

Abstract
Mutations of presenilin 1 (PS1) causing Alzheimer's disease selectively increase the secretion of the amyloidogenic βA4(1-42), whereas knocking out the gene results in decreased production of both βA4(1-40) and (1-42) amyloid peptides (De Strooper et al. 1998). Therefore, PS1 function is closely linked to the γ-secretase processing of the amyloid precursor protein (APP). Given the ongoing controversy on the subcellular localization of PS1, it remains unclear at what level of the secretory and endocytic pathways PS1 exerts its activity on APP and on the APP carboxy-terminal fragments that are the direct substrates for γ-secretase. Therefore, we have reinvestigated the subcellular localization of endogenously expressed PS1 in neurons in vitro and in vivo using confocal microscopy and fine-tuned subcellular fractionation. We show that uncleaved PS1 holoprotein is recovered in the nuclear envelope fraction, whereas the cleaved PS fragments are found mainly in post-ER membranes including the intermediate compartment (IC). PS1 is concentrated in discrete sec23p- and p58/ERGIC-53–positive patches, suggesting its localization in subdomains involved in ER export. PS1 is not found to significant amounts beyond the cis-Golgi. Surprisingly, we found that APP carboxy-terminal fragments also coenrich in the pre-Golgi membrane fractions, consistent with the idea that these fragments are the real substrates for γ-secretase. Functional evidence that PS1 exerts its effects on γ-secretase processing of APP in the ER/IC was obtained using a series of APP trafficking mutants. These mutants were investigated in hippocampal neurons derived from transgenic mice expressing PS1wt or PS1 containing clinical mutations (PS1M146L and PS1L286V) at physiologically relevant levels. We demonstrate that the APP-London and PS1 mutations have additive effects on the increased secretion of βA4(1-42) relative to βA4(1-40), indicating that both mutations operate independently. Overall, our data clearly establish that PS1 controls γ42-secretase activity in pre-Golgi compartments. We discuss models that reconcile this conclusion with the effects of PS1 deficiency on the generation of βA4(1-40) peptide in the late biosynthetic and endocytic pathways.
Keywords: Alzheimer's disease, presenilin 1, APP processing, hippocampal neuron, γ-secretase
The molecular cascades causing the complex neurodegenerative process of Alzheimer's disease (AD)1 are far from resolved. The pathological hallmarks, i.e., senile plaques and tangles, of sporadic and familial AD (FAD) are nevertheless similar, suggesting a common pathogenic pathway. This allows us to approach the complicated problem of the pathogenesis of the sporadic form by the study of the genes and gene products involved in FAD. Most of the familial forms of AD are caused by missense mutations in the genes coding for presenilin 1 (PS1) and 2. The PS1/2 genes encode proteins of 467 and 448 amino acids, respectively, and show an overall amino acid similarity of 67% (Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995). Their homology with a series of related Caenorhabditis elegans proteins indicates a conserved, albeit incompletely understood, function throughout evolution (Levitan and Greenwald 1995;Baumeister and Haass 1998). Hydropathy plots suggest up to 10 hydrophobic regions, of which most probably only 8 actually traverse the membrane (Li and Greenwald 1996, Li and Greenwald 1998). Both the amino and carboxy termini as well as the large loop domain between hydrophobic regions 6 and 8 are orientated to the cytoplasm (Doan et al. 1996; De Strooper et al. 1997; Lehmann et al. 1997; Li and Greenwald 1998).
Although PS is synthesized as a full-length protein of ∼45 kD, it undergoes proteolytic cleavage within its hydrophilic loop domain, resulting in the generation of stable amino-terminal (PS1-NTF) and carboxyl-terminal fragments (PS1-CTF) in a 1:1 stoichiometry (Mercken et al. 1996; Thinakaran et al. 1996; Capell et al. 1998). Except for the G209V PS1 mutation (Levey et al. 1997), all missense mutations studied to date do not influence endoproteolysis significantly (Thinakaran and Sisodia 1998). The deletion mutant PS1Δexon 9, in which exon 9 encoding the cleavage site is deleted, prevents endoproteolysis and also causes FAD, indicating that the clipping by itself is not a prerequisite to develop the disease (Thinakaran et al. 1996). At least one common effect of the clinical mutations is an increased secretion of the highly amyloidogenic βA4(1-42) peptide as compared with the βA4(1-40) peptide. This effect has been demonstrated in cells derived from affected individuals (Scheuner et al. 1996) as well as in transfected cells and brains of transgenic mice (Borchelt et al. 1996; Duff et al. 1996; Citron et al. 1997). The amyloid peptides are derived from the larger amyloid precursor protein (APP) through consecutive clipping by two, hitherto unknown, proteinases called β- and γ-secretase. A third enzyme, α-secretase, cleaves the protein in the amyloid region, precluding the formation of these amyloid peptides (Haass and Selkoe 1993). PS1 is directly involved in γ-secretase processing of APP, as was demonstrated using knockout models. Targeted deletion of the PS1 gene resulted in a greatly diminished production of βA4(1-42) as well as (1-40) peptides. As β- (and α-) clipping was virtually normal in neurons derived from those animals, the effect on APP processing is confined to γ-secretase activities (De Strooper et al. 1998). This observation was recently considerably extended by the demonstration that site-directed mutagenesis of either one of two aspartic residues in the transmembrane domain 6 or 7, respectively, of PS1 results in a dominant negative form of the protein (Brockhaus et al. 1998; Steiner et al. 1999; Wolfe et al. 1999). In cells overexpressing these mutant forms, a similar inhibition of γ-secretase processing is observed as in the PS1-deficient cells (Wolfe et al. 1999). These results are compatible with a catalytic role of presenilin. However, it remains difficult to explain how presenilin, which is predominantly located in the early compartments of the biosynthetic pathways, can influence the processing of APP, which is thought to occur mainly in the late compartments of this pathway, at the cell surface, and endosomes (Sambamurti et al. 1992; Sisodia 1992; De Strooper et al. 1993; Koo and Squazzo 1994; Haass et al. 1995; Selkoe 1998).
The important issue raised by all these observations is how presenilins can affect γ-secretase processing. Indeed, from a cell biological point of view at least three possibilities present themselves: (1) PS1 is indeed more widely distributed than believed until now and is located beyond the Golgi apparatus; (2) APP–carboxy-terminal stubs are redirected to the ER after their generation in the Golgi apparatus in trafficking vesicles and at the cell surface; or (3) PS1 affects γ-secretase and APP sorting to a common subcellular compartment or microenvironment, affecting γ-secretase processing of APP indirectly. To address this problem, we have reinvestigated the subcellular localization of the presenilins and the APP carboxy-terminal stubs using advanced cell fractionation and confocal microscopy and a panel of well-characterized subcellular marker proteins. In addition, a series of APP sorting mutants was used to delineate functionally the subcellular compartment(s) where PS1 exerts its activity. We focused our study on (transgenic) mouse brain and primary cultures of hippocampal neurons, thereby avoiding possible artifacts that could arise from overexpression of presenilins. Indeed, in transiently or stable transfected cell lines, overexpressed PS1 exists in the ER and Golgi compartments mainly as an uncleaved holoprotein, in contrast to the normal in vivo situation where PS1 is present as cleaved fragments (Kovacs et al. 1996; Thinakaran et al. 1996, Thinakaran et al. 1997; De Strooper et al. 1997). It is far from clear whether this overexpressed full-length protein remains biological functional and whether it remains located in its physiologically relevant compartment (Kovacs et al. 1996; Thinakaran et al. 1996; De Strooper et al. 1997; Johnston et al. 1998). In contrast, transgenic mice expressing PS1 do not accumulate full-length protein and display physiological relevant levels of the cleaved fragments (Borchelt et al. 1996; Thinakaran et al. 1996; Levesque et al. 1999). In this study, brains and neurons derived from mice expressing endogenous levels of mouse PS1, human wild-type PS1, PS1M146L or PS1L286V were used. The Semliki Forest virus (SFV) expression system allowed expression of APP as documented before (Liljestrom and Garoff 1991; De Strooper et al. 1995; Simons et al. 1996; Tienari et al. 1996b). We paid particular attention to the effects of the PS mutations on the formation of APP intermediate metabolites, such as the α- and β-cleaved stubs, since this aspect of APP metabolism has been somewhat neglected in previous studies. In addition, we addressed the question of the combined effects of PS1 and the APP London mutation on APP processing. Finally, three sorting mutants of APP were analyzed: one mainly processed in the early secretory pathway (APP/KK), one in the late secretory pathway (APP-ΔCT), and, finally, one targeted to the endosomal recycling compartment (APP/LDLR).
Materials and Methods
Viral Constructs
The cDNAs coding for human APP695, the London mutant, and human APP695ΔCT, lacking the region encoding the carboxy-terminal cytoplasmic tail of APP, were cloned blunt end in the SmaI site of pSFV-1 as described previously (Simons et al. 1996; Tienari et al. 1996b). APP695/KK was generated by exchanging the two amino acids at position 3 and 4 of APP695 to two lysines by site-directed mutagenesis (Chyung et al. 1997; Peraus et al. 1997). The chimera APP695/LDLR was constructed by exchanging the carboxy-terminal cytoplasmic domain of APP with that of the LDL receptor (cDNA provided by Walter Hunziker). In all sorting mutants, an amino-terminal c-myc epitope (EQKLISEEDL) was inserted into the KpnI site of the 5′-terminus of APP695 (Simons et al. 1995). All cDNAs were verified by sequencing.
pSFV1/APP constructs were linearized with SpeI, and run-off transcription using SP6 polymerase was performed to produce mRNA. The transcribed mix of APP and pSFV-helper were cotransfected into BHK cells by electroporation to yield recombinant SFV (Olkkonen et al. 1993). BHK cells were grown in DME/F-12 supplemented with 5% FCS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. 24 h after transfection, the culture supernatant containing infective recombinant SFV was collected. Aliquots were snap-frozen in liquid nitrogen and stored at –70°C until use.
Transgenic Animals and Neuronal Cell Culture
Transgenic FVB×C57 black mice expressing wild-type human PS1 or the clinical mutants Met146Leu (PS1M146L) or Leu286Val (PS1L286V) under the control of the prion promoter were used (Citron et al. 1997). The transgene proteins were efficiently cleaved and their subcellular localization in hippocampal neurons at different developmental stages was virtually indistinguishable from PS1 wild-type (Levesque et al. 1999).
Heterozygous transgenic mice were crossed with wild-type FVB mice, and pregnant females were killed 17–18 d postcoitum. Hippocampal neuron cultures were prepared from E17–E18-d-old fetal mice according to Goslin and Banker 1991. Single cell suspensions obtained from the hippocampi of individual embryos were plated on poly-l-lysine–coated plastic dishes (Nunc) or on poly-l-lysine–coated coverslips in minimal essential medium (MEM) supplemented with 10% horse serum. After 3–4 h, culture medium was replaced by serum-free neurobasal medium with B27 supplement (GIBCO BRL). Cytosine arabinoside (5 μM) was added 24 h after plating to prevent nonneuronal (glial) cell proliferation. Hippocampal neurons on plastic dishes were used at day 3 after plating. Hippocampal neurons on coverslips were maintained in culture for 14–16 d in the presence of a glial feeder layer to allow for full differentiation and polarization.
Cell Fractionation
Brain cortices of 1-wk-old control and transgenic PS1 wild-type mice were used as starting material. Since the transgene is under the control of the prion promoter, the human PS1 is mainly expressed in neurons. Analysis of the subcellular fractions obtained with this brain material using a human-specific antibody, therefore, reflects the distribution of PS1 in neurons in vivo. This allowed us to directly compare the fractionation data with the findings obtained in the cultured hippocampal neurons. The preparation of nuclei and nuclear envelopes was based on the method described by Otto et al. 1992 except that RNase 1 and PMSF were added before centrifugation of the nuclear envelopes.
For the isolation of the intermediate compartment (IC) fractions, the protocol of Schweizer et al. 1991 was used with modifications (Annaert et al. 1997). Gradient fractions were collected using a Büchler Auto-Densi Flow apparatus. Sucrose concentrations were calculated from the refractive index (Refractometer RF 490; Euromex) by linear regression. Highly purified intact lysosomes were prepared according to Maguire and Luzio 1985.
The protein content of each fraction was measured spectrophotometrically using a protein assay (Bio-Rad Laboratories) according to the manufacturer's instructions. Unless stated otherwise, equal amounts of protein (30 mg) were dissolved in Laemmli sample buffer, incubated for 10 min at room temperature, and resolved on 13% polyacrylamide gels (Mini-Protean II; Bio-Rad Laboratories). After SDS-PAGE and transfer of the proteins to nitrocellulose (Laemmli 1970; Towbin et al. 1979), blots were blocked in TBS containing 5% lowfat milk and 0.1% Tween 20 and incubated overnight (4°C) with the appropriate primary antibodies. Detection was done with HRP-conjugated secondary antibodies (Bio-Rad Laboratories) followed by enhanced chemiluminescence (ECL; Amersham).
Indirect Immunofluorescence Microscopy
12-d-old hippocampal neurons were fixed in 4% paraformaldehyde and 4% sucrose in 120 mM sodium phosphate buffer, pH 7.3, for 30 min at room temperature. Fixed neurons were rinsed twice in Dulbecco's PBS and transferred to ice-cold methanol (−20°C, for 4 min) followed by ice-cold acetone (−20°C, for 2 min). Coverslips were transferred to a humid chamber, washed three times with PBS, and blocked for 1 h at room temperature or overnight at 4°C (blocking solution: PBS containing 2% BSA, 2% FCS, 0.2% fish gelatin, and 4% donkey serum). Neurons were subsequently incubated with primary antibodies in blocking solution (for 2 h at room temperature). After four washes with PBS, secondary antibodies were applied (for 2 h at room temperature). CY2- and CY3- or Lissamine rhodamine-conjugated donkey anti–mouse and donkey anti–rabbit secondary antibodies (Jackson ImmunoResearch Laboratories) were used for detection. Coverslips were washed three times with PBS, once in distilled water, and finally mounted in Mowiol (Calbiochem-Novabiochem). Specimens were viewed through a Nikon Diaphot 300 (PlanApo 60/1.40 oil) connected to the MRC1024 confocal microscope (Bio-Rad Laboratories). Images were captured by LaserSharp (version 2.0) on a Compaq Prosignia 300 workstation and finally processed using Adobe Photoshop 5.0.
Antibodies
The following antibodies were used to detect PS1 (De Strooper et al. 1997): polyclonal antibody B17.2 recognizes both human and mouse PS1-CTF (peptide–antigen residues 300–315, EGDPEAQRRVSKNSKY of the human PS1 sequence); and mAb 5.2 is specific for the human CTF-PS1 (peptide–antigen residues 307–331, RRVSKNSKYNAESTERESQDTVAEN). Polyclonal antibody B14.5 detects the human PS1-NTF (peptide–antigen residues 30–44, NDNRERQEHNDRRSL); B19.2 and B32.1 are directed against the murine PS1-NTF (peptide–antigen residues 32–46, SQERQQQHGRQRLDN) and PS1-CTF (peptide–antigen residues 310–330, PKNPKYNTQRAERETQDSGSG), respectively. All polyclonal antibodies were affinity-purified against the peptide immobilized on NHS-activated Sepharose 4b (Pharmacia) according to the manufacturer's instructions. PS1 knockout brain tissue was used to confirm the specificity of the PS1 affinity-purified polyclonal antibodies.
HoloAPP, α- and/or β-cleaved carboxy-terminal fragments of APP and amyloid peptides were immunoprecipitated from cell extracts and media using polyclonal antibodies raised against the 20 carboxy-terminal amino acids of APP (B10/4 and B11/8) or against the synthetic human 1-40 βA4 peptide (B7/7) (De Strooper et al. 1995).
mAbs against lamin B1, a marker for nuclear envelopes, or against p58/Golgi, a marker for Golgi membranes, were obtained from Zymed or Sigma Chemical Co., respectively. mAbs to the transferrin receptor or to synaptobrevin II (clone 69.1) were provided by Ian Trowbridge (Salk Institute, San Diego, CA), and Reinhard Jahn (MPI-Biophysical Chemistry, Göttingen, Germany), respectively. Monoclonal anti–c-myc antibody (clone 9E10; Evan et al. 1985) was used for the detection of the carboxy-terminal–inserted c-myc epitope.
Polyclonal antibodies to rSEC61αp, calnexin, BAP31, p58/ERGIC-53, GM130, SEC23p, and TGN38 were provided by Enno Hartmann (Georg-August Universität, Göttingen), Ari Helenius (Yale University, New Haven, CT), Michael Reth (MPI-Immunologie, Freibourg, Germany), Jaakko Saraste (University of Bergen, Norway), Graham Warren (Yale University, New Haven, CT), Jean-Pierre Paccaud, and George Banting (Bristol, UK), respectively.
Viral Infection and Metabolic Labeling
Recombinant SFV was diluted 10-fold in conditioned culture medium and added to the cells (1 ml/dish). Cultures were incubated for 1 h at 37°C to allow entry of the virus, followed by incubation in conditioned medium in the absence of virus (for 2 h). Metabolic labeling was performed using methionine-free N2 medium containing 100 μCi [35S]methionine/ml (NEN). After 4 h, the conditioned medium was collected and the neuronal cell layer was washed once in PBS, and then scraped in extraction buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl containing 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS). Both the media and cell extracts were centrifuged (14,000 rpm for 10 min) to remove detached cells and aggregates. Polyclonal antibody B11/8 (1/500) was added to cell extracts and B7/7 (1/100) to the media, together with protein G–Sepharose (Pharmacia) and incubated overnight (at 4°C). The immunoprecipitates were washed five times in extraction buffer and once in TBS. To check the glycosylation status of APPwt and the APP/KK mutant (see below), the washed precipitate was treated overnight with endoH (20 mU; Boehringer) in phosphate buffer (at 37°C).
Immunoprecipitated proteins were solubilized with Tricine-SDS sample buffer (450 mM Tris-HCl, pH 8.45, 12% glycerol, 4% SDS, 0.0025% Coomassie blue G, and 0.0025% phenol red). Samples were boiled (for 5 min) and electrophoresed on 10–20% Tris-Tricine gels (Novex). Radiolabeled bands were detected by a PhosphorImager (Molecular Dynamics, Inc.) and analyzed (ImageQuaNT 4.1). Levels of α- and β-cleaved carboxy-terminal stubs and secreted total βA4 were normalized to the level of expression of APP holoprotein precipitated from the cell extracts.
Sandwich ELISA assays to measure βA4(1-40) and βA4(1-42) used the combination of antibodies G2-10/W02 and G2-11/W02, respectively (Hartmann et al. 1997; Zhang et al. 1999). Nunc MaxiSorb immunoassay plates were coated overnight at 4°C with 0.4 mg/well G2-10 in 100 mM NaHCO3, pH 9.5, or with 1 mg/well G2-11 in 100 mM Tris-HCl, pH 7.4. Subsequently, the antibody solution was removed and the wells were incubated overnight at 4°C with 5% BSA in TBS. Wells were washed with TBS plus 0.05% Tween 20 (TTBS) and were stored at 4°C (maximum 6 mo). Conditioned media were diluted with 10% BSA to yield a final concentration of 2% BSA and 100 ml of diluted media was added to each well along with 40 ng/well biotinylated W02. Biotinylation of W02 was performed with the EZ-Link TM Sulfo-NHS-LC biotinylation kit (Pierce Chemical Co.) according to the manufacturer's instructions. The plate was incubated at 4°C with gentle shaking for 12 h (for βA4(1-40) measurements) or for 24 h (for βA4(1-42) measurements). Plates were washed five times with TTBS. 100 ml of 0.5 mg/ml HRP-conjugated NeutrAvidin (Pierce Chemical Co.) was added to each well and plates were further incubated at room temperature for 1 h. The color reaction was performed with the TMB-H2O2 system (Kirkegaard & Perry Laboratories, Inc.) according to the manufacturer's instructions and absorption at 450 nm was measured. For the assessment of intracellular βA4, the cells of one 60-mm culture dish were solubilized with 450 ml buffer containing 20 mM Tris-HCl, pH 7, 2 mM EDTA, 0.2% SDS, 1% Triton X-100, 1% NP-40, and protease inhibitors (0.7 mg/ml pepstatin A, 23 mg/ml PMSF, and 11 mg/ml TPCK). 80 ml of the cell lysates was used for every measurement. The sensitivity of the assays is ∼50–100 pg/ml and values ranged between 60–800 pg/ml for intracellular βA4 and 100–3,500 pg/ml for secreted β4A. This large variation is mainly a consequence of variations in the number of cells per dish.
Results
Mouse brain and hippocampal neurons derived from wild-type mouse brain were used to localize endogenously expressed PS1 using subcellular fractionation and confocal immunofluorescence microscopy. In addition, transgenic mice expressing human PS1 under the control of the prion promoter, which drives expression of the transgene in neurons, were used for confocal microscopy and to obtain fractionation data on the distribution of PS1 in neurons in situ.
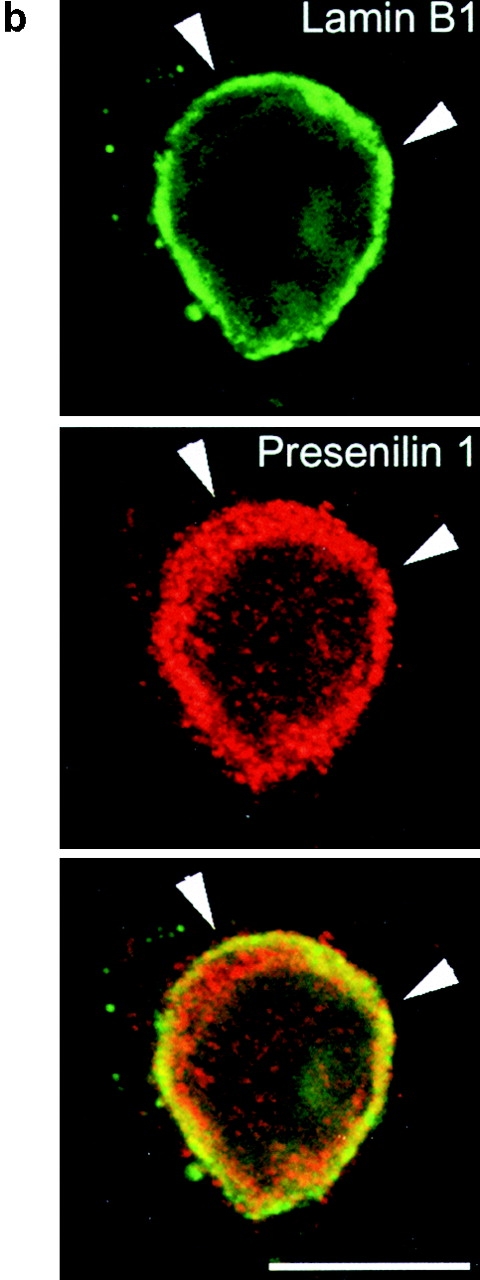
Because of their relatively high buoyant density, nuclei can be easily purified by differential centrifugation in high sucrose media. As is clear from Fig. 1, only a little PS1 staining is observed in purified nuclei, which contain mainly histones in the protein fraction (not shown). However, additional DNA digestion combined with ultracentrifugation yields a fraction of highly purified nuclear envelopes, as demonstrated by the abundant lamin B1 immunoreactivity (Fig. 1 a, NE). Interestingly, this final purification step results in a strong enrichment for PS1-CTF and -NTF immunoreactive bands (Fig. 1 a), indicating that the little amount of PS1 present in the nuclei is almost exclusively associated with the nuclear envelope. It should be noticed that this fraction also stains with calnexin antibodies, in agreement with the continuity between nuclear envelope and RER. Remarkably, in this nuclear envelope fraction, a prominent immunoreactive band representing full-length PS1 is detected with both NTF- and CTF-specific antibodies (Fig. 1 a and not shown) (see below). The presence of presenilin in the nuclear envelope was confirmed in fixed polarized hippocampal neurons using confocal microscopy. PS1 immunoreactivity colocalized with lamin B1 in confocal sections through the middle of the nucleus (Fig. 1 b, arrows). Together, these results indicate that PS1 fragments and PS1 holoprotein are present in the nuclear envelope.
Figure 1.
(a) Purification of nuclei (N) and nuclear envelopes (NE) from cortices of 5–7-d-old mice by differential centrifugation (Otto et al. 1992). Brains were homogenized in 0.32 M sucrose containing 1 mM MgCl2 and 20 mM Hepes, pH 7.4. The total homogenate (TH) was centrifuged (at 200 g av for 20 min), the resulting pellet (P1) resuspended to a final concentration of 1.8 M sucrose and centrifuged again (at 67,000 g av for 1 h). The pellet P2 was resuspended to a final concentration of 2.15 M sucrose, layered on a 2.2 M sucrose cushion and centrifuged (at 67,000 g av for 1 h). The pellet of purified nuclei (N) was, after DNAse digestion, centrifuged (at 9,500 g av for 20 min) to obtain nuclear envelopes (NE). Equal amounts of protein of each intermediate step was analyzed by SDS-PAGE and Western blotting. The final fraction (NE), which is highly enriched for lamin B1, also contains low amounts of PS1 fragments and full-length PS1. (b) Double immunofluorescent staining of fully polarized hippocampal neurons (16 d of culture) derived from nontransgenic embryos for PS1 (red) and lamin B1 (green). A merged picture shows considerable overlap of both stainings (yellow). Anti-PS1 pAb B17.2 was detected with CY3 anti–rabbit antibodies, whereas anti–lamin B1 mAb was detected with CY2-conjugated anti–mouse antibodies. Note that the focal plane is through the middle of the nucleus. The bulk of PS1 immunoreactive staining is localized close to the bottom of the cell and in the dendritic arbor (see next figure). Arrowheads point to rims of the nuclear envelope that costains for PS1-CTF (see also yellow color in overlay). Bar, 20 μm.

Next, the distribution of PS1 was investigated in pre-Golgi compartments of hippocampal neurons derived from human PS1wt transgenic mice (Fig. 2). rSEC61αp is an essential component of the mammalian ER protein translocation complex and is tightly associated with membrane-bound ribosomes in RER (Gorlich et al. 1992). PS1 immunoreactivity was found to codistribute only partially with the reticular staining pattern observed for rSEC61αp (Fig. 2, a–c, arrowheads). In contrast, a considerable fraction of PS1 staining, especially obvious in the perinuclear region, did not localize with rSEC61αp (Fig. 2, a–c, arrow). Antibodies to calnexin (Fig. 2, d–f) and BAP31 (Fig. 2, g–i), two proteins that are abundantly distributed throughout the ER, displayed a reticular staining that overlapped very well with most of the immunolabeling for PS1-CTF. However, it should be noticed that several distinct PS1-positive spots did not colocalize with calnexin immunoreactivity. Transport from the ER to the IC and Golgi apparatus occurs in specific subdomains of the ER where transported proteins are concentrated in budding transport vesicles coated by COPII coat proteins (Barlowe et al. 1994). SEC23p is one of the cytosolic components of this COPII coat complex. As shown in Fig. 2j–l, this COPII protein displayed an overall colocalization with PS1-CTF (Fig. 2 l, inset, yellow). At a higher magnification, all PS1-CTF–positive patches were also immunolabeled for SEC23p (see detailed area in Fig. 2, j–l). These export sites are often found juxtaposed to vesiculotubular clusters (VTCs) of the IC (Bannykh and Balch 1998). Indeed, immunolabeling of ERGIC-53/p58, so far the best characterized IC resident protein (Schweizer et al. 1991; Klumperman et al. 1998), showed a close colocalization with PS1-CTF (Fig. 2, m–o). Therefore, these results indicate an abundant localization of presenilin in the ER.
Figure 2.
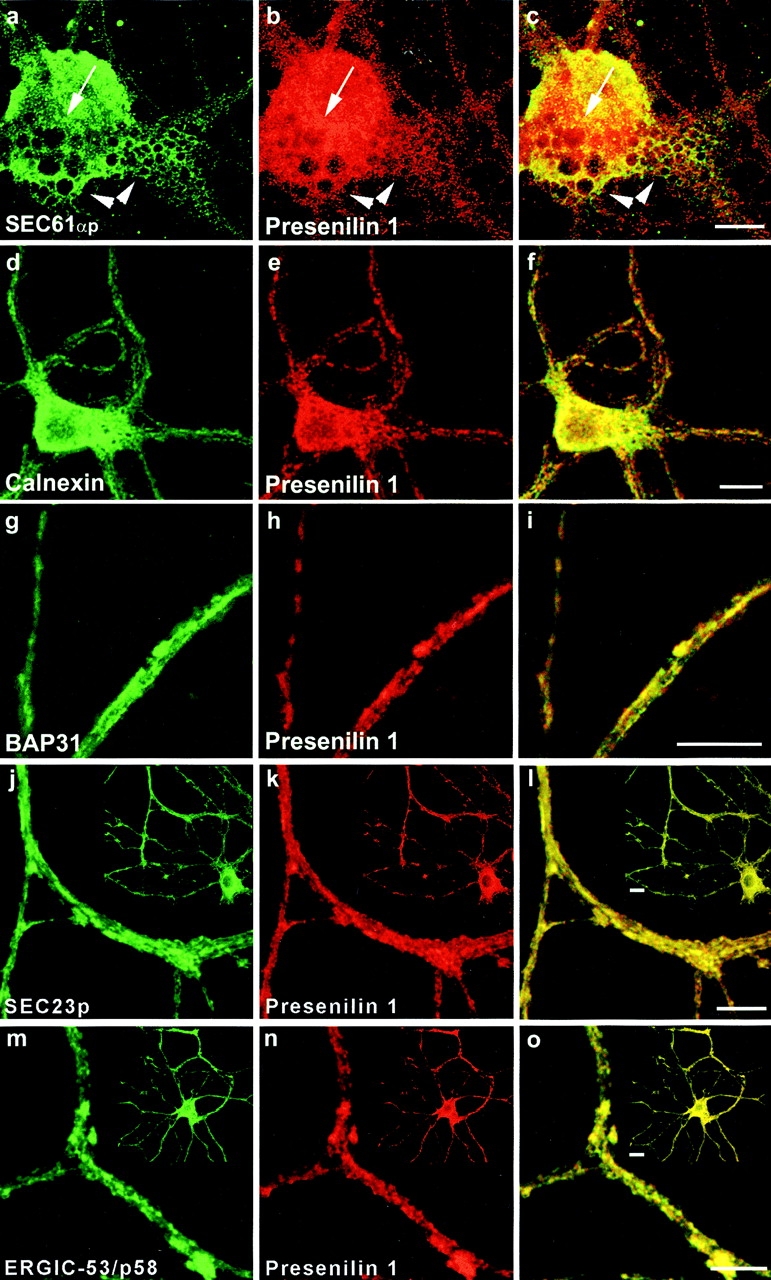
Double immunofluorescent staining of fully polarized hippocampal neurons derived from PS1wt transgenic embryos for PS1 (red in all pictures) and rSEC61αp (a–c), calnexin (d–f), BAP31 (g–i), SEC23 (j–l), and ERGIC-53/p58 (m–o). Fields c, f, i, l, and o represent color overlays. mAb 5.2 was used to label human PS1wt and detection was done with CY3 anti-mouse secondary antibodies. All other marker proteins were counterstained using CY2-conjugated anti–rabbit secondary antibodies. Notice the relative weak colocalization of PS1 with the RER marker rSEC61αp (a–c, arrowheads) mainly in the perinuclear region (a-c, arrow). PS1 colocalizes much better with other ER marker proteins such as calnexin (d–f), BAP31 (g–i), and SEC23 (j–l), or with ERGIC-53/p58, a resident protein of the IC. The pictures in (j–o) give a detailed area of the fully polarized neuron as overviewed in the insets. Bars: (a-i) 20 μm; (j-o) 10 μm.
Especially the close overlap with SEC23p and ERGIC-53 prompted us to further document the interesting localization of PS1 in the transit zone between ER and Golgi. We applied a cell fractionation scheme described by Schweizer et al. 1991 that allows one to obtain highly enriched fractions of the IC in VERO cells. This method starts with a Percoll gradient centrifugation step, resulting in a rough separation of RER and Golgi membranes. When applied on the postnuclear fraction of mouse brain, most of the PS1 immunoreactivity was detected in two peaks (Fig. 3 a). One small peak (15.1 ± 0.5% of total PS1-CTF or 19.4 ± 3.3%, n = 3, of total PS1–NTF immunoreactivity, respectively) was associated with the bottom fractions of the gradient coinciding with the distribution of the RER marker protein rSEC61αp (Fig. 2 a), confirming that a relative low amount of PS1 is associated with the RER compartment. A second, major peak (pool II: 42.7 ± 2.4% for PS1-CTF and 48.1 ± 0.5%, n = 3, for PS1–NTF) was located in the low density region coinciding with the density of Golgi membranes (Fig. 3 a and Schweizer et al. 1991). Typically, IC membranes equilibrate as a broad peak in the midportion of this self-formed gradient. Fractions 5–14, containing 61.6 ± 7.3% (n = 3) of the total ERGIC-53/p58 immunoreactivity contained 25.1 ± 2.1% of PS1-CTF and 17.9 ± 1.5% of PS1-NTF (n = 3). This fraction was pooled (Fig. 3 a, Pool I) and further purified and concentrated by an additional centrifugation step in a discontinuous Nycodenz gradient (Fig. 3 b). Three interfaces, F1, F2, and F3, are recovered. The interface F3 at 18.5–27% Nycodenz is highly enriched in IC membranes, as demonstrated by the strong immunoreactivity for p58/ERGIC-53 (Fig. 3 b). Both PS1-NTF and -CTF coenrich to a similar extent in this final F3 fraction. Unfortunately, and in contrast to the original method (Schweizer et al. 1991), immunoblotting for rSEC61αp and TRAPα, a protein associated with the translocon site (Hartmann et al. 1993), revealed contamination of F3 with RER membranes (Fig. 3 b). This discrepancy is probably a consequence of the difference in starting material, i.e., VERO cells versus mouse brain used here. Therefore, we purified the F3 fraction in an additional continuous sucrose gradient (0.8–1.6 M sucrose). Whereas the marker proteins calnexin and ERGIC-53/p58 displayed together with PS1 a bimodal distribution (Fig. 3 c), the rSEC61αp and TRAPα immunoreactivity was solely recovered in the high density region of the gradient. Thus, the PS1 immunoreactivity in the low density region (fractions 4 and 5) of the gradient cannot be explained by contaminating RER membranes. Furthermore, we quantified all marker proteins by densitometric scanning. When the results were expressed as density units per milligram of protein (Fig. 3 c, bottom), it became obvious that PS1 fragments specifically coenriched with ERGIC-53/p58 rather than with calnexin, suggesting their localization in the IC. Moreover, the enrichment factor for ERGIC-53/p58 obtained in the low density peak fraction was 35 compared with total homogenate, reaching a similar degree of enrichment as originally described for VERO cells (Schweizer et al. 1991).
Figure 3.

Analysis of PS1 in enriched fractions of the IC isolated from cortices of 5–7-d-old mice (adapted from Schweizer et al. 1991). (a) A postnuclear supernatant (PNS) was mixed with Percoll to give a final density of 1.129 g/ml and centrifuged (at 37,000 g max for 41 min). Twenty-two fractions of 1.5 ml were collected and analyzed by SDS-PAGE and Western blotting (equal amount of fraction volume). PS1 immunoreactivity was distributed bimodally with most of the immunoreactivity recovered in fractions 15–19. (b) Fractions 5–14 were pooled (Pool I), adjusted to 30% (wt/wt) Nycodenz, and overlaid with 27% and 18.5% (wt/wt) Nycodenz. After isopycnic centrifugation (85,800 g max for 20 h), three interfaces were collected (F1, F2, and F3). The 18.5–27% interface F3 is highly enriched in marker proteins for the intermediate compartment (IC), such as p58/ERGIC-53. Comparable enrichments were found for both PS1-NTF and -CTF. Equal amounts of protein were analyzed by SDS-PAGE and Western blotting. For PS1-NTF, an additional long exposure of the same blot is shown to reveal the full-length PS1. Notice that the F3 fraction is still contaminated with membranes derived from the RER as demonstrated by two specific marker proteins, rSEC61αp and TRAPα. (c) The interface fraction F3 was diluted twofold with buffer C (Schweizer et al. 1991) and loaded on a continuous sucrose gradient (0.8–1.6 M sucrose in buffer C). After isopycnic gradient centrifugation (SW41 rotor, at 250,000 g max for 4 h), 12 fractions were collected from the top and analyzed by Western blotting. Calnexin, ERGIC-53/p58 and PS1 fragments all display a bimodal distribution, whereas TRAPα and rSEC61αp are only distributed in the second half of the sucrose gradient. The bottom panel shows the distribution of the different marker proteins expressed per milligram of protein in each fraction (arbitrary units/mg protein). It should be noted that at low sucrose densities (first half of the gradient), both PS1 fragments clearly coenrich with the first pool of ERGIC-53/p58–positive membranes, whereas no clear enrichment is observed for calnexin. The data represent one out of two independent experiments.
From the initial Percoll gradient (Fig. 3 a), a major peak of PS1 immunoreactivity (Pool II) can be observed corresponding to less dense regions where Golgi-derived membranes tend to equilibrate (Schweizer et al. 1991). To analyze whether this reflected an association of PS1 with Golgi membranes, we further purified the pooled fractions 15–19 using discontinuous Nycodenz gradient centrifugation consisting of three layers of different Nycodenz concentrations. Most of the PS1 immunoreactivity was recovered in the 14–21% interface (IF3) together with marker proteins of the Golgi apparatus (such as GM130). This interface IF3 was loaded on a continuous sucrose gradient (0.3–1.7 M sucrose) and the distribution of PS1 was analyzed after ultracentrifugation (Fig. 4 A). Established Golgi marker proteins such as GM130 (Fig. 4 A) and β-cop (not shown) equilibrated at high sucrose densities in fractions 4 and 5, whereas the bulk amount of PS1-CTF and -NTF immunoreactivity was recovered at a lower buoyant density with a peak in fractions 7 and 8. The striking similar distributions of the unglycosylated form of TGN38 (Fig. 4 A, M r 45,000) and RAP with the PS1-CTF and -NTF indicate that the PS1 in this pool is mainly associated with early cis-Golgi membranes. Indeed, unglycosylated TGN38 is located in the early cis-Golgi, whereas RAP is a molecular chaperone assisting membrane proteins such as the LDL receptor in their passage through the early secretory pathway between the ER and Golgi (Bu and Schwartz 1998). Since the IC resident protein p58/ERGIC-53 did not codistribute with fractions 7 and 8, these fractions may identify a membrane-bound intermediate between the IC and cis-Golgi. Mature (glycosylated) TGN38, which is a marker for the TGN tubules, is mainly found in the bottom fraction (Fig. 4 A, fraction 1). Since this fraction is completely devoid of PS1 immunoreactivity, we can assume that PS1 does not reach the TGN in significant amounts.
Figure 4.
Analysis of PS1 immunoreactivity in the Golgi apparatus by cell fractionation (A) and confocal microscopy (B). (A) Fractions 15–19 of the Percoll gradient (see Fig. 3 a) were pooled (Pool II), adjusted to 25% (wt/wt) Nycodenz, and overlaid with 21, 14, and 10% (wt/wt) Nycodenz. After isopycnic centrifugation (SW41 rotor; at 85,800 g max for 20 h), three interfaces were collected (IF1, IF2, and IF3, not shown). Interface IF3, containing most of the PS1 immunoreactivity (not shown) was diluted twofold with buffer C (Schweizer et al. 1991) and further fractionated by continuous sucrose density gradient centrifugation (0.3–1.7 M sucrose, at 250,000 g max for 2 h). Fractions were collected from the bottom and an equal volume of each fraction was analyzed. The peak immunoreactivity for PS1-NTF and -CTF was slightly shifted to less dense regions of the gradient, thereby codistributing with BAP31, RAP, and immature TGN38. Mature TGN38 immunoreactivity was mainly recovered in the bottom fraction of the gradient. GM130, a Golgi marker, is distributed in the high density region of the gradient where no peak of PS1 immunoreactivity was observed. (B) PS1 does not distribute extensively into the Golgi apparatus. Fully polarized neurons isolated from control, nontransgene embryos were double stained for PS1 (pAb B17.2; red) and p58/Golgi (green). Detection was performed with CY3 anti-rabbit and CY2 anti-mouse secondary antibodies, respectively. In contrast to PS1 labeling (b), Golgi staining (a) is restricted to the soma and proximal part of the dendrites as can be seen in the overview of a neuron (a–c, arrow). (d–f) Enlarged area of the proximal dendrite in the same neuron stained for PS1 (red) and p58/Golgi (green). The asterisk indicates regions in the cell body with a relative low level of colocalization. Arrowheads point to areas of codistribution. (g–i) Section in the vertical plane of the neuron at the position in the proximal dendrite indicated by the arrow in (d–f). Only a partial overlap is observed and in many cases, PS1 immunoreactivity is very closely juxtaposed to p58/Golgi staining (arrowheads in g–i). c, f, and i are the merged signals. PS1 and Golgi stainings are mainly not overlapping. Bars, 10 μm.


This observation was entirely corroborated by confocal laser microscopy of control hippocampal neurons. Antibodies against the peripherally associated 58-kD Golgi protein (Fig. 4 B, p58/Golgi), detected Golgi staining restricted to the cell body and proximal dendrites of neurons (Fig. 4 B, a–c) in agreement with Krijnse-Locker et al. 1995. At first glance (Fig. 4 B, c), a high degree of overlap with the immunostaining for PS1-CTF seemed to be observed. However, when a more careful analysis was performed at a higher magnification (Fig. 4 B, d–f), only a limited overlap could be demonstrated (Fig. 4 B, d–f, asterisk and arrowhead). This becomes even more clear in the vertical plane section of a proximal dendrite (Fig. 4 B, g–i; the arrow in d–f indicates the site of the section). PS1-CTF staining was concentrated in defined stacks often capped by or juxtaposed to staining for p58/Golgi (Fig. 4 B, g–i, arrowheads). Both proteins colocalized only partially in discrete areas. This is in line with the fractionation data.
α- and β-secretase cleavage of APP occurs in the late Golgi, in transport vesicles, at the cell surface, and in endocytic compartments (Selkoe 1998), generating the α- and β-APP carboxy-terminal stubs associated with the membranes. These stubs are the direct substrates for γ-secretase. Given the restricted distribution of PS1 in the ER, IC, and early cis-Golgi compartments, the question arises how the γ-secretase cleavage of these APP stubs can be controlled by PS1.
Therefore, we analyzed the distribution of endogenous APP in the same fractions analyzed before for presenilin immunoreactivity. As shown in Fig. 5 a, both the gradient distributions of the APP holoprotein and the APP carboxy-terminal fragments were bimodal, which is similar to what was found for PS1 (Fig. 3 a). The larger amounts are found in the fractions of Pool II. Further purification resulted in the concomitant enrichment for the APP carboxy-terminal fragments in the F3 interface (Fig. 5 b), where PS1 is also recovered. Remarkably, this enrichment was much less obvious for the APP holoprotein, suggesting a selective accumulation of the carboxy-terminal APP fragments in this fraction. Further analysis of the Pool II–associated APP immunoreactivity revealed the enrichment of the APP carboxy-terminal fragments in exactly the same fractions of the sucrose gradient where PS1 fragments (together with RAP) are recovered (compare Fig. 5 c with Fig. 4 A). Interestingly, therefore, the carboxy-terminal fragments of APP are apparently at least partially codistributing with PS1-NTF and -CTF in the same subcellular membrane compartments (see Discussion). Given the existing controversy whether or not PS1 can be found in compartments beyond the ER/IC and cis-Golgi, we extended our study to some well-defined post-Golgi compartments such as lysosomes, endosomes, synaptic vesicles, the cell surface, and clathrin-coated vesicles. Both PS1 fragments clearly purify away from mature and intact lysosomes (Fig. 6, compare with the enrichment for cathepsin D), and were observed only after prolonged exposure times. The weak immunoreactivity in the pure lysosomal fractions is most likely a consequence of minor contamination of the isolated fraction.
Figure 5.

Analysis of the distribution of APP holoprotein and carboxy-terminal fragments in ER- and IC-enriched fractions isolated from cortices of 5–7-d-old mice using the same fractionation protocol as described in Fig. 3, a and b, and Fig. 4 (A). (a) Distribution of APP after Percoll gradient centrifugation of a postnuclear fraction. Like for PS1, both APP and APP stubs display a bimodal distribution with the major amount of immunoreactivity in fractions 15–20 of the gradient. (b) APP carboxy-terminal fragments enrich in the interface F3. (c) APP fragments could be further enriched after Nycodenz and sucrose gradient centrifugation of Pool II from the Percoll gradient (a). The distribution of APP carboxy-terminal fragments is identical to that of PS1-NTF and -CTF (Fig. 4 A).
Figure 6.

Purification of lysosomes from cortices of 5–7-d-old mice. Highly purified lysosomes were obtained by a combination of low speed centrifugation in 0.25 M KCl and velocity gradient centrifugation in Percoll according to the method of Maguire and Luzio 1985. The final lysosomal fraction (LYS) is clearly enriched in cathepsin D. Hardly any PS1-NTF or -CTF immunoreactivity was detected in this fraction.
We also investigated the possible colocalization of CTF-PS1 with marker proteins of post-Golgi compartments such as the transferrin receptor and synaptobrevin II in hippocampal neurons of control, nontransgenic mice (Fig. 7). In fully polarized neurons, the transferrin receptor is localized somatodendritically and, accordingly, numerous transferrin receptor–positive punctae are noticed in the dendritic arbor (Fig. 7, a–c, dendrites, and d–f, growth cone). Virtually no overlap is observed for PS1-CTF and transferrin receptor staining, indicating that PS1 is not present in recycling endosomes. Synaptobrevin II is often used as a marker for synaptic vesicles that cluster in specialized axonal regions of mature neurons referred to as synaptic boutons. Again no immunostaining for PS1 could be detected within these boutons (Fig. 7, g–l, with emphasis on the detailed area in g–i), suggesting that no PS1 is associated with the membranes of synaptic vesicles. Further analysis of highly purified clathrin-coated vesicles from nerve terminals, or double immunostaining with specific cell surface marker such as ICAM-5, and surface biotinylation experiments (not shown) did not yield any conclusive evidence that PS1 could be associated with clathrin-coated vesicles or with the cell membrane. (data not shown). It is clear that the amounts of presenilin present in the membranous compartments beyond the ER–cis-Golgi in hippocampal neurons are very limited and, in any event, below the detection limits of our current available technology. In conclusion, the data obtained from both cell fractionation and immunofluorescence studies on brain and neurons, demonstrate the abundant association of PS1 with the ER, IC, and to a lesser extent, the early cis-Golgi.
Figure 7.

PS1 is not localized in recycling endosomes and synaptic vesicles of hippocampal neurons derived from control, nontransgene embryos. (a–f) Within the somatodendritic area of 14-d-old hippocampal neurons, PS1 (pAb B17.2; red) is not colocalized with the transferrin receptor (green). A detailed area of distal parts of the dendritic arbor (a–c) and a growth cone (d–f) are shown. (d–i) Double immunofluorescent staining of hippocampal neurons for PS1 (pAb B17.2; red) and the synaptic vesicle marker synaptobrevin II (mAb 69.1, green). Axonal, presynaptic boutons are marked by synaptobrevin II immunoreactive punctae and are devoid of PS1 immunoreactivity, most clearly seen in the enlarged area displaying an axon running parallel with a dendrite while making several synaptic contacts (g–i). PS1 is distributed into the postsynaptic areas. PS1 was stained with CY3 anti-rabbit, the transferrin receptor and synaptobrevin II with CY2 anti-mouse secondary antibodies. Bars, 10 μm.
This prompted us to further investigate the relationship between APP processing and presenilin at a more functional level. Therefore, we generated a series of APP trafficking mutants that limit APP processing to specific subcellular compartments. A dilysine motif was added to the carboxy terminus of APP to retain APP/KK in pre-Golgi compartments, thereby emphasizing the βA4(1-42) production (Chyung et al. 1997). Reinternalization of cell surface APP was blocked by deleting the cytoplasmic tail (APPΔCT), limiting APP trafficking to the biosynthetic pathway (Tienari et al. 1996b), and severely impairing βA4 secretion (Perez et al. 1999). A chimerical APP/LDLR was generated by replacing the cytoplasmic domain of APP by that of the LDL receptor. This mutation increases the recycling of APP in the endosomal compartments and promotes βA4(1-40) production (see below). We also investigated the effects of the clinical APP/London mutation in combination with the PS1 mutations, to see whether these mutations operate additively or not. The APP constructs were expressed in neurons using the SFV expression system. Similar expression levels for all constructs were obtained in hippocampal cultures, except for APPΔCT (Fig. 8, CELL). For comparisons between the different experiments, the obtained phosphorimaging results were normalized to the expression levels of APP holoprotein in the cell extracts. Expression of APPΔCT led to decreased βA4 secretion concomitantly with the production of a short cell-associated β-stub. In the case of APP/KK infection, the decrease of secreted βA4 was even more pronounced (also see below). The APP/KK mutant is actively retrieved from the cis-Golgi to the ER as a consequence of the dilysine motif. This was confirmed by its complete endo-H glycosidase sensitivity (data not shown) and its codistribution with calnexin (Fig. 9 b) and endogenous PS1 (Fig. 9 c). The striking colocalization of endogenous PS1 with virally expressed APP/KK (Fig. 9 c, inset) should be noticed. As a control, SFV-APPwt is shown, which confirms the axonal delivery of this protein (Fig. 9 a) (Simons et al. 1995; Yamazaki et al. 1995; Tienari et al. 1996b). We analyzed systematically the metabolism of the expressed APP mutants (Table ). Quantitative immunoprecipitation and phosphorimaging determined the total amounts of cell-associated α- and β-stubs and secreted βA4. To standardize and to allow comparisons between different experiments, we normalized all values to cell-associated APP holoprotein. Similarly, normalization of the numbers obtained in the ELISA experiments was performed because of the relative large variations in the absolute amounts of peptide produced in separate independent experiments. Therefore, only ratios of secreted and intracellular βA4(1-42)/(1-40) are given. Except for APP/LDLR, all constructs were completely analyzed in the three types of neuronal culture, i.e., expressing human PS1 wt or the clinical mutants PS1L286V or PS1M146L. The results were compared with those obtained in cells derived from control and nontransgenic littermate embryos. Table summarizes the data obtained for APPwt (A), the London mutant (B), APPΔCT (C), and APP/KK (D).
Figure 8.

Processing of human wild-type APP (APPwt), the London mutant (APP-Lon), and the APP sorting mutants, APPΔCT, APP/KK, and APP/LDLR in SFV-infected hippocampal neurons. Immunoprecipitation of total βA4 from the culture supernatant (MEDIUM) and carboxy-terminal β-stubs and total βA4 from the cell lysates (CELL) of metabolically labeled hippocampal neurons using antibody B7/7 that recognizes an epitope on the amino terminus of the βA4 sequence. Immunoprecipitates were electrophoresed on 10–20% Tris-Tricine gels and analyzed by phosphorimaging. Lower amounts of secreted βA4 are recovered in the supernatants of cultures infected with APPΔCT and APP/KK as compared with the other virus constructs. Notice the higher expression of APPΔCT in the cell extracts. As expected, infection with the SFV-APPΔCT construct resulted in a shorter carboxy-terminal β-stub. The contribution of carboxyl-terminal fragments and βA4 from endogenous APP processing is neglectable as it is below detection level in untransfected cultures (No Virus). Antibody B7/7 used for the βA4 immunoprecipitation experiments reacts also with low affinity with APP-ectodomain, recovered in low quantities from the conditioned media (APPs).
Figure 9.
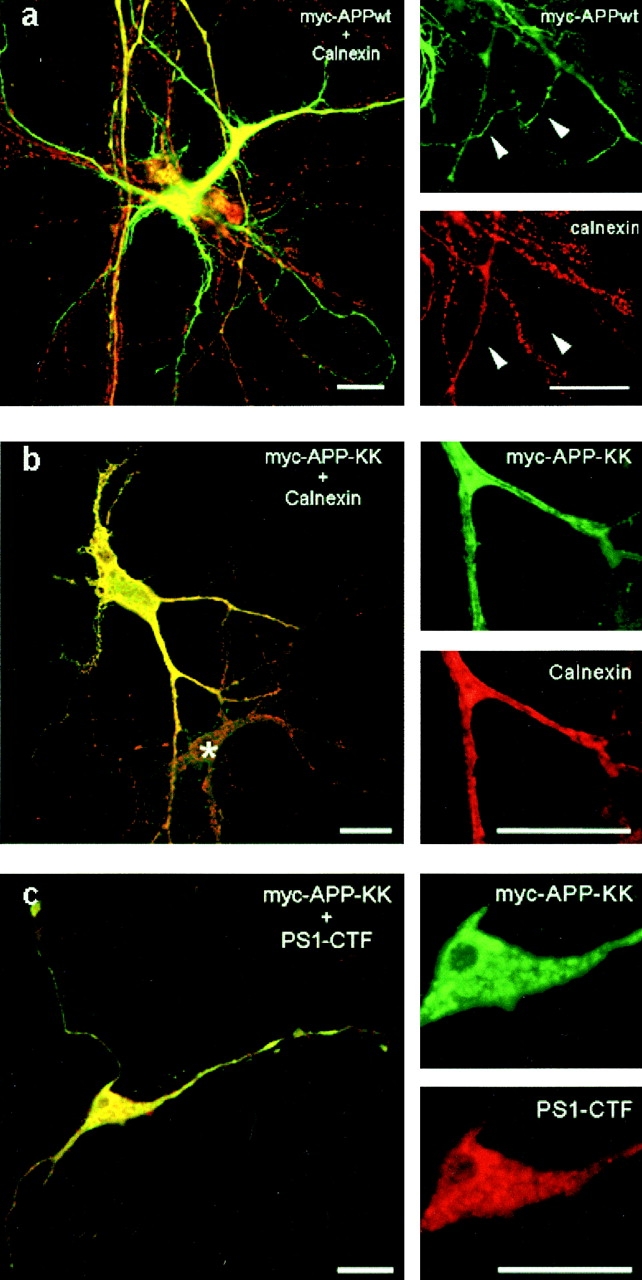
Newly synthesized APP wild-type, but not APP/KK, is sorted to axons in fully polarized hippocampal neurons. (a) Double immunofluorescent staining for APPwt (green) and calnexin (red). Newly expressed APP was detected using a myc antibody. Infection with the SFV-APPwt clearly resulted in axonal localization of the expressed protein. (b) Newly expressed APP/KK was entirely retained in the somatodendritic ER as demonstrated by the virtual complete colocalization with calnexin. APP and calnexin labeling were detected with CY2 anti-mouse and lissamine rhodamine anti-rabbit secondary antibodies, respectively. Asterisk marks a nontransfected neuron. (c) Endogenous PS1 resides in membrane-bound compartments involved in the trafficking and retrieval of dilysine-tagged proteins. The newly expressed APP/KK (green) colocalizes with endogenous PS1 (red) in polarized hippocampal neurons. The colocalization was most obvious in the somatic area (inset). Detection of primary antibodies was with CY2 anti-mouse (APP) and CY3 anti-rabbit (PS1-CTF) secondary antibodies. Bars, 20 μm.
Table 1.
Processing of APP in Hippocampal Neurons
| ↓Transgene | α-stub/APPcell | β-stub/APPcell | Secreted βA4/APPcell | Secreted 1-42/1-40 | Intracell. 1-42/1-40 |
|---|---|---|---|---|---|
| A. SFV-APP wild-type | |||||
| Control | 1.00 ± 0.04 (17) | 1.00 ± 0.04 (17) | 1.00 ± 0.04 (18) | 1.00 ± 0.02 (17) | 1.00 ± 0.05 (17) |
| PS1wt | 0.86 ± 0.04 (14) | 0.91 ± 0.03 (12) | 0.99 ± 0.07 (15) | 0.90 ± 0.17 (4) | 1.37 ± 0.20 (5) |
| PS1L286V | 1.02 ± 0.13 (4) | 0.84 ± 0.08 (5) | 0.89 ± 0.08 (6) | 1.30 ± 0.10 (6) | 1.13 ± 0.20 (6) |
| PS1M146L | 1.08 ± 0.06 (6) | 1.05 ± 0.08 (6) | 1.27 ± 0.13 (6) | 1.95 ± 0.13 (8) | 0.98 ± 0.11 (8) |
| B. SFV-APP London | |||||
| Control | 1.00 ± 0.07 (13) | 1.00 ± 0.06 (13) | 1.00 ± 0.05 (13) | 1.00 ± 0.03 (10) | 1.00 ± 0.08 (11) |
| PS1wt | 0.84 ± 0.09 (11) | 0.85 ± 0.10 (11) | 0.87 ± 0.11 (11) | 0.91 ± 0.08 (5) | 1.38 ± 0.18 (3) |
| PS1L286V | 0.64 ± 0.09 (8) | 0.74 ± 0.10 (8) | 1.09 ± 0.07 (8) | 1.38 ± 0.07 (8) | 1.44 ± 0.19 (8) |
| PS1M146L | 0.58 ± 0.08 (5) | 0.75 ± 0.12 (5) | 1.02 ± 0.34 (5) | 2.97 ± 0.65 (3) | 1.14 ± 0.04 (3) |
| C. SFV-APPΔCT | |||||
| Control | n.d. | 1.00 ± 0.08 (9) | 1.00 ± 0.06 (9) | 1.00 ± 0.05 (17) | 1.00 ± 0.05 (17) |
| PS1wt | n.d. | 1.68 ± 0.16 (3) | 1.22 ± 0.07 (3) | 1.13 ± 0.07 (3) | 1.23 ± 0.12 (3) |
| PS1L286V | n.d. | 1.04 ± 0.14 (3) | 0.93 ± 0.12 (3) | 1.24 ± 0.04 (7) | 1.11 ± 0.12 (7) |
| PS1M146L | n.d. | 0.89 ± 0.06 (6) | 1.16 ± 0.11 (6) | 1.55 ± 0.11 (7) | 1.05 ± 0.13 (7) |
| D. SFV-APP/KK | |||||
| Control | 1.00 ± 0.03 (7) | 1.00 ± 0.04 (7) | 1.00 ± 0.10 (7) | 1.00 ± 0.11 (9) | 1.00 ± 0.11 (9) |
| PS1wt | 0.96 ± 0.06 (7) | 0.88 ± 0.05 (7) | 0.92 ± 0.10 (3) | n.d. | n.d. |
| PS1L286V | 0.98 ± 0.04 (3) | 0.83 ± 0.04 (3) | 1.03 ± 0.04 (3) | 1.09 ± 0.08 (3) | 0.58 ± 0.06 (3) |
| PS1M146L | 1.01 ± 0.04 (4) | 0.92 ± 0.03 (4) | 1.90 ± 0.29 (4) | 2.83 ± 0.57 (6) | 1.07 ± 0.07 (6) |
Analysis of the processing of APP in hippocampal neurons with Semliki Forest virus expressing human APP wild-type (A), APP-London mutation (B), carboxy-terminal–truncated mice APP (APPΔCT, C) or APP/KK (D). Primary neuronal cultures are derived from transgenic mice expressing human PS1 wild-type (PS1wt) or a clinical mutation of PS1 (PS1L286V and PS1M146L).
The data represent the ratios of the α- and β-secretase cleaved carboxy-terminal fragments and the total secreted βA4 amyloid peptides to the APP-holoprotein in the cell extracts, as well as the ratios of secreted and intracellular βA4 1-42/βA4 1-40 amyloid peptides. All ratios are normalized to the ratios found in control, nontransgenic cultures (control values in the first line of each table). The number between brackets denotes the number of cultures analyzed for each condition. Data are presented as mean ± S.E.M. n.d.: not determined. For table A–D, numerical values in each column were further statistically analyzed by ANOVA and t test.
With respect to α- and β-cleaved carboxy-terminal fragments, no significant differences were noticed related to PS1 wild-type or clinical mutants except for the London mutant. The clinical mutations in PS1 do not affect α- and β-secretase activity, complementing the data obtained previously in PS1-deficient neurons (De Strooper et al. 1998). However, in the case of the London mutant, levels of α- and β-stubs were surprisingly decreased in the FAD mutant PS1 neurons. This reflects probably the increased turnover of both stubs by γ-secretase cleavage, in accordance with the strong additive effects of the London- and FAD-linked PS1 mutations (especially PS1M146L) on γ-secretase processing (see below).
To evaluate γ-secretase activities, we measured the total pool of secreted βA4 by immune precipitation as well as the ratio of the individual peptides by ELISA. A modest increase in the total amount of secreted βA4 was observed for the PS1M146L mutant. This effect was not very dramatic (except for the APP/KK, see below). In contrast, PS1 mutations caused dramatic increases in the βA4(1-42) to βA4(1-40) peptide ratio, and this effect was even more pronounced when the effect of the mild overexpression of PS1wt alone was directly compared with that of PS1 containing clinical mutations. The PS1M146L mutation was overall more effective than the PS1L286V mutation.
The London mutation on its own caused a threefold increase (3.44±0.43; n = 6) in the ratio of secreted βA4(1-42)/(1-40) versus APP wild-type in control neurons (not shown). Interestingly, an additional rise in this ratio was observed in combination with PS1 mutations (Table B). This rise ranged from 1.38±0.07 (n = 8) for the PS1L286V mutation to 2.97±0.65 (n = 3) for the PS1M146L mutation. When compared with APPwt expressed in wild-type neurons, the total raise ranged up to 12.3 ± 3.1 (n = 3). The increase appears to be solely due to an increased secretion of the β4A(1-42) peptide and not a consequence of a decreased βA4(1-40) production, explaining the decrease in α- and β-carboxy-terminal fragments as mentioned above.
APPΔCT in control neurons yielded low levels of secreted βA4. However, the ratio between the two peptides was slightly raised compared with SFV-APPwt (1.30±0.14, n = 8, versus SFV-APPwt). This ratio increased even further when this mutant was expressed in PS1M146L neurons (Table C). For APP/KK, the total secretion of βA4(1-40) was also lowered dramatically to 5.0±1.6% (n = 6) when compared with APPwt (see also Fig. 8). The βA4(1-42) secretion was not affected as strongly as the βA4(1-40) secretion and only dropped to 18.0±4.7% (n = 6) when compared with APPwt. The introduction of the ER-retrieving dilysine motif on its own, therefore, caused an appreciable increase in the 42:40 ratio of the secreted peptides to 3.98±0.70 (n = 6) times relative to APPwt. Upon expression in PS1M146L neurons, an additional raise in this ratio of 2.83±0.57 (n = 6, Table D) was observed. The increase could again solely be contributed to an increased secretion of the βA4(1-42) peptide. Interestingly, only for this construct, a sharp rise in total secreted βA4 was noticed in PS1M146L neurons. It should be noticed that the main peptide produced by APP/KK is βA4(1-42). This observation indicates that some of the presenilin mutations do not only affect the ratio of the secreted peptides, but can also increase the total production of βA4(1-42). The results obtained with this ER-retained APP/KK mutant also indicate that the clinical PS mutations are operating in the ER. This conclusion was further corroborated in an experiment comparing the amyloid peptide production from APP/LDLR in control and PS1M146L hippocampal neurons. As expected for a mutant trafficking mainly in the endocytic compartments, a relative increase in secreted βA4(1-40) of 2.36±0.18 (n = 3, SEM) fold was observed, whereas secretion of βA4(1-42) remained largely unaffected. Interestingly, the PS1M146L mutation did not cause a statistically significant effect on the ratio of secreted βA4 peptides in these neurons. Finally, we note that in our experimental system, and for all APP constructs studied, PS1 clinical mutations do not cause a significant increase in the βA4(1-42)/(1-40) ratio of intracellular peptides.
Discussion
In this study, we provide conclusive evidence that the bulk of endogenously expressed PS1 in neurons is localized in the early compartments of the secretory pathway, i.e., in the ER, the IC, and the early Golgi. Relatively limited levels of PS1 are furthermore detected in the nuclear envelope and the RER. The ER, in contrast to the Golgi apparatus, is widely distributed in the soma and the dendrites (including the most distal regions) of neurons (Krijnse-Locker et al. 1995). Therefore, our data explain the previously documented somatodendritic localization of the presenilins (Cook et al. 1996; Busciglio et al. 1997; Capell et al. 1997). Within these early compartments of the biosynthetic pathway, a clear gradient of increasing amounts of the supposedly functional PS1 fragments is observed. The PS1 immunoreactivity previously detected in the nuclear membrane (Li et al. 1997) is, therefore, a mixture of (predominantly) PS1 fragments and full-length PS1, indicating that cleavage of PS1 by the unknown presenilinase occurs very early after biosynthesis (Xia et al. 1998). Importantly, these results definitively resolve the controversy whether and where full-length presenilin can be detected, namely in nuclear envelope fractions.
Interestingly, both the PS1-NTF and -CTF codistributed and coenriched in all fractionation schemes applied, confirming at the endogenous level of protein expression that both fragments remain in a 1:1 stoichiometric relationship along the whole early biosynthetic pathway (Thinakaran et al. 1996). Our data not only settle the debate on the subcellular localization of presenilin, but also document for the first time that PS1 immunoreactivity in the ER is concentrated in discrete patches that are sec23p-positive. Sec23p is one out of the five cytosolic proteins that constitute the COPII coat complex required for vesicle budding from the ER (Barlowe et al. 1994). These COPII components bind specifically with cargo molecules destined for sorting into transport vesicles and separate them from the ER resident proteins (Campbell and Schekman 1997; Aridor et al. 1998; Kuehn et al. 1998). The formation of COPII transport vesicles occurs nonrandomly in the ER at tubular protrusions often juxtaposed to VTC, referred to as the IC (Schweizer et al. 1990, Schweizer et al. 1991; Bannykh and Balch 1998). PS1 codistributes and coenriches with markers of this intriguing compartment such as p58/ERGIC-53 (Saraste and Svensson 1991; Schweizer et al. 1991), suggesting that PS1 is operating in these compartments. A similar conclusion was recently made by Culvenor et al. 1997 who, based on temperature block experiments, suggested that PS1 is localized in the IC and cis-Golgi in stably transfected SY5Y and P19 cell lines. On the other hand, and in contrast with many earlier studies investigating overexpressed PS1 (for instance De Strooper et al. 1997), we could not confirm that PS1 at the endogenous level of expression distributes to a significant extent into the Golgi apparatus. This limits the subcellular compartment where PS1 is operating to the early compartments of the biosynthetic pathway. The earlier reported abundant Golgi localization of the presenilins is probably a consequence of overexpression and missorting of the protein. As illustrated in Fig. 4 B, f and i, colocalization studies should indeed be carefully interpreted. As we suggested before (De Strooper et al. 1997), experiments based on overexpression of presenilins may lead to erroneous conclusions since normal proteolytic maturation and also normal subcellular ER localization is apparently significantly disturbed.
This conclusion is further corroborated by recent findings that overexpression of presenilins results in their redistribution into aggresomes (Johnston et al. 1998). These structures are located at the perinuclear microtubule organizing center, where also the Golgi apparatus is located and are the signature of abnormal biosynthetic stress in the cell. In view of our new data, it is conceivable that PS1 passes from the ER, via VTCs, to cis-Golgi cisternae. The close colocalization of PS1 with sec23p further indicates that at least part of this transport is associated with the trafficking of COPII vesicles. (Schekman and Orci 1996). Since PS1 fragments exist as a stable complex with a long half-life (Ratovitski et al. 1997) and no accumulation in the Golgi apparatus was observed, PS1 is probably also retrogradely transported. Electron microscopical observations demonstrating abundant PS1 immunoreactivity in vesiculotubular structures and coated vesicles in close vicinity or in continuity with Golgi cisternae support the view that presenilins are actively transported back and forward between the ER and cis-Golgi (Lah et al. 1997). The lack of PS1 immunoreactivity in TGN-enriched fractions (Fig. 4 A) suggests that only minimal amounts of PS1, if any, exit the Golgi area. Indeed, no significant amounts of PS1 were found in recycling endosomes, lysosomes, synaptic vesicles, or clathrin-coated vesicles (not shown and Efthimiopoulos et al. 1998). Finally, all published data that support the localization of PS1 at the plasma membrane (Dewji and Singer 1997; Schwarzman et al. 1999) imply a topology of PS1 with the amino-terminal domain at the luminal side of the ER. This is not in agreement with several studies (Doan et al. 1996; Li and Greenwald 1996; De Strooper et al. 1997; Lehmann et al. 1997) that addressed this question more specifically, although it could be speculated that cell type–specific differences exist. In any event, our data in hippocampal neurons clearly indicate that the bulk of PS1 fragments are localized in compartments of the early biosynthetic pathway and that only limited amounts (if any at all) traffic beyond the Golgi apparatus, a finding that needs to be taken into account when considering the association of presenilin with γ- secretase activities.
Accumulating evidence indicate that presenilins are either part of the catalytic γ-secretase activity (Wolfe et al. 1999) or at least controlling its activity (De Strooper et al. 1998, De Strooper et al. 1999). Wolfe et al. 1999 presented evidence that presenilins are aspartate proteases that become inactivated by mutating one of the two aspartic acid residues in the sixth or seventh transmembrane region respectively. Overexpression of this mutant PS1 results in dominant negative effects on γ-secretase activity (Steiner et al. 1999; Wolfe et al. 1999). The most simple explanation is that PS1 is γ-secretase itself. This interpretation is consistent with the observation that deletion of the PS1 gene also results in the inhibition of γ-secretase (De Strooper et al. 1998, De Strooper et al. 1999). In both experiments, secretion of βA4(1-40) as well as βA4(1-42) peptides was equally affected (De Strooper et al. 1998; Wolfe et al. 1999). The subcellular localization of PS1 in the ER as we demonstrate here, is, however, difficult to reconcile with current concepts that γ-secretase processing of APP mainly occurs in the endocytic limb of the subcellular trafficking pathways.
There is clearly a spatial paradox. The first question to be addressed in this regard is whether the real substrates of γ-secretase, namely the α- and β-secretase cleaved APP carboxy-terminal stubs generally believed to be generated in the late biosynthetic compartments and the endocytic pathways, can indeed reach the subcellular compartments where PS1 resides. Unexpectedly, we could demonstrate the enrichment of the APP carboxy-terminal stubs in the ER/IC subcellular fractions where PS1 resides. From a cell biological point of view, it remains to be explained how this retrograde transport of the APP carboxy-terminal fragments to the ER/IC can occur and which sorting signals are involved, but this finding is at least consistent with the PS1–γ-secretase hypothesis (Wolfe et al. 1999). The second question is whether and how PS1 can affect the generation of amyloid peptide in the endocytic limb of the protein trafficking pathways in the cell. This problem was addressed via a functional approach by expressing a series of APP trafficking mutants in hippocampal neurons using the recombinant SFV system. In previous work, it has been extensively demonstrated that the processing and subcellular trafficking of APP expressed with this system does not differ from that observed with endogenously expressed APP (De Strooper et al. 1995; Simons et al. 1995, Simons et al. 1996; Tienari et al. 1996a,Tienari et al. 1996b; Chyung et al. 1997). Therefore, and in contrast with presenilins, the results obtained after transfection of APP with SFV under the experimental conditions used, can be considered as biologically relevant.
The results (see Table ) obtained with APP retained in the ER (APP/KK), with APP processed in the biosynthetic pathway (APPΔCT) or with APP retrieved in the endocytic pathway (APP/LDLR, not shown), corroborated the conclusion that presenilins operate in the ER/IC. Significant increases in βA4(1-42) secretion were observed with the APP/KK and to a lesser extent with the APPΔCT mutant in neurons expressing PS1M146L.. Such an effect was almost undetectable with the APP/LDL mutant. The latter most likely reflects the inability of the APP/LDLR carboxy-terminal fragments generated by α- or β-secretase to be recycled to compartments where γ-secretase resides. The APP/KK mutant was particular relevant in this regard. The dilysine Golgi–ER retrieval signal is recognized by COPI subunits in proteins such as p58/ERGIC-53 and results in their retrograde retrieval from VTCs and cis-Golgi cisternae to the ER (Cosson and Letourneur 1994; Arar et al. 1995; Lahtinen et al. 1996; Bannykh and Balch 1997). The KK motif limits the trafficking of APP/KK to those compartments where also PS1 resides. Indeed, the subcellular distribution of APP/KK was strikingly similar to that of endogenous PS1 (Fig. 9). As previously demonstrated, only very low amounts of amyloid peptide are recovered in the media with this construct (5% compared with wild-type APP). Since β-secretase is mainly operating beyond the ER/IC (Higaki et al. 1995; Chyung et al. 1997), it is not surprising that only very small amounts of the immediate precursor of βA4, i.e., the β-secretase cleaved carboxy-terminal APP fragment (Paganetti et al. 1996; De Strooper et al. 1998), are generated with APP/KK (Fig. 9). In contrast to the interpretation of Chyung et al. 1997, we do not consider the residual β-secretase activity as evidence for a novel, ER-linked β-secretase activity. Since >95% of the normal β-secretase activity is precluded with this construct, this indicates in fact that β-secretase is operating mainly beyond the cis-Golgi, and that APP/KK is only marginally processed by enzyme trailing in the cis-Golgi. This interpretation is certainly more in line with general concepts of β-secretase activity in other cell types since shown to depend on the activity of Rab1B (Dugan et al. 1995) and on O-glycosylation (Tomita et al. 1998). In any event, the residual generation of β-secretase cleaved fragments obtained with APP/KK was sufficient to demonstrate that mainly 1-42 peptide is generated in ER, IC, and early cis-Golgi, where the APP/KK carboxy-terminal stub is recycling. More importantly, the presenilin mutants have a pronounced effect on this process, which is clearly consistent with their subcellular localization.
On the other hand, γ40-secretase processing in the endocytic pathways is not at all influenced by the PS1 mutations as was most dramatically illustrated with the APP/LDLR mutant. The processing of this mutant, which selectively increases the production and secretion of the β4A(1-40) peptide (this study), was not altered by coexpression of the PS1M146L. It is generally accepted that the bulk of the βA4(1-40) peptide is generated beyond the TGN (Cook et al. 1997; Hartmann et al. 1997) and in the endocytic pathways (Koo and Squazzo 1994; Perez et al. 1996, Perez et al. 1999; Peraus et al. 1997; Wild-Bode et al. 1997; Greenfield et al. 1999) compartments where no PS1 is found (this study). This explains why PS1 mutations have no major effects on the secretion of βA4(1-40). Further evidence for the selective effect of the presenilin mutations on γ-secretase cleavage at position 42 was obtained when coexpressing APP-London together with mutant PS1 (Borchelt et al. 1997; Citron et al. 1998; this study). Both clinical mutations increase the βA4(1-42) secretion and act additively, which suggests independent or at least synergistic mechanisms operating with the two mutations. The experimental evidence presented here together with many reports in the literature suggests strongly an exclusive role for the presenilins in γ42-cleavage (as opposed to γ40 cleavage). The effect of the inactivation of PS1 (De Strooper et al. 1998) or the expression of dominant negative forms of PS1 (Wolfe et al. 1999) on γ40-secretase activity must, therefore, be indirect. One possibility is a precursor–product relationship between βA4(1-42) and βA4(1-40). In this view, only one γ-secretase is needed that generates βA4(1-42), whereas in compartments distal to the ER, carboxypeptidases convert this peptide to βA4(1-40). This would explain the above-mentioned spatial paradox, but not completely the selective increased secretion of βA4(1-42) linked to PS1 clinical mutations. In a precursor–product relationship, one should also expect a rise in the secretion of βA4(1-40), and this has not been really observed yet. Along similar lines, the fact that the APP/LDLR or APPΔCT mutations that affect APP trafficking in the endocytic compartments have a direct effect on βA4(1-40) production and secretion, support the general importance of this pathway in APP processing, and imply the existence of a γ40-secretase activity in the endocytic limb. Therefore, it is clear that further investigations are needed to explain the relationship between PS1 and γ40-secretase activity.
Unexpectedly, blocking the processing of APP in the endocytic pathway also decreases secretion of βA4(1-42) (this study; Perez et al. 1999), whereas stimulating the endocytic recycling (APP/LDLR) selectively promotes βA4(1-40) secretion. A recent study provided evidence that the cytoplasmic tail of APP contains, in addition to endocytic signals, information that influences its metabolism in the exocytotic limb (Perez et al. 1999).
Apart from the spatial paradox, it remains to be explained why mainly the immature, N-glycosylated form of APP holoprotein has been found to interact physically with PS1 (Weidemann et al. 1997; Xia et al. 1997). One would predict a preferential association with the carboxy-terminal APP fragments if PS1 was indeed γ-secretase. It could be speculated that PS1 is involved in a posttranslational modification of the newly synthesized APP, tagging it for later recognition by the γ-secretases, although any direct evidence supporting such a possibility is lacking. Other hypotheses have been proposed as well. PS1 might control trafficking of APP to its processing compartments, or alternatively, control trafficking of the γ-secretases to APP-bearing compartments. For instance, the subtle effects on the maturation of TrkB and on BDNF-inducible TrkB autophosphorylation caused by PS1 deficiency in neurons was interpreted to reflect such a function (Naruse et al. 1998). Our findings that endogenous PS1 colocalizes with marker proteins of the IC and ER, would agree with a recruiting or activating function for PS1, either as a chaperone or an adaptor molecule bringing γ-secretase, APP carboxy-terminal fragments, and maybe other substrates (De Strooper et al. 1999) together in the right subcellular microenvironment needed for the controlled and limited proteolytic cleavage of the transmembrane domain.
In conclusion, our data indicate clearly a direct role for PS1 in γ42-secretase activity in the early compartments of the biosynthetic pathway. They are compatible with the hypothesis that PS1 is γ42-secretase, and imply that γ42-secretase exerts its activity in close association with transport processes between ER and the cis-Golgi. However, some caution with this straightforward interpretation remains indicated, since several other observations remain difficult to integrate with this concept, most notoriously the relationship between PS1 deficiency and γ40-secretase processing. Therefore, the alternative possibilities discussed above have to be further explored, and further research should clarify the exact molecular relationship between PS1 and γ40- and γ42-secretase activity.
Acknowledgments
The authors wish to thank E. Hartmann, A. Helenius, R. Jahn, J.-P. Paccaud, M. Reth, J. Saraste, I. Trowbridge, and G. Warren for their gifts of polyclonal and monoclonal antibodies. We thank Dr. Philippe Cupers for stimulating discussions, Vincent Schrijvers for his excellent technical support, and Prof. Dr. Fred Van Leuven for facilities and inimitable hospitality. Lili Zhang (Department of CNS/Cardiovascular Research, Schering-Plough Research Institute, NJ) is greatly acknowledged for processing all βA4-ELISAs.
W.G. Annaert is a postdoctoral fellow and B. De Strooper is a group leader of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (F.W.O.). This work was financially supported by the Flanders Interuniversitary Institute for Biotechnology (VIB), the F.W.O., the I.U.A.P., and the Human Frontier Science Program. D. Westaway, P. Fraser, and P. St. George-Hyslop acknowledge the Medical Research Council of Canada, the Alzheimer Society of Ontario, the Scottish Rite Charitable Foundation, the EJLB Foundation and the Howard Hughes Medical Research Foundation for providing the necessary financial support.
Footnotes
1.used in this paper: βA4, β-amyloid peptide; AD, Alzheimer's disease; APP, amyloid precursor protein; ECL, enhanced chemiluminescence; FAD, familial Alzheimer's disease; IC, intermediate compartment; PS1, presenilin 1; PS1-CTF, PS1 carboxy-terminal fragment; PS1-NTF, PS1 amino-terminal fragment; SFV, Semliki Forest virus; VTC, vesiculotubular complex
References
- Annaert W.G., Becker B., Kistner U., Reth M., Jahn R. Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J. Cell Biol. 1997;139:1397–1410. doi: 10.1083/jcb.139.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arar C., Carpentier V., Le Caer J.P., Monsigny M., Legrand A., Roche A.C. ERGIC-53, a membrane protein of the endoplasmic reticulum-Golgi intermediate compartment, is identical to MR60, an intracellular mannose-specific lectin of myelomonocytic cells. J. Biol. Chem. 1995;270:3551–3553. doi: 10.1074/jbc.270.8.3551. [DOI] [PubMed] [Google Scholar]
- Aridor M., Weissman J., Bannykh S., Nuoffer C., Balch W.E. Cargo selection by the COPII budding machinery during export from the ER. J. Cell Biol. 1998;141:61–70. doi: 10.1083/jcb.141.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannykh S.I., Balch W.E. Membrane dynamics at the endoplasmic reticulum–Golgi interface. J. Cell Biol. 1997;138:1–4. doi: 10.1083/jcb.138.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannykh S.I., Balch W.E. Selective transport of cargo between the endoplasmic reticulum and Golgi compartments. Histochem. Cell Biol. 1998;109:463–475. doi: 10.1007/s004180050248. [DOI] [PubMed] [Google Scholar]
- Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M.F., Ravazzola M., Amherdt M., Schekman R. COPIIa membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Baumeister R., Haass C. Presenilin proteins and their role in development and notch signaling. In: Haass C., editor. Molecular Biology of Alzheimer's Disease. Harwood Academic Publishers; Amsterdam: 1998. pp. 219–234. [Google Scholar]
- Borchelt D.R., Thinakaran G., Eckman C.B., Lee M.K., Davenport F., Ratovitsky T., Prada C.M., Kim G., Seekins S., Yager D. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Borchelt D.R., Ratovitski T., van Lare J., Lee M.K., Gonzales V., Jenkins N.A., Copeland N.G., Price D.L., Sisodia S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Brockhaus M., Grunberg J., Rohrig S., Loetscher H., Wittenburg N., Baumeister R., Jacobsen H., Haass C. Caspase-mediated cleavage is not required for the activity of presenilins in amyloidogenesis and NOTCH signaling. Neuroreport. 1998;9:1481–1486. doi: 10.1097/00001756-199805110-00043. [DOI] [PubMed] [Google Scholar]
- Bu G., Schwartz A.L. RAP, a novel type of ER chaperone. Trends Cell Biol. 1998;8:272–276. doi: 10.1016/s0962-8924(98)01283-5. [DOI] [PubMed] [Google Scholar]
- Busciglio J., Hartmann H., Lorenzo A., Wong C., Baumann K., Sommer B., Staufenbiel M., Yankner B.A. Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J. Neurosci. 1997;17:5101–5107. doi: 10.1523/JNEUROSCI.17-13-05101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.L., Schekman R. Selective packaging of cargo molecules into endoplasmic reticulum-derived COPII vesicles. Proc. Natl. Acad. Sci. USA. 1997;94:837–842. doi: 10.1073/pnas.94.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A., Saffrich R., Olivo J.C., Meyn L., Walter J., Grunberg J., Mathews P., Nixon R., Dotti C., Haass C. Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J. Neurochem. 1997;69:2432–2440. doi: 10.1046/j.1471-4159.1997.69062432.x. [DOI] [PubMed] [Google Scholar]
- Capell A., Grunberg J., Pesold B., Diehlmann A., Citron M., Nixon R., Beyreuther K., Selkoe D.J., Haass C. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J. Biol. Chem. 1998;273:3205–3211. doi: 10.1074/jbc.273.6.3205. [DOI] [PubMed] [Google Scholar]
- Chyung A.S.C., Greenberg B.D., Cook D.G., Doms R.W., Lee V.M. Novel β-secretase cleavage of β-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J. Cell Biol. 1997;138:671–680. doi: 10.1083/jcb.138.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M., Westaway D., Xia W., Carlson G., Diehl T., Levesque G., Johnson-Wood K., Lee M., Seubert P., Davis A. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Citron M., Eckman C.B., Diehl T.S., Corcoran C., Ostaszewski B.L., Xia W., Levesque G., P. St. George Hyslop, S.G. Younkin, and D.J. Selkoe Additive effects of PS1 and APP mutations on secretion of the 42-residue amyloid beta-protein. Neurobiol. Dis. 1998;5:107–116. doi: 10.1006/nbdi.1998.0183. [DOI] [PubMed] [Google Scholar]
- Cook D.G., Sung J.C., Golde T.E., Felsenstein K.M., Wojczyk B.S., Tanzi R.E., Trojanowski J.Q., Lee V.M., Doms R.W. Expression and analysis of presenilin 1 in a human neuronal systemlocalization in cell bodies and dendrites. Proc. Natl. Acad. Sci. USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D.G., Forman M.S., Sung J.C., Leight S., Kolson D.L., Iwatsubo T., Lee V.M., Doms R.W. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Cosson P., Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Culvenor J.G., Maher F., Evin G., Malchiodi-Albedi F., Cappai R., Underwood J.R., Davis J.B., Karran E.H., Roberts G.W., Beyreuther K., Masters C.L. Alzheimer's disease-associated presenilin 1 in neuronal cellsevidence for localization to the endoplasmic reticulum-Golgi intermediate compartment. J. Neurosci. Res. 1997;49:719–731. doi: 10.1002/(SICI)1097-4547(19970915)49:6<719::AID-JNR6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- De Strooper B., Umans L., Van Leuven F., Van Den Berghe H. Study of the synthesis and secretion of normal and artificial mutants of murine amyloid precursor protein (APP)cleavage of APP occurs in a late compartment of the default secretion pathway. J. Cell Biol. 1993;121:295–304. doi: 10.1083/jcb.121.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B., Simons M., Multhaup G., Van Leuven F., Beyreuther K., Dotti C.G. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:4932–4938. doi: 10.1002/j.1460-2075.1995.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B., Beullens M., Contreras B., Levesque L., Craessaerts K., Cordell B., Moechars D., Bollen M., Fraser P., George-Hyslop P.S., Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J. Biol. Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J.S., Schroeter E.H., Schrijvers V., Wolfe M.S., Ray W.J., Goate A., Kopan R. A presenilin-1-dependent, gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Dewji N.N., Singer S.J. Cell surface expression of the Alzheimer disease-related presenilin proteins. Proc. Natl. Acad. Sci. USA. 1997;94:9926–9931. doi: 10.1073/pnas.94.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan A., Thinakaran G., Borchelt D.R., Slunt H.H., Ratovitsky T., Podlisny M., Selkoe D.J., Seeger M., Gandy S.E., Price D.L., Sisodia S.S. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- Duff K., Eckman C., Zehr C., Yu X., Prada C.M., Perez-tur J., Hutton M., Buee L., Harigaya Y., Yager D., Morgan D., Gordon M.N. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Dugan J.M., de Wit C., McConlogue L., Maltese W.A. The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J. Biol. Chem. 1995;270:10982–10989. doi: 10.1074/jbc.270.18.10982. [DOI] [PubMed] [Google Scholar]
- Efthimiopoulos S., Floor E., Georgakopoulos A., Shioi J., Cui W., Yasothornsrikul S., Hook V.Y., Wisniewski T., Buee L., Robakis N.K. Enrichment of presenilin 1 peptides in neuronal large dense-core and somatodendritic clathrin-coated vesicles. J. Neurochem. 1998;71:2365–2372. doi: 10.1046/j.1471-4159.1998.71062365.x. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D., Prehn S., Hartmann E., Kalies K.U., Rapoport T.A. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- Goslin K., Banker G.A. Rat hippocampal neurons in low density culture. In: Banker G.A., Goslin K., editors. Culturing Nerve Cells. MIT Press; Cambridge, MA: 1991. pp. 251–281. [Google Scholar]
- Greenfield J.P., Tsai J., Gouras G.K., Hai B., Thinakaran G., Checler F., Sisodia S.S., Greengard P., Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C., Selkoe D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Haass C., Lemere C.A., Capell A., Citron M., Seubert P., Schenk D., Lannfelt L., Selkoe D.J. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- Hartmann E., Gorlich D., Kostka S., Otto A., Kraft R., Knespel S., Burger E., Rapoport T.A., Prehn S. A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur. J. Biochem. 1993;214:375–381. doi: 10.1111/j.1432-1033.1993.tb17933.x. [DOI] [PubMed] [Google Scholar]
- Hartmann T., Bieger S.C., Bruhl B., Tienari P.J., Ida N., Allsop D., Roberts G.W., Masters C.L., Dotti C.G., Unsicker K., Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat. Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Higaki J., Quon D., Zhong Z., Cordell B. Inhibition of beta-amyloid formation identifies proteolytic precursors and subcellular site of catabolism. Neuron. 1995;14:651–659. doi: 10.1016/0896-6273(95)90322-4. [DOI] [PubMed] [Google Scholar]
- Johnston J.A., Ward C.L., Kopito R.R. Aggresomesa cellular response to misfolded proteins. J. Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J., Schweizer A., Clausen H., Tang B.L., Hong W., Oorschot V., Hauri H.P. The recycling pathway of protein ERGIC-53 and dynamics of the ER-Golgi intermediate compartment. J Cell Sci. 1998;111:3411–3425. doi: 10.1242/jcs.111.22.3411. [DOI] [PubMed] [Google Scholar]
- Koo E.H., Squazzo S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Kovacs D.M., Fausett H.J., Page K.J., Kim T.W., Moir R.D., Merriam D.E., Hollister R.D., Hallmark O.G., Mancini R., Felsenstein K.M., Hyman B.T., Tanzi R.E., Wasco W. Alzheimer-associated presenilins 1 and 2neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- Krijnse-Locker J., Parton R.G., Fuller S.D., Griffiths G., Dotti C.G. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol. Biol. Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn M.J., Herrmann J.M., Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lah J.J., Heilman C.J., Nash N.R., Rees H.D., Yi H., Counts S.E., Levey A.I. Light and electron microscopic localization of presenilin-1 in primate brain. J. Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahtinen U., Hellman U., Wernstedt C., Saraste J., Pettersson R.F. Molecular cloning and expression of a 58-kDa cis-Golgi and intermediate compartment protein. J. Biol. Chem. 1996;271:4031–4037. doi: 10.1074/jbc.271.8.4031. [DOI] [PubMed] [Google Scholar]
- Lehmann S., Chiesa R., Harris D.A. Evidence for a six-transmembrane domain structure of presenilin 1. J. Biol. Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- Levesque L., Annaert W., Craessaert K., Mathews P.M., Seeger M., Nixon R.A., Van Leuven F., Gandy S., Westaway D., St P., George-Hyslop B., De Strooper, Fraser P.E. Developmental expression of wild-type and mutant presenilin-1 in hippocampal neurons from transgenic miceevidence for novel species-specific properties of human presenilin-1. Mol. Med. 1999;In press [PMC free article] [PubMed] [Google Scholar]
- Levey A.I., Heilman C.J., Lah J.J., Nash N.R., Rees H.D., Wakai M., Mirra S.S., Rye D.B., Nochlin D., Bird T.D., Mufson E.J. Presenilin-1 protein expression in familial and sporadic Alzheimer's disease. Ann. Neurol. 1997;41:742–753. doi: 10.1002/ana.410410610. [DOI] [PubMed] [Google Scholar]
- Levitan D., Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E., Wasco W., Poorkaj P., Romano D.M., Oshima J., Pettingell W.H., Yu C.E., Jondro P.D., Schmidt S.D., Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus [see comments] Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Li J., Xu M., Zhou H., Ma J., Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Li X., Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- Li X., Greenwald I. Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc. Natl. Acad. Sci. USA. 1998;95:7109–7114. doi: 10.1073/pnas.95.12.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljestrom P., Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology. 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- Maguire G.A., Luzio J.P. The presence and orientation of ecto-5′-nucleotidase in rat liver lysosomes. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1985;180:122–126. doi: 10.1016/0014-5793(85)80244-1. [DOI] [PubMed] [Google Scholar]
- Mercken M., Takahashi H., Honda T., Sato K., Murayama M., Nakazato Y., Noguchi K., Imahori K., Takashima A. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodiesevidence that Alzheimer mutations affect proteolytic processing. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1996;389:297–303. doi: 10.1016/0014-5793(96)00608-4. [DOI] [PubMed] [Google Scholar]
- Naruse S., Thinakaran G., Luo J.J., Kusiak J.W., Tomita T., Iwatsubo T., Qian X., Ginty D.D., Price D.L., Borchelt D.R., Wong P.C., Sisodia S.S. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Olkkonen V.M., Liljestrom P., Garoff H., Simons K., Dotti C.G. Expression of heterologous proteins in cultured rat hippocampal neurons using the Semliki Forest virus vector. J. Neurosci. Res. 1993;35:445–451. doi: 10.1002/jnr.490350412. [DOI] [PubMed] [Google Scholar]
- Otto H., Buchner K., Beckmann R., Hilbert R., Hucho F. GTP-binding proteins in bovine brain nuclear membranes. Neurochem Int. 1992;21:409–414. doi: 10.1016/0197-0186(92)90192-t. [DOI] [PubMed] [Google Scholar]
- Paganetti P.A., Lis M., Klafki H.W., Staufenbiel M. Amyloid precursor protein truncated at any of the gamma-secretase sites is not cleaved to beta-amyloid. J. Neurosci. Res. 1996;46:283–293. doi: 10.1002/(SICI)1097-4547(19961101)46:3<283::AID-JNR1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Peraus G.C., Masters C.L., Beyreuther K. Late compartments of amyloid precursor protein transport in SY5Y cells are involved in beta-amyloid secretion. J. Neurosci. 1997;17:7714–7724. doi: 10.1523/JNEUROSCI.17-20-07714.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez R.G., Squazzo S.L., Koo E.H. Enhanced release of amyloid beta-protein from codon 670/671 “Swedish” mutant beta-amyloid precursor protein occurs in both secretory and endocytic pathways. J. Biol. Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- Perez R.G., Soriano S., Hayes J.D., Ostaszewski B., Xia W., Selkoe D.J., Chen X., Stokin G.B., Koo E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including abeta42. J. Biol. Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Ratovitski T., Slunt H.H., Thinakaran G., Price D.L., Sisodia S.S., Borchelt D.R. Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J. Biol. Chem. 1997;272:24536–24541. doi: 10.1074/jbc.272.39.24536. [DOI] [PubMed] [Google Scholar]
- Rogaev E.I., Sherrington R., Rogaeva E.A., Levesque G., Ikeda M., Liang Y., Chi H., Lin C., Holman K., Tsuda T. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Sambamurti K., Refolo L.M., Shioi J., Pappolla M.A., Robakis N.K. The Alzheimer's amyloid precursor is cleaved intracellularly in the trans-Golgi network or in a post-Golgi compartment. Ann. NY. Acad. Sci. 1992;674:118–128. doi: 10.1111/j.1749-6632.1992.tb27481.x. [DOI] [PubMed] [Google Scholar]
- Saraste J., Svensson K. Distribution of the intermediate elements operating in ER to Golgi transport. J. Cell Sci. 1991;100:415–430. doi: 10.1242/jcs.100.3.415. [DOI] [PubMed] [Google Scholar]
- Schekman R., Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schwarzman A.L., Singh N., Tsiper M., Gregori L., Dranovsky A., Vitek M.P., Glabe C.G., St P.H., George-Hyslop, Goldgaber D. Endogenous presenilin 1 redistributes to the surface of lamellipodia upon adhesion of jurkat cells to a collagen matrix. Proc. Natl. Acad. Sci. USA. 1999;96:7932–7937. doi: 10.1073/pnas.96.14.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A., Fransen J.A., Matter K., Kreis T.E., Ginsel L., Hauri H.P. Identification of an intermediate compartment involved in protein transport from endoplasmic reticulum to Golgi apparatus. Eur. J. Cell Biol. 1990;53:185–196. [PubMed] [Google Scholar]
- Schweizer A., Matter K., Ketcham C.M., Hauri H.P. The isolated ER–Golgi intermediate compartment exhibits properties that are different from ER and cis-Golgi. J. Cell Biol. 1991;113:45–54. doi: 10.1083/jcb.113.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Sherrington R., Rogaev E.I., Liang Y., Rogaeva E.A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Simons M., Ikonen E., Tienari P.J., Cid-Arregui A., Monning U., Beyreuther K., Dotti C.G. Intracellular routing of human amyloid protein precursoraxonal delivery followed by transport to the dendrites. J. Neurosci. Res. 1995;41:121–128. doi: 10.1002/jnr.490410114. [DOI] [PubMed] [Google Scholar]
- Simons M., De Strooper B., Multhaup G., Tienari P.J., Dotti C.G., Beyreuther K. Amyloidogenic processing of the human amyloid precursor protein in primary cultures of rat hippocampal neurons. J. Neurosci. 1996;16:899–908. doi: 10.1523/JNEUROSCI.16-03-00899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia S.S. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA. 1992;89:6075–6079. doi: 10.1073/pnas.89.13.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H., Duff K., Capell A., Romig H., Grim M.G., Lincoln S., Hardy J., Yu X., Picciano M., Fechteler K. A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and notch signaling. J. Biol. Chem. 1999;In press doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Borchelt D.R., Lee M.K., Slunt H.H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Harris C.L., Ratovitski T., Davenport F., Slunt H.H., Price D.L., Borchelt D.R., Sisodia S.S. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Sisodia S. The molecular biology of presenilin 1. In: Haass C., editor. Molecular Biology of Alzheimer's Disease. Harwood Academic Publishers; Amsterdam: 1998. pp. 193–206. [Google Scholar]
- Tienari P.J., De Strooper B., Ikonen E., Ida N., Simons M., Masters C.L., Dotti C.G., Beyreuther K. Neuronal sorting and processing of amyloid precursor proteinimplications for Alzheimer's disease Cold Spring Harbor Symp. Quant. Biol 61 1996. 575 585a [PubMed] [Google Scholar]
- Tienari P.J., De Strooper B., Ikonen E., Simons M., Weidemann A., Czech C., Hartmann T., Ida N., Multhaup G., Masters C.L., Van Leuven F., Beyreuther K., Dotti C.G. The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein EMBO (Eur. Mol. Biol. Organ.) J. 15 1996. 5218 5229b [PMC free article] [PubMed] [Google Scholar]
- Tomita S., Kirino Y., Suzuki T. Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J. Biol. Chem. 1998;273:6277–6284. doi: 10.1074/jbc.273.11.6277. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheetsprocedure and some applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A., Paliga K., Durrwang U., Czech C., Evin G., Masters C.L., Beyreuther K. Formation of stable complexes between two Alzheimer's disease gene productspresenilin-2 and beta-amyloid precursor protein. Nat. Med. 1997;3:328–332. doi: 10.1038/nm0397-328. [DOI] [PubMed] [Google Scholar]
- Wild-Bode C., Yamazaki T., Capell A., Leimer U., Steiner H., Ihara Y., Haass C. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J. Biol. Chem. 1997;272:16085–16088. doi: 10.1074/jbc.272.26.16085. [DOI] [PubMed] [Google Scholar]
- Wolfe M.S., Xia W., Ostaszewski B.L., Diehl T.S., Kimberly W.T., Selkoe D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Xia W., Zhang J., Perez R., Koo E.H., Selkoe D.J. Interaction between amyloid precursor protein and presenilins in mammalian cellsimplications for the pathogenesis of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W., Zhang J., Ostaszewski B.L., Kimberly W.T., Seubert P., Koo E.H., Shen J., Selkoe D.J. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 1998;37:16465–16471. doi: 10.1021/bi9816195. [DOI] [PubMed] [Google Scholar]
- Yamazaki T., Selkoe D.J., Koo E.H. Trafficking of cell surface β-amyloid precursor proteinretrograde and transcytotic transport in cultured neurons. J. Cell Biol. 1995;129:431–442. doi: 10.1083/jcb.129.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Song L., Parker E.M. Calpain inhibitor I increases beta-amyloid peptide production by inhibiting the degradation of the substrate of gamma-secretase. Evidence that substrate availability limits beta-amyloid peptide production. J. Biol. Chem. 1999;274:8966–8972. doi: 10.1074/jbc.274.13.8966. [DOI] [PubMed] [Google Scholar]