

Abstract
Efficiency of synaptic transmission within the neocortex is regulated throughout life by experience and activity. Periods of correlated or uncorrelated presynaptic and postsynaptic activity lead to enduring changes in synaptic efficiency [long-term potentiation (LTP) and long-term depression (LTD), respectively]. The initial plasticity triggering event is thought to be a precipitous rise in postsynaptic intracellular calcium, with higher levels inducing LTP and more moderate levels inducing LTD. We used a pairing protocol in visual cortical brain slices from young guinea pigs with whole-cell recording and calcium imaging to compare the kinetic profiles of calcium signals generated in response to individual pairings along with the cumulative calcium wave and plasticity outcome. The identical pairing protocol applied to layer 2/3 pyramidal neurons results in different plasticity outcomes between cells. These differences are not attributable to variations in the conditioning protocol, cellular properties, inter-animal variability, animal age, differences in spike timing between the synaptic response and spikes, washout of plasticity factors, recruitment of inhibition, or activation of different afferents. The different plasticity outcomes are reliably predicted by individual intracellular calcium transients in the dendrites after the first few pairings. In addition to the differences in the individual calcium transients, the cumulative calcium wave that spreads to the soma also has a different profile for cells that undergo LTP versus LTD. We conclude that there are biological differences between like-type cells in the dendritic calcium signals generated by coincident synaptic input and spiking that determine the sign of the plasticity response after brief associations.
Keywords: cortex, imaging, calcium, synaptic plasticity, LTP, LTD, pairing
Introduction
Covariance or pairing of synaptic input with postsynaptic depolarization leads to strengthening of synaptic transmission [long-term potentiation (LTP)] (Harsanyi and Friedlander, 1997; Chen et al., 1999). If the pairing epoch is weak or poorly correlated, no change or weakening [synaptic long-term depression (LTD)] (Mulkey and Malenka, 1992; Artola and Singer, 1993) occurs. Considerable evidence supports the central role played by intracellular calcium for triggering these forms of plasticity (Yasuda and Tsumoto, 1996; Hansel et al., 1997; Zucker, 1999; Lisman, 2001). Moreover, the different thresholds for inducing LTP versus LTD generally relate to the levels of calcium, particularly at sites of activated synapses (Mulkey and Malenka, 1992; Hansel et al., 1997; Yang et al., 1999; Cormier et al., 2001), with higher levels leading to LTP and lower levels leading to LTD. Powerful imaging techniques for visualizing calcium in dendritic spines have demonstrated that large rapid increases do occur in response to various protocols (Petrozzino et al., 1995; Koester and Sakmann, 1998; Wang et al., 2000). These increases are initially localized to spines that may act as diffusion barriers (Svoboda et al., 1996; Majewska et al., 2000), although similar calcium localization can occur in neurons with smooth dendrites (Goldberg et al., 2003). Intracellular calcium levels can also increase over larger spatial and temporal scales. This amplification, often through release from intracellular stores (Jaffe and Brown, 1994; Nishiyama et al., 2000; Zufall et al., 2000), can affect cellular processes such as gene expression (West et al., 2001; W. G. Chen et al., 2003) that contribute to more persistent synaptic plasticity (Chen W. G. et al., 2003). Thus, it is of interest to visualize intracellular calcium dynamics within a neuron while it undergoes induction and expression of synaptic plasticity.
Advances in calcium imaging technology (Petrozzino et al., 1995; Koester and Sakmann, 1998; Yuste et al., 1999; Cormier et al., 2001; Oertner et al., 2002) have led to the ability to perform combined electrophysiology-imaging experiments in individual neurons; however, the technologies that provide the most exquisite imaging can interfere with induction of plasticity. For example, optical considerations make use of cultured neurons or organotypic slice cultures favorable for imaging (Connor et al., 1994), but such reduced preparations (Gahwiler et al., 1997) are not identical to brain slices. Also, higher concentrations of calcium indicators necessary to label finer dendritic processes buffer calcium required for plasticity induction (Takahashi et al., 1999), whereas repeated excitation at a single dendritic spine using laser scanning confocal microscopy may hinder cellular physiology throughout conditioning protocols. Thus, we used acute brain slices with intracellular delivery of a relatively low concentration of a ratiometric calcium dye (100 μm fura-4F) (Sorensen et al., 2002) with a sufficiently high KD (∼1 μm) that is permissive for the induction of plasticity. We found that conditioning (consisting of synaptic activation paired with postsynaptic depolarizations) of layer 2/3 pyramidal neurons in visual cortex of neonatal guinea pigs leads to a tripartite outcome [LTP, LTD or no change (NC)] in different cells, with each group having characteristic peak amplitude and kinetic profile “calcium signatures” that manifest after a few pairings.
Materials and Methods
Brain slice preparation. Acute neocortical slices were prepared from 6- to 12-d-old pigmented tricolor guinea pigs. Animals were housed and handled in accordance with the approved University Institutional Animal Care and Use Committee and National Institutes of Health guidelines. Briefly, the animals were deeply anesthetized (0.85 mg/kg ketamine and 0.15 mg/kg xylazine) and decapitated, and the brains were quickly removed. Slices of ∼300 μm thickness were cut on a Vibratome 1000 (Technical Products International, St. Louis, MO) from the occipital portion of the cortex, bathed with an ice-cold solution containing (in mm): 124 NaCl, 2 KCl, 0.2 CaCl2, 3.5 MgCl2, 26 NaHCO3, 10 d-glucose, and saturated with a 95% O2-5% CO2 mixture. Before recording, slices were incubated for at least 1 hr at room temperature (∼25°C) in ACSF containing (in mm): 124 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, and 10 d-glucose. The solution was bubbled continuously with a mixture of 95% O2-5% CO2.
Electrophysiological recordings. For the somatic whole-cell patch-clamp recording, slices were transferred to a recording chamber and perfused continuously (2-3 ml/min) with oxygenated ACSF that in most experiments contained 1 μm bicuculline (a series of control experiments without bicuculline with synaptic inhibition intact were also performed). Layer 2/3 pyramidal neurons were visually identified using infrared differential interference contrast video microscopy on an upright microscope (Axioskope FS2, Carl Zeiss, Oberkochen, Germany) equipped with an Achroplan 63× 0.9 numerical aperture (NA) W (Zeiss part #440067) water immersion lens. Patch electrodes with an open tip resistance of 3-4 MΩ were used. The standard pipette filling solution contained (in mm): 110 K-gluconate, 10 KCl, 10 HEPES, 10 Na2-phosphocreatine, 2 Mg1.3-ATP, 0.2 Na3-GTP; pH was adjusted to 7.2 and osmolarity to 290 mOsm using a Wescor 5500 vapor pressure osmometer (Wescor, Logan, UT). For the perforated-patch recordings (Hamill et al., 1981; Horn and Marty, 1988; Rae et al., 1991), the pipettes were first tip filled with the standard intracellular solution and then backfilled with a solution containing 200 μg/ml amphotericin B. Membrane potentials were recorded in whole-cell current-clamp mode (I = 0) using an Axopatch 200B (Axon Instruments, Foster City, CA) amplifier after breaking through the tight-seal (>2 GΩ resistance) patches. Mean input resistance was 157 ± 38 MΩ SD and was monitored continuously with 180 msec-0.2 nA hyperpolarizing pulses delivered 1 sec after the afferent stimulus (except during the plasticity-inducing protocol). Access resistance was checked initially after breakthrough and through 30 min of recording stabilization before the experimental protocols commenced with values ranging from 10 to 20 MΩ and was not compensated. Recordings in which a significant increase in the input resistance (>20%) or a deviation of resting membrane potential (more than ±2 mV) had occurred were discarded. Liquid junction potentials were not subtracted from the data presented. The electrical signals were filtered on-line at 5 kHz and digitized at 10 kHz. Recordings were done at room temperature (∼25°C).
Synaptic responses were evoked with a bipolar stimulating electrode positioned in layer 6 on beam with the recording electrode in most experiments or in layer 4 in some experiments. Stimuli to afferents consisted of constant-current square-wave pulses of 50-100 μA in amplitude and 50 μsec in duration. The evoked EPSP amplitudes were measured as the differences between baseline and peak (determined using a 3 msec event duration threshold and a 1 mV event amplitude threshold). Synaptic plasticity was induced by a single train of 60 low-frequency (0.1 Hz) pairings of afferent stimulation coincident with direct depolarization of the postsynaptic cell to approximately -10 mV (100 msec in duration, onset 10 msec before the afferent stimulus). The time, plots (see Figs. 1, 3, and 5, Normalized EPSP amplitude) of the synaptic responses were constructed by plotting the ratio of each individual EPSP peak value over the average of the 30 EPSP peak values during the last 5 min of the control period. Synaptic plasticity (see Figs. 2, 4, 6, and 7, Normalized EPSP amplitude after pairing) was quantified by calculating the ratio of the average EPSP amplitude of the 30 consecutive responses collected between the 25th and 30th min after pairing to the average EPSP amplitude of 30 consecutive responses during the last 5 min of the control period. Evaluation of the significance of the change in synaptic response attributable to the application of the pairing protocol was accomplished for each cell by application of a two-tailed paired t test to the EPSP peak response (and to the slope of the rising phase of the EPSP) in the 30 trials during the 25th to 30th min after pairing versus the 30 trials in the last 5 min of the control period before pairing. Criteria of a change of at least + or -15% in the average peak amplitude and a significance level of p < 0.01 were used to classify a response as LTP or LTD, respectively. For the grouped results, the normalized responses were analyzed for the samples of the entire group of cells tested with a particular protocol and for the groups of cells that were assigned to one of the plasticity outcome groups (LTP, LTD, NC, or the cells in the control group that received depolarizing pulses only without pairing with the synaptic input), also using a two-tailed paired t test and significance level criterion of p < 0.01. All results are reported as significant (Sig) or nonsignificant (NS) according to these criteria.
Figure 1.
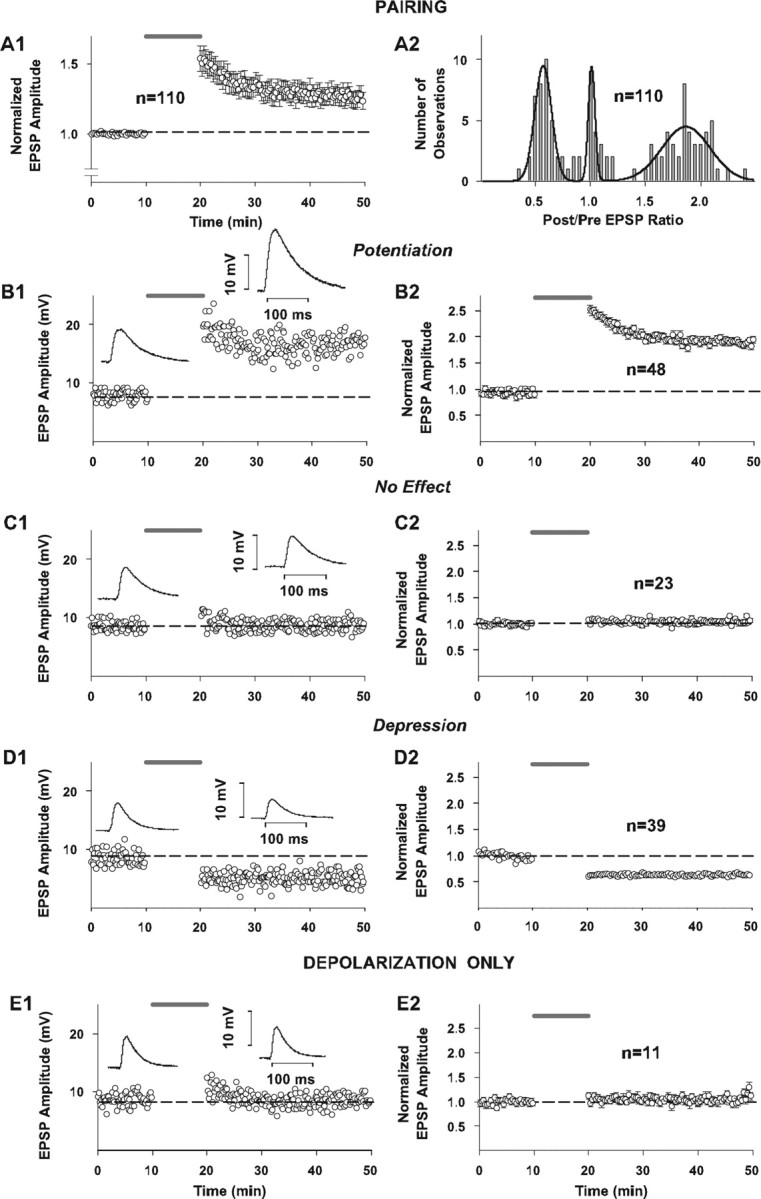
Effect of pairing on synaptic strength in whole-cell configuration with fura-4F in the pipette solution. In all panels in which grouped data are illustrated (A1, B2, C2, D2, E2), every other data point is illustrated for graphical clarity. Dashed horizontal lines indicate average baseline peak amplitude responses normalized to 1.0 for all grouped results (A1, B2, C2, D2, E2) or average raw baseline peak amplitude responses for individual example records (B1, C1, D1, E1). The shaded line above records indicates time of application of 10 min pairing protocol (A1, B1, B2, C1, C2, D1, D2) or time of application of depolarizing pulses alone (E1, E2). A1, Summary time plot of EPSP changes in response to pairing. Synaptic responses to the constant-current square-wave pulses (50-100 μA in amplitude and 50 μsec in duration) delivered at 0.1 Hz to a bipolar electrode positioned in layer VI on a beam with the recording electrode were acquired in a whole-cell current-clamp mode (I = 0). Standard pipette solution contained (in mm): 110 K-gluconate, 10 KCl, 10 HEPES, 10 Na2-phosphocreatine, 2 Mg1.3-ATP, 0.2 Na3-GTP, 0.1, pH 7.2, osmolarity = 290 mOsm, and was supplemented with 100 μm fura-4F. Synaptic plasticity was induced by a single train of 60 pairings of afferent stimulation coincident with intracellular depolarization of the postsynaptic cell to approximately -10 mV. Symbols and error bars represent means ± SEM (n = 110) of the EPSP amplitudes (measured as a difference between baseline and peak) normalized for the average amplitude of 30 consecutive evoked synaptic responses collected during the last 5 min of recording before pairing. A2, Distribution of normalized EPSP amplitude changes in response to pairing. Bars in the graph represent binned changes in synaptic efficacy in all 110 cells, measured as a ratio of the average EPSP amplitude of 30 consecutive responses to afferent stimulation collected between 25th and 30th min after pairing to the average amplitude of 30 consecutive EPSPs during the last 5 min of recording before pairing. Bin width is equal to 0.05. Line in the graph represents a trimodal Gaussian fit of the data with the following values: peaks (M ± m) 0.57 ± 0.08 (N1max = 9.5); 1.01 ± 0.03 (N2max = 9.5); 1.86 ± 0.22 (N3max = 4.5); R2 = 0.79; DOF-R2 = 0.75; SEE = 1.28. B1, C1, D1, Recordings of the EPSP peak amplitude before and after pairing in three individual cells, each exhibiting potentiation, no change in synaptic strength, and depression, respectively. Shown in the insets next to the control and postpairing parts of the time plots are the average EPSP traces from 30 consecutive responses collected during the last 5 min before pairing and between the 25th and 30th min of recording after pairing in the same cells, respectively. B2, C2, D2, Time plot of normalized changes in EPSP peak amplitude in response to pairing in the groups of cells exhibiting potentiation (n = 48), no change in synaptic strength (n = 23), and depression (n = 39), respectively, from the sample of 110 cells shown in A1 and A2. Symbols and error bars represent means ± SEM of the EPSP amplitudes measured as a difference between baseline and peak and normalized for the average EPSP amplitude of 30 consecutive responses collected during the last 5 min of recording before pairing for the respective numbers of observations. E1, E2, Recordings of the EPSP peak amplitude in an individual neuron and in a sample of 11 cells subjected to a 0.1 Hz train of 60 intracellular depolarizations to approximately -10 mV (without afferent stimulation), respectively. Shown in the insets in E1 are the average EPSP traces from 30 consecutive responses collected during the last 5 min of control recording and between 25th and 30th min of recording after the experimental paradigm.
Figure 3.

Effect of pairing on synaptic strength in response to layer 6 stimulation in the conventional whole-cell configuration (without fura-4F in the pipette solution) (panels in A) and in the perforated-patch configuration (also without fura-4F in the pipette solution) (panels in B). For the conventional whole-cell recordings, standard pipette solution was used except that fura-4F was omitted (see Fig. 1 legend). For perforated-patch experiments, the pipettes were first tip-filled with the standard pipette solution and then backfilled with the same solution supplemented with 200 μg/ml amphotericin B. The afferent stimulation in layer 6 and the pairing protocol were the same as described in Fig. 1. In all panels in which grouped data are illustrated (A1, A3-A5, B1, B3-B5), every other data point is illustrated for graphical clarity. Dashed horizontal line indicates average baseline peak amplitude responses normalized to 1.0 for all grouped results. Shaded line above records indicates time of application of 10 min pairing protocol. A1 (n = 39)-B1 (n = 21), Summary time plots of EPSP changes in response to pairing for entire samples tested with these protocols. Here and elsewhere in the figure, symbols and error bars represent means ± SEM of the EPSP amplitudes measured as the difference between baseline and peak and normalized for the average EPSP amplitude of 30 consecutive responses collected during the last 5 min of recording before pairing, for the respective number of experiments.A2-B2, Distributions of normalized EPSP amplitude changes in response to pairing in all 39 cells studied in the conventional whole-cell configuration (without fura-4F) and in all 21 cells studied in the perforated-patch configuration (also without fura-4F). Bars in the graph represent binned (bin width 0.05) changes in synaptic efficacy measured as a ratio of the postpairing response (average EPSP amplitude of 30 consecutive responses to afferent stimulation collected between the 25th and 30th min after pairing) to the prepairing response (average amplitude of 30 consecutive EPSPs during the last 5 min of recording before pairing). Line in the graphs represents a trimodal Gaussian fit of the data. For the conventional whole-cell data distribution, parameters of the fit were as follows: peaks (M ± m) 0.64 ± 0.10 (N1max = 2.91); 0.99 ± 0.04 (N2max = 4.02); 1.92 ± 0.12 (N3max = 2.82); R2 = 0.78; DOF-R2 = 0.74; SEE = 0.61. For the perforated-patch histogram, parameters of the fit were as follows: peaks (M ± m) 0.57 ± 0.51 (N1max = 4.02); 1.03 ± 0.05 (N2max = 1.99); 1.92 ± 0.06 (N3max = 2.69); R2 = 0.83; DOF-R2 = 0.80; SEE = 0.45. A3-B3, Summary time plots of EPSP changes in response to pairing for the subsample of cells that underwent LTP only for the whole-cell (n = 17) and perforated-patch (n = 9) experiments (without fura-4F). A4-B4, Summary time plots of EPSP changes in response to pairing for the subsample of cells that did not change in response to the pairing protocol for the whole-cell (n = 8) and perforated-patch (n = 4) experiments. A5-B5, Summary time plots of EPSP changes in response to pairing for the subsample of cells that underwent LTD only for the whole-cell (n = 14) and perforated-patch (n = 8) experiments.
Figure 5.
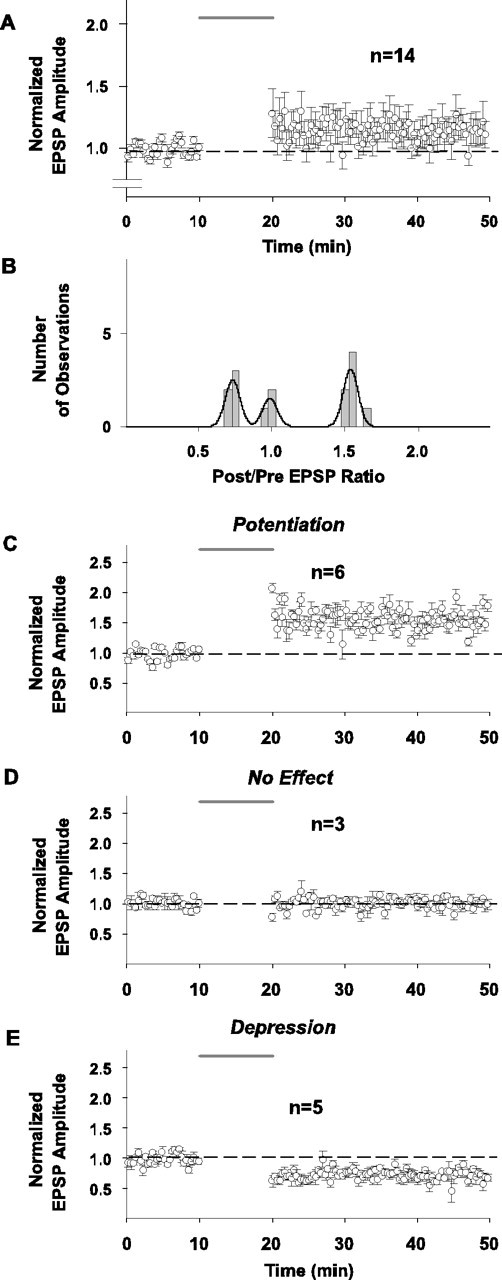
Effect of pairing on synaptic strength in response to stimulation in layer 4 on beam with the recorded cell (recordings done in the conventional whole-cell configuration without fura-4F in the pipette solution and with bicuculline omitted from the bathing solution). In all panels with grouped data (A, C-E), every other data point is illustrated for graphical clarity. Dashed horizontal lines indicate average baseline peak amplitude responses normalized to 1.0 for all grouped results. Shaded line above records indicates time of application of 10 min pairing protocol. A, Summary time plot of EPSP changes in response to pairing for entire group of 14 cells tested with this protocol. Symbols and error bars represent means ± SEM of the EPSP amplitudes measured as the difference between baseline and peak and normalized for the average EPSP amplitude of 30 consecutive responses collected during the last 5 min of recording before pairing. B, Distribution of normalized EPSP amplitude changes plotted as postpairing/prepairing ratios in response to pairing for 14 cells in response to layer 4 stimulation. Bars in the graph represent binned (bin width 0.05) changes in synaptic efficacy measured as a ratio of the average EPSP amplitude of 30 consecutive responses to afferent stimulation collected between the 25th and 30th min after pairing to the average amplitude of 30 consecutive EPSPs during the last 5 min of recording before pairing. Line in the graphs represents a trimodal Gaussian fit of the data. The parameters of the fit were as follows: peaks (M ± m) 0.73 ± 0.05 (N1max = 2.5); 0.99 ± 0.05 (N2max = 1.52); 1.53 ± 0.05 (N3max = 3.08); R2 = 0.80; DOF-R2 = 0.76; SEE = 0.41. C-E, Time plot of normalized changes in EPSP peak amplitude in response to pairing in the groups of cells exhibiting potentiation (C; n=6), no change in synaptic strength (D; n = 3), and depression (E; n = 5), respectively, from the sample of 14 cells shown in A and B. Symbols and error bars represent means ± SEM of the EPSP amplitudes measured as a difference between baseline and peak and normalized for the average EPSP amplitude of 30 consecutive responses collected during the last 5 min of recording before pairing for the respective numbers of observations.
Figure 2.
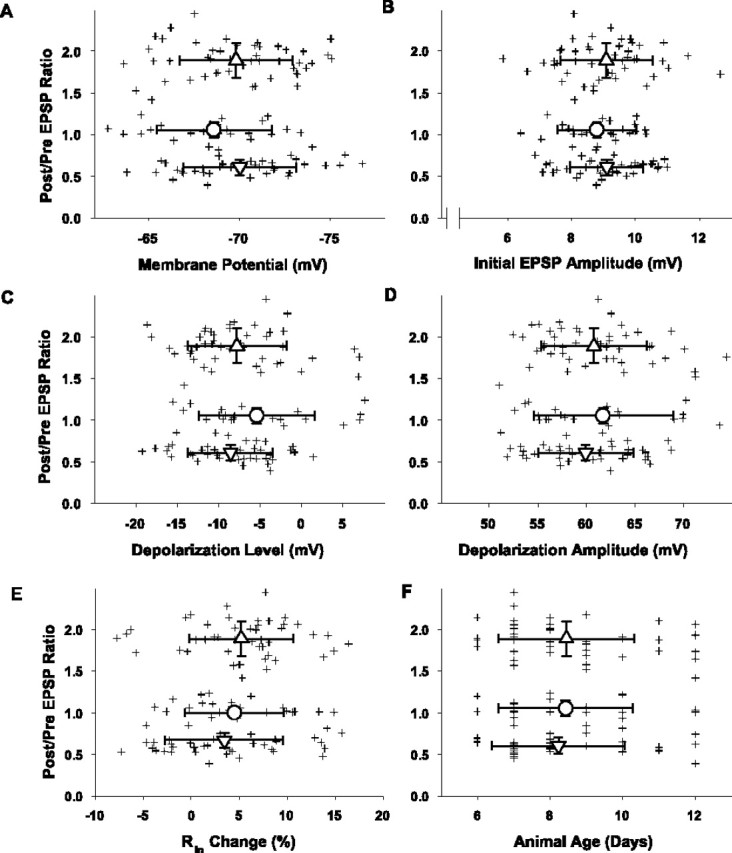
Synaptic plasticity outcome as a function of the resting membrane potential (A), the initial EPSP amplitude (B), the level of postsynaptic depolarization (C), the magnitude of postsynaptic depolarization (D), the change in input resistance (Rin) (E), and the age of the animal from which the slices were obtained (F). Crosshair symbols represent the ratio of average EPSP postpairing to prepairing amplitude responses (ordinate) and the value of the indicated parameter (abscissa) for each cell (n = 110) subjected to pairing. The mean ± SD values for the relative change in the EPSP peak amplitudes for the three plasticity outcome groups plotted along the ordinate are 1.89 ± 0.21 for the LTP group, 1.06 ± 0.09 for the no change group, and 0.61 ± 0.09 for the LTD group. A, Resting membrane potential was calculated as a mean voltage during 1 sec of recording (collected 5 sec after each 0.1 Hz afferent stimulation) averaged for 30 consecutive interstimulus intervals within the period between the 5th and 10th min of recording before pairing and for the 30 consecutive interstimulus intervals between the 25th and 30th min after pairing. The mean membrane potentials ± SD = -69.8 ± 3.1 mV (LTP group; n = 48; upward triangle), -68.6 ± 3.2 mV (NC group; n = 23; circle), and -70.0 ± 3.1 mV (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p > 0.1). B, Initial EPSP amplitudes were measured as a difference between baseline and peak and averaged for 30 consecutive synaptic responses during the last 5 min of recording before pairing. The means ± SD for the initial EPSP amplitudes = 9.0 ± 1.3 mV (LTP group; n = 48; upward triangle), 8.8 ± 1.1 mV (no change group; n = 23; circle), and 9.1 ± 1.1 mV (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p > 0.1). C, Level of depolarization was determined as a mean voltage of a steady state achieved in each depolarizing pulse (usually the last 10-15 msec of the pulse) and averaged between all 60 responses of a given cell to pairings. The means ± SD for the level of postsynaptic depolarization = -7.8 ± 6.0 mV (LTP group; n = 48; upward triangle), -5.4 ± 7.0 mV (no change group; n = 23; circle), and 8.2 ± 5.8 mV (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p > 0.1). D, Depolarization amplitude was calculated as a difference between the resting membrane potential and the depolarization level in each given cell. The means ± SD for the depolarization amplitudes = 60.8 ± 5.4 mV (LTP group; n = 48; upward triangle), 61.7 ± 7.2 mV (no change group; n = 23; circle), and 60.2 ± 5.6 mV (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p > 0.1). E, The input resistance change was calculated as the ratio of Rin 25-30 min after pairing over that during the 5 min period before pairing. The calculation of each Rin value was obtained from Ohm's law based on the measurement of the level of steady-state membrane hyperpolarization at the end of a 180 msec, -0.2 nA current pulse applied 1 sec after the afferent stimulus. The means ± SD for the Rin changes = +5.21 ± 5.43% (LTP group; n = 48; upward triangle), +4.51 ± 5.13% (no change group; n = 23; circle), and +3.44 ± 6.14% (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p>0.1). F, The animals' ages were expressed in postnatal days (PND). The means ± SD for the animals' ages = 8.46 ± 1.88 d (LTP group; n = 48; upward triangle), 8.43 ± 1.85 d (no change group; n = 23; circle), and 8.23 ± 1.84 d (LTD group; n = 39; downward triangle). No groups were significantly different from the others (p > 0.1).
Figure 4.

Analysis of initial (during prepairing control period) EPSP waveform and latency of the EPSPs for the entire group (n = 110) with whole-cell recording inresponse to layer 6 stimulation and with fura-4F present. A, Overlay of 30 individual traces of 60 total evoked EPSPs during 10 min prepairing control period for a typical cell. The stimulus artifact indicating layer 6 stimulation is evident at time 0.Em = -67 mV. EPSPs are 9-12 mV peak amplitude. B, Example of analysis of an individual EPSP (1 of the 30 traces shown in A). The solid line that overlays the individual raw trace is the fitted function described by the following: Em = Em0 + a * (1 - exp(-x/t1))n * exp(-x/t2) with the following fitting parameters Em0 = -65.7; a = 53.2; t1 = 3.3; t2 = 45.3; n = 2.5 × 107. Triangle indicates EPSP onset; squares indicate 20 and 80% of peak values for calculation of EPSP rise time; circle indicates EPSP peak amplitude; all values were obtained from the fitted averaged EPSP. These parameters for this example EPSP are as follows: peak time = 21.7 msec; EPSP peak = -53.7 mV; resting Em = -65.7 mV; one SD from the resting Em = 0.21 mV; EPSP onset = resting Em + 2 SDs = -65.3 mV; onset latency = 8.0 msec; 20% of EPSP peak = -63.3 mV; time to 20% of EPSP peak = 11.0 msec; 80% of EPSP peak = -56.1 mV; time to 80% of EPSP peak = 16.0 msec (all times are referenced to the afferent stimulation); 20-80% slope = 1.46 mV/msec.C, Scatter plot of individual (crosses) mean EPSP onset latencies (abscissa) versus the relative changes in EPSP peak amplitude (ordinate) for all cells in the three plasticity outcome groups. Means ± SD of ratios of postpairing to prepairing average EPSP peak amplitudes plotted along the ordinate are the same as those in Figure 2. Means ± SDs for the onset latencies are as follows for the three plasticity outcome groups: LTP =6.37 ± 0.76 msec (n = 48), upward triangle; no change = 6.64 ± 0.75 msec (n= 23), circle; LTD = 6.43 ± 0.72 msec (n = 39), downward triangle. None of the groups are significantly different from each other (p > 0.1). D, Scatter plot of individual (crosses) variances (SDs, abscissa) of the onset latencies versus the relative changes in EPSP peak amplitude (ordinate) for all cells in the three plasticity outcome groups. Mean variances for each plasticity outcome group are indicated ± SD; LTP = 0.63 ± 0.09 msec (n = 48), upward triangle; no change = 0.64 ± 0.10 msec (n = 23), circle; LTD = 0.65 ± 0.10 msec (n = 39), downward triangle. None of the groups are significantly different from the others (p > 0.1). E, Scatter plot of the individual (crosses) mean latencies to the EPSP peak versus the relative changes in EPSP peak amplitude (ordinate) for all cells in the three plasticity outcome groups. Mean latencies for each plasticity outcome group are indicated ± SD; LTP = 19.26 ± 3.40 msec (n = 48), upward triangle; no change = 17.56 ± 3.57 msec (n = 23), circle; LTD = 18.26 ± 3.10 msec (n = 39), downward triangle. None of the groups are significantly different from the others (p > 0.1). F, Scatter plot of the individual (crosses) variances (SDs, abscissa) of the peak latencies versus the relative changes in EPSP peak amplitude (ordinate) for all cells in the three plasticity outcome groups. Mean variances for each plasticity outcome group are indicated ± SD; LTP = 1.83 ± 0.74 msec (n = 48), upward triangle; no change = 2.03 ± 1.01 msec (n = 23), circle; LTD = 1.84 ± 0.94 msec (n = 39), downward triangle. None of the groups are significantly different from the others (p > 0.1).
Figure 6.
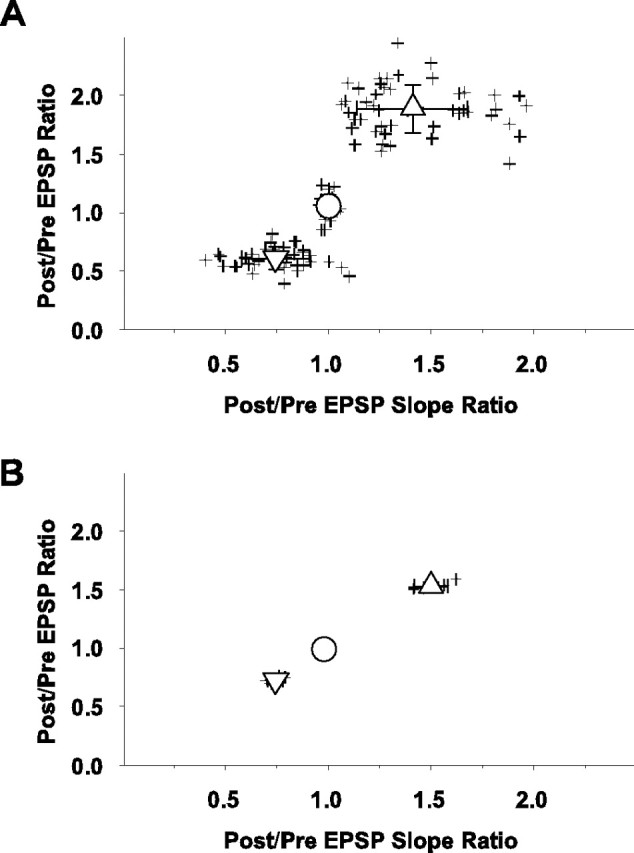
Scatter plots of individual (crosses) postpairing/prepairing EPSP peak amplitude ratios (ordinate) versus the ratios of postpairing/prepairing slopes of the rising phases of the EPSPs (abscissa). Slope ratios were taken as the 20-80% rise time at 25-30 min postpairing divided by the 20-80% rise time during the last 5 min of the prepairing control period, as described in Figure 4.A, Cells evaluated with layer 6 stimulation in which fura-4F was used (n = 110). As in previous figures, upward triangle indicates LTP group, circle indicates no change group, and downward triangle indicates LTD group. The mean ± SD for the slopes for the three outcome groups are 1.41 ± 0.27 mV/msec for the LTP group, 1.00 ± 0.03 mV/msec for the no change group, and 0.75 ± 0.17 mV/msec for the LTD group. The slopes between the three groups are significantly different (p < 0.001 for the LTD vs NC group, and p < 0.001 for the NC vs LTD group). B, Cells evaluated with layer 4 stimulation in which fura-4F was not used (n = 14). Upward triangle indicates LTP group, circle indicates no change group, and downward triangle indicates LTD group. The mean ± SD for the slopes for the three outcome groups are 1.50 ± 0.08 mV/msec for the LTP group, 0.98 ± 0.03 mV/msec for the no change group, and 0.75 ± 0.04 mV/msec for the LTD group. The slopes between the three groups are significantly different (p < 0.001 for the LTD vs NC group, and p < 0.001 for the NC vs LTD group).
Figure 7.

Analysis of potential contribution of spike timing to plasticity. A, Examples of 30 overlaid traces of sequential alternate depolarizations applied to a cell during the pairing procedure (for clarity purposes, only 30 of the 60 traces evoked during the control period are illustrated). Five spikes occurred during most applied depolarizations. Arrows indicate the time of the peaks of the first and second spikes for one depolarization in each group; dashed lines extend these time points to abscissa for comparison of spike time occurrences to evoked EPSP illustrated in B. B, Average of 60 control evoked EPSPs (30 of the 60 individual records used to construct this average are shown in A) to indicate timing relationship between spikes elicited during the pairing of the applied depolarizing pulses and the EPSP onset and peak. The triangle and dashed line indicate the time of the EPSP onset as derived from the averaged fitted functions (Fig.4); the circle and dotted line indicate the time of the EPSP peak as derived from the averaged fitted functions (Fig. 4). For this example, the EPSP onset latency = 7.3 msec and the latency to peak = 21.1 msec. The results illustrated below in C-F in which the timing relationship between the spikes and the EPSP onset and peak are plotted are derived following the procedure for each cell. C, Scatter plot of individual (crosses) ratios of EPSP peak amplitudes (postpairing/prepairing ratio; ordinate) versus time between the first spike elicited during the depolarizing pulse and the EPSP onset (abscissa). Large upward triangle, circle, and downward triangle indicate mean for the LTP, NC, and LTD groups, respectively ± SD (n = 110). Mean ± SDs for the three plasticity outcome groups are -7.09 ± 2.54 msec for the LTP group, -7.50 ± 2.03 msec for the no change group, and -7.41 ± 2.40 msec for the LTD group. None of these groups are significantly different from each others (p > 0.1). D, Scatter plot of individual (crosses) ratios of EPSP peak amplitudes (postpairing/prepairing ratio; ordinate) versus time between the second spike elicited during the depolarizing pulse and the EPSP onset (abscissa). Large upward triangle, circle, and downward triangle indicate mean for the LTP, NC, and LTD groups, respectively, ± SD (n=110). Mean ± SDs for the three plasticity outcome groups are 3.64 ± 3.77 msec for the LTP group, 4.25 ± 3.89 msec for the no change group, and 4.12 ± 3.32 msec for the LTD group. None of these groups are significantly different from each other (p > 0.1). E, Scatter plot of individual (crosses) ratios of EPSP peak amplitudes (postpairing/prepairing ratio; ordinate) versus time between the first spike elicited during the depolarizing pulse and the EPSP peak (abscissa). Large upward triangle, circle, and downward triangle indicate mean for the LTP, NC, and LTD groups, respectively, ± SD (n = 110). Mean ± SDs for the three plasticity outcome groups are -19.98 ± 4.01 msec for the LTP group, -18.42 ± 3.88 msec for the no change group, and -19.24 ± 3.94 msec for the LTD group. None of these groups are significantly different from each other (p > 0.1). F, Scatter plot of individual (crosses) ratios of EPSP peak amplitudes (postpairing/prepairing ratio; ordinate) versus time between the second spike elicited during the depolarizing pulse and the EPSP peak (abscissa). Large upward triangle, circle, and downward triangle indicate grand mean for the LTP, NC, and LTD groups, respectively, ± SD (n = 110). Mean ± SDs for the three plasticity outcome groups are -9.26 ± 4.95 msec for the LTP group, -6.67 ± 5.19 msec for the no change group, and -7.70 ± 4.56 msec for the LTD group. None of these groups are significantly different from each other (p > 0.1).
For the analyses of EPSP latencies and rise times, each EPSP was first smoothed by fitting to the following function: Em = Em0 + a * (1 - exp(-x/t1))n * exp(-x/t2), where Em = membrane potential, Em0 = average baseline membrane potential during the 100 msec before the stimulus, a and n are fitting coefficients, t1 = time constant for the EPSP rising phase, and t2 = time constant for the decay phase of the EPSP. Common goodness of fit statistics was used in all fitting procedures, including the fitting of the EPSPs (as described above) and for additional fitting operations for Gaussian functions fitted to plasticity outcome histograms (see below and results in Figs. 1 and 3) and to analysis of calcium imaging data describing kinetics of calcium transients (see below and results in Fig. 10) and cumulative calcium signals (see below and results in Fig. 11). The common goodness of fit statistics uses include the following: (1) coefficient of determination R2 = (1 - SSE)/SSM, where SSM is the sum of squares about the mean and SSE is the sum of squared errors; (2) degree of freedom adjusted coefficient of determination DOF R2 = 1 - SSE × (n - 1)/SSM × (DOF - 1), where the degree of freedom DOF = n - m, with n being the total number of data values and m being the number of coefficients in the model; and (3) standard error of estimate SEE = √SSE/DOF.
Figure 10.
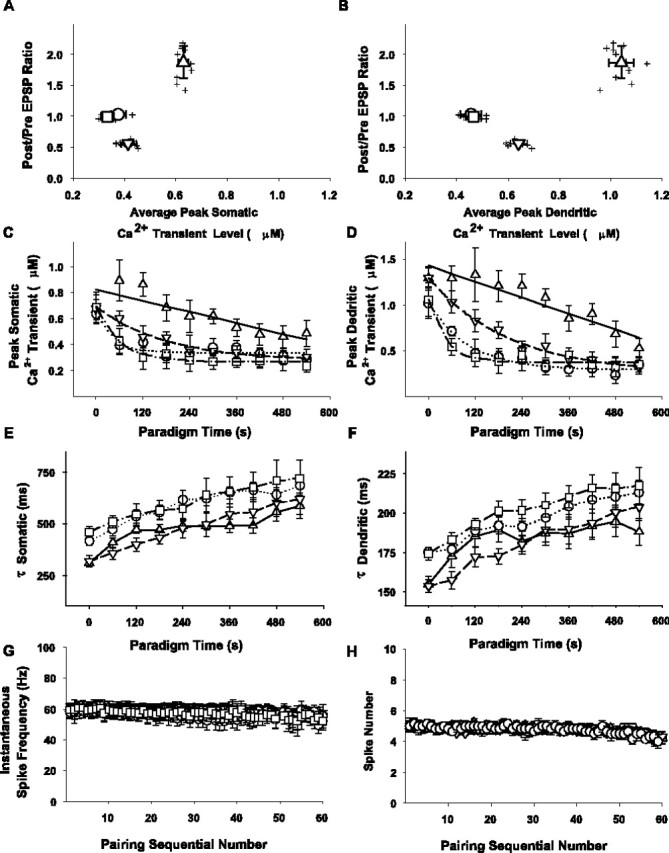
Summary of calcium transients during sequential pairings or application of depolarizing pulses alone and their relationship to electrophysiological responses. A, B, Scatter plot of the ratios of the average EPSP peak amplitude postpairing/prepairing (ordinate) versus the average peak somatic (A) and dendritic (B) calcium transient peak levels (abscissa) for cells subjected to pairing with fura-4F in the pipette solution in response to layer 6 stimulation as measured using the fast-frame calcium imaging protocol (n = 26). Shown is the average peak amplitude of all 10 Ca2+ transients (in response to every 6th successive pairing), as determined in the proximal apical dendrite (25-100 μm away from the cell body) and in the soma of each given cell. Before averaging, absolute peak parameters of individual calcium transients were determined using a third-order piecewise polynomial interpolation of the data. Symbols and bidirectional error bars represent means ± SDs for the respective sets of data in the LTP (upward triangle; n = 11), no change (circle; n = 6), and LTD (downward triangle; n = 9) groups and the depolarization only group (square; n = 11). C, D, The absolute peak levels of individual Ca2+ transients (ordinate) in response to every sixth consecutive pairing (or depolarization) over the course of the protocol (abscissa) were determined in the soma (C) and apical dendrite (D) as described for A1 and A2. Symbols represent means ± SEMs as follows: upward triangle = LTP group, circle = NC group, and downward triangle = LTD group. Responses to respective successive pairings averaged between 11 cells exhibiting potentiation, 6 cells that displayed no change in synaptic strength, and 9 cells that underwent LTD, respectively, are illustrated. Squares and error bars represent mean ± SEM of the responses to respective consecutive depolarizations alone averaged for the eight cells subjected to a 0.1 Hz train of 60 intracellular depolarizations without afferent stimulation. Solid lines in the graphs represent linear fits of the data in the LTP group with slopes for the somatic (C) and dendritic (D) measurements, respectively. Dotted, dashed, and mixed dashed-dotted lines represent the average of the individual single exponential fits of the time course of the decline of the dendritic and somatic calcium transients over the course of the conditioning protocol in the no change, LTD, and depolarization only groups, respectively. E, F, Symbols and error bars represent the mean ± SEM of the time constants (ordinate) derived from single exponential fits of the decay phase of the individual calcium transients in response to every sixth pairing or depolarization over the course of the conditioning protocol for the LTP (upward triangle), no change (circle), LTD (downward triangle), and depolarization only (square) groups as described in A and B. G, H, Symbols and error bars represent the mean ± SEM of instantaneous frequency and number of spikes elicited in the respective groups with regard to the pairing-induced plasticity outcomes (upward triangles, LTP; circles, no change; downward triangles, LTD) or in the depolarization only group (squares).
Figure 11.
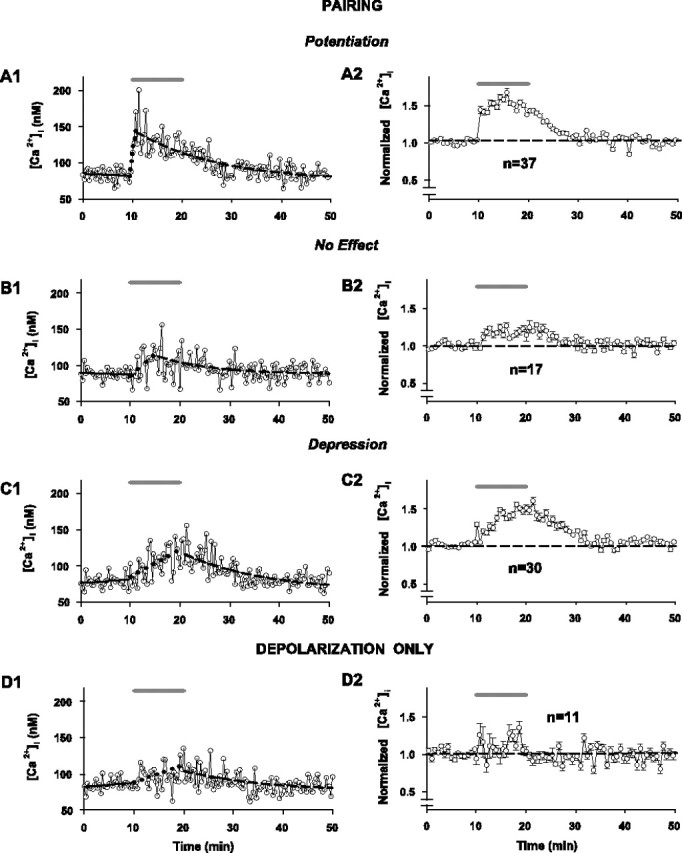
Dynamics of cumulative intracellular [Ca2+] associated with the development of synaptic plasticity changes for cells subjected to pairing with Fura-4F in the whole-cell recording pipette solution in response to layer 6 stimulation as measured using the slow-imaging method (n = 84). Shaded bars above all graphs represent 10 min pairing period (A1, A2, B1, B2, C1, C2) or period of application of depolarizing pulses alone (D1, D2). A1-C1, Data points represent cumulative somatic [Ca2+] in three individual cells, exhibiting potentiation, no change in synaptic strength, and depression, respectively. Free cytosolic Ca2+ concentrations were calculated from the background-subtracted ratiometric images collected once every 20 sec throughout the experiment within a single ROI placed on the soma (see Materials and Methods and Fig. 8 for details of image acquisition and calculation of [Ca2+]i). For the associated recordings of EPSP amplitude in the same three individual cells, see Figure 1 (B1-D1, respectively). Solid lines in the graph represent a linear regression of the data during control recording (10 min before pairing). Dotted and dashed lines represent a linear and a single exponential fit of the rise and decay phases of the recordings, respectively. For all fits and for calculation of the numeric values for the time and level of peak sustained intracellular [Ca2+]i (determined using a third-order piecewise polynomial interpolation), the data were first smoothed by a three-point moving average routine. A2-C2, Normalized changes in sustained somatic [Ca2+]i in response to pairing in the groups of cells exhibiting potentiation (A2; n = 37), no change in synaptic strength (B2; n = 17), and depression (C2; n = 3), respectively, from the sample of 84 cells (every other data point is illustrated for graphical clarity). Time plots of grouped data (symbols and error bars indicate means ± SEM) were constructed using all data points in the individual recordings of intracellular Ca2+ normalized for the mean value of intracellular [Ca2+] at rest (last 5 min of control recording) and averaged for the respective number of cells. D1, D2, Recordings of the sustained somatic [Ca2+] in an individual neuron and in a sample of 11 cells subjected to a 0.1 Hz train of 60 intracellular depolarizations to approximately -10 mV (without afferent stimulation), respectively. For the EPSP data in the same individual cell, see Figure 1 E1. The horizontal dashed lines indicate the normalized baseline response = 1.0 for the grouped results.
Latencies to EPSP onset and peak were calculated for individual fitted EPSPs using a third-order piecewise polynomial interpolation (the XlXtrFun.xll freeware for MS Excel developed by Scott Allen Rauch at http://www.xlxtrfun.com). Onset latency for each individual EPSP was defined as the time after the stimulus when the fitted function of the membrane potential exceeded 2 SDs of the actual baseline Em recording as measured during the 100 msec before afferent stimulation. The EPSP peak for each individual EPSP was defined as the maximum of the second-order parabola defined by the following three points: (1) the maximum tested point, (2) the tested point to the immediate left of the maximum tested point, and (3) the tested point to the immediate right of the maximum tested point. The EPSP rise times were defined as the slope of the 20-80% peak amplitude of the rising phase of the EPSP.
Analysis of timing relationships between the evoked EPSPs and the spikes elicited by direct depolarization of the postsynaptic cell during individual pairings was done by measuring the time from the peak of the first and second spikes elicited by the depolarization to the fitted EPSP onset and peak. Spike peaks were determined as the maxima of the second-order parabola defined by the following three points: (1) the maximum tested point, (2) the tested point to the immediate left of the maximum tested point, and (3) the tested point to the immediate right of the maximum tested point. Latency was determined using a third-order piecewise polynomial interpolation of the fitted curves (XlXtrFun.xll). All pyramidal neurons recorded were regular spiking (vs the rare bursting nonpyramidal cells that were occasionally encountered but not included in the study).
Electrophysiological parameters used for the correlational analyses of parameters as a function of plasticity outcome (see Fig. 2) were calculated as follows: the resting membrane potential was calculated as a mean voltage during 1 sec of recording (collected 5 sec after each 0.1 Hz afferent stimulation) averaged for the 30 consecutive interstimulus intervals within the period between the 5th and 10th min of recording before pairing and for the 30 consecutive interstimulus intervals between the 25th and 30th min after pairing; the level of depolarization was determined as a mean voltage of a steady state achieved in each depolarizing pulse (measured after the last spike in the burst elicited by the depolarizing pulse usually during the last 5-10 msec of the pulse) and averaged between all 60 responses of a given cell to pairings. The depolarization amplitude was calculated as a difference between the resting membrane potential and the depolarization level in each given cell.
Optical recording. Cells were loaded with the ratiometric Ca2+ indicator fura-4F (Molecular Probes, Eugene, OR) added to the pipette filling solution at a final concentration of 100 μm. The indicator was allowed to equilibrate between the pipette and cytoplasm for at least 30 min after breaking into whole-cell mode. Fluorescence was excited with monochromatic light (bandwidth of 8-15 nm and a homogeneity of >10%) of appropriate wavelengths using a rapidly tunable (<1.5 msec wavelength change) galvanometric scanner-mounted grating (Polychrome IV; TILL Photonics, Gräfelfing, Germany), with a 150 W xenon short-arc lamp as a light source. Fluorescence detection was accomplished using a 12-bit CCD camera (Quantix EEV57-10; Photometrics, Tucson, AZ) mounted on an Axioscope 2FS equipped with an Achroplan 63× 0.9 NA W water immersion lens, a 400 DCLP dichroic mirror (Chroma Technology, Brattleboro, VT), D510/40m emitter filter (Chroma Technology). Synchronized acquisition of electrophysiological and optical data, afferent stimulus triggering, and control of the monochromator were all performed via a PCI-1200 interface (National Instruments, Austin, TX) using a single 800 MHz G4 Power PC computer (Apple Computer, Cupertino, CA) running custom-developed software.
The timing diagram for optical data acquisition with respect to the conditioning paradigm is shown in Results (see Fig. 8). At the beginning of every experiment, the baseline intracellular Ca2+ level was determined (while establishing the baseline EPSP amplitude during the control recording). It was achieved by acquiring the fluorescence images in rapid succession in pairs at the calibrated excitation maxima of Ca2+-free (F1 = 354 nm) and Ca2+-bound (F2 = 374 nm) forms of fura-4F once every 20 sec. This translates into generation of the ratios at 650 msec before every other stimulation, 9.35 sec after the previous stimulation, or pairing. We refer to this calcium-imaging protocol as a slow-frame method, which captures the cumulative concentration of intracellular calcium periodically throughout our entire 50 min experimental protocol.
Figure 8.

Timing diagram for the experimental paradigm and the optical data acquisition protocol. A, Red lines in the graph indicate the 0.1 Hz afferent stimuli to evoke an EPSP. The superimposed black lines indicate timing of the 100 msec depolarizing pulses coincident with synaptic stimulation (onset of depolarization 10 msec before the afferent stimulus) for the 10 min pairing epoch. B, Determination of the resting baseline intracellular Ca2+ level was made by taking the fluorescence images in pairs during the control period (10-20 msec frame readout interval) at the calibrated excitation maxima of Ca2+-bound and Ca2+-free forms of fura-4F (354 and 374 nm, respectively), thus generating ratios once every 20 sec. In addition, when continued throughout the entire experiment, this mode of optical data acquisition allowed us to monitor slow dynamics of sustained intracellular Ca2+ during development and expression of synaptic plasticity. C, To record fast intracellular Ca2+ dynamics in response to individual pairings, a string of 60 images was taken in a sequence at a single Ca2+-sensitive wavelength (374 nm) every 30 msec starting 200 msec before the onset of depolarization and lasting for 1.8 sec. A pair of F2 frames at calibrated isoasbestic wavelength of 358 nm was taken before and after this string to generate ratiometric images. To minimize photo damage to the cells, only responses to every sixth pairing (once every 60 sec) were studied. Expanded time-scale diagrams (B, C) illustrate the timing of ratiometric image capture with respect to the afferent stimulus (and/or depolarization), with the pink line indicating the exposure of the cell to the monochromatic light of appropriate wavelengths. D, E, Representative pseudocolor background-subtracted ratiometric images of a fura-4F-filled neuron from each method are shown below the respective timing diagrams. Colored rectangles in the images over the somata and apical dendrites indicate the ROIs, which is where calcium measurements were typically made. The white rectangles outside the labeled cell in each image represent typical placement of an ROI to determine background fluorescence. Scale bars, 10 μm. Pseudocolor scale indicates calcium concentration.
Imaging the intracellular Ca2+ signaling elicited by individual pairings of depolarizations plus synaptic input or to depolarizing pulses alone was accomplished by acquiring the images in a fast sequence (33.3 Hz frame succession rate) at a single calibrated Ca2+-sensitive wavelength (F1, 374 nm), with the two F2 frames at the calibrated isoasbestic wavelength (F2, 358 nm) taken before and after this sequence to generate the ratiometric images. We refer to this experiment as the fast-frame method, which captures a rapid series of measures of intracellular calcium in a time period surrounding an individual pairing of synaptic input with postsynaptic depolarization, providing a detailed profile of the calcium waveform associated with that individual pairing, starting 200 msec before the onset of depolarization and ending 1.5 sec after the end of the depolarizing pulse. The CCD controller firmware was allowed to manage the integration time and readout interval in this mode (these parameters varied depending on the number of pixels in the acquired subarray and on its position on the chip but were kept constant within a given experiment). Regions of interest (ROIs) were selected at four different locations on the soma and on the apical dendrites at various distances (25-100 μm) from the soma. Background fluorescence was estimated by measuring the fluorescence of an equivalent position in the slice that contained no injected neurons and was subtracted. A two-tailed paired t test with a significance criterion of p < 0.01 was used to compare [Ca2+]i levels in ROIs along the apical dendrites and in the somata during individual pairings in the conditioning phase of the experiment (with the fast-frame method described above) and throughout the entire protocol (with the slow-frame method described above). All results in the text are reported as Sig or NS, according to these criteria.
Care was taken to ensure that the baseline electrophysiological properties, baseline synaptic transmission, and induction of synaptic plasticity were not affected by phototoxicity as compared with our controls with no light exposure. In the slow-imaging mode, the preparation was exposed to the UV light for a total time of 110 msec every 20 sec (0.5 sec before every second pairing). Thus, the total time of UV light exposure during a 50 min experimental run with this form of data acquisition is 16.5 sec (0.6%). The total time of UV exposure with the fast-imaging method was 18 sec plus 13.2 sec for the slow frame images collected during the baseline and post pairing periods for a total exposure time of 31.2 sec out of the total 50 min (3000 sec) protocol (or 1.0% exposure time).
Calibration of Ca2+ indicators. Ca2+ calibrations were done in the solutions containing known concentrations of free Ca2+, 1 mm free Mg2+ (Calcium Calibration Buffer kit with Magnesium #2, Molecular Probes) and 100 μm fura-4F trapped in a synthetic fused silica rectangular capillary (300 μm outer diameter, 50 μm inner diameter, both measured flat-to-flat; Polymicro Technologies, Phoenix, AZ) and imaged with the Achroplan 63× 0.9 NA W. At each Ca2+ concentration the fluorescence intensity data were collected for the wavelengths ranging from 320 to 440 nm in 2 nm increments. The excitation wavelengths corresponding to the peak fluorescence of Ca2+-bound and Ca2+-free forms of fura-4F (354 and 374 nm, respectively; mean values of at least 15 individual measurements, rounded to the nearest integer) and to the isoasbestic point (358 nm; mean value of at least 15 individual measurements, rounded to the nearest integer) were determined using a third-order piecewise polynomial interpolation of the experimental data (the XlXtrFun.xll freeware for MS Excel developed by Scott Allen Rauch at http://www.xlxtrfun.com). Ca2+ dose-response curves were constructed using the fluorescence intensity data of Ca2+-bound and Ca2+-free forms of the dyes determined by a third-order piecewise polynomial interpolation of the experimental data (the XlXtrFun.xll freeware for MS Excel) at the excitation wavelengths corresponding to peak fluorescence and rounded to the nearest integer. A dissociation constant (KD) of ∼1 μm was calculated from the fit of the experimental data obtained at either wavelength to a first-order Langmur equation.
Free cytosolic Ca2+ concentrations were calculated from the background-subtracted average fluorescence intensities within ROIs by the ratio method using KD effective, Rmax and Rmin values calculated by fitting the experimentally determined ratio dose-response curve to the first-order Langmur equation (mean values resultant from at least 15 individual measurements).
In each experiment, the baseline [Ca2+]i was determined first as a mean of 15 measurements during the last 5 min of control recording. Cells displaying [Ca2+]i at rest >100 nm were excluded from further experimentation. Peak parameters in each individual Ca-transient in response to every sixth pairing (or depolarization) were determined using a third-order piecewise polynomial interpolation of the data and corrected for the baseline [Ca2+]i level in each transient by subtracting the mean value of [Ca2+]i calculated from the first five (taken before the respective pairing or depolarization) ratiometric images in each 60-frame string. The numeric value of the peak sustained intracellular [Ca2+] level was determined using a third-order piecewise polynomial interpolation of the data smoothed by a three-point moving average routine. To calculate the amplitude of the peak sustained [Ca2+]i rise during the induction period, the baseline [Ca2+]i was subtracted from the peak sustained level. The mean value of the 15 [Ca2+]i measurements made between the 25th and 30th min after pairing was used to estimate the residual [Ca2+]i level. The time necessary to reach the peak sustained [Ca2+]i level in each cell also was determined using a third-order piecewise polynomial interpolation of the three-point moving average smoothed data. A single exponential fit of the three-point moving average smoothed data, after the peak sustained [Ca2+]i, was used to calculate the time necessary to reach a steady state of the residual [Ca2+]i level in each cell.
A potentially problematic issue, common in all fluorescence studies of living cells and taken into consideration when our imaging experiments were designed, is that of phototoxicity. This process is thought to be caused by highly reactive byproducts such as singlet oxygens and free radicals, all of which are generated because of photochemical breakdown of a fluorophore (for review, see Picciolo and Kaplan, 1984), and is potentially detrimental to cells. In addition, shorter wavelengths (UV) are capable of eliciting autofluorescence of intracellular constituents, thus raising even more concerns about long-term viability of the cells, especially when repeated images are collected during lengthy neurophysiological experiments. Therefore, the time of exposure of the neurons to UV (and therefore the number of images collected) was limited to the empirically established values that ensure that the baseline electrophysiological properties, baseline synaptic transmission, and induction of synaptic plasticity were not affected by photo toxicity (as compared with the controls with no dye and/or light exposure). We reasoned that these maneuvers should bring the rate of production of potentially toxic products down to the levels that can be eliminated by the intrinsic scavenging mechanisms of living tissue. The aggregate time of UV exposure during a 50 min experimental run was 3.3 sec during the control recording (110 msec once every 20 sec for collecting the baseline [Ca2+]i data); for the slow-frame paradigm, it was 12.2 sec during the plasticity induction and expression periods (110 msec to collect a single pair of images, once every 20 sec), and for the fast-frame paradigm, it was 18.6 sec during the plasticity induction period (1.86 sec to collect a single string of 62 images at 33.3 Hz succession rate once every 60 sec over the 10 min pairing epoch).
Results
Individual cells respond differently to pairing
Figure 1 depicts the effect of conditioning on the peak amplitude of the evoked EPSPs in response to stimulation of layer 6 and with the calcium dye fura-4F in the recording pipette. A net potentiation of the EPSPs (1.26 ± 0.06 SEM; Sig) occurs (Fig. 1A1). Interestingly, when plotted as a frequency distribution histogram (Fig. 1A2), distinct groupings in outcomes are apparent. The histogram is best fit with a trimodal Gaussian distribution (Fig. 1A2). Of the 110 cells, the EPSPs of 48 (44%) significantly potentiated (Fig. 1B1, time plot of an individual example; Fig. 1B2, time plot of grouped results for all of the cells that potentiated) (mean = 1.89 ± 0.03 SEM; Sig), 23 (21%) did not change (Fig. 1C1, time plot of an individual example; Fig. 1C2, time plot of the grouped results for all of the cells that did not change) (mean = 1.06 ± 0.02 SEM; NS), and 39 (35%) depressed (Fig. 1D1, time plot of an individual example; Fig. 1D2, time plot of the grouped results for all of the cells that depressed) (mean = 0.61 ± 0.02 SEM; Sig). An additional control group (n = 11) (Fig. 1E1,E2) received only the depolarizing pulses, with no effect on their EPSPs (mean = 1.07 ± 0.02 SEM; NS).
Differential plasticity outcomes are biologically based
This tripartite outcome suggests either experimentally induced variability or some intrinsic difference between cells. To assess the former, we analyzed the outcomes with respect to the resting membrane potentials, the strength of afferent stimulation (using the initial EPSP amplitude as an index), the level to which the postsynaptic membrane was depolarized during application of the pairing pulses, the magnitude of the depolarizing pulses, any changes in the input resistance throughout the protocol, and the age of the individual animal from which each recording was obtained. The results of these analyses are shown in Figure 2. None of these parameters differ among the groups according to plasticity outcome (NS for all group comparisons; see legend for details).
Other factors that may account for the outcome include variable susceptibility to washout of plasticity factors caused by whole-cell recording, variation in calcium buffering by fura-4F, activation of different synaptic inputs, or variable effects of blocking synaptic inhibition. We evaluated these possibilities in a series of control experiments. In experiments that used whole-cell recording without fura-4F (Fig. 3A1-A5) or perforated-patch recording (also without fura-4F) (Fig. 3B1-B5), similar plasticity outcome distributions occurred (Fig. 3A2,B2), as they did in the whole-cell experiments with fura-4F (Fig. 1A2). With whole-cell recording and no fura-4F in the pipette, a net potentiation of the EPSPs occurs (mean = 1.20 ± 0.08 SEM; n = 39; Sig) (Fig. 3A1), as it does with the perforated-patch recording, (mean = 1.20 ± 0.14 SEM; n = 21) (Fig. 3B1). The fractions of cells exhibiting each plasticity outcome were as follows: LTP with whole-cell recording and no fura-4F = 17 of 39 or 44% (mean = 1.72 ± 0.03 SEM; Sig) (Fig. 3A3); LTP with perforated-patch recording = 9of 21 or 43% (mean = 1.92 ± 0.05 SEM; Sig) (Fig. 3B3); NC with whole-cell recording and no fura-4F = 8 of 39 or 21% (mean = 1.05 ± 0.02 SEM; NS) (Fig. 3A4); NC with perforated-patch recording = 4 of 21 or 19% (mean = 1.05 ± 0.01 SEM; NS) (Fig. 3B4); LTD with whole-cell recording and no fura-4F = 14 of 39 or 36% (mean = 0.70 ± 0.02 SEM; Sig) (Fig. 3A5), and LTD with perforated-patch recording = 8 of 21 or 38% (mean = 0.68 ± 0.02 SEM; Sig) (Fig. 3B5). Thus, the different outcomes between individual cells are not caused by either variable calcium buffering by the fura-4F or variable washout of plasticity factors attributable to the whole-cell recording condition.
Another possibility that may explain the different outcomes is potential activation of different populations of inputs to different cells. Thus, we analyzed the individual responses of the EPSPs evoked by layer 6 stimulation for each cell (Fig. 4) and did an additional series of control experiments in which the stimulating electrode was positioned directly below the recorded cell in layer 4 (Fig. 5) to restrict the population of afferents activated. A series of 30 individual control EPSPs elicited from an individual cell are illustrated in Figure 4A; their average and the fitted smoothing function (see Materials and Methods) are illustrated in Figure 4B, indicating how the EPSP onset, peak amplitude, latencies to EPSP onset and peak, rising phase slope, and decay-phase time constant were measured. The individual measures of latency and its associated variance (SD) for each cell in the three outcome groups also are illustrated (Fig. 4C,E and D,F, respectively). These results make several points. There is very little jitter in the individual EPSPs for a single cell (example in Fig. 4A and plot of variances for EPSP onset latencies for each cell and each outcome group are illustrated in Fig. 4D), consistent with a major monosynaptic input component; the mean latencies to the EPSP onset for the individual cells (5-7.5 msec) (Fig. 4C) are consistent with monosynaptic inputs when the conduction distance of ∼1.5 mm for afferents and synaptic delay at 25°C are taken into account; the decay of the EPSP is well fitted by a single exponential (Fig. 4B), which is indicative of an EPSP that is not “contaminated” by an underlying IPSP; and the differences in the plasticity outcomes (LTP vs NC vs LTD) are not attributable to variance in the latency (as might be expected if one outcome group represented a monosynaptic set of inputs and another represented a disynaptic pathway) (Fig. 4D). Because our stimulating electrode is placed in layer 6 and the concentration of bicuculline used may only partially block GABAA-mediated synaptic inhibition, however, we also performed a series of experiments with stimulation in layer 4, directly below the layer 2/3 neuron being recorded and without bicuculline. These results are illustrated in Figure 5, with the grouped data for the entire sample illustrated in Figure 5A (mean = 1.13 ± 0.10 SEM; NS) and the outcome distribution represented in Figure 5B. With stimulation in layer 4, the net change in EPSPs for the entire sample is only +13% and not significant; however, although this is a small sample, the distribution forms three clusters (see details of trimodal Gaussian fit in legend) with an LTP group, an NC group, and an LTD group, in which the plasticity for the individual EPSPs in the LTP and LTD groups is significant, as are the grouped results for the LTP and LTD subsamples. The fractions of EPSPs exhibiting each plasticity outcome are as follows: LTP = 6 of 14 or 43% (mean = 1.53 ± 0.01 SEM; Sig) (Fig. 5C); NC = 3 of 14 or 27% (mean = 1.00 ± 0.01 SEM; NS) (Fig. 5D); LTD = 5 of 14 or 36% (mean = 0.73 ± 0.01 SEM; Sig) (Fig. 5E).
The responses from the layer 6 and layer 4 stimulation experiments also were analyzed for effects of the pairing on the slope of the EPSP rise (Fig. 6). Interestingly, the relative changes in both parameters for the three outcome groups are similar for both experimental groups; however, the variance in the postpairing versus prepairing EPSP slope ratio is less for the layer 4 stimulation condition, suggesting that in this experiment a more homogeneous group of synapses is activated. Also, in support of an individual cellular basis for the differential plasticity outcomes is the observation that the different outcomes (LTP, LTD, or NC) occur in different slices from the same animal as well as in neighboring cells from the same slice.
Another possible explanation for the different plasticity outcomes between cells is a difference in the timing of the evoked postsynaptic spike during the applied depolarization with respect to the synaptic response. Indeed, spike-timing-dependent plasticity is a potentially important variable. Thus, we analyzed all of our results with respect to the temporal relationship of the evoked spikes to the EPSPs. Figure 7 illustrates this analysis. A series of 30 of the 60 traces (every other trial) during the 10 min pairing period for an example cell is overlaid (Fig. 7A). Note the regularity of the first and second spikes in each depolarization, with subsequent spikes in the five spike train showing some increasing variable latency. The averaged evoked EPSP (obtained during the last 5 min of the control period) for this cell is illustrated in Figure 7B, with the EPSP onset (triangle and dashed line) and peak (circle and dotted line) indicated for analysis of spike timing relative to these parts of the EPSP. These results (Fig. 7C-F) show that the spike timing with respect to the EPSP was consistent between cells and not related to the plasticity outcome, whether evaluated for the first (Fig. 7C,E) or second spike (Fig. 7D,F) and with respect to the EPSP onset (Fig. 7C,D) or peak (Fig. 7E,F). Similar results (data not shown) occur for the subsequent spikes. Thus, reasons for the different plasticity outcome groups must be found elsewhere.
Calcium imaging strategies and responses to individual pairings
Considering the pivotal role of [Ca2+]i for induction of plasticity (Mulkey and Malenka, 1992; Yasuda and Tsumoto, 1996; Hansel et al., 1997; Yang et al., 1999; Zucker, 1999; Cormier et al., 2001; Lisman, 2001), we investigated the calcium signals generated during the pairing protocol in the three different outcome groups. We used two approaches: recording of [Ca2+]i transients in response to individual pairings during plasticity induction (fast imaging) and recording of cumulative sustained [Ca2+]i levels over the course of the entire experiment (slow imaging). The timing diagram of the optical data acquisition (blue and green lines) for both approaches are shown with respect to the electrophysiological stimulation paradigm (red and black lines) in Figure 8A and with an expanded time scale in B and C. The pseudocolor ratiometric images taken with the slow and fast-imaging methods are also illustrated (Fig. 8, D and E, respectively). We randomly applied our fast calcium imaging method to approximately one-fourth of the sample (n = 26 of 110 cells). LTP occurred in 11 of 26 of those cells (42%), LTD in occurred in 9 of 26 of those cells (35%), and there was no change in 6 of 26 of those cells (23%), a distribution similar to the entire sample of 110 cells tested. In addition, we applied this fast calcium imaging protocol to another eight control cells that received only the depolarizing pulses during the conditioning protocol. Examples (Fig. 9) of the calcium transients (top rows) and electrophysiological responses (bottom rows) during individual pairings are illustrated for a representative cell from each group [LTP (Fig. 9A), NC (Fig. 9B), LTD (Fig. 9C), depolarizing pulses only (Fig. 9D)]. The magnitude of the calcium rise is highest in response to the first pairing-depolarization and decays over the course of conditioning.
Figure 9.

Intracellular Ca2+ responses to individual pairings during the induction of synaptic plasticity as measured with fura-4F with whole-cell recording in response to layer 6 stimulation. For each pair of traces, the top rows are recordings of fast [Ca2+] dynamics in the dendrites of an example cell that underwent LTP (A), no change (B), or LTD (C), respectively, or received only depolarizing pulses without synaptic stimulation (D). To minimize photo damage to the cells, only responses to every sixth pairing (once every 60 sec) were studied. Bottom rows depict recordings of electrophysiological responses to respective pairings-depolarizations in the same cells.
Amplitude and time course of dendritic calcium transients during individual pairings predict plasticity outcome
The analysis of the [Ca2+]i transients in the dendrites and somata in response to individual pairings-depolarizations is summarized with respect to plasticity outcome (Fig. 10A,B). The peak transient [Ca2+]i during the pairings is greater both in the apical dendrites (1040 ± 50 nm) and in the somata (630 ± 20 nm) for the LTP group (n = 11; upward triangles) versus the LTD group (641 ± 33 nm in the dendrite, Sig; 414 ± 32 nm in the somata, Sig; n = 9; downward triangles) and versus the no change group (454 ± 41 nm in the apical dendrites, Sig; 376 ± 28 nm in the soma, Sig; n = 6; circles). Furthermore, the average peak Ca2+ levels in the LTD group in both the apical dendrites and the somata are different (Sig) from the no change group. The peak transient [Ca2+]i during pairing for the NC group does not differ from the depolarizing pulses only group (465 ± 32 nm in the apical dendrites, NS; 334 ± 26 nm in the somata, NS; n = 8; squares).
The larger intracellular Ca2+ signal in the LTP group can be partly ascribed to the differences in time course of the responses over the conditioning period. In the LTP group (upward triangles; n = 11), the peak amplitudes of the sequential somatic (Fig. 10C) and apical dendritic (Fig. 10D) Ca2+ signals decline in a nearly linear manner with slopes of -6.7 ± 0.8 and -14.0 ± 0.8 pm/sec, respectively. The decline in amplitude of the sequential peaks of the individual somatic and apical dendritic Ca2+ transients over the course of the pairing protocol in the LTD group (downward triangles) and NC group (circles) are each fitted by a single exponential decay (Fig. 10C,D). The average of the decay time constants that characterizes the progressive diminution of the peak calcium transients in response to successive pairings is slower for the LTD group (τdendritic = 360 ± 52 sec SEM; τsomatic = 320 ± 18 sec SEM; n = 9) than for the no change group (τdendritic = 95 ± 2 sec SEM; τsomatic = 32 ± 13 sec SEM; n = 6). Thus, the magnitude of the individual Ca2+ transients in the apical dendrites and somata remains elevated in cells that will undergo LTP versus those that will not change or undergo LTD. The average of the decay time constants that characterize the progressive diminution of the peak calcium transients in response to successive pairings in response to application of the depolarizing pulses alone (squares) also is best fitted to a single exponential decay with time constants comparable with those in the cells that exhibit no change in synaptic efficacy in response to the pairing (τdendritic = 44 ± 6 sec SEM; τsomatic = 63 ± 6 sec SEM; n = 8).
Over the course of the 60 pairings, the time constants that fit the decaying phase of the individual calcium transients of each cell associated with each sequential pairing (or depolarizations alone) lengthen for each group (Fig. 10E,F). Notably, the decay time constants of the individual calcium transients during the very first pairing for the cells in the groups that undergo plasticity (τdendritic for the LTP group = 155 ± 5 msec SEM; τdendritic for the LTD group = 154 ± 4 msec SEM; τsomatic for the LTP group = 310 ± 21 msec SEM; τsomatic for the LTD group = 319 ± 25 msec SEM) are significantly shorter (p < 0.01 for the LTP vs NC group, for the LTP vs depolarizing pulses only group, for the LTD vs NC group, and for the LTD versus depolarizing pulses only group) than for those that did not undergo plasticity. Interestingly, the decay time constants of the first calcium transients are not different between the LTP versus LTD groups (p > 0.1). Moreover, although the decay time constants of the individual calcium transients increase with successive pairings (or depolarizing pulses alone) throughout the course of the protocol for all groups, those of the LTP and LTD groups remain below the level of the NC and depolarizing pulse only groups over the full 10 min of conditioning.
Individual 0.1 Hz trials of afferent stimulation alone do not cause any detectable calcium rises as evaluated with our fast-frame imaging method (data not shown). Thus, our measurements are not sensitive enough to detect the individual EPSP-mediated calcium signaling that is likely confined to dendritic spines; however, the calcium signals that occur in the proximal apical dendrite and soma in response to pairing in cells that subsequently undergo plasticity are greater than those elicited by depolarizing pulses alone or in response to pairing that does not induce plasticity. These signals thus correlate with the plasticity induction cascade and are suggestive of an intrinsic difference of the responses of those cells to the pairing. We also tested the possibility that the decline in the sequential Ca2+ transient peaks and the concomitant increase in the decay time constants of each transient over the course of the pairing protocol might reflect a change in the electrophysiological activation of the neuron by analyzing the spiking response from each cell in the different outcome groups; however, neither the number of spikes nor the average instantaneous firing frequency changes for the sequential pairings-pulses in the conditioning train for each of the groups (Fig. 10G,H), and there is no difference between groups (NS).
Kinetics of the cumulative somatic calcium rise predict plasticity outcome
In fast-frame calcium imaging experiments, it was apparent that the baseline intracellular calcium levels on which the sequential transient Ca2+ rises are imposed (Fig. 9, dashed lines connecting transients) also change for the LTP and LTD groups but not for the no change group. Thus, we measured these cumulative intracellular calcium signals with the slow-imaging method for 95 cells (84 conditioned with the pairing protocol and 11 with depolarizing pulses alone). The time-plot analyses of the sustained somatic [Ca2+] dynamics in the 84 cells (of 110 total) grouped according to synaptic plasticity outcome are illustrated in Figure 11 A-C. Records from a cell within each of the three outcome groups, respectively, are shown (Fig. 11 A1,B1,C1); the corresponding EPSPs from the same cells are shown in Figure 1, B1, C1, and D1. The time plots of the cumulative somatic calcium grouped results normalized to baseline [Ca2+]i level are illustrated in Figure 11, B2, C2, and D2, respectively (LTP group, n = 37; no change group, n = 17; LTD group, n = 30). A consistent rise in the cumulative somatic [Ca2+]i during the period of induction occurs only in the LTP and LTD groups. The trains of 60 depolarization pulses without synaptic pairing induce low and short-lived Ca2+ responses (Fig. 11 D1,D2), similar to those in the groups exhibiting no change in the EPSP amplitude in response to pairing (Fig. 11 B1,B2). The analysis of the relative EPSP amplitudes as a function of time course and level of the cumulative somatic calcium signals in response to the pairing is shown in Figure 12. The peak and amplitude (mean ± SD) of the sustained [Ca2+]i rise (Fig. 12 A, B) in both the LTP group (128 ± 24 and 55 ± 11 nm, respectively; Sig; n = 37) and the LTD group (123 ± 23 and 52 ± 12 nm, respectively; Sig; n = 30) are significantly higher than in the no change group (95 ± 28 and 28 ± 12 nm, respectively; Sig; n = 17); however, the magnitude of these cumulative [Ca2+]i responses does not differ between the LTP and LTD groups (NS). The absolute values (mean ± SD) of the initial or residual [Ca2+]i levels (Fig. 12C,D) also do not differ among groups (NS) (LTP group: 73 ± 14 and 72 ± 14 nm, n = 37; NC group: 67 ± 19 and 68 ± 20 nm, n = 17; LTD group: 71 ± 13 and 76 ± 14 nm, n = 30, for initial and residual [Ca2+], respectively). The average time to peak (mean ± SD) for the cumulative [Ca2+]i rise from baseline (Fig. 12 E) is less in the LTP group (190 ± 127 sec; Sig) than in the LTD group (579 ± 22 sec), however, and both differ from the no change group (417 ± 197 sec; Sig). The average time constants of decay (mean ± SD) (Fig. 12 F) are not different (NS) among the groups (576 ± 111 sec in the LTP group, 438 ± 185 sec in the no change group, and 520 ± 94 sec in the LTD group).
Figure 12.
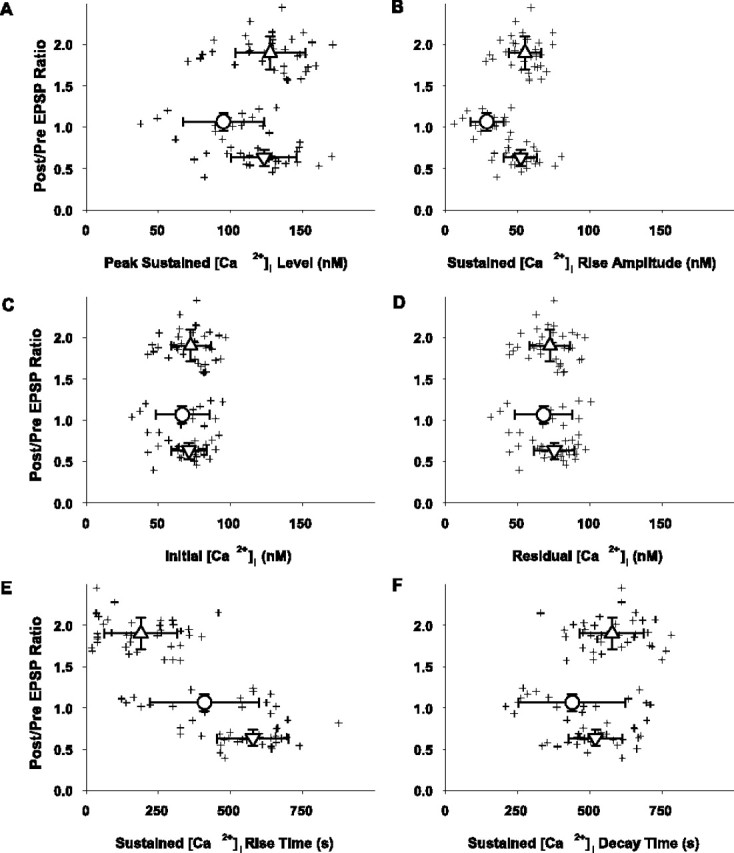
Synaptic plasticity outcome as a function of sustained intracellular Ca2+ levels measured over the course of the protocol (n = 84). Crosshair symbols represent the sustained [Ca2+]i parameters (abscissa) as determined for each cell versus the ratio of the postpairing/prepairing EPSP peak amplitude (ordinate). Symbols and bidirectional error bars represent means ± SD (values given in Results) for the respective sets of data in the LTP (upward triangle; n = 37), no change (circle; n = 17), and LTD (downward triangle; n = 30) groups for all panels in this figure. A, The peak sustained [Ca2+]i level was determined using a third-order piecewise polynomial interpolation of the data that were smoothed by a three-point moving average routine. B, The peak sustained [Ca2+]i rise amplitude was calculated as the difference in the peak sustained calcium level (A) and the initial calcium level (C). C, The initial [Ca2+]i was determined as a mean of the 15 measurements during the last 5 min of control recording. D, The residual [Ca2+]i was determined as the mean value of the 15 consecutive [Ca2+]i measurements made between the 25th and 30th min after pairing. E, The sustained rise time of [Ca2+]i is the time necessary to reach the peak sustained [Ca2+]i level in each cell and was determined using a third-order piecewise polynomial interpolation of the three-point moving average smoothed data. F, The decay time of the sustained [Ca2+]i is the time necessary to reach a residual steady-state [Ca2+]i level in each cell and was determined by using a single exponential fit of the three-point moving average smoothed data after the peak sustained [Ca2+]i.
Discussion
Plasticity outcomes
We have shown that in pyramidal neurons of visual cortex, repetitive low-frequency conjunctions of synaptic input and brief postsynaptic membrane depolarization produce a tripartite outcome (LTP, LTD, or NC) in different cells from the same animal at the same age. In addition, our results demonstrate that distinct spatiotemporal profiles of [Ca2+]i are predictive of plasticity outcome in response to the same conditioning protocol.
Although synaptic input correlated with strong postsynaptic depolarization is generally considered to lead to LTP (Fregnac et al., 1994; Harsanyi and Friedlander, 1997; Chen et al., 1999; Bear, 2003), there are reports of heterogeneous outcomes (Debanne et al., 1999; Lei et al., 2003). In our study, these differences are not attributable to any of the following: variance in intrinsic electrophysiological properties or strength of conditioning stimuli (Fig. 2A-D), the response during conditioning (Figs. 9, 10G,H), cell-to-cell differences in calcium buffering by the dye (Fig. 3A), differential washout of plasticity factors (Fig. 3B), variable contributions of synaptic inhibition (Fig. 5), activation of different sets of synaptic inputs between cells (Figs. 4, 5, 6), timing differences between the EPSP and evoked spikes (Fig. 7), or cell class-slice-individual animal or age (Fig. 2F) differences. These results are consistent with a differential state (metaplasticity) (Abraham and Bear, 1996) for neurons determining their responses to conditioning stimuli. They may result from the history of the neuron or synapse before slice preparation (Abraham and Bear, 1996), unrecognized differences in neuronal subtypes (e.g., intrinsic calcium buffering) (Lee et al., 2000) among pyramidal, neurons and/or variable maturation between neighboring neurons at a chronological age (Murchison and Griffith, 1999).
The synaptic responses that we recorded were likely compound, reflecting a mixture of excitatory inputs from various afferent sources activated from layer 6, including monosynaptic geniculocortical afferents, disynaptic inputs routed through layer 4, and monosynaptic inputs from axon collaterals of infragranular neurons. Thus, it is possible that the differential plasticity outcomes reflect variable activation of these pathways for different cells; however, we consider this unlikely because analysis of the initial slopes of the EPSPs (reflecting monosynaptic inputs) (Fig. 6) results in the same outcome as layer 4 stimulation (Fig. 5). Moreover, similar outcomes, regardless of whether bicuculline is used, suggest that variable synaptic inhibition is unlikely to account for the differences. Because the dye used for calcium imaging buffers calcium, variable loading of the cell may contribute to different plasticity outcomes (although access resistances were low and similar between cells); however, this interpretation is negated by the results from the experiments without fura-4F during which similar plasticity outcomes occur.
There is considerable evidence demonstrating the importance of timing of spikes with respect to EPSPs for determining plasticity (Song et al., 2000; Sjostrom et al., 2001; Celikel et al., 2004; Kobayashi and Poo, 2004; Tzounopoulos et al., 2004; Yao et al., 2004) (for review, see Sjostrom and Nelson, 2002). Thus, during depolarization, the spike timing may have varied between cells; however, this was not the case (Fig. 7), implicating other factor(s). This is not to say that spike timing is not important, and indeed, if the experimental protocol were designed to alter spike timing, it might further modify the plasticity outcome.
Calcium transients in LTP versus LTD
Our results provide direct measurements of [Ca2+]i during plasticity induction and maintenance. To our knowledge, this is the first demonstration of distinct combined amplitude and kinetic calcium profiles during induction of LTP versus LTD in the same cell type in response to the same induction protocol. Our observation that the amplitude component of the calcium transients is predictive of the outcome is consistent with previous theoretical considerations (Bienenstock et al., 1982; Bear et al., 1987), biochemical correlates for activation of kinase and phosphatase signaling pathways, respectively (Otmakhov et al., 1997; D'Alcantara et al., 2003), and experimental measures of relative changes in calcium levels during plasticity (Artola et al., 1990; Hansel et al., 1997; Yang et al., 1999; Cormier et al., 2001). The larger peaks and more rapid decay of the calcium transients during individual pairings in cells that undergo plasticity may reflect a more primed status of calmodulin or effective capture by target protein cascades (Mermelstein et al., 2001), more efficient calcium buffering-pumping by those cells causing a calcium profile that is optimally tuned (Murchison and Griffith, 1999), or more efficient calcium uptake that releases substantial calcium triggering plasticity after the pairings (Blaustein and Golovina, 2001). Although the amplitudes of calcium transients in the dendrites are predictive (Artola et al., 1990; Hansel et al., 1997; Yang et al., 1999; Cormier et al., 2001) and the kinetics of the calcium rises at synapses have been suggested potentially to play a role in the polarity of plasticity (Kato et al., 1999; Mizuno et al., 2001; Helmchen, 2002), such a distinct relationship between the rate of rise of the centripetal calcium signal and the plasticity outcome has not been demonstrated experimentally. Because we recorded at 25°C, the kinetics of the calcium responses do not precisely reflect the in vivo situation. There is the possibility that at elevated temperatures, different effects on synaptic responses may occur because of effects on certain currents; however, there are at least three reasons for the validity of the analysis at room temperature: (1) strong correlations of differences in the calcium profiles with respect to plasticity outcome (for individual calcium transients and for cumulative calcium rises); (2) distinct differences in the calcium profiles during the control experiments where depolarizing pulses alone are applied and when pairings do not induce plasticity; and (3) a significant difference in the individual calcium transient decay time constants (dendritic and somatic) that occur between the plasticity groups versus the NC groups on the very first pairing. Moreover, previous studies in slices of both basic synaptic physiology-plasticity (Keller et al., 1998; Yoshimura et al., 2000; Gonzalez-Islas and Hablitz, 2003; Hegg et al., 2003; Rae and Irving, 2004; Yamada et al., 2004) and calcium signaling using imaging methods (Miller et al., 1996; Pozzo-Miller et al., 1999; Kaiser et al., 2004) have been performed at room temperature of 22-25°C and are consistent with results from other studies at elevated temperatures ranging from 30°C to 37°C in many respects including synaptic properties, plasticity outcomes, and calcium signaling.
Differences in calcium profiles throughout the conditioning protocol
It is notable that the differences in the peaks of the calcium transients (in proximal dendrites and somata) manifest between plasticity outcome groups within the first few pairings (Fig. 10C,D). It is equally striking that on the very first pairing the transients decay significantly faster in those cells that will undergo either type of plasticity (LTP or LTD) (Fig. 10E,F) versus in the NC or depolarization only groups. These results suggest that the calcium signal that leaves the sites of synaptic input after correlation of neurotransmitter binding and postsynaptic depolarization has already achieved the requisite amplitude and kinetic profile for induction of plasticity through interaction with other downstream calcium amplifying events (e.g., release of calcium from intracellular stores). The decline in the peak calcium transients with sequential pairings or depolarizing pulses alone suggests rundown of the calcium signals, probably through voltage-gated calcium channels (VGCCs) solely in the case of the depolarizing pulses alone. Our control experiments (Figs. 9, 10G,H) demonstrate that these changes are not caused by a declining level of depolarization of the neuron or its spike-generating capacity, so that the level of either passive depolarization or actively back-propagated spikes that travel to the synaptic sites is not the source of the change. It is more likely that this declination is caused by incomplete recovery of VGCCs (Patil et al., 1998) and/or enhanced sequestration-removal of calcium (Lee et al., 2000).
Our results are obtained from the calcium signals in dendritic shafts and somata versus dendritic spines; however, recent reports on synaptic inputs from layer 4 neurons onto layer 2/3 pyramidal neurons (Silver et al., 2003) demonstrate that many of these synapses are also on dendritic shafts, including apical and basilar dendrites (Feldmeyer et al., 2002; Silver et al., 2003), and located within 70 μm of the soma (Feldmeyer et al., 2002; Silver et al., 2003), where our calcium measurements were made. Thus, although many synapses are activated and the exact location of those inputs cannot be ascertained, the intradendritic calcium measures that have characteristic signatures for different outcomes are within the range of the monosynaptic input from layer 4 and may reflect either primary or secondary changes in [Ca2+]i. Interestingly, Goldberg et al. (2003) demonstrated that calcium signals in dendritic shafts also are spatially constrained. The difference between the LTP and LTD groups in the kinetics of the cumulative somatic calcium rise in the first pairings is striking (Fig. 11A1,A2,C1,C2). Although the significance of this difference is unclear, specific temporal profiles of somatic calcium may contribute to selective modification of gene expression (West et al., 2001) implicated in longer term plasticity (Frey et al., 1993) requiring transcription and translation with delivery of newly synthesized proteins to “tagged” synaptic sites (Steward and Worley, 2001).
Footnotes
This work was supported by National Institutes of Health Grant EY012782 to M.J.F. We thank Felecia Hester for expert technical assistance, Dr. John Hablitz for valuable discussions and advice on experiments, and Dr. Sergey Mirov for his assistance with calibrating the Till monochromator output.
Correspondence should be addressed to Michael J. Friedlander, Civitan International Research Center 516, 1719 Sixth Avenue South, Department of Neurobiology, University of Alabama at Birmingham, Birmingham, AL 35294-0021. E-mail: mjf@nrc.uab.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249847-15$15.00/0
References
- Abraham WC, Bear MF (1996) Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19: 126-130. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W (1993) Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci 16: 480-487. [DOI] [PubMed] [Google Scholar]
- Artola A, Brocher S, Singer W (1990) Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature 347: 69-72. [DOI] [PubMed] [Google Scholar]
- Bear MF (2003) Bidirectional synaptic plasticity: from theory to reality. Philos Trans R Soc Lond B Biol Sci 358: 649-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF (1987) A physiological basis for a theory of synapse modification. Science 237: 42-48. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW (1982) Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci 2: 32-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Golovina VA (2001) Structural complexity and functional diversity of endoplasmic reticulum Ca(2+) stores. Trends Neurosci 24: 602-608. [DOI] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE (2004) Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci 7: 534-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, Gilliam TC, Kandel ER (2003) Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron 39: 655-669. [DOI] [PubMed] [Google Scholar]
- Chen HX, Otmakhov N, Lisman J (1999) Requirements for LTP induction by pairing in hippocampal CA1 pyramidal cells. J Neurophysiol 82: 526-532. [DOI] [PubMed] [Google Scholar]
- Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME (2003) Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci 23: 2572-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Miller LD, Petrozzino J, Muller W (1994) Calcium signaling in dendritic spines of hippocampal neurons. J Neurobiol 25: 234-242. [DOI] [PubMed] [Google Scholar]
- Cormier RJ, Greenwood AC, Connor JA (2001) Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J Neurophysiol 85: 399-406. [DOI] [PubMed] [Google Scholar]
- D'Alcantara P, Schiffmann SN, Swillens S (2003) Bidirectional synaptic plasticity as a consequence of interdependent Ca2+-controlled phosphorylation and dephosphorylation pathways. Eur J Neurosci 17: 2521-2528. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM (1999) Heterogeneity of synaptic plasticity at unitary CA3-CA1 and CA3-CA3 connections in rat hippocampal slice cultures. J Neurosci 19: 10664-10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Silver RA, Sakmann B (2002) Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signaling within a cortical column. J Physiol (Lond) 538: 803-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregnac Y, Burke JP, Smith D, Friedlander MJ (1994) Temporal covariance of pre- and postsynaptic activity regulates functional connectivity in the visual cortex. J Neurophysiol 71: 1403-1421. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER (1993) Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260: 1661-1664. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM (1997) Organotypic slice cultures: a technique has come of age. Trends Neurosci 20: 471-477. [DOI] [PubMed] [Google Scholar]
- Goldberg JH, Tamas G, Aronov D, Yuste R (2003) Calcium microdomains in aspiny dendrites. Neuron 40: 807-821. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ (2003) Dopamine enhances EPSCs in layer II III pyramidal neurons in prefrontal cortex. J Neurosci 23: 867-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391: 85-100. [DOI] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W (1997) Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci 9: 2309-2322. [DOI] [PubMed] [Google Scholar]
- Harsanyi K, Friedlander MJ (1997) Transient synaptic potentiation in the visual cortex. I. Cellular mechanisms. J Neurophysiol 77: 1269-1283. [DOI] [PubMed] [Google Scholar]
- Hegg CC, Greenwood D, Huang W, Han P, Lucero MT (2003) Activation of purinergic receptor subtypes modulates odor sensitivity. J Neurosci 23: 8291-8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F (2002) Raising the speed limit—fast Ca(2+) handling in dendritic spines. Trends Neurosci 25: 438-441. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A (1988) Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol 92: 145-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe DB, Brown TH (1994) Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol 72: 471-74. [DOI] [PubMed] [Google Scholar]
- Kaiser KMM, Lubke J, Zilberter Y, Sakmann B (2004) Postsynaptic calcium influx at single synaptic contacts between pyramidal neurons and bitufted interneurons in layer 2/3 of rat neocortex is enhanced by backpropagating action potentials. J Neurosci 24: 1319-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Tanaka T, Yamamoto K, Isomura Y (1999) Distinct temporal profiles of activity-dependent calcium increase in pyramidal neurons of the rat visual cortex. J Physiol (Lond) 519: 467-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Yagodin S, Aroniadou-Anderjaska V, Zimmer LA, Ennis M, Sheppard NF Jr, Shipley MT (1998) Functional organization of rat olfactory bulb glomeruli revealed by optical imaging. J Neurosci 18: 2602-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Poo MM (2004) Spike train timing-dependent associative modification of hippocampal CA3 recurrent synapses by mossy fibers. Neuron 5: 445-454. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B (1998) Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA 95: 9596-9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Rosenmund C, Schwaller B, Neher E (2000) Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol (Lond) 525: 405-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Pelkey KA, Topolnik L, Congar P, Lacaille JC, McBain CJ (2003) Depolarization-induced long-term depression at hippocampal mossy fiber-CA3 pyramidal neuron synapses. J Neurosci 23: 9786-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (2001) Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol (Lond) 532: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska A, Brown E, Ross J, Yuste R (2000) Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J Neurosci 20: 1722-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW (2001) Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci USA 98: 15342-15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LD, Petrozzino JJ, Golari G, Connor JA (1996) Ca2+ release from intracellular stores induced by afferent stimulation of CA3 pyramidal neurons in hippocampal slices. J Neurophysiol 76: 554-562. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kanazawa I, Sakurai M (2001) Differential induction of LTP and LTD is not determined solely by instantaneous calcium concentration: an essential involvement of a temporal factor. Eur J Neurosci 14: 701-708. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9: 967-975. [DOI] [PubMed] [Google Scholar]
- Murchison D, Griffith WH (1999) Age-related alterations in caffeine-sensitive calcium stores and mitochondrial buffering in rat basal fore-brain. Cell Calcium 25: 439-452. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K (2000) Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408: 584-588. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K (2002) Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci 5: 657-664. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Griffith LC, Lisman JE (1997) Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci 17: 5357-5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT (1998) Preferential closed-state inactivation of neuronal calcium channels. Neuron 20: 1027-1038. [DOI] [PubMed] [Google Scholar]
- Petrozzino JJ, Pozzo Miller LD, Connor JA (1995) Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron 14: 1223-1231. [DOI] [PubMed] [Google Scholar]
- Picciolo GL, Kaplan DS (1984) Reduction of fading of fluorescent reaction product for microphotometric quantitation. Adv Appl Microbiol 30: 197-234. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Inoue T, Murphy DD (1999) Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J Neurophysiol 81: 1404-1411. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M (1991) Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods 37: 15-26. [DOI] [PubMed] [Google Scholar]
- Rae MG, Irving AJ (2004) Both mGluR1 and mGluR5 mediate Ca2+ release and inward currents in hippocampal CA1 pyramidal neurons. J Neuropharmacol 46: 1057-1069. [DOI] [PubMed] [Google Scholar]
- Silver RA, Lubke J, Sakmann B, Feldmeyer D (2003) High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science 302: 1981-1984. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Nelson SB (2002) Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol 12: 305-314. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2001) Rate, timing and cooperativity jointly determine cortical synaptic plasticity. Neuron 32: 1149-1164. [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF (2000) Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci 3: 919-926. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J (2002) The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci USA 99: 1627-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Worley PF (2001) A cellular mechanism for targeting newly synthesized mRNAs to synaptic sites on dendrites. Proc Natl Acad Sci USA 98: 7062-7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W (1996) Direct measurement of coupling between dendritic spines and shafts. Science 272: 716-719. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Camacho P, Lechleiter JD, Herman B (1999) Measurement of intracellular calcium. Physiol Rev 79: 1089-1125. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO (2004) Cell-specific, spike-timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci 7: 719-725. [DOI] [PubMed] [Google Scholar]
- Wang SS, Denk W, Hausser M (2000) Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3: 1266-1273. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024-11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Takechi H, Kanchiku I, Kita Y, Kato N (2004) Small-conductance Ca2+-dependent K+ channels are the target of spike-induced Ca2+ release in a feedback regulation of pyramidal cell excitability. J Neurophysiol 91: 2322-2329. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS (1999) Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol 81: 781-787. [DOI] [PubMed] [Google Scholar]
- Yao H, Shen Y, Dan Y (2004) Intracortical mechanisms of stimulus-timing-dependent plasticity in visual cortical orientation tuning. Proc Natl Acad Sci USA 101: 5081-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Tsumoto T (1996) Long-term depression in rat visual cortex is associated with a lower rise of postsynaptic calcium than long-term potentiation. Neurosci Res 24: 265-274. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Sato H, Imamura K, Watnabe Y (2000) Properties of horizontal and vertical inputs to pyramidal cells in the superficial layers of the cat visual cortex. J Neurosci 20: 1931-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W (1999) Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci 19: 1976-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS (1999) Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9: 305-313. [DOI] [PubMed] [Google Scholar]
- Zufall F, Leinders-Zufall T, Greer CA (2000) Amplification of odor-induced Ca(2+) transients by store-operated Ca(2+) release and its role in olfactory signal transduction. J Neurophysiol 83: 501-512. [DOI] [PubMed] [Google Scholar]