

Abstract
Allosteric modulators for adenosine receptors (ARs) are of an increasing interest and may have potential therapeutic advantage over orthosteric ligands. Benzoylthiophene derivatives (including PD 81,723), 2-aminothiazolium salts, and related allosteric modulators of the A1 AR have been studied. The benzoylthiophene derivatives were demonstrated to be selective enhancers for the A1 AR, with little or no effect on other subtypes of ARs. Allosteric modulation of the A2A AR has also been reported. A3 allosteric enhancers may be predicted to be useful against ischemic conditions. We have recently characterized two classes of A3 AR allosteric modulators: 3-(2-pyridinyl)isoquinolines (e.g. VUF5455) and 1H-imidazo-[4,5-c]quinolin-4-amines (e.g. DU124183), which selectively decreased the agonist dissociation rate at the human A3AR but not at A1 and A2A ARs. DU124183 left-shifted the agonist conc.-response curve for inhibition of forskolin-stimulated cAMP accumulation in intact cells expressing the human A3AR with up to 30% potentiation of the maximal efficacy. The increased potency of A3 agonists was evident only in the presence of an A3 antagonist, since VUF5455 and DU124183 also antagonized, i.e. displaced binding at the orthosteric site, with Ki values of 1.68 and 0.82 μM, respectively. A3AR mutagenesis studies implicated F1825.43 and N2747.45 in the action of the enhancers and was interpreted using a rhodopsin-based A3AR molecular model, suggesting multiple binding modes. Amiloride analogues, SCH-202676 (N-(2,3-diphenyl-1,2,4-thiadiazol-5(2H)-ylidene)methanamine), and sodium ions were demonstrated to be common allosteric modulators for at least three subtypes (A1, A2A, and A3) of ARs.
INTRODUCTION
Four subtypes of adenosine receptors (ARs) have been cloned, termed A1, A2A, A2B and A3 ARs [1]. Activation of A1 and A3 ARs induces the inhibition of the enzyme adenylate cyclase, whereas activation of A2A and A2B receptors leads to the stimulation of this enzyme. All four subtypes belong to the largest category, termed Group 1, within the superfamily of G protein-coupled receptors (GPCRs), which possess seven membrane-spanning α-helices [1]. The therapeutic areas for which there is growing interest in these receptors include: immune function and inflammation, CNS disorders, pulmonary and cardiovascular diseases. While there are not yet highly potent and selective agents for any of these receptors in use as therapeutic agents, a means of indirectly modulating the action at these receptors by pharmacological means seems appealing.
It has been suggested that allosteric modulators may provide therapeutic advantages over orthosteric agonists [2,3]. Such advantages may include greater subtype selectivity and fewer side effects. For example, diazepam and other benzodiazepines, which act as allosteric enhancers of the ion channel-coupled GABAA receptor, have acceptable side effects and are used clinically. In contrast, directly acting GABAA agonists have widespread side effects and are not used clinically. In the ligand gated ion channel nicotinic receptors, the alkaloid galanthamine acted as an acetylcholinesterase inhibitor as well as an allosteric modulator at nicotinic receptor sites potentiating nicotinic cholinergic neurotransmission. Galanthamine has recently been extensively and successfully used in the clinic and also showed satisfactory therapeutic effects in Alzheimer’s disease [4].
Thus, the presence of allosteric site(s) on GPCRs has provided new targets for drug discovery. The effects of an allosteric enhancer on an organ or tissue might be event-specific due to an increase in the local concentration of the endogenous agonist [3,5]. For example, hypoxic conditions increase the local production of cyto-protective adenosine. Compounds that either augment the concentration of adenosine or enhance its action, locally, may have a better therapeutic profile than the agonists. Additionally, neurotransmitter receptors have been reported to be less sensitive to desensitization or down-regulation by allosteric enhancers than by exogenous agonists [3]. Thus, allosteric modulators could offer a control of receptor function not found with competitive agonists.
A1 ARs
Allosteric enhancers, in theory, are such a means of indirectly enhancing the action of a native agonist such as adenosine. The allosteric modulators act at a site distinct from the agonist binding site, and their effect is evident only in the presence of exogenously added agonist. Bruns and colleagues introduced the first allosteric modulators of the A1AR in 1990 [6,7]. These initial reports described benzoylthiophene derivatives (Fig. (1)), such as PD 81,723 (2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]-methanone) 1, as A1AR allosteric enhancers. These allosteric enhancers were identified while screening chemical libraries in a binding assays at the rat A1AR. Bruns made the initial discovery of this effect based on small increases (~25%) in the level of agonist binding.
Fig. 1.

Benzoylthiophenes as A1AR allosteric modulators.
PD 81,723 has now been extensively modified structurally, leading to a variety of substituted derivatives. In one of the seminal papers by Bruns and coworkers [7] three compounds (PD 71,605, PD 81,723 and PD 117,975) were tested in substantial detail (Fig. (1)). They have since served as source of inspiration for further synthetic efforts, leading to a variety of analogs. Their structure-activity relationships will be discussed here. Table 1, based on the work of Van der Klein et al. [8] and Kourounakis et al. [9], shows that appropriate substitution of the benzoyl ring is essential for high activity. In the largest series of PD 71,605 analogues the potency order for substitution at the benzoyl ring was 3,4-Cl2 > 4-CH3 = 4-t-Bu > 4-CF3 ≥ 4-Br ≥ 4-Cl ≥3-CF3 > 3-Cl > 2-Cl > H > 4-NO2, with the latter three substantially less active as enhancers than PD 81,723. All analogues appeared to possess some degree of antagonistic activity, potentially compromising their enhancing effectiveness. However, the SAR (structure activity relationship) for allosteric enhancement and antagonism appeared to be different. In this series, LUF 5484 (2-amino-3-(3,4-dichlorobenzoyl)-4, 5, 6, 7-tetrahydrobenzo[b]thiophene) was 2.4 times more potent than PD 81,723 as an enhancer, while showing comparable antagonistic activity. In the series of 6-benzyl-2-amino-3-benzoyl-4, 5, 6, 7-tetrahydrothieno[2.3-c] pyridines the compound with a 3,4-dichloro substituted benzoyl moiety 18 was again the most potent allosteric enhancer (Table 2). Substitution of the 6-benzyl group generally lowered enhancing activity, although a 3-chloro substituent 19 was well tolerated. All compounds showed antagonistic activity to varying extents.
Table 1.
Adenosine A1 Receptor Enhancement and Antagonism by 2-Amino-3-Benzoylthiophenes in Rat Brain Membranes
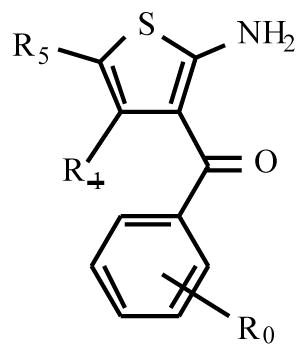
| |||||||
|---|---|---|---|---|---|---|---|
| Enhancement | Antagonism | ||||||
| compound | R0 | R1 | R5 | %* | EC50 (μM) | %** | Ki (μM) |
| PD81,723 | 3-CF3 | CH3 | CH3 | 100 | 15 | 40 | 4.7 |
| 2 | H | CH3 | CH3 | 8 | 14 | 18 | |
| 3 | 3,4-Cl | CH3 | CH3 | 116 | 50 | 3.2 | |
| 4 | 4-tBu | CH3 | CH3 | 125 | 47 | 4.4 | |
| RS74,513 | 3-CF3 | CH2CH3 | CH3 | 112 | 29 | 10 | |
| 5 | H | CH2CH3 | CH3 | 31 | 13 | 15 | |
| 6 | 3-Cl | CH2CH3 | CH3 | 30 | 22 | 13 | |
| 7 | 3-CF3 | -(CH2)4- | 122 | 6.0 | 32 | ||
| 8 | H | -(CH2)4- | 47 | 35 | 5.5 | ||
| 9 | 2-Cl | -(CH2)4- | 73 | 35 | |||
| PD71,605 | 3-Cl | -(CH2)4- | 93 | 11 | 51 | 4.2 | |
| 10 | 4-Cl | -(CH2)4- | 123 | 6.8 | 40 | ||
| 11 | 4-CF3 | -(CH2)4- | 131 | 4.7 | 57 | ||
| 12 | 4-CH3 | -(CH2)4- | 137 | 35 | 30 | ||
| 13 | 4-NO2 | -(CH2)4- | 34 | 20 | |||
| LUF 5484 | 3,4-Cl | -(CH2)4- | 151 | 6.2 | 35 | ||
| 14 | 4-tBu | -(CH2)4- | 137 | 40 | |||
| 15 | 4-Br | -(CH2)4- | 128 | 16 | 42 | ||
Enhancing activity by 10 μM of test compound is expressed as percent decrease in [3H]CCPA dissociation over control (0%) and that of 10 μM PD81,723 (100%). For some compounds EC50 values were determined, defined as the ligand concentration causing half maximal enhancement
Antagonistic activity is expressed as percent displacement of 0.4 nM of [3H]DPCPX by 10 μM of test compound. For some compounds Ki values were determined.
Table 2.
Adenosine A1 Receptor Enhancement and Antagonism by 2-amino-3-benzoyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridines in Rat Brain Membranes
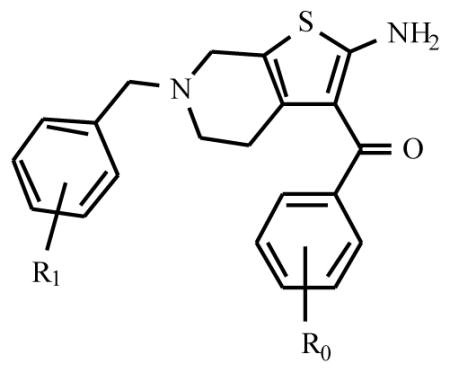
| |||||
|---|---|---|---|---|---|
| Enhancement | Antagonism | ||||
| R0 | R1 | %* | EC50 (μM) | %** | |
| PD117,975 | H | H | 53 | 67 | |
| 4-Cl | H | 132 | 11.3 | 61 | |
| 3,4-Cl | H | 174 | 9.2 | 51 | |
| H | 3-Cl | 106 | 15.1 | 80 | |
| H | 4-Cl | 69 | 52 | ||
| H | 3,4-Cl | 57 | 4 | ||
Enhancing and antagonistic activity were expressed as described in Table 1.
In a similar manner, Baraldi et al. [10] developed a series of PD 81,723 analogs. The assay used was quite different from the above (cAMP determinations in CHO cells expressing human adenosine A1 receptors vs. radioligand dissociation experiments on rat brain membranes). However, similar findings were obtained, although none of the compounds synthesized was more active at 10 μM than PD 81,723. Among the more potent derivatives, when tested at 0.1 μM, were three compounds in Table 1, i.e. 2, 10 and 15. In some cases in which compounds with both enhancing and substantial antagonistic activity were tested, no significant effects on cAMP production were noted.
Tranberg et al. [11] also performed a systematic survey of the PD 81,723 lead structure. When studying tetrahydrobenzo derivatives such as PD 71,605, they obtained findings similar to the ones in Table 1, now with a radioligand dissociation assay on human rather than rat adenosine A1 receptors. Interestingly, extending the tetrahydrobenzo moiety with one methylene group also yielded potent enhancers (Fig. (2)). The 3-OMe derivative 22 had high enhancing activity and relatively low antagonistic potency, 99% and 13% at 100 μM, respectively.
Fig. 2.

Extension of the tetrahydrobenzo moiety of analogues of PD 81,723 1 with methylene groups.
Replacing the aroyl for a naphthoyl moiety also yielded potent enhancers. Baraldi and coworkers [12] identified five compounds more potent than PD 81,723 (e.g. 23, 24, see Fig. (2)) in increasing radiolabeled agonist binding to both human and rat adenosine A1 receptors.
Slightly more exotic derivatives stem from a number of patents on allosteric enhancers, filed by Medco Research, Inc. (presently King Pharmaceuticals) [13–16]. Fig. (3) shows the structures of some typical examples (25 – 28). T28 proved a potent enhancer of the binding of [3H]CCPA (2-chloro-N6-cyclopentyladenosine) to membranes of CHO cells expressing the human A1 receptor. Its maximum effect (≈ 185% of control values) was reached at a low concentration of 100 nM only. T7 enhanced the binding over a small range of concentrations (50 – 500 nM) with modest efficacy (maximum of 120%), whereas it acted as a receptor antagonist at higher concentrations. T13 also enhanced agonist binding, with a maximum effect (130%) at 500 nM.
Fig. 3.

Representative A1AR allosteric modulators from a series of recent patents [13–16].
Thus, allosteric enhancement of agonist binding and activity on adenosine A1 receptors has been convincingly demonstrated in vitro for a series of 3-substituted 2-aminothiophenes. The activity of the compounds is relatively modest, and is sometimes compromised by a far from negligible antagonism. From the SAR it appears that lipophilicity of the 3-substituent is important for activity, which is a drawback to the solubility of the materials. These are all aspects that need to be addressed in order to arrive at chemical entities with utility in whole animal studies.
A new class of allosteric enhancers for the A1AR was recently reported [17]. 2-Aminothiazolium salts (Fig. (4)), such as the catechol derivative 29, the acetate ester 30, and the cyclopentanoate ester 31, which decreased the agonist dissociation rates from the A1AR with EC50 values of 1.2, 3.8, and 4.5 μM respectively. These enhancers appear to be more chemically stable that the benzoylthiophenes and therefore may prove to be more useful in vivo.
Fig. 4.

2-A minothiazolium salts as A1AR allosteric modulators.
The potassium sparing diuretic amiloride has been shown to act as both an antagonist and an allosteric modulator for a number of GPCRs [41–45]. Other allosteric modulators (Fig. (5)) for A1ARs include amiloride derivatives [21], the nonselective modulator of GPCRs SCH-202676 (35, N-(2,3-diphenyl-1,2,4-thiadiazol-5(2H)-ylidene)methanamine) [20,47], and sodium ions [22]. These are nonselective allosteric modulators, as they also modulate other adenosine receptor subtypes and other GPCRs [3].
Fig. 5.

A miloride derivatives and other allosteric modulators of A1 and A2A ARs.
The allosteric site(s) on the A1 AR have not been explored in detail. However, several residues have been demonstrated to be critical to the modulation of either PD 81,723 or sodium ions. The mutation of Thr277 (7.42) to Ala not only decreased agonist affinity but also inhibited the effects of PD 81,723 [23]. Studies of the D55A2.50 mutant A1 AR revealed that Asp55 is responsible for allosteric regulation of binding by sodium because the affinity for [3H]CCPA did not change over broad ranges of sodium concentrations [22].
The unique pharmacological properties of the prototypical A1 AR allosteric enhancer PD 81,723 1, in comparison to agonists, have been characterized in additional detailed studies. PD 81,723 is less likely to cause desensitization and down-regulation of receptors than are selective A1 AR agonists [18]. The behavior of PD 81,723 on functional effects of A1AR activation in CHO cells was studied [19]. Additionally, it has been demonstrated that PD 81,723 possesses some degree of tissue selectivity, modulating neuronal but not adipocyte adenosine receptors [5].
The cardiovascular effects of PD 81,723 have been studied extensively [24–30]. The slowing effects of PD 81,723 on nodal conduction mimicked those of adenosine acting at the A1 AR. Adenosine is also known to be a cardioprotective mediator, in case of ischemia, which has been demonstrated through the application of exogenous A1 AR selective agonists. PD 81,723 also has cardioprotective properties in vivo [28].
Activation of A1 ARs in vivo produces anti-nociception [31] and also reduces hypersensitivity in models of inflammatory and nerve-injury pain [32]. The allosteric enhancer T62 (28, Fig. (3)) was injected intrathecally to produce a similar anti-nociceptive effect in a model of spinal nerve ligation in the rat. This anti-nociceptive effect was blocked using a selective A1 AR antagonist, thus supporting the interpretation that enhanced activation of the A1 receptor in the brain in the presence of T62 reduces pain. Similar conclusions were reached in a spinal model of neuropathic pain [32]. Positive allosteric modulation of the A1 AR by T62 reduced hypersensitivity, suggesting of the use of such modulators in the treatment of chronic pain associated with hyperalgesia and allodynia.
A2A and A2B ARs
Isolated examples of allosteric modulation of the A2AAR have been described [20,33,34]. PD 120,918 (36, 4-methyl-7-([methylamino]carbonyl)oxy)-2H-1-benzopyran-2-one)was reported to enhance agonist radioligand binding to the rat striatal A2AAR, but functional enhancement was not demonstrated [33]. PD 120,918 also appeared to slow the dissociation of agonist from the rat brain A1AR, but had no effect on A1AR responses in FRTL-5 thyroid cells.
Other substances appeared to have allosteric effects on antagonist binding to the A2AAR without selectivity for this subtype. Both amiloride analogues [21,34] and SCH-202676 [20,47] increased the dissociation rate of the antagonist [3H]ZM241385 from the A2AAR, while they did not show any effect on the dissociation rate of the agonist [3H]CGS21680. The effects of these compound classes on dissociation kinetics were more pronounced at the A2AAR than at other AR subtypes.
In A2A AR mutagenesis studies, amiloride displayed different binding characteristics compared with the competitive antagonists [36], suggesting different modes of binding. At V84L3.32 mutant receptors, AR antagonists CPX (8-cyclopentyl-1,3-dipropylxanthine) and XAC (8-(2-aminoethyl)aminocarbonylmethyloxy-1,3-dipropylxanthine) had reduced affinity (4–6-fold) compared with wild type receptor. However, amiloride displayed wild type affinity for V84L3.32 mutant receptors. At H250N6.52 mutant receptors, competitive antagonists displayed slightly decreased or approximately wild type affinity, whereas amiloride displayed a 4-fold gain in affinity [36]. A E13Q1.39 mutation, which decreased agonist binding, influenced the affinity of neither the classic A2A receptor antagonists nor amiloride [37]. The E13Q1.39 and H278Y7.43 mutations did not significantly influence the effect of GTP on agonist binding but reduced the effects of sodium ions. This suggested these two residues are partly responsible for the allosteric regulation by sodium ions [35,37].
There are no known allosteric modulators of the A2B AR. However, it is speculated that amiloride analogues, SCH-202676, and sodium ions may be allosteric modulators for A2B receptors from the fact that they are allosteric modulators for the other three subtypes of ARs and several other GPCRs.
A3ARs
In a series of recent studies, the first allosteric modulators of the human A3 ARs have been characterized [21,38,39]. The first chemical series shown to act in this manner consisted of derivatives of 3-(2-pyridinyl) isoquinoline. These derivatives, originally synthesized as A3 AR antagonists by IJzerman and colleagues, were investigated as allosteric enhancers [38] by examining their effects on the dissociation of a high affinity A3 AR agonist radioligand, [125I]N6-(4-amino-3-iodobenzyl)-5′-N-methyl-carboxamidoadenosine (I-AB-MECA). Several 3-(2-pyridinyl) isoquinoline derivatives (Table 3), including VUF5455, VUF8502, VUF8504, and VUF8507, slowed the dissociation of the agonist radioligand [125I] I-AB-MECA in a concentration-dependent manner, suggesting an allosteric interaction. An EC50 of ~ 10 μM was observed for this allosteric effect of VUF5455 37, which displayed a relatively low degree of antagonism. These compounds had no effect on the dissociation of the radiolabeled antagonist [3H]PSB-11 (8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2.1-i]purin-5-one) [40] from the A3 AR, suggesting a selective enhancement of agonist binding. By comparison, compounds of similar structure (VUF8501, VUF8503, VUF8505), the classical AR antagonist CGS15943 (5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline) and the A1 receptor allosteric enhancer PD 81,723 did not significantly influence the dissociation rate of [125I]I-AB-MECA.
Table 3.
Effects of Pyridyl Isoquinoline Derivatives on Kinetic and Binding Parameters at the Human A3AR

| |||||
|---|---|---|---|---|---|
| R4 = | R7 = | X = | Compound | %Changea in k−1 | Ki (binding, μM) |
| 37 O-CH3 | CH3 | O | VUF5455 | −43% | 1.68 |
| 38 H | H | NH | VUF8501 | N.E. | 0.74 |
| 39 O-H | H | O | VUF8502 | −43% | 0.096 |
| 40 O-H | H | NH | VUF8503 | N.E. | 0.58 |
| 41 O-CH3 | H | O | VUF8504 | −48% | 0.017 |
| 42 O-CH3 | H | NH | VUF8505 | N.E. | 0.31 |
| 43 H | H | O | VUF8507 | −50% | 0.20 |
in presence of 10 μM
N.E. no effect
Functional effects of these allosteric enhancers were also studied. The effect of the A3 AR agonist Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide) on forskolin-induced cAMP production was significantly enhanced by VUF5455. The functional enhancement by VUF5455 was only observed in the presence of an extremely high concentration of A3 receptor antagonist to overwhelm its antagonistic activity. When the subtype-selectivity of the allosteric enhancement was tested the compounds had no effect on the dissociation of either the agonist [3H]N6-[(R)-phenylisopropyl]adenosine ([3H]R-PIA) from the A1 AR or the agonist [3H]CGS21680 from the A2A AR.
Probing of SAR suggested that an amide carbonyl group is essential for allosterism but preferred only for competitive antagonism. The presence of a 7-methyl group decreased the competitive binding affinity without a major loss of the allosteric enhancing activity, suggesting that the structural requirements for allosteric enhancement might be distinct from those for competitive antagonism.
A second series of A3 AR allosteric modulators was a group of 1H-imidazo-[4,5-c]quinolines [39] (Table 4), which acted as selective allosteric enhancers of human A3 ARs. Similar to the 3-(2-pyridinyl)isoquinoline derivatives [38], several of these compounds selectively decreased the dissociation of the agonist [125I]I-AB-MECA from human A3 ARs. There was no effect on the dissociation of the antagonist [3H]PSB-11 from the A3 AR, as well as [3H]PIA from rat brain A1 AR and [3H]CGS21680 from rat striatal A2A AR, suggesting the selective enhancement of agonist binding at A3 ARs. The analogs were tested as antagonists of competitive binding at the human A3 AR, and Ki values ranging from 120 nM to 101 μM were observed; as for many allosteric modulators of GPCRs, an orthosteric effect was also present. Some members of this series also bound competitively at the A1 AR. The most promising leads from the present set of analogs seem to be the 2-cyclopentyl-1H-imidazo[4,5-c]quinoline derivatives, of which the 4-phenylamino analog DU124183 45 had the most favorable degree of allosteric modulation versus receptor antagonism.
Table 4.
Effects of Imidazoquinoline Derivatives on Kinetic and Binding Parameters at the Human A3AR
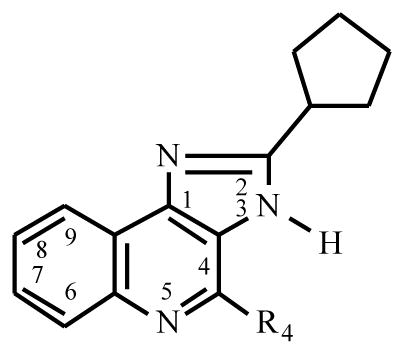
| |||
|---|---|---|---|
| R4 = | Compound | %Changea in k−1 | Ki (binding, μM) |
| 44 O-Ph | DU124182 | −45% | 0.31 |
| 45 NH-Ph | DU124183 | −46% | 0.82 |
| 46 NH-Cp | DU124184 | −25% | 0.32 |
in presence of 10 μM
Functional effects of the imidazo-[4,5-c]quinolines were also studied [39]. The inhibition of forskolin-stimulated cyclic AMP accumulation in intact cells that express the human A3 AR was employed as a functional index of A3 AR activation. The enhancer DU124183 caused a marked leftward shift of the concentration-response curve of the A3 AR agonists in the presence of antagonist and, surprisingly, a potentiation of the maximum agonist efficacy by approximately 30%. The functional potentiation of A3 agonists was evident only in the presence of the A3 antagonist MRS 1220 or upon stimulation of the receptor by 100 μM 2-chloroadenosine, an extremely high agonist concentration. Thus, we have identified a novel structural lead for developing allosteric enhancers of A3 ARs. Based on studies of agonists as cited in a recent review [1], such enhancers may be useful for treating brain ischemia and other hypoxic conditions.
Recently, it was shown that amiloride and amiloride analogues (Fig. (5)) are allosteric modulators for the A2A AR [34]. In a subsequent study it was demonstrated that amiloride analogues are allosteric modulators for agonist binding at A3 but not A1 and A2A ARs and that they are allosteric inhibitors for antagonist binding to A1, A2A and A3 ARs [21]. The binding modes of the amiloride analogues at agonist-occupied and antagonist-occupied AR subtypes are markedly different [21].
Specifically, amiloride analogues (Fig. (5)) increased the dissociation rates of the antagonist radioligands, [3H]DPCPX and [3H]PSB-11, from the human A1 and A3 ARs, respectively. Amiloride (32, 3,5-diamino-N-(aminoiminomethyl)-6-chloro-pyrazinecarboxamide hydrochloride) and 5-(N,N-dimethyl)amiloride (33, DMA) were more potent in competitive binding at the A1 AR than at the A3 AR, while 5-(N,N-hexamethylene)amiloride (34, HMA) was more potent in binding at the A3 AR. In contrast to their effects on antagonist-occupied receptors, amiloride analogues did not affect the dissociation rates of the A1 agonist [3H]R-PIA from the A1 AR or the A2A agonist [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarb-oxamidoadenosine ([3H]CGS21680) from the human A2A AR. The dissociation rate of the A3 agonist radioligand [125I]I-AB-MECA from the human A3 receptor was significantly decreased by amiloride analogues. Thus, amiloride analogues are allosteric inhibitors of antagonist binding at A1, A2A and A3 AR subtypes. The binding modes of amiloride analogues at agonist-occupied and antagonist-occupied receptors differed markedly, which was demonstrated in all three subtypes of ARs tested in this study. The effects of the amiloride analogues on the action of the A3 AR agonist were further explored using a cyclic AMP functional assay in intact CHO cells expressing the human A3 AR. Both binding and functional assays support the allosteric interactions of amiloride analogues with A3 ARs.
Curiously, the nonselective GPCR allosteric modulator SCH-202676 35 [20,47] increased the dissociation rate of the agonist [125I]I-AB-MECA from the human A3 AR, while it did not show any effect on the dissociation rate of a selective A3 AR antagonist.
A3 AR Mutagenesis Studies
The possible location of allosteric site(s) on the A3 AR was explored using site-directed mutagenesis [46]. D582.50 and D1073.49 were each mutated to an uncharged asparagine, and other residues in transmembrane domains (TMs) 1, 2, 3, 5, 6 and 7 were mutated to alanine. We first examined the effects of various allosteric modulators on the dissociation rates of the agonist radioligand, [125I]I-AB-MECA, from wild-type (WT) and mutant A3ARs. The N30A1.50 and D58N2.50 mutations abolished the effects of the imidazoquinoline DU124183 45 and the pyridinylisoquinoline VUF5455 37, but not the amiloride analogue HMA, on the dissociation rate of [125I]I-AB-MECA. In contrast, the D107N3.49 mutation abolished the effect of DU124183, but not HMA or VUF5455. The N274A7.45 mutation eliminated the effects of all of these allosteric modulators. The F182A5.43 mutation eliminated the effects of DU124183, but had no effect on the binding of A3AR agonist or antagonist. The T94A3.36, H95A3.37, K152AEL2, W243A6.48, L244A6.49 and S247A6.52 mutations did not significantly influence the effects of any of the allosteric modulators tested. The M177A5.38, V178A5.39, S271A7.42 and H272A7.43 mutations lost both agonist and antagonist high affinity binding, and could not be studied further.
We next examined the effects of sodium ions in mutant A3ARs on slowing the dissociation rate of the antagonist radioligand, [3H]PSB-11. The D58N2.50, but not L244A6.49 or S247A6.52 mutations abolished this effect of sodium ions. We further examined the effects of sodium ions on the equilibrium binding of the agonist, [125I]I-AB-MECA. Sodium ions (100 mM) caused an approx. 80% inhibition of [125I]I-AB-MECA binding in WT. The D58N2.50, D107N3.49 and F182A5.43 mutant receptors were completely insensitive to 100 mM sodium ions. In contrast, 100 mM sodium ions induced a modest but significant increase of agonist binding in N30A1.50 and N274A7.45 mutant receptors. Previous studies have implicated the aspartic acid in TM2 in the sodium modulatory effect [48]. At the A3AR, mutation of residues other than (D58) clearly had major effects on the ability of sodium ions to influence ligand recognition allosterically.
Thus, nonequivalent sets of amino acid residues were found to be involved in the three actions at the human A3 AR: allosteric modulation by heterocyclic derivatives, competitive binding at A3 AR of the same derivatives (which resembles closely the pattern previously discerned for pure antagonists, such as MRS 1220), and the allosteric modulation of A3 AR agonist binding by 100 mM sodium ions.
Molecular Modeling of A3 AR
The results were interpreted using a rhodopsin-based A3AR molecular model, suggesting multiple binding modes of the allosteric modulators [46]. First a minimized family of conformations of VUF 5455 37 was calculated using a semi-empirical PM3 method, and similar calculations were carried out for MRS 1220 and the other allosteric modulators. Calculations aimed at defining two separate pharmacophores, at the putative orthosteric and allosteric sites on the receptor, were conducted using the Sybyl® (Tripos Associates) module “DISCO”, which includes distance correlation. In comparison among the allosteric modulators alone using DU124183 45 as reference, VUF5455 displayed a high predicted overlap. In a separate comparison, the overlay of the allosteric modulator VUF5455 and the antagonist MRS 1220 suggested commonality of binding features, supporting the hypothesis of binding of the modulator at the orthosteric site, in addition to an allosteric site. Docking of the modulator molecules separately in the unoccupied and agonist-occupied receptor supported this view. A favorable binding mode identified for the modulator molecules in the agonist-occupied A3 AR (Cl-IB-MECA) suggested that a possible allosteric site in the upper (towards the cytoplasmic loops) regions of TM7 (Fig. (6)). Thus, the agonist and the allosteric modulator would bind in adjacent regions, involving mainly different amino acid residues, but be within distance to make direct contact between the two molecules possible. Thus, according to the model, docking of the allosteric modulator VUF5455 to the unoccupied receptor might take place either at the orthosteric ligand binding site (involving TMs 3, 5, 6, and 7) or on a putative allosteric binding site at the extracellular end of TM7. A network of H-bonds in the TM regions proximal to the cytoplasmic side involving residues known to affect the receptor activation and modulation by sodium and/or VUF5455 was characterized. Many of these H-bonds are common features within the Group 1 GPCR family, but others (e.g. those involving Glu1.39, Ser3.39, His7.43, Asn7.45, and Ser7.46) were unique to the ARs.
Fig. 6.

The putative binding site of the A3AR with Cl-IB-MECA as an A3-selective agonist represented by atom type color with ball and stick model and VUF5455 37 as an allosteric modulator in dark shading with capped sticks. The side chains of amino acids in the binding site were shown in line model. The backbone of A3AR was displayed by tube. The amino acids in the putative allosteric binding site were S155, H158, Q167, S170 in EL2, L246, I249, N250, I253 in TM6, V259, P260 in EL3, and V263, L264 in TM7.
SUMMARY
A disadvantage of this approach is that the current allosteric modulators for A1 and A3 ARs have both an allosteric effect and apparent antagonistic activity. Thus, when administered in vivo they would be expected to have opposing effects on receptor activation. Through structure-activity studies, compounds with more potent enhancing effect but with lower or no antagonistic activity might be obtained. No AR-specific, or subtype-selective, A2A allosteric modulators have been reported. The mutagenesis and modeling studies might help the rational design of more effective allosteric enhancers.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Christopoulos A. Nature Rev Drug Disc. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 3.Christopoulos A, Kenakin T. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 4.Olin J, Schneider L. Cochrane Database System Review. 2002:CD001747. doi: 10.1002/14651858.CD001747. [DOI] [PubMed] [Google Scholar]
- 5.Jarvis MF, Gessner G, Shapiro G, Merkel L, Myers M, Cox BF, Martin GE. Brain Res. 1999;840:75–83. doi: 10.1016/s0006-8993(99)01747-3. [DOI] [PubMed] [Google Scholar]
- 6.Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, Tinney FJ. Mol Pharmacol. 1990;38:950–958. [PubMed] [Google Scholar]
- 7.Bruns RF, Fergus JH. Mol Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- 8.van der Klein PA, Kourounakis AP, IJzerman AP. J Med Chem. 1999;42:3629–3635. doi: 10.1021/jm991051d. [DOI] [PubMed] [Google Scholar]
- 9.Kourounakis AP, van der Klein PAM, IJzerman AP. Drug Dev Res. 2000;49:227–237. [Google Scholar]
- 10.Baraldi PG, Zaid AN, Lampronti I, Fruttarolo F, Pavani MG, Tabrizi MA, Shryock JC, Leung E, Romagnoli R. Bioorg Med Chem Lett. 2000;10:1953–1957. doi: 10.1016/s0960-894x(00)00379-6. [DOI] [PubMed] [Google Scholar]
- 11.Tranberg CE, Zickgraf A, Giunta BN, Luetjens H, Figler H, Murphree LJ, Falke R, Fleischer H, Linden J, Scammells PJ, Olsson RA. J Med Chem. 2002;45:382–389. doi: 10.1021/jm010081p. [DOI] [PubMed] [Google Scholar]
- 12.Baraldi PG, Romagnoli R, Pavani MG, Del Carmen Nunez M, Tabrizi MA, Shryock JC, Leung E, Moorman AR, Uluoglu C, Iannotta V, Merighi S, Borea PA. J Med Chem. 2003;46:794–809. doi: 10.1021/jm0210212. [DOI] [PubMed] [Google Scholar]
- 13.Medco Research, Inc. WO9921617. 1999
- 14.Medco Research, Inc. US5,939.432. 1999
- 15.Medco Research, Inc. US6,177,444B1. 2001
- 16.Medco Research, Inc. US6,194,449B1. 2001
- 17.Chordia MD, Murphree LJ, Macdonald TL, Linden J, Olsson RA. Bioorg Med Chem Lett. 2002;12:1563–1566. doi: 10.1016/s0960-894x(02)00236-6. [DOI] [PubMed] [Google Scholar]
- 18.Bhattacharya S, Linden J. Mol Pharmacol. 1996;50:104–111. [PubMed] [Google Scholar]
- 19.Kollias-Baker CA, Ruble J, Jacobson M, Harrison JK, Ozeck M, Shryock JC, Belardinelli L. J Pharmacol Exp Ther. 1997;281:761–768. [PubMed] [Google Scholar]
- 20.Gao ZG, Gross AS, Jacobson KA. Life Sci. 2004;74:3173–3180. doi: 10.1016/j.lfs.2003.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao ZG, Melman N, Erdermann A, Kim SG, Müller CE, IJzerman AP, Jacobson KA. Biochem Pharmacol. 2003;65:525–534. doi: 10.1016/s0006-2952(02)01556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbhaiya H, McClain R, IJzerman A, Rivkees SA. Mol Pharmacol. 1996;50:1635–1642. [PubMed] [Google Scholar]
- 23.Kourounakis A, Visser C, de Groote M, IJzerman AP. Biochem Pharmacol. 2001;61:137–144. doi: 10.1016/s0006-2952(00)00536-0. [DOI] [PubMed] [Google Scholar]
- 24.Mudumbi RV, Montamat SC, Bruns RF, Vestal RE. Am J Physiol. 1993;264:1017–1022. doi: 10.1152/ajpheart.1993.264.3.H1017. [DOI] [PubMed] [Google Scholar]
- 25.Kollias-Baker C, Xu J, Pelleg A, Belardinelli L. Circ Res. 1994;75:972–980. doi: 10.1161/01.res.75.6.972. [DOI] [PubMed] [Google Scholar]
- 26.Kollias-Baker C, Ruble J, Dennis D, Bruns RF, Linden J, Belardinelli L. Circ Res. 1994;75:961–971. doi: 10.1161/01.res.75.6.961. [DOI] [PubMed] [Google Scholar]
- 27.Dennis DM, Raatikainen MJ, Martens JR, Belardinelli L. Circulation. 1996;94:2551–2559. doi: 10.1161/01.cir.94.10.2551. [DOI] [PubMed] [Google Scholar]
- 28.Mizumura T, Auchampach JA, Linden J, Bruns RF, Gross GJ. Circ Res. 1996;79:415–423. doi: 10.1161/01.res.79.3.415. [DOI] [PubMed] [Google Scholar]
- 29.Brandts B, Bunemann M, Hluchy J, Sabin GV, Pott L. Br J Pharmacol. 1997;121:1217–1223. doi: 10.1038/sj.bjp.0701254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musser B, Mudumbi RV, Liu J, Olson RD, Vestal RE. J Pharmacol Exp Ther. 1999;288:446–454. [PubMed] [Google Scholar]
- 31.Li X, Conklin D, Ma W, Zhu X, Eisenach JC. Pain. 2002;97:117–125. doi: 10.1016/s0304-3959(02)00011-8. [DOI] [PubMed] [Google Scholar]
- 32.Pan HL, Xu Z, Leung E, Eisenach JC. Anesthesiology. 2001;95:416–420. doi: 10.1097/00000542-200108000-00025. [DOI] [PubMed] [Google Scholar]
- 33.Bruns RF, Lu GH. In: Adenosine Receptors in the Nervous System. Ribeiro JA, editor. Taylor and Francis; London: 1989. p. 192. [Google Scholar]
- 34.Gao ZG, IJzerman AP. Biochem Pharmacol. 2000;60:669–676. doi: 10.1016/s0006-2952(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 35.Gao ZG, Jiang Q, Jacobson KA, IJzerman AP. Biochem Pharmacol. 2000;60:661–668. doi: 10.1016/s0006-2952(00)00357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA. J Med Chem. 1997;40:2588–2595. doi: 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.IJzerman AP, von Frijtag Drabbe Künzel JK, Kim J, Jiang Q, Jacobson KA. Eur J Pharmacol. 1996;310:269–272. doi: 10.1016/0014-2999(96)00495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao ZG, van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA. Mol Pharmacol. 2001;60:1057–1063. [PMC free article] [PubMed] [Google Scholar]
- 39.Gao ZG, Kim SG, Soltysiak KA, Melman N, IJzerman AP, Jacobson KA. Mol Pharmacol. 2002;62:81–89. doi: 10.1124/mol.62.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller CE, Diekmann M, Thorand M, Ozola V. Bioorg Med Chem Lett. 2002;12:501–503. doi: 10.1016/s0960-894x(01)00785-5. [DOI] [PubMed] [Google Scholar]
- 41.Howard MJ, Hughes RJ, Motulsky HJ, Mullen MD, Insel PA. Mol Pharmacol. 1987;32:53–58. [PubMed] [Google Scholar]
- 42.Blankesteijn WM, Siero HL, Rodrigues de Miranda JF, van Megen YJ, Russel FG. Eur J Pharmacol. 1993;244:21–27. doi: 10.1016/0922-4106(93)90055-e. [DOI] [PubMed] [Google Scholar]
- 43.Hoare SRJ, Coldwell MC, Armastrong D, Strange PG. Br J Pharmacol. 2000;130:1045–1059. doi: 10.1038/sj.bjp.0703370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garritsen A, Beukers MW, IJzerman AP, Cragoe EJ, Jr, Soudijn W. Neurochem Int. 1992;20:207–213. doi: 10.1016/0197-0186(92)90169-r. [DOI] [PubMed] [Google Scholar]
- 45.Leppik RA, Lazareno S, Mynett A, Birdsall NJ. Mol Pharmacol. 1998;53:916–925. [PubMed] [Google Scholar]
- 46.Gao ZG, Kim SK, Gross AS, Chen A, Blaustein JB, Jacobson KA. Mol Pharmacol. 2003;63:1021–1031. doi: 10.1124/mol.63.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fawzi AB, Macdonald D, Benbow LL, Smith-Torhan A, Zhang H, Weig BC, Ho G, Tulshian D, Linder ME, Graziano MP. Mol Pharmacol. 2001;59:30–37. doi: 10.1124/mol.59.1.30. [DOI] [PubMed] [Google Scholar]
- 48.Horstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ, Jr, Limbird LE. J Biol Chem. 1990;265:21590–21595. [PubMed] [Google Scholar]