

Abstract
Activation of volume-regulated anion current (VRAC) plays a key role in the maintenance of cellular volume homeostasis. The mechanisms, however, that regulate VRAC activity are not fully understood. We have examined whether VRAC activation is modulated by the cholesterol content of the membrane bilayer. The cholesterol content of bovine aortic endothelial cells was increased by two independent methods: (a) exposure to a methyl-β-cyclodextrin saturated with cholesterol, or (b) exposure to cholesterol-enriched lipid dispersions. Enrichment of bovine aortic endothelial cells with cholesterol resulted in a suppression of VRAC activation in response to a mild osmotic gradient, but not to a strong osmotic gradient. Depletion of membrane cholesterol by exposing the cells to methyl-β-cyclodextrin not complexed with cholesterol resulted in an enhancement of VRAC activation when the cells were challenged with a mild osmotic gradient. VRAC activity in cells challenged with a strong osmotic gradient were unaffected by depletion of membrane cholesterol. These observations show that changes in membrane cholesterol content shift VRAC sensitivity to osmotic gradients. Changes in VRAC activation were not accompanied by changes in anion permeability ratios, indicating that channel selectivity was not affected by the changes in membrane cholesterol. This suggests that membrane cholesterol content affects the equilibrium between the closed and open states of VRAC channel rather than the basic pore properties of the channel. We hypothesize that changes in membrane cholesterol modulate VRAC activity by affecting the membrane deformation energy associated with channel opening.
Keywords: anion channels, volume regulation, cholesterol
INTRODUCTION
A variety of physiological and pathophysiological factors increase the intracellular osmolarity and challenge cellular volume homeostasis (McManus and Churchwell 1994; Waldegger et al. 1998). To maintain the volume within a narrow range, cells decrease the intracellular concentrations of K+, Cl−, and small organic osmolytes (Hoffman 1992; Law 1994). The critical step in the regulation of cellular volume is activation of volume-regulated anion current (VRAC), which is responsible for the efflux of Cl− and organic osmolytes (for reviews, see Nilius et al. 1996; Strange et al. 1996). The current is activated by the expansion of cell volume that can be induced by an increase in the intracellular osmolarity or a decrease in the osmolarity of the extracellular medium (Levitan and Garber 1995; Zhang and Lieberman 1996). The mechanisms of VRAC regulation, however, are not well understood. One hypothesis suggests that cell swelling is coupled to VRAC activation by mechanical forces that develop in the lipid bilayer of the membrane (Hoffman 1992). In this study, we examine the role of the lipid composition of the membrane bilayer in VRAC activation by modulating the cholesterol content of the plasma membrane.
Transitions between the closed and open states of ion channels are thought to result from conformational changes of the channel proteins (Miller and White 1984; Catterall 1986). If these transitions perturb the surrounding lipid bilayer, then the overall energetic cost of an ion channel opening includes the contributions of the intrinsic protein activation energy and the membrane deformation energy (Huang 1986; Helfrich and Jacobsson 1990; Lundbaek et al. 1996; Lundbaek and Andersen 1999). The contribution of membrane deformation energy has been shown to be significant in the regulation of the activity of several types of ion channels, such as gramicidin channels (Huang 1986; Lundbaek and Andersen 1994), alamethicin channels (Keller et al. 1993), and N-type calcium channels (Lundbaek et al. 1996). Membrane deformation energy is a function of the unperturbed bilayer thickness and spontaneous monolayer curvature, as well as of the mechanical properties of the membrane lipid bilayer, such as membrane surface tension and bending and the compressibility moduli (Huang 1986; Helfrich and Jacobsson 1990; Nielsen et al. 1998). Alterations of membrane deformation energy, therefore, have been suggested to constitute a general mechanism by which changes in the mechanical properties of the membrane bilayer may regulate the activity of ion channels (Lundbaek et al. 1996). We hypothesize that membrane deformation energy may play an important role in the mechanism by which VRAC channels sense osmotic gradients across the cell membrane.
If membrane deformation energy constitutes a significant contribution to the energetic cost of VRAC activation, then changes in the mechanical properties of the membrane bilayer that affect membrane deformation energy would be expected to alter VRAC activity. Membrane deformation energy can been approximated by a phenomenological spring model in which bilayer mechanical properties are characterized by a bilayer stiffness coefficient A, defined as A = ΔG 0 def/μ2, where μ is the sum of the deformation depths of the monolayers of the membrane (Mouritsen and Bloom 1984; Huang 1986; Nielsen et al. 1998; Lundbaek and Andersen 1999). Elevation of membrane cholesterol content increases the bilayer stiffness (Lundbaek et al. 1996), and therefore can be used as a tool to determine whether the contribution of membrane deformation energy to the energetic cost of VRAC activation is significant.
In the present study, we show that enriching endothelial cells with cholesterol using two independent methods suppresses VRAC activation, while depletion of membrane cholesterol enhances activation of the current. These observations support our hypothesis that membrane deformation energy is involved in the regulation of VRAC activity. Further studies, however, are needed to discriminate between the roles of membrane deformation energy and of specific sterol–protein interactions in the regulation of VRAC activity.
METHODS
Tissue Culture
Bovine endothelial cells were grown in DMEM culture medium (Cell Grow; Fisher Scientific/Mediatech) supplemented with 10% fetal bovine serum (GIBCO BRL). Cell cultures were maintained in a humidified incubator at 37°C with 5% CO2. The cells were fed and split every 3–4 d. We used cells between passages 10 and 30. There was no difference in VRAC activation between the earlier and later passages.
Cholesterol Enrichment
Two methods have been used to enrich the cells with cholesterol: exposure of cells to methyl-β-cyclodextrin (MβCD) solution saturated with cholesterol and exposure of cells to cholesterol-enriched lipid dispersions.
MβCD-cholesterol solution was prepared as described previously (Christian et al. 1997). In brief, cholesterol was stored as a stock solution in chloroform:methanol (1:1 vol:vol), and a small volume of the stock solution was added to a glass tube and the solvent was evaporated. Then, 5 mM MβCD solution in DMEM medium without serum was added to the dried cholesterol. The tube was vortexed, sonicated, and incubated overnight in a rotating bath at 37°C. MβCD was saturated with cholesterol at a CD:cholesterol molar ratio of 8:1; the saturation limit was determined as described previously (Christian et al. 1997). In preparation for an experiment, cells were washed three times with serum-free DMEM to remove the serum of the growth medium. Cells were then incubated with MβCD saturated solution or with MβCD solution containing no cholesterol (empty MβCD) for 30, 60, or 120 min. During the incubation, cells were maintained in a humidified CO2 incubator at 37°C. Control cells were treated similarly and incubated with serum-free DMEM solution without any MβCD. After exposure to MβCD, cells were washed three times with serum free DMEM and left in the incubator overnight in the same solution. MβCD was a generous gift from Cerestar USA, Inc., and cholesterol was purchased from Sigma Chemical Co.
Cholesterol-rich liposomes (dispersions) were prepared as described previously (Gleason et al. 1991). In brief, unesterified (free) cholesterol (FC) and egg phosphatidylcholine (PL) were mixed in 2:1 molar ratio to prepare cholesterol-rich liposomes. Lipids were added to sterile endotoxin-free buffer solution, sonicated, centrifuged to remove undispersed lipids, and mixed with DMEM solution containing 1% FBS to a final concentration of 500 μg/ml cholesterol. Cells were exposed to cholesterol-rich liposomes for 48 h. Liposomes with an FC:PL ratio of 1:2 and cholesterol-free liposomes were prepared by a similar procedure and used as control treatment.
Cholesterol Measurement
Lipid was extracted from the washed cell monolayer using isopropanol as previously described (McClosky et al. 1987). Total and free cholesterol mass analysis was done by gas-liquid chromotography as previously described (Ishikawa et al. 1974; Klansek et al. 1995). Esterified cholesterol mass was calculated to be the difference between total and free cholesterol mass determinations. Cell protein was determined on the lipid-extracted monolayer using a modification (Markwell et al. 1978) of the method of Lowry et al. 1951. All mass values were normalized on the basis of cell protein.
Electrophysiological Recording
Solutions.
External recording solution contained (mM): 150 NaCl, 1 EGTA, 2 CaCl2, 10 HEPES, pH 7.2. Internal solutions contained (mM): 120 CsGlut or CsAsp, 10 HEPES, 4 ATP, pH 7.2 (CsOH) with free [Ca2+] ∼10 nM (0.1 CaCl2, 1.1 EGTA). Intracellular solutions contained Cs+ to decrease possible contamination of Cl− outward current (inward flux) by outward K+ currents (outward flux). Chemicals were obtained from Fisher Scientific or Sigma Chemical Co. The osmolarities of all solutions were determined immediately before recording with a vapor pressure osmometer (Wescor Inc.) and were adjusted by the addition of sucrose, as required.
Recording.
The current was activated by challenging cells with a transmembrane hyposmotic gradient between the intracellular and extracellular solutions. The gradient was created by membrane rupture. Current development was monitored by 500-ms linear voltage ramps from a holding potential of −60 to +60 mV at an interpulse interval of 10 s. Normal cellular current convention is used when referring to the direction of current; i.e., outward current refers to inward Cl− ion flow. Ionic currents were measured using the whole cell configuration of the standard patch clamp technique (Hamill et al. 1981). Pipettes were pulled from N51A glass (Garner Glass), coated with sylgard (Dow Corning Corp.), and fire polished to give a final resistance of 2–6 MΩ, using the above recording solutions. Fire polishing was necessary for N51A glass to create high resistance seals. Part of the experiments were performed using SGI glass from Richland Glass Co. These pipettes generated high resistance seals without fire polishing. NaCl agar bridge was used for a reference electrode. Currents were recorded using an EPC9 amplifier (HEKA Electronik) and accompanying acquisition and analysis software (Pulse & PulseFit; HEKA Electronik) running on a Macintosh Quadra 700 or PowerCenter 150. Pipette and whole-cell capacitance was automatically compensated. Whole cell capacitance and series resistance were monitored throughout the recording. In recordings using cells with large current amplitudes (>500 pA), series resistance compensation (95% compensation with a 100-μs lag) was used. Occasionally, a lower percentage compensation was required to prevent current oscillation. Cells exhibiting small current amplitudes (<500 pA) did not require series resistance compensation. In these cases, the voltage error involved was <5%.
Statistical Analysis
Statistical analysis of the data was performed using a standard two-sample Student's t test assuming unequal variances of the two data sets. Statistical significance was determined using a two-tail distribution assumption and was set at 5% level (P < 0.05).
RESULTS
Exposure to MβCD:Cholesterol Modulates Cholesterol Level in Endothelial Cells
Recent studies have demonstrated that cyclic oligosaccharides β-cyclodextrins provide a precise and reproducible method for modulating membrane cholesterol content in several cell types (Christian et al. 1997). In this study, we applied this method to modulate the cholesterol content in bovine aortic endothelial cells (BAEC). Total cholesterol level in BAEC that were not exposed to MβCD was 29 ± 11 μg/mg protein (n = 9), similar to total cholesterol levels reported earlier for rabbit endothelial cells (Kim et al. 1991) and for other cell types (Christian et al. 1997; Gimpl et al. 1997). Fig. 1, inset, shows that total cell cholesterol was predominantly in the form of free cholesterol, indicating that BAEC accumulate free cholesterol rather than esterified cholesterol, in agreement with previous results (Kim et al. 1991).
Figure 1.
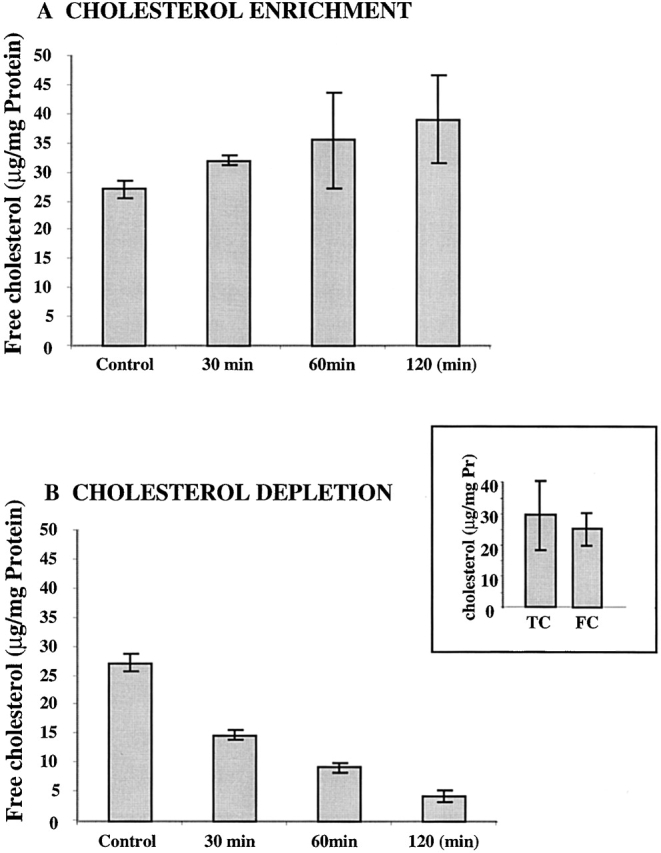
Modulation of free cholesterol content in BAEC by MβCD. (A) Cells were exposed to 5 mM MβCD: cholesterol (8:1 mol:mol) in DMEM, pH 7.2, with no serum. (B) Cells were exposed to 5 mM MβCD that was not complexed with cholesterol. Control cells were treated with serum-free medium. The bars show means ± SD (n = 3). Free cholesterol content in cells exposed to 30 min of saturated MβCD; cholesterol was significantly different from that in control cells (P < 0.05). Longer exposures in this experiment do not show statistical significance because of the high variation between the samples. Therefore, this experiment (120-min exposure) was repeated two more times (n = 3 in each experiment). In both experiments, the difference was statistically significant at the level of P < 0.01. The difference between the cholesterol content in cells exposed to free MβCD for 30, 60, or 120 min was significantly different from that in control for all exposure times (P < 0.01).
Exposure of BAEC to MβCD saturated with cholesterol for time periods between 30 and 120 min resulted in a gradual increase of cholesterol content in these cells (Fig. 1 A). A 45% increase that was observed after 120 min is similar to the degree of cholesterol enrichment in endothelial cells exposed to β–very low density lippoprotein (β-VLDL) (Kim et al. 1991) and in smooth muscle cells isolated from atherosclerotic arteries (Chen et al. 1995). Exposure of BAEC to empty MβCD (MβCD that was not complexed with cholesterol) resulted in a drastic decrease of cholesterol level (Fig. 1 B).
Modulation of Cholesterol Content Affects Activation of VRAC
Cholesterol enrichment with saturated MβCD.
Activation of VRAC was tested by challenging the cells either with a mild osmotic gradient (extracellular:intracellular osmotic ratio of 0.85) or with a strong osmotic gradient (0.70 extracellular:intracellular osmotic ratio). Osmotic gradients were maintained throughout the experiment. Enrichment of the cells with cholesterol resulted in suppression of VRAC activation when the cells were challenged with a mild osmotic gradient (Fig. 2A and Fig. B), but had no effect on VRAC activation when the cells were challenged with a strong osmotic gradient (Fig. 2C and Fig. D).
Figure 2.

VRAC activation in cells enriched with cholesterol. (A and C) Families of current traces recorded from individual cells that were challenged with a mild osmotic gradient (extracellular/intracellular osmotic ratio of 0.85; A) or with a strong osmotic gradient (extracellular/intracellular osmotic ratio of 0.70; C). Currents were elicited by linear voltage ramps from a holding potential of −60 to +60 mV, and recorded 50, 200, 350, 500, and 650 s after the establishment of the whole cell configuration. (B and D) Average time courses of VRAC current densities that developed in cells exposed to saturated MβCD:cholesterol solutions (120 min) and in control cells in response to a mild osmotic gradient (B) or to a strong osmotic gradient (D). Current densities were calculated by normalizing the maximal current amplitudes of each individual ramp by the cell capacitance. Since cell capacitance does not change significantly during the experiment, the currents were normalized to the initial cell capacitance measured immediately after the establishment of the whole-cell configuration. Plateau VRAC current densities (calculated by averaging the current densities between 350 and 600 s after the establishment of the whole cell configuration) in cells enriched with cholesterol were significantly smaller from those in control cells when the cells were challenged with a mild osmotic gradient (P < 0.01; B). There was no significant difference between the plateau VRAC current densities in cholesterol-enriched and control cells when the cells were challenged with a strong osmotic gradient (D).
Fig. 2 A shows typical VRAC currents that developed in a cell that was exposed to MβCD:cholesterol solution for 120 min (bottom) and in a control cell (top) when both cells were challenged with a mild osmotic gradient and recorded on the same day. Currents that developed in response to a strong osmotic gradient are shown in Fig. 2 C. All currents were elicited by voltage ramps from a holding potential of −60 to +60 mV and recorded 50, 200, 350, 500, and 650 s after challenging the cells with an osmotic gradient. Amplitudes of the currents elicited by the same voltage ramps increase as VRAC develops. An outward rectification of current traces results from an endogenous rectification typical for VRAC currents and from a nonsymmetric anion composition of the recording solutions.
The experimental conditions allow recording of VRAC with minimal contamination from cation currents (see methods). Therefore, activation of VRAC is measured as an increase in outward current amplitude (net inward Cl− flux) under hyposmotic conditions. Average time courses of VRAC development in cells exposed to saturated MβCD:cholesterol solution and in control cells are compared in Fig. 2B and Fig. D. The currents are normalized for cell capacitance to eliminate the variability of VRAC amplitudes that may result from the differences in the sizes of individual cells. In all our experiments, VRAC activity in cells exposed to MβCD:cholesterol (or cholesterol-free MβCD) solutions was compared with VRAC activity in control cells recorded on the same day. Since stable VRAC recording could be achieved only from four to five cells a day, in a typical experiment the currents were recorded from two to three cells enriched with cholesterol and from two to three control cells. The time courses of VRAC development were then averaged over at least three experimental days. This protocol allowed us to minimize the variability in VRAC activity between the experiments that could arise from uncontrollable fluctuations in the cell culture conditions.
Similar to our earlier observations (Levitan et al. 1995; Levitan and Garber 1998), VRAC develops gradually over a period of several minutes until the current reaches a plateau level. Enrichment of cells with cholesterol resulted in a decrease of maximal VRAC current densities in cells that were challenged with a mild osmotic gradient (Fig. 2 C), but not in cells challenged with a strong osmotic gradient (D). These observations show that an increase in membrane cholesterol makes VRAC less sensitive to the osmotic challenge.
Membrane cholesterol depletion.
To test whether the suppression of VRAC activation by MβCD:cholesterol is due to cholesterol enrichment and not to an effect of MβCD itself, current activation was determined in cells exposed to cholesterol-free MβCD. Exposure of cells to cholesterol-free MβCD for 120 min resulted in a marked increase in VRAC amplitudes that developed in response to a mild osmotic gradient (Fig. 3A and Fig. B). A few cells in this experimental group developed currents in the 300 pA/pF range, currents that we never observed in control cells recorded on the same days when challenged with a mild osmotic gradient. Exposure to empty MβCD, however, had no effect on VRAC currents that developed in response to a strong osmotic gradient (Fig. 3C and Fig. D), indicating that a decrease in membrane cholesterol increases the VRAC sensitivity to the osmotic challenge.
Figure 3.

VRAC activation in cells depleted of cholesterol. The experimental protocol is as described for Fig. 2. (A and C) Current traces from individual cells challenged with a mild (A) or strong (C) osmotic gradient. (B and D) Average time courses of VRAC current densities in response to a mild (B) or strong (D) osmotic gradient. Note that average VRAC time courses in control cells in this experiment are different from those in the previous experiment for both osmotic gradients. This difference reflects the variability between the experimental batches of cells that results from uncontrollable fluctuations in the conditions of cell culture. To minimize this difference, VRAC activity in cells depleted of cholesterol is compared with VRAC activity in control cells recorded on the same day, as described in detail in results. Similar to the previous experiment, plateau VRAC current densities in cells depleted of cholesterol were significantly different from those in control cells when the cells were challenged with a mild osmotic gradient (P < 0.01; B), but were not different in cells challenged with a strong osmotic gradient (D).
Fig. 4 shows mean peak VRAC current densities measured in cells challenged with a mild osmotic gradient after the cells were exposed to MβCD:cholesterol or to empty MβCD solutions for variable periods of time (30–120 min). Mean peak currents decrease gradually with an increase in cell cholesterol content. Linear regression analysis yielded a correlation coefficient of 0.98. Enhancement of VRAC activation in cells exposed to empty MβCD confirmed that VRAC activation is sensitive to the cholesterol content of the membrane. The opposite effects of saturated MβCD:cholesterol complexes and of cholesterol-free MβCD on VRAC activation exclude the possibility that suppression of VRAC development in cells enriched with cholesterol is due to an effect of the MβCD alone.
Figure 4.

The dependence of mean peak current density on membrane cholesterol content. The points represent mean peak VRAC current densities plotted as a function of average levels of free cholesterol in cells exposed to various experimental conditions. All cells were challenged with a mild osmotic gradient. The conditions are specified near each point. The data is described by linear fit with a correlation coefficient of 0.98. (The fit was calculated using Igor WaveMetrics software.) Peak current densities in cells enriched with cholesterol and cells depleted from cholesterol were significantly different from those in control cells (P < 0.05).
Cholesterol-enriched lipid dispersions.
Dependence of VRAC activation on cholesterol content of the membrane was further confirmed using an alternative method to enrich the cells with cholesterol. Cholesterol-enriched lipid dispersions with a cholesterol:phospholipid molar ratio of 2:1 have been previously used to study the effects of cholesterol enrichment on function of smooth muscle cells (Gleason et al. 1991) and endothelial cells (Luo, Z., L. Laury-Kleintop, K. Pratt, and T.N. Tulenko, manuscript submitted for publication). Exposure of BAEC to cholesterol-enriched lipid dispersions for 48 h increased membrane cholesterol content approximately twofold (data not shown). Fig. 5 compares the development of VRAC in cells that were exposed to cholesterol-enriched lipid dispersions and in cells that were exposed to control lipid dispersions.
Figure 5.

VRAC activation in BAEC is suppressed by cholesterol-enriched lipid dispersions. (A) Families of current traces recorded from cells exposed to cholesterol-free liposomes (0:1 FC:PL molar ratio), to liposomes with 1:2 FC:PL ratio and to cholesterol-rich lipid dispersions with 2:1 FC:PL ratio. The experimental protocol was similar to that described in Fig. 2. (B) Average time courses of VRAC development for cells exposed to 0:1; 2:1, and 1:2 FC:PL lipid dispersions. Average plateau current densities in cells exposed to 2:1 FC:PL lipid dispersions were significantly smaller than those in cells exposed to 0:1 FC:PL (P < 0.05). There is no difference between cells exposed to 0:1 or 1:2 FC:PL.
Cells that were exposed to cholesterol-enriched lipid dispersions developed much smaller currents than cells that were exposed to control lipid dispersions (Fig. 5). There was no difference in VRAC activity between cells that were exposed to 0:1 and 1:2 lipid dispersions, but cells that were not exposed to any lipid dispersions developed significantly larger currents (Fig. 5, inset). One hypothesis to explain this observation is that the cell membrane equilibrates with liposomal lipids during the relatively long exposure (48 h) and that leads to an alteration of cholesterol distribution in the cellular membranes. Since cholesterol is distributed nonrandomly in the plasma membranes (Yeagle 1985), it is possible that an alteration of its distribution in the membrane affects activation of VRAC. The difference between VRAC activation in cells exposed to cholesterol-enriched liposomes and to control liposomes supports our observation that cholesterol enrichment results in suppression of VRAC activation.
Modulation of the Membrane Cholesterol Content Has No Effect on VRAC Anion Selectivity
Earlier studies have shown that VRAC channels are permeable not only to inorganic anions but organic anions, such as glutamate and aspartate (Banderali and Roy 1992; Levitan and Garber 1998). This study shows that modulation of membrane cholesterol has no effect on the glutamate−/Cl− and aspartate−/Cl− permeability ratios (Fig. 6). Permeability ratios were calculated from measured reversal potentials, using the Goldman-Hodgkin-Katz equation (Hille 1992):
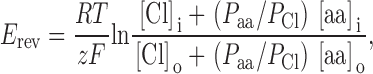 |
where P aa/P Cl is the ratio of the permeability of the relevant amino acid to that of Cl−. A lack of cholesterol effect on the anion permeability ratios suggests that the basic pore properties of the channel are not affected by the changes in membrane cholesterol.
Figure 6.

Modulation of membrane cholesterol has no effect on VRAC anion permeability ratios. Permeability ratios for glutamate−/Cl− (▪) and aspartate−/Cl− (□) were calculated from the values of measured reversal potentials. Reversal potentials were measured in at least five cells for each experimental group. Data points represent means ± SEM. There was no significant difference between VRAC permeability ratios in cells enriched with or depleted from cholesterol.
Modulation of the Membrane Cholesterol Content Has No Effect on VRAC Inactivation Properties
Voltage-dependent inactivation is a hallmark of volume-regulated anion current in a variety of cell types (Levitan and Garber 1995; Jackson and Strange 1995). Voltage-dependent inactivation of VRAC develops only at voltages more positive than +40 or +60 mV and therefore is not likely to be physiological. It provides, however, a characteristic of the biophysical properties of VRAC and can be used for further characterization of the nature of a cholesterol effect on VRAC.
Voltage-dependent inactivation of VRAC in cells exposed either to MβCD:cholesterol complexes, cholesterol-free MβCD, as well as control cells are shown in Fig. 7. A two-pulse voltage protocol with a conditioning pulse ranging from −60 to +140 mV followed by a test pulse to +100 mV was used to determine the voltage sensitivity and kinetic properties of the inactivation. Voltage-dependent inactivation is apparent from the accelerated decay of the anion current at depolarization more positive than +80 mV, as shown in Fig. 7 A. The inactivation ratio was defined as the ratio between the amplitude of a test pulse delivered after a conditioning prepulse to the amplitude of a control test pulse (Fig. 7 B). The steepness of the decrease of the inactivation ratio provides a measure of the amount of charge that has to move for a channel to change its conformation from an open to an inactivated state. A similarity between the inactivation curves in cells enriched with, or depleted from, cholesterol suggests that changes in cholesterol content do not affect the transition between the open and inactivated states of the channel. A differential effect of membrane cholesterol on activation and inactivation of the same channels has been reported previously for N-type calcium channels (Lundbaek et al. 1996), where an increase in membrane cholesterol affected inactivation but not the activation of the channels.
Figure 7.

Voltage-dependent inactivation of VRAC is not altered by the modulation of membrane cholesterol. (A) Families of traces recorded in response to a two-pulse voltage protocol with a 500-ms conditioning pulse from −60 to +140 mV, followed immediately with a short 10-ms test pulse to +100 mV. (B) Inactivation ratios were calculated as Itest,V/I control test, where Itest,V is the current amplitude of a test pulse after a conditioning pulse to voltage V, and Icontrol test is the current amplitude of a test pulse applied from a holding potential of −60 mV. The points represent means ± SEM (n = 7 for each experimental group). There was no significant difference in the voltage dependence of the inactivation between cells enriched with or depleted from cholesterol.
Modulation of Cholesterol Content Has No Effect on Membrane Capacitance
Earlier studies have shown that a two- to threefold increase of membrane cholesterol results in a 10–20% increase in membrane bilayer thickness in aortic smooth muscle cells (Chen et al. 1995) and in model membrane vesicles prepared from bovine cardiac phosphatidylcholine and cholesterol (Tulenko et al. 1998). In the latter study, however, a saturation point for cholesterol was demonstrated in model membranes, beyond which phase separation occurred, resulting in the formation of discrete cholesterol domains (34 Å) within the bilayer and restoration of bilayer width to control dimensions (56–57 Å). Similar effects appeared to occur in smooth muscle cell membranes as well (Tulenko et al. 1998).
In the present study, we have evaluated the thickness of endothelial membranes in cholesterol-depleted and cholesterol-enriched cells by measuring electrical capacitance of the membrane. Since the specific capacitance of the membrane is inversely proportional to its width (Hille 1992), cholesterol enrichment is expected to decrease the membrane capacitance. If we assume that the membrane bilayer is homologous to a simple capacitor formed by two parallel plates of area A (membrane surface) and separated by an insulator of thickness d (thickness of the insulating hydrophobic bilayer of the membrane), then the membrane capacitance C is (Hille 1992):
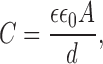 |
where ε is the dielectric constant of the membrane and ε0 is the polarizability of the free space. In this case, a 20% increase in membrane thickness may result in up to a 20% decrease in specific membrane capacitance. In the present study, however, changes in cell cholesterol had no effect on membrane capacitance of endothelial cells (Fig. 8 A), suggesting that thickness of the membrane was not altered significantly.
Figure 8.

Cell capacitance of BAEC is not altered by modulation of membrane cholesterol. (A) Cell capacitance was measured immediately after the establishment of the whole cell configuration. The bars represent the average cell capacitance of cells exposed to MβCD:cholesterol (n = 19), cells exposed to empty MβCD (n = 16) for at least 60 min, and control cells (n = 18). There is no significant difference in cell capacitance between these experimental groups. (B) Cell capacitance does not change during cell swelling. Cells were challenged by a transmembrane osmotic gradient and capacitance measured every 10 s for the duration of the experiment. The values of cell capacitance at each time point were normalized to the initial capacitance measured at time 0.
Fig. 8 B shows that endothelial cell capacitance did not increase when the cells were challenged osmotically, indicating that there is no increase in membrane area during cell swelling. This observation is in agreement with our earlier studies in B-lymphocytes (Levitan and Garber 1997). It suggests that cells swell by unfolding membrane invaginations rather than by adding membrane from the intracellular stores.
DISCUSSION
The main finding of this study is that VRAC activation in endothelial cells is modulated by the cholesterol content of the membrane when the cells are challenged with a mild osmotic gradient, but not when the cells are challenged with a strong osmotic gradient. These observations provide direct evidence that the lipid environment of the plasma membrane plays a role in the regulation of VRAC. The effect of cholesterol is specific for VRAC activation in the sense that elevation of membrane cholesterol reduces the steady state (plateau) level of VRAC current density without having any effect on VRAC ion selectivity or its inactivation properties.
One mechanism that may be responsible for the suppression of VRAC activity in response to the elevation of membrane cholesterol is a shift in the equilibrium between the open and closed states of the channel towards the closed state. Alternatively, it may alter the basic pore properties of the channel, decreasing the single-channel conductance. Earlier studies have shown that an increase in membrane cholesterol content reduced the open probability of Ca2+-dependent K+ channels (Bolotina et al. 1989; Chang et al. 1995) while having no (Bolotina et al. 1989) or little effect on single-channel conductance (Chang et al. 1995). A lack of the effect on single-channel conductance suggests that membrane cholesterol affects the equilibrium between the open and closed states of these channels rather than their basic pore properties. In endothelial cells, we could not detect VRAC single channels either in cell-attached or excised configurations even when the outside-out configuration was created after VRAC was fully developed. We have used, therefore, an alternative approach to discriminate between the effect of cholesterol on the equilibrium between the open and closed conformation states of VRAC channels versus altering the channel pore properties.
To determine whether VRAC suppression by an increase in membrane cholesterol results from a shift in the equilibrium between VRAC conformational states or from a decrease in its single channel conductance, we have compared the effects of membrane cholesterol on VRAC activation in cells challenged with mild or strong osmotic gradients. The rationale of this approach is that if elevation of membrane cholesterol affects the equilibrium between the channel conformational states, then membrane cholesterol is expected to shift the sensitivity of VRAC to osmotic stimuli so that the channel becomes less sensitive to the osmotic challenge. In this case, the saturating level of stimulus is expected to invoke the same responses at low and high membrane cholesterol. By contrast, if cholesterol decreases VRAC single channel conductance, then VRAC activity is expected to be suppressed at all levels of the osmotic stimulus. Our study shows that an increase in membrane cholesterol suppresses VRAC activation when the cells are challenged with a mild osmotic gradient (0.85 osmotic ratio), whereas it had no effect on VRAC development when the cells were challenged with a strong osmotic gradient (0.70 osmotic ratio). These observations suggest that increasing membrane cholesterol affects the equilibrium between the open and closed states of VRAC channel rather than its single channel conductance. A lack of cholesterol effect on glutamate−:Cl− and aspartate−:Cl− permeability ratios supports the conclusion that membrane cholesterol content has no effect on the basic pore properties of VRAC and that its effect on VRAC activity is through a shift in the equilibrium between the channel conformational states.
How can changes in membrane cholesterol shift the equilibrium between the closed and open states of the channel? We suggest that suppression of VRAC by an increase in membrane cholesterol is due to an increase in membrane deformation energy that is associated with the transition between the closed and open states of the channel. Membrane deformation energy has been described as a function of three major components: (a) a compression–expansion component that depends on membrane thickness, (b) a splay–distortion component that depends on the orientation of the phospholipid hydrocarbon chains of the lipid molecules, and (c) a surface tension component that depends on the density of the polar head groups along the surface of the membrane (Huang 1986; Helfrich and Jacobsson 1990; Nielsen et al. 1998). The compressibility coefficient, defined as K = ΔT/(ΔA/A), where ΔT is a change in membrane tension accompanied by a relative increase of membrane area of ΔA/A, is considered a measure of membrane stiffness (Evans and Neeham 1987). If a transition from a closed to an open state of an ion channel perturbs membrane lipid bilayer, by a change in the protein hydrophobic length, then the stiffer the membrane the larger the energetic cost of the transition (Lundbaek et al. 1996). Elevation of membrane cholesterol level alters the mechanical properties of the membrane bilayer, including lipid ordering, spontaneous monolayer curvature, and the modulus of membrane compressibility (Yeagle 1985; Evans and Neeham 1987; Seddon 1990). This results in an increase in membrane stiffness and, consequently, is expected to increase membrane deformation energy associated with channel opening (Lundbaek et al. 1996). Therefore, if membrane deformation energy constitutes a significant contribution to the overall energetic cost of VRAC activation, an increase in membrane cholesterol is expected to suppress activation of the current, as was observed in this study. An increase in membrane deformation energy, however, is not expected to affect the maximal number of VRAC channels. Therefore, increasing the osmotic shock to the point where all VRAC channels are open overrides the effect of cholesterol. A differential effect of membrane cholesterol on VRAC activation and on its inactivation suggests that membrane deformation energy may be involved in some but not all of the conformational changes of the same membrane protein.
Is it possible to determine which of the mechanical properties of the membrane bilayer that are affected by membrane cholesterol content are responsible for the effect of cholesterol on VRAC activation? A complete separation of different mechanical properties is not likely possible because of the interdependence between these properties (Nielsen et al. 1998). It has been pointed out, however, that membrane fluidity, defined as “the ease of movement of a liquid,” while affecting the rates of protein conformation changes, cannot be responsible for the changes in the equilibrium properties of protein function regardless of the nature of the underlying structural change (Lee 1991; Andersen et al. 1999). Our experiments show that an increase in membrane cholesterol reduces the plateau levels of VRAC activation. As discussed above, this effect suggests that elevation of membrane cholesterol shifts the equilibrium between the closed and open states of the channel. We suggest, therefore, that changes in membrane fluidity per se are not sufficient to account for the observed effect of cholesterol on the VRAC activity and that changes in the material properties of the membrane, defined by membrane stiffness, are of primary importance.
Alternatively, membrane cholesterol may affect the intrinsic activation energy of the VRAC protein through specific sterol–protein interactions. In many cases, there is no strong specificity between intrinsic proteins and surrounding lipids (Devaux and Seigneuret 1985; Andersen et al. 1999), but specific sterol–protein interactions have been described for several membrane proteins (Yeagle 1985), including amphotericin B ion channels (Mickus et al. 1992). Therefore, we cannot exclude this possibility and it will be addressed in our further studies by the structural analysis of the effects of cholesterol analogues on VRAC activation.
We propose a model for the role of membrane deformation energy in the mechanism by which VRAC channels may sense osmotic gradients across the plasma membrane. We suggest that cell swelling may alter membrane deformation energy that is associated with VRAC activation by altering the physical properties of the membrane bilayer. Specifically, membrane tension may alter the thickness of the membrane, consequently modulating a hydrophobic mismatch between the channel hydrophobic exterior and the thickness of the bilayer hydrophobic core (Goulian et al. 1998). A change in the hydrophobic mismatch affects membrane deformation energy of the protein conformation change (Nielsen et al. 1998) and, therefore, may shift the equilibrium between the closed and open states of the VRAC channel protein. An increase in membrane tension has been shown to activate a bacterial mechanosensitive channel inserted into planar lipid bilayers (Sukharev et al. 1994, Sukharev et al. 1999). Sensitivity to lipid tension was also observed in ion channels formed by an antibiotic protein alamethicin inserted into a lipid bilayer (Opsahl and Webb 1994). Changes in the hydrophobic mismatch between these channel proteins and the hydrophobic core of the membrane bilayer may constitute a general mechanism by which mechanical signals activate mechanosensitive ion channels.
Elevation of plasma cholesterol levels is associated with an increased risk of atherosclerosis (Kannel et al. 1979). Impairment of endothelial cells is thought to constitute one of the earliest stages of the development of the disease (Ross 1995; Li et al. 1993). Since VRAC is implicated in regulation of cell growth and proliferation in several cell types (Voets et al. 1997; Rouzaire-Dubois and Dubois 1998), including endothelial cells (Voets et al. 1995), suppression of VRAC activity by cholesterol may contribute to the impairment of endothelial function during development of atherosclerosis. Further studies are needed to establish the mechanism by which cholesterol affects activation of endothelial ion channels.
Acknowledgments
We thank Dr. Sarah Garber for her support during the initial stages of these experiments and for many interesting and helpful discussions. We thank Drs. Peter F. Davies, Olaf S. Andersen, John M. Russell, Michal Bental-Roof, and Michael M. White for critical reading of the manuscript. We are grateful to Ms. Liz Cannon for excellent technical assistance in preparing lipid dispersions and lipid measurements and to Ms. Rebecca Riley for her help with cell culture.
This work was supported by National Institutes of Health grants NIDDK47762 (S. Garber), HL 30496 (T.N. Tulenko), Program Project Grant HL 22633 (G.H. Rothblat), and Training Grant HL 07443 (A.E. Christian).
Footnotes
Abbreviations used in this paper: BAEC, bovine aortic endothelial cells; FC, unesterified (free) cholesterol; MβCD, methyl-β-cyclodextrin; PL, unesterified egg phosphatidylcholine; VRAC, volume-regulated anion current.
References
- Andersen O.S., Nielsen C., Maer A.M., Lundbaek J.A., Goulian M., Koeppe H. Ion channels as tools to monitor lipid bilayer-membrane protein interactionsgramicidin channels as molecular force transducers. Methods Enzymol. 1999;294:208–224. doi: 10.1016/s0076-6879(99)94013-2. [DOI] [PubMed] [Google Scholar]
- Banderali U., Roy G. Anion channels for amino acids in MDCK cells. Am. J. Physiol. Cell Physiol. 1992;263:C1200–C1207. doi: 10.1152/ajpcell.1992.263.6.C1200. [DOI] [PubMed] [Google Scholar]
- Bolotina V., Omelyanenko V., Heyes B., Ryan U., Bregestovski P. Variations of membrane cholesterol alter the kinetics of Ca2+-dependent K+ channels and membrane fluidity in vascular smooth muscle cells. Pflügers Arch. 1989;415:262–268. doi: 10.1007/BF00370875. [DOI] [PubMed] [Google Scholar]
- Catterall W.A. Voltage-dependent gating of sodium channelscorrelating structure and function. Trends Neurosci. 1986;9:7–10. [Google Scholar]
- Chang H.M., Reitstetter R., Mason R.P., Gruener R. Attenuation of channel kinetics and conductance by cholesterolan interpretation using structural stress as a unifying concept. J. Membr. Biol. 1995;143:51–63. doi: 10.1007/BF00232523. [DOI] [PubMed] [Google Scholar]
- Chen M., Mason R.P., Tulenko T.N. Atherosclerosis alters composition, structure and function of arterial smooth muscle plasma membrane. Biochim. Biophys. Acta. 1995;1272:101–112. doi: 10.1016/0925-4439(95)00073-d. [DOI] [PubMed] [Google Scholar]
- Christian A.E., Haynes M.P., Phillips M.C., Rothblat G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Devaux P.F., Seigneuret M. Specificity of lipid–protein interactions as determined by spectroscopic techniques. Biochim. Biophys. Acta. 1985;822:63–125. doi: 10.1016/0304-4157(85)90004-8. [DOI] [PubMed] [Google Scholar]
- Evans E., Neeham D. Physical properties of surfactant bilayer membranesthermal transition, elasticity, rigidity, cohesion and colloidal interactions. J. Phys. Chem. 1987;91:4219–4228. [Google Scholar]
- Gimpl G., Burger K., Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry. 1997;36:10959–10974. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- Gleason M.M., Medow M.S., Tulenko T.N. Excess membrane cholesterol alters calcium movements, cytosolic calcium levels and membrane fluidity in arterial smooth muscle cells. Circ. Res. 1991;69:216–227. doi: 10.1161/01.res.69.1.216. [DOI] [PubMed] [Google Scholar]
- Goulian M., Mesquita O.N., Fygenson D.K., Nielsen C., Andersen O.S., Libchaber A. Gramicidin channel kinetics under tension. Biophys. J. 1998;74:328–337. doi: 10.1016/S0006-3495(98)77790-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Helfrich P., Jacobsson E. Calculation of deformation energies and conformations in lipid membranes containing gramicidin channels. Biophys. J. 1990;57:1075–1084. doi: 10.1016/S0006-3495(90)82625-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels in Excitable Membranes 2nd ed 1992. Sinauer Associates Inc; Sunderland, MA: pp. 607 [Google Scholar]
- Hoffman E.K. Cell swelling and volume regulation. Can. J. Physiol. Pharmacol. 1992;70:S310–S313. doi: 10.1139/y92-277. [DOI] [PubMed] [Google Scholar]
- Huang H.W. Deformation free energy of bilayer membrane and its effects on gramicidin channel lifetime. Biophys. J. 1986;50:1061–1070. doi: 10.1016/S0006-3495(86)83550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T.T., MacGee J., Morrison J.A., Glueck C.J. Quantitative analysis of cholesterol in 5 to 20 μL of plasma. J. Lipid Res. 1974;15:286–291. [PubMed] [Google Scholar]
- Jackson P.S., Strange K. Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J. Gen. Physiol. 1995;105:661–671. doi: 10.1085/jgp.105.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel W.B., Castelli W.P., Gordon T. Cholesterol in the prediction of atherosclerotic disease. Ann. Intern. Med. 1979;90:825–836. doi: 10.7326/0003-4819-90-1-85. [DOI] [PubMed] [Google Scholar]
- Keller S.L., Bezrukov S.M., Gruner S.M., Tate M.W., Vodyanoy I., Parsegian V.A. Probability of alamethicin conductance states varies with nonlamellar tendency of bilayer phospholipids. Biophys. J. 1993;65:23–27. doi: 10.1016/S0006-3495(93)81040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.A., Maxwell K., Hajjar D.P., Berliner J.A. b-VLDL increases endothelial cell plasma membrane cholesterol. J. Lipid Res. 1991;32:1125–1131. [PubMed] [Google Scholar]
- Klansek J., Yancey P., St. Clair R.W., Fisher R.T., Johnson W.J., Glick J.M. Cholesterol quantification by GLCartifactual formation of short-chain steryl esters. J. Lipid Res. 1995;36:2261–2266. [PubMed] [Google Scholar]
- Law R.O. Regulation of mammalian brain cell volume. J. Exp. Zool. 1994;268:90–96. doi: 10.1002/jez.1402680204. [DOI] [PubMed] [Google Scholar]
- Lee A.G. Lipids and their effects on membrane proteinsevidence against a role for fluidity. Prog. Lipid Res. 1991;30:323–348. doi: 10.1016/0163-7827(91)90002-m. [DOI] [PubMed] [Google Scholar]
- Levitan I., Almonte C., Mollard P., Garber S.S. Modulation of a volume-regulated chloride current by F-Actin. J. Membr. Biol. 1995;147:283–294. doi: 10.1007/BF00234526. [DOI] [PubMed] [Google Scholar]
- Levitan I., Garber S.S. Voltage-dependent inactivation of volume regulated Cl− current in T84 and Myeloma cells. Pflügers Arch. 1995;431:297–299. doi: 10.1007/BF00410203. [DOI] [PubMed] [Google Scholar]
- Levitan I., Garber S.S. Volume regulated anion current and cytoskeletal interaction. In: Latorre R., editor. From Ion Channels to Cell-to-Cell Conversations. Plenum Press; Centro de Estudios Cientificos de Santiago, Chile: 1997. pp. 245–268. [Google Scholar]
- Levitan I., Garber S.S. Anion competition for a volume-regulated current. Biophys J. 1998;75:226–235. doi: 10.1016/S0006-3495(98)77509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Cybulsky M.I., Gimbrone M.A., Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb. 1993;13:197–204. doi: 10.1161/01.atv.13.2.197. [DOI] [PubMed] [Google Scholar]
- Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lundbaek J.A., Andersen O.S. Lysiphospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J. Gen. Physiol. 1994;104:645–673. doi: 10.1085/jgp.104.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbaek J.A., Andersen O.S. Spring constants for channel-induced lipid bilayer deformations estimates using gramicidin channels. Biophys. J. 1999;76:889–895. doi: 10.1016/S0006-3495(99)77252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbaek J.A., Birn P., Hansen A.J., Andersen O.S. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- Markwell M.A.K., Haas S.M., Bieber L.L., Tolbert N.E. A modification of the Lowry procedure to simplify protein determination in the membrane and lipoprotein samples. Anal. Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- McClosky H.M., Rothblat G.H., Glick J.M. Incubation of acetylated low-density lipoprotein with cholesterol-rich dispersions enhances cholesterol uptake by macrophages. Biochim. Biophys. Acta. 1987;921:320–332. doi: 10.1016/0005-2760(87)90033-6. [DOI] [PubMed] [Google Scholar]
- McManus M.L., Churchwell K.B. Clinical significance of cellular osmoregulation. In: Strange K., editor. Cellular and Molecular Physiology of Cell Volume Regulation. CRC Press; Boca Raton, FL: 1994. pp. 63–80. [Google Scholar]
- Mickus D.E., Levitt D.G., Rychnovsky S.D. Enantiomeric cholesterol as a probe for ion-channel structure. J. Am. Chem. Soc. 1992;114:359–360. [Google Scholar]
- Miller C., White M.M. Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. USA. 1984;81:2772–2775. doi: 10.1073/pnas.81.9.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouritsen O.G., Bloom M. Mattress model of lipid–protein interactions in membranes. Biophys. J. 1984;46:141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C., Goulian M., Andersen O.S. Energetics of inclusion-induced bilayer deformations. Biophys. J. 1998;74:1966–1983. doi: 10.1016/S0006-3495(98)77904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B., Eggermont J., Voets T., Droogmans G. Volume-activated Cl− channels. Gen. Pharmacol. 1996;27:1131–1140. doi: 10.1016/s0306-3623(96)00061-4. [DOI] [PubMed] [Google Scholar]
- Opsahl L.R., Webb W.W. Transduction of membrane tension by the ion channel alamethicin. Biophys. J. 1994;66:71–74. doi: 10.1016/S0006-3495(94)80751-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- Rouzaire-Dubois B., Dubois J.M. K+ channel block-induced mammalian neuroblastoma cell swellinga possible mechanism to influence proliferation. J. Physiol. 1998;510:93–102. doi: 10.1111/j.1469-7793.1998.093bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon J.M. Structure of the inverted hexagonal (HII) phase, and non-lammelar phase transitions of lipids. Biochim. Biophys. Acta. 1990;1031:1–69. doi: 10.1016/0304-4157(90)90002-t. [DOI] [PubMed] [Google Scholar]
- Strange K., Emma F., Jackson P.S. Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol. Cell Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Sukharev S.I., Blount P., Martinac B., Blattner F.R., Kung C. A large-conductance mechanosensitive channel in E. coli encoded by mscL alone. Nature. 1994;368:265–268. doi: 10.1038/368265a0. [DOI] [PubMed] [Google Scholar]
- Sukharev S.I., Sigurdson W.J., Kung C., Sachs F. Energetic and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J. Gen. Physiol. 1999;113:525–539. doi: 10.1085/jgp.113.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulenko T.N., Chen M., Mason P.E., Mason R.P. Physical effects of cholesterol on arterial smooth muscle membranesevidence of immiscible cholesterol domains and alterations in bilayer width during atherogenesis. J. Lipid Res. 1998;39:947–956. [PubMed] [Google Scholar]
- Voets T., Szucs G., Droogmans G., Nilius B. Blockers of volume-activated Cl− currents inhibit endothelial cell proliferation. Pflügers Arch. 1995;431:132–134. doi: 10.1007/BF00374387. [DOI] [PubMed] [Google Scholar]
- Voets T., Wei L., De Smet P., Van Driessche W., Eggermont J., Droogmans G., Nilius B. Downregulation of volume-activated Cl− currents during muscle differentiation. Am. J. Physiol. Cell Physiol. 1997;272:C667–C674. doi: 10.1152/ajpcell.1997.272.2.C667. [DOI] [PubMed] [Google Scholar]
- Waldegger S., Steuer S., Risler T., Heidland T., Capasso G., Massry S., Lang F. Mechanisms and clinical significance of cell volume regulation. Nephrol. Dial. Transpl. 1998;13:867–874. doi: 10.1093/ndt/13.4.867. [DOI] [PubMed] [Google Scholar]
- Yeagle P.Y. Cholesterol and the cell membrane. Biochim. Biophys. Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- Zhang J., Lieberman M. Chloride conductance is activated by membrane distension of cultures chick heart cells. Cardiovasc. Res. 1996;32:168–179. [PubMed] [Google Scholar]