release was significantly delayed in the synaptotagmin-2-deficient neurons than in littermate control neurons. The most dramatic change was a more than twofold increase in the time constant of the slow constituent of the IPSC. At the same time, the kinetics of spontaneous “mini” IPSCs was unchanged, demonstrating that the delayed time course of evoked release in striatal synapses is not attributable to a change in receptor kinetics (Fig. 6H–K). Thus, synaptotagmin-2 is not essential for neurotransmitter release in striatal neurons, but contributes dramatically to accelerate the release process, possibly because it is coexpressed with a second Ca2+ sensor for fast release that almost completely compensates for the loss of synaptotagmin-2.
Synaptic transmission in NMJs from synaptotagmin-2-deficient mice
NMJ function exhibited the following six major changes in synaptotagmin-2-deficient mice.
(1) Spontaneous release is increased >10-fold, independent of whether Ca2+ was chelated in the NMJ terminals or not (Fig. 7). This change is similar to the change we previously observed in mutant mice containing a point mutation in the synaptotagmin-2 gene (Pang et al., 2006), demonstrating that this phenotype is not attributable to a particular strain or expression level of synaptotagmin-2.
(2) The amplitude and quantal content of action potential-evoked synaptic release is dramatically decreased in mutant NMJs (Fig. 8). The scatter diagram in Figure 8 shows that the variability among samples in the size of the synaptic responses was large; nevertheless, the average size of the amplitude and quantal content is decreased more than twofold. Moreover, similar to the cultured striatal neurons, the speed with which synaptic responses develop is decreased significantly in synaptotagmin-2-deficient NMJs.
(3) In the mutant NMJs, the decrease in evoked release is accompanied by an increase in the paired-pulse ratio of synaptic responses (Fig. 9), indicating a reduction in synaptic release probability.
(4) High-frequency stimulus trains (10, 20, or 50 Hz) produce a reliable synaptic response in wild-type mice, with only moderate facilitation or depression under the conditions used here (Fig. 10). In contrast, the same stimulus trains produce dramatic facilitation in synaptotagmin-2-deficient NMJs at 10 or 20 Hz, whereas at 50 Hz no facilitation is observed, presumably because the NMJ membrane trafficking machinery becomes exhausted. These results are consistent with the notion that the release probability is reduced in synaptotagmin-2-deficient NMJs, but that the buildup of Ca2+ during the stimulus trains partially rescues the impairment of synaptic release (Fig. 10).
(5) During high-frequency stimulus trains, vesicle release becomes highly asynchronous in synaptotagmin-2-deficient NMJs when residual Ca2+ accumulates (Fig. 11). This process becomes manifest by the large increase in the number of “mini” events during the stimulus train (Fig. 11A,B), and the dramatic scattering of rise and decay times of EPPs during the train as a function of the number of stimuli applied (Fig. 11C–E).
(6) After a stimulus train, delayed release persists for several seconds in mutant NMJs, manifested as an increased number of spontaneous release events (Fig. 11F,G), which is similar to what we previously observed in cultured synaptotagmin-1-deficient cortical neurons (Maximov and Südhof, 2005). These results indicate that similar to synaptotagmin-1, synaptotagmin-2 restricts delayed asynchronous release induced by residual Ca2+ after action potential trains.
In summary, our results demonstrate that synaptotagmin-2 plays an important role in the Ca2+ triggering of neurotransmitter release in synapses formed by striatal neurons and in NMJ synapses, but that the loss of synaptotagmin-2 from these structures does not lead to a complete loss of release in these synapses. Although the reason for the coexpression of multiple Ca2+-sensing synaptotagmins in a particular synapse such as the NMJ or the synapses formed by striatal neurons remains unclear, it seems likely that the evolutionary diversification of synaptotagmin-1 into multiple similar isoforms that include synaptotagmin-2 served a regulatory role, possibly mediated by different regulatory properties of the different synaptotagmins. Future experiments will have to address this interesting and important question.
Footnotes
This work was supported by The University of Texas M. D. Anderson Cancer Center Physician Scientist Program (R.A.) and National Institutes of Health Grant HL072984 (B.F.D.). We thank Andrea Roth, Jason Mitchell, and Nicky Hamlin for assistance in animal care and genotyping. We also thank members of the Südhof lab for insightful discussions.
References
- Atluri PP, Regehr Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram D, Adachi R, Medalia O, Tuvim M, Dickey BF, Mekori YA, Sagi-Eisenberg R. Synaptotagmin II negatively regulates Ca2+-triggered exocytosis of lysosomes in mast cells. J Exp Med. 1999;189:1649–1658. doi: 10.1084/jem.189.10.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol (Lond) 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffelli M, Burgess RW, Feng G, Lobe CG, Lichtman JW, Sanes JR. Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature. 2003;424:430–434. doi: 10.1038/nature01844. [DOI] [PubMed] [Google Scholar]
- Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem. 1985;260:9280–9288. [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium-dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks W, Wirth M, Hauser H. Dicistronic transcription units for gene expression in mammalian cells. Gene. 1993;128:247–249. doi: 10.1016/0378-1119(93)90569-o. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Geppert M, Archer BT, III, Südhof TC. Synaptotagmin II. A novel differentially distributed form of synaptotagmin. J Biol Chem. 1991;266:13548–13552. [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci USA. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhamer DJ, Faerman A, Shani M, Emerson CP., Jr Regulatory elements that control the lineage-specific expression of myoD. Science. 1992;256:538–542. doi: 10.1126/science.1315077. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Janz R, Goda Y, Geppert M, Missler M, Südhof TC. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron. 1999;24:1003–1016. doi: 10.1016/s0896-6273(00)81046-6. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The timing of calcium action during neuromuscular transmission. J Physiol (Lond) 1967;189:535–544. doi: 10.1113/jphysiol.1967.sp008183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Reisinger E, Fakler B, Jonas PM. Synaptotagmin 1 is not required for fast, synchronous synaptic transmission at inhibitory synapses between parvalbumin-expressing basket cells and granule cells in hippocampus. Soc Neurosci Abstr. 2006 337.2/I15. [Google Scholar]
- Liley AW. The quantal components of the mammalian end-plate potential. J Physiol (Lond) 1956;133:571–587. doi: 10.1113/jphysiol.1956.sp005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/s0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Upregulation of preprodynorphin and preproenkephalin mRNA expression by selective activation of group I metabotropic glutamate receptors in characterized primary cultures of rat striatal neurons. Mol Brain Res. 2001;86:125–137. doi: 10.1016/s0169-328x(00)00276-x. [DOI] [PubMed] [Google Scholar]
- Marqueze B, Boudier JA, Mizuta M, Inagaki N, Seino S, Seagar M. Cellular localization of synaptotagmin I, II, and III mRNAs in the central nervous system and pituitary and adrenal glands of the rat. J Neurosci. 1995;15:4906–4917. doi: 10.1523/JNEUROSCI.15-07-04906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin-1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sorensen JB. Different effects on fast exocytosis induced by synaptotagmin-1 and 2 isoforms and abundance, but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci. 2004;24:6127–6132. doi: 10.1523/JNEUROSCI.1563-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13–1. J Cell Biol. 2002;158:577–590. doi: 10.1083/jcb.200202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR, Murphy TH. Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J Neurosci. 2004;24:420–433. doi: 10.1523/JNEUROSCI.4452-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC. Genetic analysis of synaptotagmin-2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Synaptotagmins: why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin O-H, Han W, Lao Y, Südhof TC. Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich B, Li C, Zhang JZ, McMahon H, Anderson RG, Geppert M, Südhof TC. Functional properties of multiple synaptotagmins in brain. Neuron. 1994;13:1281–1291. doi: 10.1016/0896-6273(94)90415-4. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]