

Abstract
The development of head and neck squamous cell carcinoma (HNSCC) involves the accumulation of genetic and epigenetic alterations in tumor-suppressor proteins, together with the persistent activation of growth-promoting signaling pathways. The activation of epidermal growth factor receptor (EGFR) is a frequent event in HNSCC. However, EGFR-independent mechanisms also contribute to the activation of key intracellular signaling routes, including signal transducer and activator of transcription-3 (STAT3), nuclear factor κB (NFκB), and Akt. Indeed, the autocrine activation of the gp130 cytokine receptor in HNSCC cells by tumor-released cytokines, such as IL-6, can result in the EGFR-independent activation of STAT3. In this study, we explored the nature of the molecular mechanism underlying enhanced IL-6 secretion in HNSCC cells. We found that HNSCC cells display an increased activity of the IL-6 promoter, which is dependent on the presence of an intact NFκB site. Furthermore, NFκB inhibition downregulated IL-6 gene and protein expression, and decreased the release of multiple cytokines. Interestingly, interfering with NFκB function also prevented the autocrine/paracrine activation of STAT3 in HNSCC cells. These findings demonstrate a cross-talk between the NFκB and the STAT3 signaling systems, and support the emerging notion that HNSCC results from the aberrant activity of a signaling network.
Keywords: Oral cancer, IL-6, cytokine, gene expression regulation, transcription factor
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world and accounts for 90% of malignant neoplasias of the upper respiratory system [1]. Despite recent advances in the management of locally advanced HNSCC, the overall survival of patients has improved only marginally over the past three decades [2]. In this regard, HNSCC progression often involves the accumulation of a number of genetic and epigenetic alterations in tumor-suppressor proteins, such as p53, p16, and RB, concomitant with the aberrant activity of signaling molecules that drive the unrestricted growth of HNSCC cells [3]. Thus, the recent development of novel molecular-targeted therapies may now afford the rational selection of treatment modalities for HNSCC patients based on specific molecular mechanisms whose deregulated activity contributes to the initiation, development, and metastatic spread of this cancer type.
Roughly 90% of all HNSCC cases exhibit an enhanced expression of epidermal growth factor receptor (EGFR) [4–6], and approximately 50% of all advanced HNSCC cases present an elevated activity of this polypeptide growth factor tyrosine kinase receptor [7]. This provided the molecular basis for current efforts aimed at evaluating the clinical activity of EGFR inhibitors in HNSCC [3,8]. As expected, many of the downstream intracellular targets of EGFR, such as signal transducer and activator of transcription-3 (STAT3), Ras-ERK, and PI3K-Akt pathways are also often activated in HNSCC [9–11]. However, emerging evidence suggests that both EGFR-dependent and EGFR-independent mechanisms contribute to the persistent activation of these key signaling routes in HNSCC [12,13]. The mechanisms responsible for the EGFR-independent unregulated function of these signaling routes are still poorly understood.
Of interest, whereas the activation of EGFR leads to the rapid tyrosine phosphorylation of STAT3 in tyrosine705 and the consequent activation of STAT3-dependent gene expression, we have recently observed that STAT3 tyrosine phosphorylation and the formation of active STAT3 DNA-binding complexes are insensitive to the inhibition of EGFR in a large fraction of HNSCC cell lines [13]. Indeed, 9 of 10 cell lines form a representative panel of HNSCC-derived cells showing increased tyrosine phosphorylation and activity of STAT3, but constitutive activity of EGFR was present in only 3 of them [13]. In search for the mechanism responsible for the EGFR-independent activation of STAT3 in HNSCC cells, we observed that the activation of the gp130 cytokine receptor subunit promoted the phosphorylation of STAT3 in tyrosine705 through the activation of intracellular tyrosine kinases of the JAK family [13]. Surprisingly, the activation of gp130 was found to be primarily initiated by IL-6, which, on its secretion and release by HNSCC cells, acts on the IL-6 receptor-gp130 complex on the cell surface of HNSCC cells in an autocrine fashion [13]. These findings, together with recently published reports [14,15], suggest that the persistent activation of STAT3 in HNSCC can result from the deregulated activity of EGFR or from the autocrine activation of STAT3 by tumor-released cytokines in an EGFR-independent fashion.
These observations prompted us to explore the nature of the molecular mechanism underlying the enhanced production and secretion of IL-6 in HNSCC cells. In this study, we found that overexpression of IL-6 in HNSCC cells involves increased transcription from the IL-6 promoter, which is dependent on the presence of an intact nuclear factor κB (NFκB) response element located 63 to 75 bp upstream of the IL-6 transcriptional initiation site. Furthermore, inhibition of NFκB led to a remarkable down regulation of IL-6 gene and protein expression, concomitant with a decreased release of other inflammatory cytokines, such as IL-8, IL-10, granulocyte-macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor (G-CSF). Surprisingly, the blockade of NFκB led also to a drastic inhibition of the constitutive STAT3 activity in HNSCC cells, as reflected by the reduced tyrosine phosphorylation of STAT3. Interestingly, interfering with NFκB function also prevented the ability of HNSCC cell supernatants to promote the activation of STAT3 in nontumorigenic epithelial cells in a paracrine fashion. These findings support the emerging notion that the aberrant activity of a network of interrelated signaling pathways, rather than a single deregulated biochemical route, contributes to squamous carcinogenesis. They also provide an interesting example of a cross-talk between the NFκB and the STAT3 signaling systems. This cross-talk is initiated by the release of IL-6 as a consequence of the NFκB-dependent activation of the IL-6 promoter, and the subsequent tyrosine phosphorylation of STAT3 by the autocrine/paracrine activation of IL-6 receptors in tumor cells.
Materials and Methods
DNA Constructs and Reagents
The dominant-negative IκB A32/36S super-repressor mutant (kindly provided by Dr. Siebenlist) [16] was cloned in-frame with a green fluorescent protein (GFP) coding sequence into the pCEFL expression vector, generating a pCEFL-GFP-IκB A32/36S plasmid. The lentivirus-encoding dominant-negative GFP-IκB and GFP genes were generated using the gateway system from Invitrogen Life Technologies (Carlsbad, CA) and CSCG-based retroviral vectors, as previously reported [17,18]. The full-length IL-6 promoter and the point mutations in the AP-1 and NFκB regulatory elements of the IL-6 promoter (kindly provided by Dr. Libermann) [19] were inserted into the KpnI-XhoI sites of the pGL3 luciferase reporter vector by polymerase chain reaction (PCR), generating pGL3-IL-6-Luc and its mutants. For report assays, PGL3-NFκB-Luc, in which luciferase expression is controlled by five NFκB response elements, was also used. The plasmid pcDNA3-β-galactosidase (BD Biosciences, San Jose, CA) and pCEFL-GFP were used as controls. Recombinant human IL-6 was purchased from PeproTech, Inc. (Rocky Hill, NJ), and tumor necrosis factor α (TNF-α) was from Roche (Indianapolis, IN). Cells were treated with a single dose of IL-6 or TNF-α (10 ng/ml) for 10 minutes.
Cell Lines and Culture Conditions
The HNSCC cell lines HN6, HN12, HN13, and HN30 [20]; HaCaT cells [21]; an immortalized nontumorigenic human skin keratinocyte cell line; and HEK293T and HEK293FT cells were cultured in DMEM supplemented with 10% fetal calf serum, penicillin, and streptomycin. The cells were maintained in a 5% CO2-humidified incubator. Stable cell lines were established by lentiviral infection using CSCG-based retroviral vectors and 293FT cells as packaging cells. HN13 and HN30 cells were infected with viral supernatants for 24 hours at 37°C in the presence of 8 µg/ml polybrene (hexadimethrine bromide; Sigma, St. Louis, MO). Conditioned media (CM) from HaCaT and HNSCC cell lines were prepared by incubating subconfluent cultured cells for 24 hours in DMEM without serum and supplements. Harvested CM were then filtered through a 0.22-µm low-protein-binding polyethylsulfonate membrane filter.
Transcription Factor Analysis
Nuclear extracts were prepared using a NE-PER Nuclear and Cytoplasmic Extraction reagent (Pierce Biotechnology, Inc.). The double-stranded oligonucleotide consensus sequence for NFκB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and activating protein 1 (AP1; 5′-CGCTTGATGACTCAGCCGGAA-3′) (Santa Cruz Biotechnology, Inc., Santa Cruz Biotechnology, CA) was labeled with [γ32P]ATP using T4 polynucleotide kinase (Invitrogen Life Technologies), purified, and added to the reactions (20,000 cpm/reaction) for 15 minutes. Complexes were analyzed on nondenaturing (4.5%) polyacrylamide gels. For supershift assays, 1 µg of anti-NFκB (C-20) antibody (Santa Cruz Biotechnology, Inc.) was added to the binding reaction before the addition of a radiolabeled probe. In separate experiments, to identify the NFκB subunit proteins in the complex, 2 µl of p65 antibody was incubated with nuclear proteins before the addition of the radiolabeled probe to visualize any supershift-retarded bands in the NFκB complex. Protein-DNA complexes were separated by electrophoresis on 4% polyacrylamide gel in Tris-borate-EDTA buffer. NFκB DNA-binding activity assays were also performed using Trans-AM enzyme-linked immunosorbent assay (ELISA)-based kit from Active Motif (Carlsbad, CA), according to the manufacturer's protocol. Briefly, cell extracts were incubated in a 96-well plate coated with an oligonucleotide containing the NFκB consensus-binding site. Activated transcription factors from extracts specifically bound to the respective immobilized oligonucleotides were detected using antibodies to NFκB p65, followed by a secondary antibody conjugated to horseradish peroxidase (HRP) in an ELISA-like assay.
IκB Kinase (IKK) Knockdown
Cells were seeded in 24-well or 6-well plates and, on reaching 70% confluence, the cells were washed twice in serum-free medium and transfected with 12.5 nM double-stranded RNA oligonucleotides directed against human IKKα (NM_001278, SI00605115; Qiagen, Valencia, CA) and with 25 nM against IKKβ (NM_001556, SI02777376; Qiagen) using HyperFect (Qiagen), following the manufacturer's instructions. Optimal concentrations of siRNA and time points were determined by performing a dilution curve of siRNA for each target, and knockdown was determined by Western blot analysis. After 72 hours, the cells were treated as indicated, and nuclear extracts were isolated as described above. The sequences of the negative siRNA (Qiagen) oligonucleotides used as controls were as follows: 5′-UUCUCCGAACGUGUCACGUdTdT-3′ and 5′-ACGUGACACGUUCGGAGAAdTdT-3′.
Immunoblotting
Cultured cells were harvested at the times indicated in each experiment, washed with phosphate-buffered saline, and lysed in 62.5 mM Tris, 2% sodium dodecyl sulfate (SDS), and 10% glycerol (SDS sample buffer) with aprotinin, leupeptin, pepstatin, and 4-(2-aminoethyl) benzenesulfonyl fluoride. Cell suspensions were briefly sonicated, and the protein concentration for each cell lysate was determined using DC protein assay (Bio-Rad, Hercules, CA). Twenty to 40 µg of total protein from whole cell lysates was loaded onto each lane for gel electrophoresis. Immunoblotting was performed using 0.1 M Tris (pH 7.5), 0.9% NaCl, 0.05% Tween20 with 5% nonfat dry milk as a blocking and antibody-dilution buffer, and working antisera for STAT3, phospho-STAT3 (pSTAT3)-tyrosine 705, IKKα, IKKβ, phospho-IKKα/β (Cell Signaling Technology, Beverly, MA), GFP (Covance, Inc., Denver, PA), and tubulin (Santa Cruz Biotechnology, Inc.). We used 62.5 mM Tris (pH 6.8), 2% SDS, and 100 mM β-mercaptoethanol to strip probed filters, as indicated in each experiment.
Immunohistochemistry
Individual paraffin blocks of formalin-fixed tissues from human squamous cell carcinomas of the oral cavity and HNSCC tissue arrays containing approximately 460 cases of oral SCC and normal control tissues were obtained from the National Institute of Dental and Craniofacial Research, National Institutes of Health Oral Cancer Tissue Array Initiative (Bethesda, MD) (Molinolo et al., in preparation). Briefly, individual paraffin-embedded HNSCC tissues from the oral cavity were arrayed and processed following the standard methodology of the Tissue Array Research Program (www.cancer.gov/tarp). Immunohistochemistry was performed as previously described [22]. In brief, the slides were dewaxed in xylene and hydrated through graded alcohols. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0) placed in a microwave oven for 20 minutes (2 minutes at 100% power and 18 minutes at 10% power). The slides were allowed to cool down for 30 minutes at room temperature, rinsed twice with tris-buffered saline solution (TBST), and incubated in 3% hydrogen peroxide for 30 minutes to quench the endogenous peroxidase. The sections were then incubated in blocking solution (5% bovine serum albumin) for 1 hour at room temperature, followed by treatment with anti-IL-6 primary antibody (clone 6708.11; Sigma), NFκB p65 (A) antibody (Santa Cruz Biotechnology, Inc.), and pSTAT3Y705-specific antibody (Santa Cruz Biotechnology, Inc.), where indicated, overnight at 4°C. After washing with TBST, the slides were incubated with the labeled streptavidin biotin reagent (LSAB+system HRP; DAKO Corporation, Carpinteria, CA), following the manufacturer's instructions. The slides were developed in 3,3-diaminobenzidine (Sigma FASTDAB tablet; Sigma) and counterstained with Mayer's hematoxylin. Images were taken using a SPOT digital camera attached to a Zeiss Axiophot microscope (Carl Zeiss, Thornwood, NY). All immunostainings were assessed by two experienced pathologists. The tumors were classified as positive or negative (no staining). Statistical analysis was performed by two-tailed Fisher's exact test, and P ≤ .05 was considered statistically significant.
Reporter Assays
The NFκB and IL-6 promoter reporter constructs containing the firefly luciferase cDNA (0.1–0.3 µg/well) and the pRL-null normalization construct (0.01–0.03 µg/well) containing Renilla luciferase from Renilla reniformis were transfected into HaCaT, HNSCC, and HEK293T cells (40% confluent in six-well plates). The total amount of plasmid DNA was adjusted with pcDNA3-β-galactosidase. When indicated, the cells were also transfected with pCEFL-p65 plasmid and pretreated with 10 ng/ml TNF-α or left untreated for 10 minutes. The measurement of firefly and Renilla luciferase activities present in cellular lysates was carried out using the dualluciferase reporter assay (Promega, Madison, WI) 24 hours after adding DNA. Light emission was quantitated using a Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA). Data were presented as firefly luciferase activity normalized by the Renilla luciferase activity present in each sample, and the values plotted were the average ± SE of triplicate samples from typical experiments, which were repeated at least three to five times with nearly identical results.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed using the ChIP Assay Kit (Upstate Biotechnology, Waltham, MA). Briefly, HN cancer cells were plated on two 100-mm dishes; cross-linking was performed by adding formaldehyde directly to a tissue culture medium to a final concentration of 1% and by incubating for 10 minutes at room temperature; and nuclear extracts were isolated and sonicated to generate DNA fragments. Transcription factors bound to chromatin were immunoprecipitated with the specific antibody anti-NFκB (C-20; Santa Cruz Biotechnology, Inc.), protein-DNA cross-linking was reversed, and isolated genomic DNA was amplified by PCR, using specific primers encompassing the NFκB region of the IL-6 promoter. PCR reactions were performed using 2 µl of a 50-µl DNA extraction in Tris-EDTA buffer with Hi-Fi Taq polymerase (Invitrogen Life Technologies). PCR mixtures were amplified for 1 cycle at 94°C for 2 minutes; followed by 32 cycles at 94°C for 30 seconds, annealing temperature of 55°C for 30 seconds, and 68°C for 1 minute; and then subjected to a final elongation at 68°C for 5 minutes. The primers used were hIL-6P-NFκB-F3 (5′-GCTAGCCTCAATGACGACCT-3′) and hIL-6P-NFκB-R3 (5′-GCCTCAGACATCTCCAGTCC-3′), which amplify 227 bp of the hIL-6 promoter surrounding the NFκB site. hGAPDH (sense: 5′-CCCCACACACATGCACTTACC-3′; antisense: 5′-CCTAGTCCCAGGGCTTTGATT-3′), which amplified a fragment with an expected size of 96 bp, served as a control.
Protein Secretion Analysis
CM from stably infected HNSCC cell lines and HaCaT cells were prepared by incubating subconfluent cultured cells for 24 hours in DMEM without supplements. Harvested CM were then filtered through a 0.22-µm low-protein-binding polyethylsulfonate membrane filter. IL-2, IL-5, IL-6, IL-8, IL-10, IL-12(p70), IL-13, IL-17, GM-CSF, and G-CSF proteins in the supernatant were assayed by the customized quantitative multiplexed sandwich ELISA Searchlight (Pierce Biotechnology, Inc.)
Results
HNSCC Cell Lines Constitutively Express IL-6: Correlation with NFκB Activity
To begin exploring the mechanism(s) responsible for the elevated IL-6 release from HNSCC, we first examined a representative set of HNSCC for IL-6 secretion. As shown in Figure 1A, only a limited amount of IL-6 was found in CM from an immortalized nontumorigenic epithelial cell line, HaCaT. In contrast, three representative HNSCC lines, HN12, HN13, and HN30, secreted remarkable levels of IL-6. Only one HNSCC from the many cells studied did not secrete IL-6 to the cultured medium HN6, thus serving as an internal control. IL-6 mRNA levels paralleled the pattern of IL-6 secretion in these cells (not shown). Because of the proposed role of NFκB in IL-6 expression and secretion in other cellular systems [23], we next determined the activity of NFκB in these cell lines. Under normal cultured conditions, the nuclear extracts from HN12, HN13, and HN30 cells exhibited constitutively active NFκB activity, as judged by its ability to bind to NFκB consensus oligonucleotides (Figure 1B), whereas HaCaT and HN6 showed nearly undetectable levels of protein-DNA complexes. As a control, the treatment of HaCaT and HN6 cells with TNF-α led to enhanced NFκB activity. However, only a slight activation was observed in HN12, HN13, and HN30 on TNF-α stimulation, aligned with the fact that these cells exhibit elevated levels of NFκB activity already under basal conditions. To confirm these observations, we assessed the activity of NFκB in each cell line. Indeed, as shown in Figure 1C, using an electrophoretic mobility shift assay (EMSA), we observed that nuclear extracts from HN12, HN13, HN30, and TNF-α-treated HaCaT retarded the mobility of NFκB consensus oligonucleotides in acrylamide gels. The identity of NFκB in these protein-DNA complexes was confirmed by the supershift in the apparent mobility of the complex through the addition of antisera against the p65 subunit of NFκB (Figure 1C). Thus, the remarkable secretion of IL-6 in this panel of HNSCC cell lines correlated with their constitutive activity of NFκB.
Figure 1.

HNSCC cells secrete IL-6 and exhibit enhanced NFκB DNA-binding activity. (A) IL-6 levels in CM from the indicated cell lines were measured and presented as bar graphs. Limited amounts of IL-6 were produced and secreted by HaCaT and HN6; in contrast, high levels were detected in CM from the HN12, HN13, and HN30 cell lines. The data are presented as the mean ± SE of triplicate measurements. (B) Levels of activated NFκB contained in nuclear extracts were assessed by their ability to bind to a consensus NFκB-binding site oligonucleotide, and an NFκB-DNA binding complex was revealed by chemiluminescence on incubation with antip65 antiserum followed by a secondary antibody conjugated to HRP. Bar graphs represent the mean ± SE of quantitative measurements of NFκB in arbitrary units, which were high in HN12, HN13, and HN30, but not in HaCaT and HN6, under normal culture conditions. Cell lines were also stimulated with TNF-α (10 ng/ml for 10 minutes), which stimulates NFκB activation. HaCaT cells served as positive control. Data are presented as the mean ± SE of triplicate measurements. (C) EMSA was performed using a 32P-labeled consensus oligonucleotide containing the sequence for NFκB-binding sites. Nuclear extracts were harvested from each cell line. Bands (black arrow) represent shifted protein-DNA complexes, which are found in HN12, HN13, and HN30, but not in HaCaT and HN6, under regular culture conditions. HaCaT was also stimulated with TNF-α as a control, thus confirming the binding and shifting of protein-DNA complexes. Preincubation of nuclear extracts with anti-NFκB (p65) resulted in the appearance of a slower migrating (supershifted) band (open arrow), confirming the presence of active NFκB in protein-DNA complexes.
Deregulated IL-6 Secretion in HNSCC Cells Correlates with Enhanced Activity of the IL-6 Promoter: A Role for the NFκB and AP1 Response Elements
To investigate whether the underlying mechanism leading to deregulated IL-6 expression involves enhanced transcription from the IL-6 promoter, we inserted the wild-type (WT) 1.2-kb IL-6 promoter (Figure 2A) and an IL-6 promoter containing a point mutation in its NFκB response element (Figure 2B) upstream of the firefly luciferase gene in the pGL3 reporter plasmid. As a control, we also used an IL-6 promoter, including a point mutant in its AP1 site and in both NFκB and AP1 sites. When we transfected these constructs in HEK293T cells, the IL-6 promoter was greatly stimulated by the coexpression of the p65 NFκB subunit. In contrast, as expected, the IL-6 promoter harboring a point mutation in its NFκB site did not respond to p65. Of interest, the AP1mutated IL-6 promoter still responded to p65, but the level of luciferase expression was slightly reduced, suggesting a potential interaction between these two transcriptional response elements.
Figure 2.
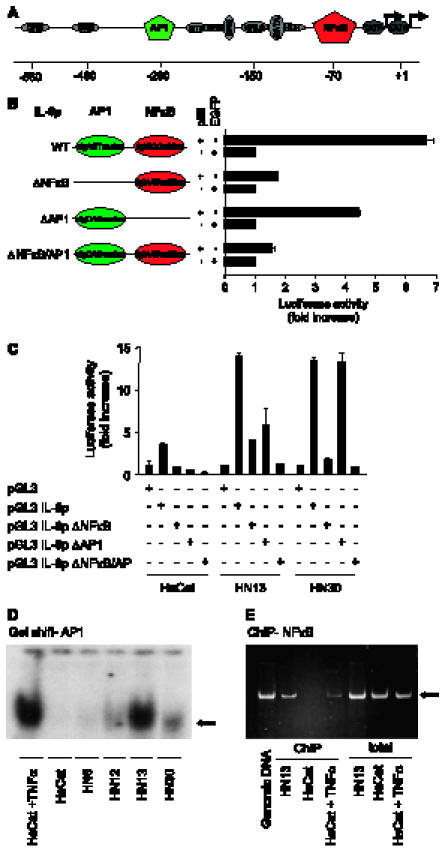
Enhanced activity of the IL-6 promoter in HNSCC. A key role for NFκB. (A) Graphic representation of the human IL-6 promoter (IL-6p) and its regulatory elements depicting their approximate locations relative to the transcription start site (+1). This promoter region includes response elements for the NFκB, AP1, and helix-loop-helix transcription factors, multiple responsive element, a cAMP-responsive element, and glucocorticoid-responsive element. (B) Site-direct mutations within the IL-6 promoter. Full-length IL-6p WT and site-direct mutations within the NFκB (ΔNFκB) and ΔAP1 (DAP1) sites were inserted into the luciferase vector (pGL3-Luc). Sequences of NFκB (red circle) and AP1 (green circle) transcriptional factor-binding sites and their point mutants (capital letters) are indicated. Full-length IL-6p WT and its mutants (0.1 µg) were transiently transfected into HEK293T cells with p65 (1 µg) and the pRL-null construct (0.01 µg), and were cultured in serum-free conditions. Dual-luciferase activity was determined, as described in Materials and Methods section. Data were presented as firefly luciferase activity normalized by the Renilla luciferase activity present in each sample, expressed as fold increase relative to control. Values plotted are the average ± SE of triplicate samples from a typical experiment. (C) The HNSCC cell lines HN13 and HN30, and immortalized keratinocytes (HaCaT) were transiently transfected with pGL3-Luc, pGL3 IL-6p WT and pGL3 IL-6p-ΔNFκB, and pGL3 IL-6p-ΔAP1 (0.3 µg), along with the pRL-null construct (0.01 µg), and cultured in serum-free conditions. Data are presented as firefly luciferase activity normalized by the Renilla luciferase activity present in each sample, expressed as fold increase relative to the vector reporter control (pGL3-Luc). Values plotted were the average ± SE of triplicate samples from a typical experiment that was repeated three to five times with nearly identical results. (D) AP1 activity in HNSCC. EMSA was performed using a 32P-labeled consensus oligonucleotide containing the sequence for AP1-binding sites. Nuclear extracts were harvested from serum-deprived cells. Bands (black arrow) represent shifted protein-DNA complexes, which are highly active only in HN13 under regular culture conditions. HaCaT was also stimulated with TNF-α as a control. (E) In vivo binding of endogenous NFκB to the IL-6 promoter. Chromatin proteins were cross-linked to DNA by formaldehyde, and purified nucleoprotein complexes were immunoprecipitated by anti-NFκB p65 antibody. The precipitated DNA and total nuclear extracts were analyzed by PCR for the presence of NFκB IL-6 promoter region corresponding to a fragment of 217 bp (arrow), which was revealed by staining agarose gels with ethidium bromide. Genomic DNA and TNF-α-stimulated HaCaT were used as a positive control confirming the binding of NFκB to the IL-6 promoter in vivo.
The reporter plasmids under the control of the WT IL-6 promoter and its mutants were then transiently transfected in the HNSCC lines. For these experiments, we selected HN13 and HN30 HNSCC cells, as HN12 cells behaved very similarly to HN30, and we used HaCaT cells as controls. When compared with luciferase expression from the reporter plasmid without the IL-6 promoter, HaCaT cells showed a fourfold activation of the IL-6 promoter. In contrast, HN13 and HN30 both showed a 16-fold activation of the IL-6 promoter. Mutations in the NFκB-binding site drastically reduced IL-6 promoter activity in all cell lines, indicating a critical role for this transcription factor in IL-6 promoter activity. A less pronounced reduction in IL-6 promoter activity was observed with the point mutant in the AP1 site, particularly in HN13. These observations suggested that NFκB is critical for the activation of IL-6 in all cells, but that, in HN13cells, AP1 may cooperate with NFκB to regulate IL-6 expression (Figure 2C). To examine this possibility, we analyzed the activity of AP1 in HNSCC cell lines by EMSA. AP1 activity was particularly highly active in HN13 cells, as demonstrated by its ability to bind to AP1 consensus oligonucleotides, whereas most cells showed low levels of AP1 protein-DNA complexes. As a positive control, HaCaT cells showed a high AP1 nuclear activity on TNF-α stimulation (Figure 2D).
As both NFκB and AP1 were required to stimulate the IL-6 promoter in HN13 cells, we wanted to confirm whether endogenous NFκB indeed binds to the IL-6 promoter in vivo in these cells. Therefore, we immunoprecipitated NFκB cross-linked to chromatin and amplified the human IL-6 promoter using PCR primers surrounding the NFκB-binding site (ChIP assay). As shown in Figure 2E, whereas PCR amplification of total genomic DNA from control, HaCaT, and HN13 cells yielded a fragment of 217 bp, no amplification was observed in NFκB immunoprecipitates from HaCaT cells, unless they were stimulated with TNF-α. In contrast, IL-6 promoter sequences were readily amplified in NFκB immunoprecipitates from HN13 cells (Figure 2E), demonstrating that this transcription factor is constitutively bound to the IL-6 promoter in vivo.
IKKα and IKKβ Contribute to p65 Constitutive Activation in HNSCC
The basal activity of p65 NFκB is repressed by its association with IκB, an ankyrin repeat-containing protein that binds to NFκB and masks its nuclear localization signal, thus retaining NFκB in its inactive state in the cytosol. The activation of NFκB involves the phosphorylation of IκB on two serine residues, Ser32 and Ser36, which triggers the rapid ubiquitination and degradation of phosphorylated IκB in the proteosome, and the consequent nuclear translocation and activation of NFκB [16]. IKK includes a regulatory subunit, NEMO (IKKγ), and two catalytic kinases subunits, IKKα (IKK1) and IKKβ (IKK2), which are lysates readily detected from HaCaT, HN12, HN13, and HN30 cells (Figure 3A). Whereas no phosphorylated IKKα/β could be detected in HaCaT cells, the levels of phosphorylated (active) IKKα/β increased dramatically in these cells on TNF-α stimulation (Figure 3A). In contrast, HN12, HN13, and HN30 showed an expression of phosphorylated IKKα/β under basal conditions. To analyze the contribution of IKKs in the constitutive activation of p65 in these cells, we interfere with endogenous IKKa and IKKβ expression using siRNA. Using the interference in HN13 as an example, we observed that IKKα- and IKKβ-specific siRNA effectively knocked down the expression of corresponding kinases without altering the expression of the untargeted IKK isoform (Figure 3B). Similar results were obtained in other cell lines (not shown). When siRNA were transfected into HaCaT cells, the inhibition of IKKα and IKKβ expression drastically reduced p65 activity (P < .001). Similarly, the knockdown of these IKKs reduced the activation of p65 by TNF-α (P < .05) (not shown). Furthermore, the constitutive activation of NFκB was also repressed in the HNSCC cell lines HN13 and HN30 by the knockdown of IKKs, although, in general, the most striking results were obtained by the interference of IKKβ. Indeed, IKKβ knockdown reduced the constitutive activation of p65 in both HN13 (P < .05) and HN30 (P < .01) (Figure 3C).
Figure 3.

The role of IKKα and IKβK in NFκB regulation in HNSCC cells. Serum-starved cell lines were immunoblotted with anti-IKKα, IKKβ, and phospho-IKKα/β antibodies. Tubulin was used as a loading control (A and B). (A) HaCaT, HN12, HN13, and HN30 immunoblot analysis shows the level of expression of IKKα and IKKβ. Elevated levels of active phospho-IKKα/β in HN12, HN13, and HN30 cells were detected under basal conditions, using TNF-α-stimulated HaCaT cells as positive control. (B) A time-course analysis was performed to assess the effective and selective knockdown of IKKα and IKKβ on transfection with their corresponding siRNA. Representative Western blot analyses depict the expression levels of IKKα/β and tubulin as loading controls in each cellular lysate. Negative siRNA oligonucleotides were used as control (C). (C) Nuclear extracts from HaCaT, HN13, and HN30, with the use of indicated siRNA, were assayed for activated NFκB levels, as assessed by their ability to bind to a consensus NFκB-binding site oligonucleotide, and an NFκB-DNA binding complex was revealed by chemiluminescence on incubation with anti-p65 antiserum followed by a secondary antibody conjugated to HRP. Bar graphs represent the mean ± SE of quantitative measurements of NFκB in arbitrary units. Asterisks denote a significant inhibition of NFκB activity by each specific siRNA interference (*P < .05, **P < .01, and ***P < .001; analysis of variance and Bonferroni's multiple comparison test).
Inhibition of NFκB Diminishes IL-6 Promoter Activity and IL-6 Protein Expression
To further analyze the contribution of NFκB to IL-6 secretion from HNSCC cells, we asked whether interference with NFκB function affected the production and release of IL-6 from these cells. As both IKKα and IKKβ can contribute to NFκB signaling in HNSCC, and (as previously described) [24–26] as IKKα and IKKβ have unique activities but can partially compensate for each other's function, we chose to explore the role of NFκB through the use of the super repressor IκB. The super repressor IκB harbors a substitutionκ of Ser32 and Ser36 for alanine, causing the blockade of IκB proteolysis and, consequently, the sequestration of NFκB in the cytoplasm, thereby preventing the translocation of NFκB to the nucleus and its transcriptional activity [16]. Initially, we observed that this IκB repressor inhibited transcription from an NFκB reporter plasmid in HaCaT cells in a doseαdependent matter, both under basal conditions or on exposure to TNF-α, using a plasmid encoding the β-galactosidase gene as a control (Figure 4A). The IκB repressor also inhibited the activation of the IL-6 promoter by TNF-α in HaCaT cells (Figure 4B). Similarly, this repressor of NFκB inhibited the expression of luciferase when driven by the isolated NFκB response element or the WT IL-6 promoter in both HN13 and HN30 cells (Figure 4, C and D).
Figure 4.

Inhibition of NFκB decreases the activity of the IL-6 promoter. HaCaT (A and B) and HNSCC cell lines HN13 (C) and HN30 (D) were transiently transfected with pNFκB-Luc (0.3 µg) (A; □ in C and D), pIL-6p-Luc (0.3 µg) (B; ■ in C and D), the pRL-null construct (0.01 µg), and increased concentrations of pCEFL IκB S32/36A, and cultured in serum-free conditions. The total amount of plasmid DNA was adjusted to 1 µg with pcDNA3-β-galactosidase. HaCaT was stimulated with TNF-α (10 ng/ml for 10 min), where indicated (A). Lysates were assayed for dual-luciferase activities. Data were presented as firefly luciferase activity normalized by the Renilla luciferase activity present in each sample, expressed as fold increase relative to control with only pcDNA3-β-galactosidase (A and B) or as percentage (C and D) of the activity without NFκB inhibition bypCEFL IκB S32/36A. Values plotted were the average ± SE of triplicate samples from typical experiments, which were repeated at least three to five times with nearly identical results.
We next engineered lentiviral constructs encoding the IκB super repressor fused to GFP (Figure 5A) to analyze the consequences of stably inhibiting NFκB in HNSCC cells. As depicted in Figure 5B, the protein product of the GFP-IκB super repressor was readily detected in HaCaT cells infected with corresponding lentiviruses, using GFP virus as control. Similarly, these constructs were highly expressed in HNSCC cells, as visualized by the fluorescent detection of GFP and its fusion protein (Figure 5, C–F). As expected, GFP-IκB S32/36A was restricted primarily to the cytoplasm, whereas GFP also exhibited strong nuclear distribution. Infection of HNSCC cells with the lentivirus encoding the super suppressor IκB S32/36A dramatically diminished the level of IL-6 secreted in the supernatants of both HN30 and HN13 HNSCC cell lines, further supporting the notion that NFκB is crucial for IL-6 promoter activity and protein expression.
Figure 5.

IL-6 production and secretion are dependent on NFκB activation. Lentiviral vectors were engineered for GFP and the full-length IκB harboring a substitution of Ser32 and Ser36 for alanine (A, top), which acts as an NFκB super repressor by blocking IκB proteolysis, tagged with a GFP-IκB. The expression levels of GFP-IκB (B, empty arrow) and GFP (B, black arrow) in HaCaT cells 72 hours after infection with these lentiviruses were documented by immunoblotting with anti-GFP antibody. Similar results were observed in the HNSCC cell lines HN13 and HN30. GFP-IκB (C and D) and GFP (E) were visualized in vivo in HN13 cells, as well as in HaCaT and HN30 cells (not shown), by fluorescence microscopy at the indicated magnification. High magnification (original magnification, x 63) revealed a cytoplasmatic expression of GFP-IκB (D) and a mostly nuclear expression of GFP (E, inset). After serum starvation, CM from the HNSCC cell lines were analyzed for IL-6 (F). Data were presented as a percentage of IL-6 secreted in GFP-IκB-expressing cells with respect to GFP-infected controls. Values plotted were the average ± SE of triplicate samples.
Inhibition of NFκB Diminishes Cytokine Release from HNSCC Cells
As NFκB plays a key role in the regulation of the expression of numerous inflammatory cytokines, we next explored the contribution of NFκB to the release of cytokines known to be expressed in HNSCC cells, in addition to IL-6. Indeed, on infection with GFP-IκB, we observed a dramatic decrease in the release of key cytokines from HNSCC cells, including IL-2, IL-6, IL-8, IL-10, IL-12, GM-CSF, and G-CSF (Figure 6). Interestingly, there appears to be a more marked inhibition of certain cytokines in HN13 cells than in HN30 cells, as, for example, IL-8, IL-12, and GM-CSF were inhibited by less than 30% in HN30 cells, whereas they were greatly reduced in HN13 cells.
Figure 6.

NFκB-dependent production and secretion of multiple cytokines in HNSCC cells. CM from the HNSCC cell lines HN13 and HN30 stably expressing IκB S32/36A (GFP-IκB) and GFP were analyzed for the secretion of IL-2, IL-8, IL-10, IL-12, G-CSF, and GM-CSF using a proteomic array. Bar graphs represent the quantitative measurement of proteins secreted in the CM. Values plotted were the average ± SE of triplicate samples.
Inhibition of NFκB Blocks the Autocrine/Paracrine Activation of STAT3
The remarkable effect of NFκB inhibition on the secretion of IL-6 and other cytokines from HNSCC cells prompted us to examine the contribution of NFκB to the potential autocrine/paracrine activation of the STAT3 signaling pathway in HNSCC cells. As shown in Figure 7A, inhibition of NFκB did not affect the expression levels of STAT3 in these cells, but diminished dramatically the levels of tyrosine-phosphorylated active STAT3 (pSTAT3Y705). Furthermore, whereas CM from HN13 and HN30 promote the accumulation of activated pSTAT3Y705 when added to HaCaT cells, we observed that the inhibition of NFκB in HN13 and HN30 HNSCC cells and the consequent reduction in IL-6 and other cytokine expressions diminished the ability of their CM to activate STAT3 (Figure 7B).
Figure 7.

Blockade of NFκB inhibits the autocrine/paracrine activation of STAT3. The HaCaT and HNSCC cell lines HN13 and HN30 were immunoblotted with anti-pSTAT3Y705 antibody and anti-STAT3. Tubulin was used as a loading control. (A) Immunoblot analysis shows a decreased level of active STAT3Y705 in HN13 and HN30 cells stably expressing GFP-IκB S32/36A with respect to control cells expressing GFP. (B) HaCaT cells were serumstarved and stimulated for 10 minutes with IL-6 (10 ng/ml), or CM from HN13 and HN30 stably expressing GFP-IκB S32/36A and GFP, or CM from HaCaT cells (C) as a control. The addition of IL-6 and CM from HN13 and HN30 cells induces remarkable STAT3 activation (STAT3Y705). The effects of CM on HN13 and HN30 were prevented by the inhibition of NFκB.
Nuclear Localization of NFκB Correlates with IL-6 Expression and STAT3 Activation in HNSCC
These findings prompted us to explore whether the activation status of NFκB correlates with the expression of IL-6 and the phosphorylation of STAT3 in HNSCC tumors using an immunohistochemical approach. We detected a robust immunohistochemical stain for p65 in the cytoplasm and the nucleus (Figure 8A), which is more clearly seen at a higher magnification (Figure 8B). The nuclear staining of p65 was present in all areas of the carcinoma, including invading islands. Indeed, NFκB was present in most of the cases analyzed (n = 196) (Figure 8F). pSTAT3Y705 was also expressed in the majority of tumor cells (Figure 8C). A close-up view demonstrates the specific nuclear staining of their phosphorylated form of STAT3 (Figure 8D). IL-6 presented a generalized cytoplasmic immunoreactivity (Figure 8E), which was positive in a large fraction of cases (n = 144) (Figure 8F). Immunohistochemical analyses of human HNSCC tissue array samples demonstrated an excellent correlation in vivo between the expressions of NFκB, IL-6, and pSTAT3 in the same tissue tumor cores (Table 1). Indeed, a significant correlation was present between NFκB and IL-6 (P ≤ .02), between IL-6 and pSTAT3Y705 (P ≤ .001), and between NFκB and pSTAT3Y705 (P ≤ .005).
Figure 8.

NFκB, pSTAT3, and IL-6 are highly expressed in head and neck (HN) clinical samples. Human oral SCC individual samples and tissue arrays were stained for NFκB p65, pSTAT3Y705, and IL-6. Photographs show representative tumorareas. (A) NFκB is predominantly observed in tumor cells. (B) Cytoplasmatic and nuclear stainings for NFκB are seen at a higher magnification. (C) pSTAT3Y705 is present in the nucleus of tumor cells. (D) Higher magnification. (E) IL-6 is strongly expressed in the cytoplasm of tumor cells. (F) The bar graphs summarize the expression of NFκB, pSTAT3Y705, and IL-6 in the HNSCC tissue array. The number of positive cases is presented as the percentage of tumor tissue samples (n = total number of samples analyzed).
Table 1.
Correlation between the Expressions of NFκB, IL-6, and pSTAT3Y705 in HNSCC [n (%)].
| NFκB- | NFκB+ | P | pSTAT3Y705- | pSTAT3Y705+ | P | |
| IL-6- | 8 (6.7) | 30 (25.0) | ≤ .02 | 12 (14.3) | 18 (21.4) | ≤ .001 |
| IL-6+ | 4 (3.3) | 78 (65) | 4 (4.7) | 50 (59.6) | ||
| NFκB- | 6 (6.3) | 8 (8.3) | ≤ .005 | |||
| NFκB+ | 8 (8.3) | 74 (77.1) |
Together, these data are aligned with our observations in HNSCC cells and support the existence of the autocrine/paracrine activation of STAT3, which can be initiated by the NFκB-dependent release of IL-6 and other inflammatory cytokines from HNSCC cells (Figure 9).
Figure 9.

Proposed mechanism of cross-talk between the NFκB and the STAT3 pathways. Constitutive activation of NFκB leads to the production and secretion of cytokines such as IL-6, which acts on the gp130 cytokine receptor family in an autocrine/paracrine manner and causes the consequent activation of STAT3 in an EGFR-independent fashion. Inhibition of NFκB by IκB S32/36A diminishes the secretion of those molecules, thereby blocking the activation of the STA T3 pathway.
Discussion
A better understanding of the molecular mechanisms underlying the development of HNSCC may help identify novel targets for pharmacological intervention in this disease [27]. In this regard, the nature of signal transduction pathways whose aberrant activity promotes the unregulated growth and survival of HNSCC cells has just begun to be elucidated. These include the overexpression and enhanced phosphorylation of EGFR [10], the elevated activity of the Akt-mTOR pathway [11,12,28], and the persistent activation of the NFκB and STAT3 transcription factors [9,29,30]. Whereas EGFR can stimulate NFκB and STAT3, recent observations indicate that HNSCC can also exhibit EGFR-independent activation of these key signaling routes [13]. We now show that the aberrant function of the transcription factor NFκB can lead to the stimulation of STAT3 by an autocrine/paracrine mechanism that involves the release of IL-6, thus providing evidence of the existence of a molecular cross-talk between the NFκB and STAT3 signaling systems in HNSCC.
IL-6 is a pleiotropic cytokine expressed by a variety of cell types, including macrophages, fibroblasts, endothelial cells, T cells, and B cells (reviewed in Oppenheim et al. [31]). It plays an important role in immune responses, acute-phase reactions, hematopoiesis, and regeneration, and its production is induced by different stimuli, such as T-cell mitogens, antigenicstimulation, viral infection, and peptide factors, such as IL-1, TNF-α, IL-2, interferon α, and PGDF [32]. IL-6 is expressed in psoriatic skin and in cancerous epithelial cells in HNSCC [33–37]; however, what leads to IL-6 secretion in human HNSCC is still poorly understood.
The human IL-6 gene, located on chromosome 7p21, is approximately 5 kb, and its promoter contains important cis-acting response elements, including NFκB and AP1 sites, located between nucleotides 75–63 and 286–265 upstream of the IL-6 mRNA cap site, respectively [38]. In this study, we found that the high expression of IL-6 in HNSCC cells correlates with the enhanced activity of the IL-6 promoter, whereas in certain HNSCC cells, the AP1 transcription factor, whose activity is associated with malignant transformation of squamous cell epithelia [39], also contributed partially. This situation is not unique, as NFκB and AP1 have an additive effect on the activation of a number of genes associated with metabolic and inflammatory responses [40,41], which may enable rapid changes in gene expression in response to the stimulation of EGFR and the Ras/Raf signal transduction pathway, which controls AP1 activity [42], in coordination with proinflammatory cytokines that stimulate NFκB [43]. In particular, for IL-6 expression, AP1 can cooperate with NFκB, but the latter appears to have a more prominent and general role. Indeed, NFκB was found to be bound to the endogenous IL-6 promoter, as judged by chromatin inmmunoprecipitation assays, and mutations in the NFκB site reduce IL-6 promoter activity in all HNSCC cells tested. Furthermore, inhibition of this transcription factor is sufficient to downregulate IL-6 gene and protein expression. Thus, available evidence suggests that NFκB plays a central role in promoting IL-6 secretion from HNSCC cells.
Constitutive activation of NFκB is a frequent event in a variety of neoplasias, including melanoma [44], breast and prostate carcinoma [45,46], T-cell leukemia [47], Hodgkin's and B-cell lymphomas [48,49], multiple myeloma [50], and HNSCC [29], and the aberrant activity of NFκB has been shown to contribute to tumor cell survival, proliferation, migration, and radiation resistance. As previously reported [51], we observed constitutive NFκB activation in the majority of HNSCC cells examined, as reflected by the presence of elevated NFκB DNA-binding activity. The activation of NFκB in HNSCC depends on IKKα and IKKβ activity, as suggested by knockdown experiments using kinase specific-siRNA. However, in general, IKKa is less efficient in activating NFκB B and cannot be substituted by IKKβ in animal knockout studies [24–26,52–54]. Aligned with these observations, our results suggest that IKKβ plays a more prominent role in NFκB activation in HNSCC tissues. This observation may facilitate future efforts aimed at elucidating which of the many molecular events that converge in the phosphorylation and activation of IKKβ are in turn activated in HNSCC cells.
The persistent activation of NFκB can lead to the elevated expression and secretion of IL-6 in HNSCC, similar to what was recently described in prostate cancer cells [23]. Furthermore, the transcription factor NFκB also regulates the expression of other cytokines involved in both inflammatory and immune responses in HNSCC. In particular, we observed high levels of secretion of IL-2, IL-6, IL-8, IL-10, IL12, GM-CSF, and G-CSF, whose expressions were reduced by NFκB inhibition. Other cytokines were also analyzed, demonstrating elevated levels of IL-13 followed by lower levels of IL-5 and IL-17 (data not shown) secreted by HNSCC cells. Some of these cytokines have been associated with increased tumor growth and metastasis, and overall decrease of survival [55,56].
IL-6 is readily detected in the serum, tissues, and saliva of HNSCC patients [36,37,57]; once released, IL-6 secretion may contribute to HNSCC tumor progression and metastasis, as well as to inflammatory and angiogenic responses [58,59] that characterize this tumor type. An interesting possibility is that IL-6 secretion may also facilitate the immune evasion of HNSCC cancer cells. Patients with HNSCC exhibit a number of functional defects in their tumor-infiltrating and circulating T cells, which may compromise their antitumor immune responses (reviewed in Whiteside [60]). In particular, recent attention has focused on CD4+ T helper (Th) cells, one of whose functions is to prime and stimulate the cell-mediated antitumor immunity initiated by CD8+ cytotoxic T lymphocytes [61]. In this regard, the release of IL-6 from HNSCC cells can inhibit the differentiation of CD4+ Th cells into type 1 cells (which is a subset of CD4+ Th cells that promote the tumoricidal activity of CD8+ cytotoxic T lymphocytes) while favoring the differentiation of CD4+ Th cells into type 2 cells (which inhibit cell-mediated immune response to tumor cells but stimulate a less effective humoral response) [62]. The IL-6-initiated autocrine activation of STAT3 in HNSCC cells may in turn influence the overall pattern of cytokines released by tumor cells, which can inhibit the maturation and activation of dendritic cells [63], thus providing an additional mechanism that facilitates HNSCC cells to evade the immune surveillance system.
Remarkably, we observed here that blocking NFκB diminished the accumulation of active STAT3 in HNSCC cells, thus suggesting the existence of cross-talk between the NFκB and the STAT3 pathways through the release of IL-6 and other cytokines and the autocrine/paracrine activation of cytokine receptors expressed in HNSCC cells. Indeed, most of the HNSCC clinical samples that exhibit NFκB also expressed IL-6 and exhibited nuclear accumulation of pSTAT3. Only a few tumors that exhibit pSTAT3 do not show elevated levels of IL-6 expression, which may represent a subgroup of HNSCC patients in which the activation of STAT3 is dependent on EGFR or on another tumor or stromal-released cytokine [64]. Together, our findings support the emerging view that a deregulated signaling network, rather than the alteration of a single biochemical route, underlies the aberrant growth of HNSCC cells. Conversely, considering that STAT3 is constitutively activated in oral HNSCC (but not in the normal oral epithelium) [9,10] and that inhibition of STAT3 function leads to the growth inhibition of HNSCC [65], the observation that NFκB can in turn enhance the activity of STAT3 in HNSCC cancer cells may now provide a molecular framework for the future clinical evaluation of targeting NFκB and/or IL-6 in HNSCC patients, together with EGFR inhibitors, as a combined treatment modality. This would be particularly important in HNSCC patients displaying elevated local and serum levels of IL-6, in which their enhanced tyrosine phosphorylation of STAT3 could be resistant to EGFR inhibition.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127–135. doi: 10.1038/nrc1549. [DOI] [PubMed] [Google Scholar]
- 3.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 4.Dassonville O, Formento JL, Francoual M, Ramaioli A, Santini J, Schneider M, Demard F, Milano G. Expression of epidermal growth factor receptor and survival in upper aerodigestive tract cancer. J Clin Oncol. 1993;11:1873–1878. doi: 10.1200/JCO.1993.11.10.1873. [DOI] [PubMed] [Google Scholar]
- 5.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 6.Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 7.Kong A, Leboucher P, Leek R, Calleja V, Winter S, Harris A, Parker PJ, Larijani B. Prognostic value of an activation state marker for epidermal growth factor receptor in tissue microarrays of head and neck cancer. Cancer Res. 2006;66:2834–2843. doi: 10.1158/0008-5472.CAN-05-2994. [DOI] [PubMed] [Google Scholar]
- 8.Pomerantz RG, Grandis JR. The epidermal growth factor receptor signaling network in head and neck carcinogenesis and implications for targeted therapy. Semin Oncol. 2004;31:734–743. doi: 10.1053/j.seminoncol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, Tweardy DJ. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin Grandis J, Zeng Q, Drenning SD. Epidermal growth factor receptor-mediated stat3 signaling blocks apoptosis in head and neck cancer. Laryngoscope. 2000;110:868–874. doi: 10.1097/00005537-200005000-00016. [DOI] [PubMed] [Google Scholar]
- 11.Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, Sausville EA, Molinolo AA, Gutkind JS. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004;10:4029–4037. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 12.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, Molinolo AA, Gutkind JS. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65:9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 13.Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948–2956. [PubMed] [Google Scholar]
- 14.Quadros MR, Peruzzi F, Kari C, Rodeck U. Complex regulation of signal transducers and activators of transcription 3 activation in normal and malignant keratinocytes. Cancer Res. 2004;64:3934–3939. doi: 10.1158/0008-5472.CAN-04-0214. [DOI] [PubMed] [Google Scholar]
- 15.Siavash H, Nikitakis NG, Sauk JJ. Abrogation of IL-6—mediated JAK signalling by the cyclopentenone prostaglandin 15dPGJ(2) in oral squamous carcinoma cells. Br J Cancer. 2004;91:1074–1080. doi: 10.1038/sj.bjc.6602055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 17.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basile JR, Afkhami T, Gutkind JS. Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase-Akt pathway. Mol Cell Biol. 2005;25:6889–6898. doi: 10.1128/MCB.25.16.6889-6898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dendorfer U, Oettgen P, Libermann TA. Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol Cell Biol. 1994;14:4443–4454. doi: 10.1128/mcb.14.7.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel V, Jakus J, Harris CM, Ensley JF, Robbins KC, Yeudall WA. Altered expression and activity of G1/S cyclins and cyclindependent kinases characterize squamous cell carcinomas of the head and neck. Int J Cancer. 1997;73:551–555. doi: 10.1002/(sici)1097-0215(19971114)73:4<551::aid-ijc16>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 21.Patel V, Senderowicz AM, Pinto D, Jr, Igishi T, Raffeld M, Quintanilla-Martinez L, Ensley JF, Sausville EA, Gutkind JS. Flavopiridol, a novel cyclin-dependent kinase inhibitor, suppresses the growth of head and neck squamous cell carcinomas by inducing apoptosis. J Clin Invest. 1998;102:1674–1681. doi: 10.1172/JCI3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Squarize CH, Castilho RM, Santos Pinto D., Jr Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J Oral Pathol Med. 2002;31:379–384. doi: 10.1034/j.1600-0714.2002.00142.x. [DOI] [PubMed] [Google Scholar]
- 23.Zerbini LF, Wang Y, Cho JY, Libermann TA. Constitutive activation of nuclear factor kappaB p50/p65 and Fra-1 and JunD is essential for deregulated interleukin 6 expression in prostate cancer. Cancer Res. 2003;63:2206–2215. [PubMed] [Google Scholar]
- 24.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 25.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 26.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 27.Nagpal JK, Das BR. Oral cancer: reviewing the present understanding of its molecular mechanism and exploring the future directions for its effective management. Oral Oncol. 2003;39:213–221. doi: 10.1016/s1368-8375(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 28.Nathan CA, Amirghahari N, Abreo F, Rong X, Caldito G, Jones ML, Zhou H, Smith M, Kimberly D, Glass J. Overexpressed eIF4E is functionally active in surgical margins of head and neck cancer patients via activation of the Akt/mammalian target of rapamycin pathway. Clin Cancer Res. 2004;10:5820–5827. doi: 10.1158/1078-0432.CCR-03-0483. [DOI] [PubMed] [Google Scholar]
- 29.Ondrey FG, Dong G, Sunwoo J, Chen Z, Wolf JS, Crowl-Bancroft CV, Mukaida N, Van Waes C. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog. 1999;26:119–129. doi: 10.1002/(sici)1098-2744(199910)26:2<119::aid-mc6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 30.Duffey DC, Chen Z, Dong G, Ondrey FG, Wolf JS, Brown K, Siebenlist U, Van Waes C. Expression of a dominant-negative mutant inhibitor-kappaBalpha of nuclear factor-kappaB in human head and neck squamous cell carcinoma inhibits survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res. 1999;59:3468–3474. [PubMed] [Google Scholar]
- 31.Oppenheim JJ, Feldmann M, Durum SK. Cytokine Reference: A Compendium of Cytokines and Other Mediators of Host Defense. London; San Diego: Academic Press; 2001. p. 2 v. (xxx, 2260p) [Google Scholar]
- 32.Hirano T. The biology of interleukin-6. Chem Immunol. 1992;51:153–180. [PubMed] [Google Scholar]
- 33.Krueger JG, Krane JF, Carter DM, Gottlieb AB. Role of growth factors, cytokines, and their receptors in the pathogenesis of psoriasis. J Invest Dermatol. 1990;94:135S–140S. doi: 10.1111/1523-1747.ep12876121. [DOI] [PubMed] [Google Scholar]
- 34.Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT, Kupper TS, Sehgal PB, Gottlieb AB. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA. 1989;86:6367–6371. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshizaki K, Nishimoto N, Matsumoto K, Tagoh H, Taga T, Deguchi Y, Kuritani T, Hirano T, Hashimoto K, Okada N, et al. Interleukin 6 and expression of its receptor on epidermal keratinocytes. Cytokine. 1990;2:381–387. doi: 10.1016/1043-4666(90)90069-6. [DOI] [PubMed] [Google Scholar]
- 36.Woods KV, El-Naggar A, Clayman GL, Grimm EA. Variable expression of cytokines in human head and neck squamous cell carcinoma cell lines and consistent expression in surgical specimens. Cancer Res. 1998;58:3132–3141. [PubMed] [Google Scholar]
- 37.Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW, Enamorado I, Yeh NT, Kroog GS, Rudy S, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369–1379. [PubMed] [Google Scholar]
- 38.Ray A, Tatter SB, May LT, Sehgal PB. Activation of the human “beta 2-interferon/hepatocyte-stimulating factor/interleukin 6” promoter by cytokines, viruses, and second messenger agonists. Proc Natl Acad Sci USA. 1988;85:6701–6705. doi: 10.1073/pnas.85.18.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Domann FE, Jr, Levy JP, Finch JS, Bowden GT. Constitutive AP-1 DNA binding and transactivating ability of malignant but not benign mouse epidermal cells. Mol Carcinog. 1994;9:61–66. doi: 10.1002/mc.2940090202. [DOI] [PubMed] [Google Scholar]
- 40.Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol. 1994;56:554–558. [PubMed] [Google Scholar]
- 41.Thomas RS, Tymms MJ, McKinlay LH, Shannon MF, Seth A, Kola I. ETS1, NFkappaB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene. 1997;14:2845–2855. doi: 10.1038/sj.onc.1201125. [DOI] [PubMed] [Google Scholar]
- 42.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 43.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 44.Huang S, DeGuzman A, Bucana CD, Fidler IJ. Level of interleukin-8 expression by metastatic human melanoma cells directly correlates with constitutive NF-kappaB activity. Cytokines Cell Mol Ther. 2000;6:9–17. doi: 10.1080/13684730050515868. [DOI] [PubMed] [Google Scholar]
- 45.Mukhopadhyay A, Bueso-Ramos C, Chatterjee D, Pantazis P, Aggarwal BB. Curcumin downregulates cell survival mechanisms in human prostate cancer cell lines. Oncogene. 2001;20:7597–7609. doi: 10.1038/sj.onc.1204997. [DOI] [PubMed] [Google Scholar]
- 46.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA. 2004;101:10137–10142. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori N, Fujii M, Ikeda S, Yamada Y, Tomonaga M, Ballard DW, Yamamoto N. Constitutive activation of NF-kappaB in primary adult T-cell leukemia cells. Blood. 1999;93:2360–2368. [PubMed] [Google Scholar]
- 48.Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin's disease tumor cells. J Clin Invest. 1997;100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi NC, Richardson PG, et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21:5673–5683. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- 51.Wolf JS, Chen Z, Dong G, Sunwoo JB, Bancroft CC, Capo DE, Yeh NT, Mukaida N, Van Waes C. IL (interleukin)-1 alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res. 2001;7:1812–1820. [PubMed] [Google Scholar]
- 52.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 53.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 54.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith DR, Polverini PJ, Kunkel SL, Orringer MB, Whyte RI, Burdick MD, Wilke CA, Strieter RM. Inhibition of interleukin 8 attenuates angiogenesis in bronchogenic carcinoma. J Exp Med. 1994;179:1409–1415. doi: 10.1084/jem.179.5.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda T, Shimada E, Urakawa T. Serum levels of cytokines in patients with colorectal cancer: possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis. J Gastroenterol. 1994;29:423–429. doi: 10.1007/BF02361238. [DOI] [PubMed] [Google Scholar]
- 57.Rhodus NL, Ho V, Miller CS, Myers S, Ondrey F. NF-kappaB dependent cytokine levels in saliva of patients with oral preneoplastic lesions and oral squamous cell carcinoma. Cancer Detect Prev. 2005;29:42–45. doi: 10.1016/j.cdp.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 59.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 60.Whiteside TL. Immunobiology of head and neck cancer. Cancer Metastasis Rev. 2005;24:95–105. doi: 10.1007/s10555-005-5050-6. [DOI] [PubMed] [Google Scholar]
- 61.Sparano A, Lathers DM, Achille N, Petruzzelli GJ, Young MR. Modulation of Th1 and Th2 cytokine profiles and their association with advanced head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg. 2004;131:573–576. doi: 10.1016/j.otohns.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 62.Lathers DM, Young MR. Increased aberrance of cytokine expression in plasma of patients with more advanced squamous cell carcinoma of the head and neck. Cytokine. 2004;25:220–228. doi: 10.1016/j.cyto.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 63.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 64.Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A, Ferris RL, Grandis JR. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene. 2005;24:4442–4449. doi: 10.1038/sj.onc.1208635. [DOI] [PubMed] [Google Scholar]
- 65.Xi S, Gooding WE, Grandis JR. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene. 2005;24:970–979. doi: 10.1038/sj.onc.1208316. [DOI] [PubMed] [Google Scholar]