

Abstract
To better understand the basis for human immunodeficiency virus type 1 (HIV-1) persistence and latency, the form in which viral DNA exists in the peripheral T lymphocyte reservoir of infected individuals was investigated. In asymptomatic individuals, HIV-1 was harbored predominantly as full-length, unintegrated complementary DNA. These extrachromosomal DNA forms retained the ability to integrate upon T cell activation in vitro. In patients with acquired immunodeficiency syndrome (AIDS), there was an increase in integrated relative to extrachromosomal DNA forms. By analysis of DNA from patient lymphocyte subpopulations depleted of human lymphocyte antigen–Dr receptor–positive cells, quiescent T cells were identified as the source of extrachromosomal HIV-1 DNA. Thus quiescent T lymphocytes may be a major and inducible HIV-1 reservoir in infected individuals.
The major reservoir for HIV-1 in the peripheral blood compartment of infected individuals is the CD4+ T lymphocyte (1, 2). The high percentage of cells (1 to 0.01%) within this reservoir that contain viral DNA (2) is difficult to reconcile with low percentage of infected cells (0.01 to 0.001%) that express viral RNA at levels detectable by in situ hybridization (3). In addition, the gradual depletion of CD4+ T lymphocytes during disease progression (4) contrasts with the acute cytocidal nature of HIV-1 infection of permissive T lymphocytes in vitro (5). These features suggest that a small population of infected cells is permissive for virus replication, whereas the majority of host cells harbor HIV-1 in a minimally replicative yet inducible state.
The life cycle of retroviruses can be separated into pre- and postintegration stages. Integration of HIV-1 DNA with the host cell genome, which depends on the activated state of the host cell (6), must occur for a productive virus infection (6, 7). Thus, although HIV-1 retains the capacity to bind and infect quiescent T lymphocytes in vitro (6–8), inefficient reverse transcription (7) and a block to integration of full-length HIV-1 DNA (6) restrict the viral life cycle in these cells to preintegration events.
Current in vitro models for HIV-1 persistence and latency have focused on agents that activate HIV-1 gene expression in cells harboring latent integrated provirus (9). However, initial integration requires T cell activation (6, 7), and, because the number of activated T cells in asymptomatic individuals is low, it has been hypothesized that quiescent T cells form a latent and inducible reservoir for HIV-1 in vivo (6, 7).
To identify molecular events during HIV-1 replication in vivo, we analyzed the arrangement of proviral DNA in single infected cells. This approach allowed us to determine whether the presence of extrachromosomal HIV-1 DNA within patient lymphocytes is due to restricted integration in quiescent cells or to superinfection (10, 11) of permissive cells by HIV-1. By initially determining HIV-1 proviral DNA copy number and the percentage of cells harboring HIV-1 within each infected individual included in this study (Table 1), we could analyze multiple (10 to 40) cell fractions, each containing not more than one HIV-1–infected cell (12). DNA was extracted from multiple replicate cell fractions and separated into high molecular weight DNA (containing integrated provirus) and low molecular weight DNA (containing extrachromosomal HTV-1 DNA forms) by agarose gel electrophoresis, and HIV-1 DNA was identified in high and low molecular weight fractions by polymerase chain reaction (PCR) (12).
Table 1.
Integrated and extrachromosomal viral DNA in total and purified lymphocyte populations of HTV-1–seropositive individuals. After determination of the percentage of HIV-1–infected cells for each patient (12), a series of replicate (10 to 40) lymphocyte fractions was prepared. Each replicate fraction contained sufficiently few cells so that there was not more than one infected cell per fraction. The number of replicates containing HIV-1 DNA and the number of fractions analyzed are listed for each patient. The number of fractions containing exclusively integrated viral DNA, exclusively unintegrated viral DNA, or both DNA forms, were calculated as a percentage of the total number of replicates harboring viral DNA. Fractions not containing viral DNA were excluded from the analysis. For examination of viral DNA in purified lymphocyte populations, the percentages of HLA-Dr–positive cells was determined by flow cytofluorimetry after affinity cell sorting with HLA-Dr antibody–coated magnetic beads (27). (Asym., asymptomatic; ND, not done.)
| (A) Total lymphocyte populations | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Patient | Status | CD4/CD8 | Lymphocytes per fraction | Infected/analyzed | HIV-1–infected lymphocytes (%) | Integrated DNA (%) | Extrachromosomal DNA (%) | Both forms DNA (%) |
| E32 | AIDS | 0.15 | 250 | 17/21 | 0.40 | 59 | 35 | 6 |
| B3 | AIDS | <0.03 | 250 | 17/20 | 0.40 | 66 | 17 | 17 |
| B5 | AIDS | 0.08 | 250 | 25/30 | 0.40 | 60 | 12 | 28 |
| B10 | AIDS | 0.39 | 100 | 8/20 | 1.00 | 75 | 12.5 | 12.5 |
| B12 | AIDS | 0.28 | 250 | 20/20 | 1.00 | 40 | 20 | 40 |
| B12 | 100 | 16/20 | 62.5 | 18.75 | 18.75 | |||
| B12 | 50 | 5/10 | 80 | 20 | 0 | |||
| B1 | Asym. | 0.68 | 250 | 10/20 | 0.40 | 30 | 50 | 20 |
| B4 | Asym. | 0.76 | 250 | 19/30 | 0.40 | 21 | 58 | 21 |
| B7 | Asym. | 0.52 | 250 | 6/10 | 0.12 | 33 | 67 | 0 |
| B9 | Asym. | 0.09 | 400 | 21/40 | 0.15 | 14 | 86 | 0 |
| B11 | Asym. | 0.12 | 400 | 12/20 | 0.10 | 25 | 50 | 25 |
| B20 | Asym. | 0.25 | 250 | 5/20 | 0.25 | 20 | 80 | 0 |
| B23 | Asym. | 0.87 | 250 | 7/20 | 0.20 | 29 | 71 | 0 |
| B24 | Asym. | 0.30 | 400 | 11/20 | 0.21 | 18 | 55 | 27 |
| B25 | Asym. | 1.79 | 400 | 17/20 | 0.15 | 0 | 88 | 12 |
| B34 | Asym. | 0.97 | 400 | 6/20 | 0.20 | 33 | 50 | 17 |
| B35 | Asym. | 0.17 | 400 | 5/20 | 0.07 | 0 | 80 | 20 |
| (B) Purified lymphocyte populations | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Patient | Lymphocyte population | Cells per fraction | HLA-Dr+ cells (%) | HIV-1–infected lymphocytes (%) | Integrated DNA (%) | Extra-Chromosomal DNA (%) | Both forms DNA (%) |
| B28* | Total | 400 | 25 | 0.30 | 65 | 35 | 0 |
| B28 | HLA-Dr+ | 400 | ND | 80 | 20 | 0 | |
| B28 | HLA-Dr+ | 400 | 6 | 0 | 80 | 20 | |
| B30* | Total | 400 | 32 | 0.125 | 60 | 20 | 20 |
| B30 | HLA-Dr+ | 400 | <1 | 0 | 100 | 0 | |
| B4 | Total in vitro activated | 250 | 0.40 | 0.40 | 20 | 60 | 20 |
| B4 | 250 | 100 | 0 | 0 | |||
AIDS patient.
Application of our protocol to 8E5 cells (13), which contain one noninfectious provirus per cell and completely lack unintegrated viral DNA, demonstrated that, if there were fewer than eight infected cells in each sample analyzed, mechanical shearing did not lead to separation of detectable amounts of integrated viral DNA in the low molecular weight fraction (Fig. 1A). Because our protocol (12) ensured that there was one infected cell in each fraction, we were able to distinguish integrated and unintegrated forms of HIV-1 DNA. Analysis of DNA isolated from cells infected with an HIV-1 mutant which, because of deletions in the HIV-1 intergrase coding region, is unable to associate with host-cell DNA (6, 14), demonstrated that trapping of low molecular weight extrachromosomal HIV-1 DNA forms in the genomic DNA fraction during gel electrophoresis did not lead to detectable amounts of unintegrated HIV-1 DNA in the high molecular weight genomic fraction (Fig. 1B).
Fig. 1.
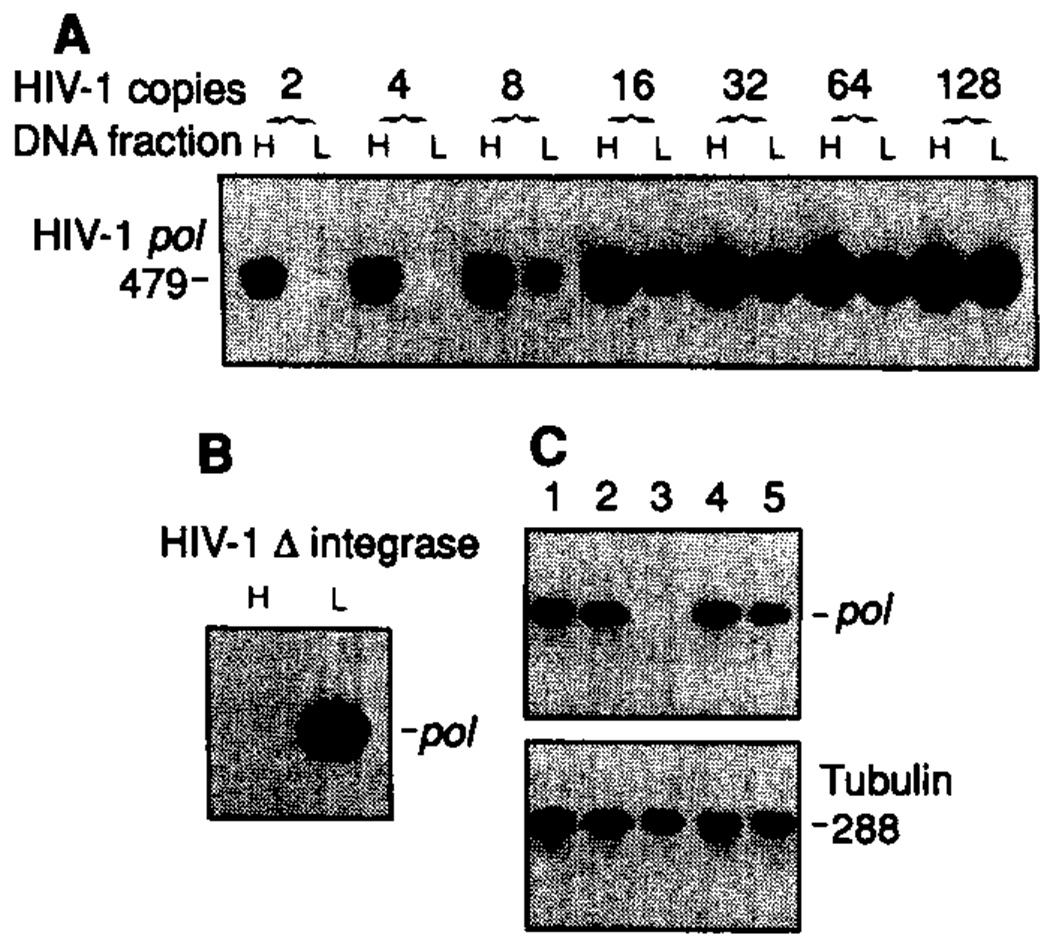
Discrimination between extrachromosomal and integrated HIV-1 DNA forms. (A) Doubling dilutions (from 128 to 2) of 8E5 cells (containing one defective provirus per cell) were mixed with 400 uninfected lymphocytes. After isolation of total cellular DNA, fractionated high (H) and low (L) molecular weight DNA (12) were analyzed by PCR with primers specific for HIV-1 pol (25). (B) HIV-1 PCR analysis of high and low molecular weight DNA fractions from 2000 MT-4 cells (human CD4+ T cell line) infected with an integration minus HIV-1 mutant (6, 14). (C) Four 8E5 lymphocytes were mixed with 2000 uninfected T lymphocytes and distributed in five replicate fractions (1 to 5). Total cellular DNA was isolated, and HIV-1 and tubulin DNA were amplified by 30 and 20 cycles of PCR, respectively. PCR product sizes are in base pairs.
The PCR protocol we used could detect a single HIV-1 provirus. Four HIV-1–infected cells (containing one provirus per cell) were mixed with 2 × 103 uninfected lymphocytes and distributed into five fractions to be analyzed by PCR. Four of five fractions gave positive amplifications with primers specific for HIV-1 pol, whereas primers to the α-tubulin gene indicated the presence of equivalent amounts of genomic DNA in each fraction (Fig. 1C).
Table 1 summarizes the results of our analysis on the arrangement of HIV-1 DNA within individual infected T lymphocytes from eleven asymptomatic and seven AIDS/ARC (AIDS-related complex) patients. The number of peripheral blood lymphocytes (PBL) harboring HIV-1 genome (Table 1) varied among individuals within asymptomatic and AIDS groups; however, the increased virus DNA load in AIDS patients is in agreement with other studies (2, 15). The HIV-1 pol primers detect both complete and incomplete products of reverse transcription. However, use of primers [long terminal repeat (LTR)–gag] spanning the primer binding site and 5′ LTR-gag junction—synthesized after both template switching events (16)—identified late products in reverse transcription and gave identical results to those with the pol primers (Fig. 2).
Fig. 2.
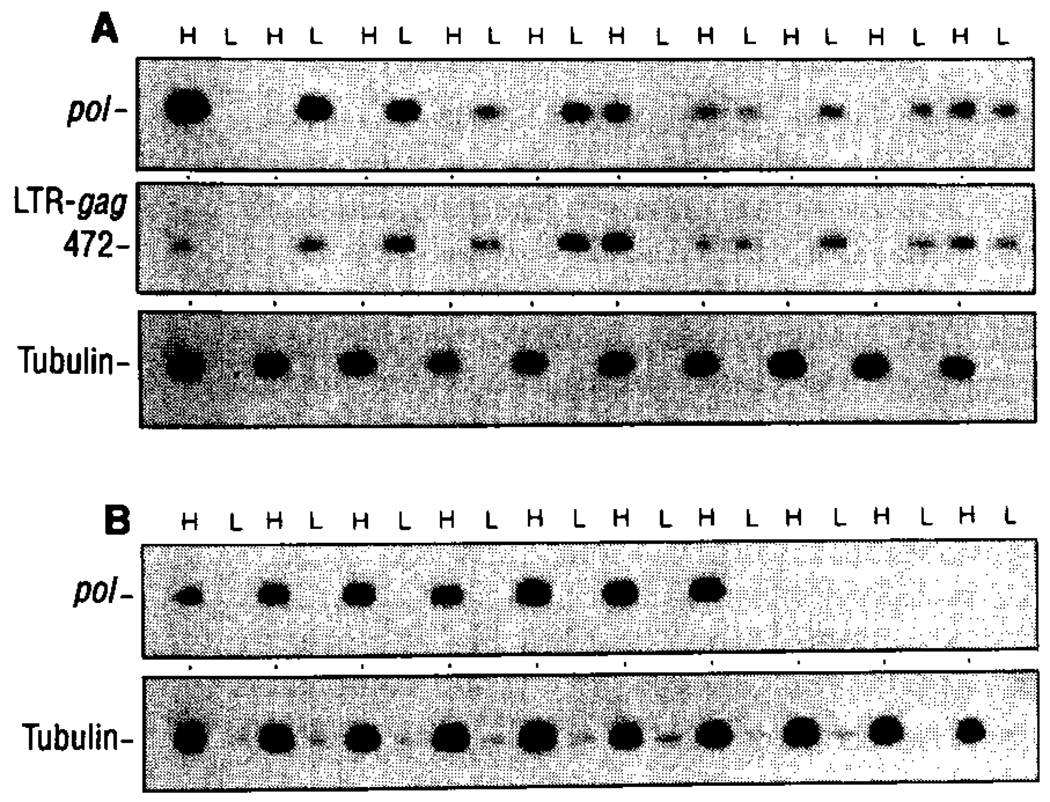
Arrangement of HIV-1 DNA before and after T cell activation in vitro. Replicate macrophage-depleted cell samples (400 cells per sample) were prepared from lymphocyte cultures of patient B4 (asymptomatic) before (A) and after (B) activation of the culture in vitro with PHA. Presence of HIV-1 DNA in high and low molecular weight fractions was determined by PCR with primers specific for HTV-1 pol or primers directed to a region spanning the 5′ LTR-gag terminus (25). The separation of genomic DNA in high and low molecular weight fractions was confirmed using α-tubulin–specific primers.
In all patients analyzed, the presence of integrated and unintegrated HIV-1 DNA forms within the same fraction was rarely observed. When both forms were detected in the same fraction, this was due to the presence of more than one infected cell in that fraction, and preparation of a second dilution series containing fewer cells in each fraction resolved this. For example, in AIDS patient B12, analysis of replicate cell fractions in aliquots of 250 cells per fraction revealed integrated and unintegrated HIV-1 DNA in 40% of the replicate samples (Table 1). Repeat analyses of the same lymphocyte preparation with 100 or 50 cells per fraction resulted in the presence of both DNA forms in 18.75% and 0% of the replicates, respectively (Table 1). Limiting dilution and discrimination of integrated and extrachromosomal HIV-1 DNA in all patients in this study demonstrated that superinfection did not appear widespread in lymphocytes of individuals infected with HIV-1. This is not surprising because infected quiescent cells are not productive for virions and thus do not provide the conditions for superinfection. On the other hand, superinfection of a permissive (activated) T lymphocyte may be restricted because of receptor interference (10, 11, 17) or may be a rare and rapid event, difficult to detect in vivo. In any event, because of the low numbers of infected cells within patients (≤ 1%), superinfection is statistically unlikely.
Monoclonal antibody to the major histocompatibility complex class II human lymphocyte antigen (HLA)–Dr, which is expressed on macrophages and activated T lymphocytes, but not quiescent T lymphocytes (18), was used to deplete patient lymphocyte populations of activated T cells. After depletion of activated T cells, the proportion of cells harboring exclusively unintegrated viral DNA increased from 35 to 80% for patient B28 and from 20 to 100% for patient B30 (Table 1). Depletion of interleukin-2 receptor–positive lymphocytes of asymptomatic patient B24 demonstrated a similar striking predominance of extrachromosomal HIV-1 DNA forms in quiescent lymphocytes (19).
The biological activity of extrachromosomal HIV-1 DNA forms in infected individuals was assessed after T cell activation in vitro. Lymphocytes from patient B4 (HIV-1–positive, asymptomatic) were isolated; half of these were activated with phytohemagglutinin (PHA) and half of these were untreated. After 24 hours, 20 replicate cell fractions were prepared from the quiescent and activated cultures, and each replicate fraction was then analyzed for the presence of integrated and extrachromosomal viral DNA (12). High concentrations (7 μg/ml) of soluble CD4 (sCD4), which has been shown to completely inhibit HIV-1 binding and infection (20), were incorporated in the culture medium to prevent new rounds of infection by virus released from permissively infected (activated) lymphocytes. Of ten infected replicate cell aliquots (400 cells per fraction) from patient B4, six had exclusively extrachromosomal HIV-1 DNA forms (Fig. 2 and Table 1), as detected with the use of HIV-1 pol primers or HIV-1 LTR-gag primers that detect late products in reverse transcription. Two cell aliquots displayed exclusively integrated provirus, and two aliquots had both DNA forms (Fig. 2 and Table 1). After T cell activation in vitro and fractionation, all replicate cell samples containing HIV-1 genome displayed exclusively integrated provirus (Fig. 2 and Table 1). Thus, a large proportion of HIV-1 genome in asymptomatic individuals exists as full-length, extrachromosomal DNA, which retains the ability to integrate upon activation of the host cell.
To provide additional verification for the presence of full-length, extrachromosomal HIV-1 DNA with quiescent PBLs, we isolated total cellular DNA from HLA-Dr–depleted lymphocytes of AIDS patients B28 and B29 and analyzed this DNA with a ligation-mediated PCR protocol (21). The method was adopted so that free blunt 3’ LTR termini could be identified within enriched quiescent T lymphocytes of HIV-1–infected individuals. The blunt 3′ LTR terminus, which comprises full-length (−) strand (complementary to genomic viral RNA) and (+) strand (same polarity as genomic viral’RNA) viral DNA, exists only after completion of reverse transcription (22). The presence of free HIV-1 DNA termini in cells infected with an integration-defective HIV-1 mutant (HIV-1 ΔIN) was evident (Fig. 3C, upper panel), whereas no amplification products, even after two rounds of amplification (Fig. 3C, lower panel), were detectable in DNA isolated from 8E5 cells, which contain one integrated provirus per cell and completely lack extrachromosomal HIV-1 DNA (13). Integrated provirus and circular forms of unintegrated HIV-1 DNA (containing one or two LTRs) (23) are not detected by this approach because these viral DNA forms do not provide free 3’ LTR termini for attachment of the common linker that is required for subsequent PCR amplification (Fig. 3). Application of this ligation-mediated PCR method to DNA from HLA-Dr–depleted lymphocytes from patients B28 and B29 confirmed the presence of complete HIV-1 (−) strand cDNA terminating at nucleotide 9718 (Fig. 3A, step 1), as evidenced by the amplification of a 155–base pair (bp), HIV-1 LTR terminus–specific product (Fig. 3D). Use of HIV-1–specific primers directed to the 5’ LTR-gag junction demonstrated the abundance of HIV-1 DNA in each sample analyzed (Fig. 3B, upper panel). Sequence analysis of ligation-mediated PCR products (two rounds of amplification) by means of the nested LTR R primer (Fig. 3A, step 4) confirmed the amplification of complete 3’ LTR ends containing a GCAGT terminus (19). In a second series of PCR reactions, double-stranded DNA extracted from HLA-Dr–depleted lymphocytes from patients B28 and B29 was ligated directly to the common linker as in Fig. 3A, step 3 (essentially bypassing DNA denaturation and primer extension steps in Fig. 3A, steps 1 and 2). PCR amplification with the nested R primer and longer oligomer of the common linker resulted in amplification of a 155-bp product for DNA from both patients’ lymphocytes and from CD4+ cells infected with the HIV-1 integrase mutant, but not for 8E5 cell DNA (Fig. 3C). This evidence points to the presence of full-length (+) strand in extrachromosomal HIV-1 DNA in quiescent T lymphocytes of infected individuals.
Fig. 3.
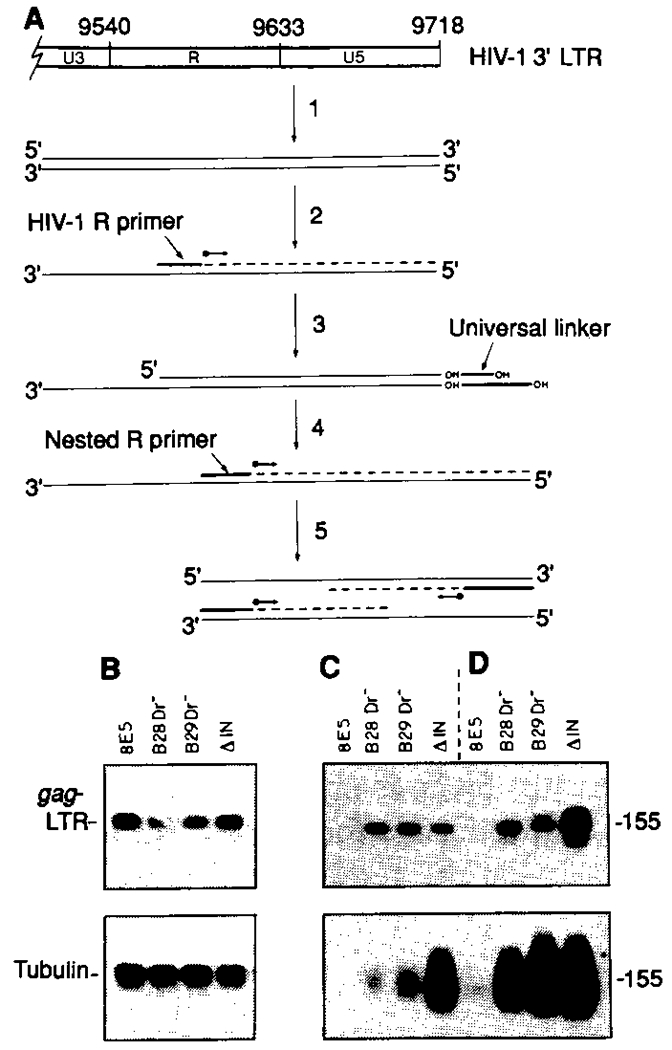
Ligation-mediated PCR analysis of 3′ LTR termini in enriched quiescent lymphocyte populations. (A) The ligation PCR protocol was essentially as described (21). A representation of the HIV-1 3′ LTR and locations of the U3-R, and R-U5 junctions (26) are shown at the top of the figure. Total cellular DNA from HLA-Dr–depleted quiescent lymphocytes of HIV-1–infected individuals is denatured by alkali (1) and annealed to an HIV-1 R-specific LTR primer (2) complementary to the (−) strand of the HIV-1 LTR. DNA polymerase extension from the R primer (2) results in formation of double-stranded blunt-end 3′ LTR terminus, the end of which is defined by the U5 terminus. The newly created blunt-end 3′ U5 LTR terminus provides a substrate for ligation of a universal linker (3). The composition of the universal linker is as described (21). Annealing and extension from the R-specific LTR primer results in a blunt-end LTR terminus only in linear extrachromosomal HIV-1 DNA forms. A second polymerase extension step (4) from a nested (−) strand R-specific primer results in formation of a new (+) strand that is complementary to the LTR (−) strand and that incorporates the sequence of the longer oligomer component of the universal linker. The resultant double-stranded products provide suitable substrates for PCR (5) by means of the nested R-specific LTR primer and the longer oligomer component of the universal linker. The expected PCR product size of 155 bp includes 130 bp of HIV-1 LTR [extending from nucleotide 9591 (26) at the HIV-1 R-specific primer binding site to the 3′ GCAGT terminus of U5 at nucleotide 9720] and 25 bp of incorporated common linker sequence. (B through D) PBLs from two HIV-1–infected individuals with ARC (B28 and B29) were depleted of macrophages and activated T cells by magnetic affinity sorting with HLA-Dr antibody–conjugated magnetic particles (27). Total cellular DNA from the HLA-Dr− lymphocyte population was subject to direct PCR (B) with primers to the HIV-1 5′ LTR-gag junction (upper panel) and to the human α-tubulin gene (lower panel) or to a ligation-mediated PCR approach (C and D), as outlined in (A), using a single round (upper panel) or double round (lower panel) of PCR. In (D), samples were processed exactly as outlined in (A), in that the substrate for ligation of the universal linker was provided after DNA denaturation and polymerase extension from an annealed HIV-1 R-specific primer. In a second series of reactions (C), lymphocyte DNA was ligated directly to the universal linker, essentially bypassing the DNA denaturation and primer extension steps [steps 1 and 2 in (A)]. 8E5 is a human CD4+ T cell line that contains one integrated noninfectious HIV-1 provirus per cell and completely lacks extrachromosomal HIV-1 DNA (13). ΔIN represents DNA from CD4+ lymphocytes infected with an integration minus mutant of HIV-1 (6, 14).
Our results demonstrate the existence of a quiescent T cell reservoir where HIV-1 integration is restricted and HIV-1 DNA is harbored in an extrachromosomal state. The demonstration that a large fraction of HIV-1 DNA can exist in an extrachromosomal state in infected individuals is in agreement with other reports (24). However, the results presented in this study attribute the extrachromosomal DNA pool in HIV-1–infected individuals to infection of quiescent T cells, restricted integration, and establishment of a latent and inducible HIV-1 reservoir. Because T cell activation is a prerequisite for viral DNA integration, the contribution of factors affecting T cell activation (mitogenic agents, opportunistic infection) to reactivation of extrachromosomal HIV-1 DNA forms thus becomes apparent.
Acknowledgments
We thank R. Gallo, J. Coffin, G. Tarpley, and J. Giam for helpful comments and discussion; J. Goldsmith, J. Pierson, and R. Lovely for providing clinical samples; R. Axel and R. Sweet for CD4; C. Kuszynski for fluorescence cytometry analysis; G. Pallas and S. Diaz for artwork; and K. Hansen and M. Notley for manuscript preparation. The 8E5 cell line was obtained through the AIDS Research and Reference Reagent Program of the NIH. Supported by grants A124481 and A130386 from the NIH, the United Kingdom AIDS-directed program (E30/376), and funds from the Nebraska Research Initiative (M.S.).
Contributor Information
M. I. Bukrinsky, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198
T. L. Stanwick, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198
M. P. Dempsey, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198
M. Stevenson, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198, and Division of Virology, National Institute for Medical Research, The Ridgeway, Mill Hill, London, NW7 1AA.
REFERENCES AND NOTES
- 1.McElrath MJ, Pruett JE, Cohn ZA, Proc. Natl. Acad. Sci. U.S.A 86, 675 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Psallidopoulos MC et al. , J. Virol 63, 4626 (1989); [DOI] [PMC free article] [PubMed] [Google Scholar]; Schnittman SM et al. , Science 245, 305 (1989). [DOI] [PubMed] [Google Scholar]
- 3.Harper ME, Marselle LM, Gallo RC, Wong-Staal F, Proc. Natl. Acad. Sci. U.S.A 83, 772 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho DD, Pomerantz RJ, Kaplan JC, N. Engl. J. Med 317, 278 (1987); [DOI] [PubMed] [Google Scholar]; Goedert J et al. , J. Am. Med. Assoc 257, 331 (1987); [Google Scholar]; Lang W et al. , ibid, p. 326. [Google Scholar]
- 5.Sodroski J, Goh WC, Rosen C, Campbell K, Haseltine WA, Nature 322, 470 (1986); [DOI] [PubMed] [Google Scholar]; Lifson JD et al. , ibid 323, 725 (1986); [Google Scholar]; Somasundaran M and Robinson HL, J. Virol 61, 3114 (1987); [DOI] [PMC free article] [PubMed] [Google Scholar]; Popovic M, Sarangadharan MG, Read E, Gallo RC, Science 224, 497 (1984). [DOI] [PubMed] [Google Scholar]
- 6.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA, EMBO J. 9, 1551 (1990a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zack JA et al. , Cell 61, 213 (1990). [DOI] [PubMed] [Google Scholar]
- 8.Zagury D et al. , Science 231, 850 (1986); [DOI] [PubMed] [Google Scholar]; McDougal JS et al. , Immunology 135, 3151 (1985). [PubMed] [Google Scholar]
- 9.Pomerantz RJ, Trono D, Feinberg MB, Baltimore D, Cell 61, 1271 (1990); [DOI] [PubMed] [Google Scholar]; Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB, Proc. Natl. Acad. Sci. U.S.A 86, 5974 (1989); [DOI] [PMC free article] [PubMed] [Google Scholar]; Fauci AS, Science 239, 617 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Stevenson M, Meier C, Mann AM, Chapman N, Wasiak A, Cell 53, 483 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weller SK, Joy AE, Temin HM, J. Virol 33, 494 (1980); [DOI] [PMC free article] [PubMed] [Google Scholar]; Weiss RA, in RNA Tumor Viruses, Weiss RA, Teich NM, Varmus HE, Coffin J, Eds. (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1984), vol. I, pp. 209–260; [Google Scholar]; Keshet E and Temin H, J. Virol 31, 376 (1979); [DOI] [PMC free article] [PubMed] [Google Scholar]; Mullins JI, Chen CS, Hoover EA, Nature 319, 333 (1986); [DOI] [PubMed] [Google Scholar]; Pauza CD, Galindo JE, Richman DD, J. Exp. Med 172, 1035 (1990); [DOI] [PMC free article] [PubMed] [Google Scholar]; Robinson HL and Zinkus DM, J. Virol 64, 4836 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.All patients from whom PBLs were derived for this study were registered in the Clinical AIDS Unit at the University of Nebraska Medical Center, and only patients not undergoing azidothymidine (AZT) treatment were recruited. CD4 and CD8 cell counts were determined at the time blood was drawn for analysis. Isolation of PBL and depletion of monocytes and macrophages was performed essentially as described (6). Analysis and quantitation of PCR-amplified products was as described (6). The autoradiographic signal of PCR-amplified products from doubling dilutions of DNA from patient lymphocytes was compared with that of PCR-amplified products from doubling dilutions of DNA extracted from 8E5 cells that contain one noninfectious HIV-1 provirus per cell (13). HIV-1 DNA copy number in each patient was quantitated after normalization of the autogradiographic signal of HTV-1–specific products to those generated from PCR amplification of primers specific for α-tubulin on parallel dilutions of 8E5 and patient lymphocyte DNA. After quantitation of proviral DNA copy number, lymphocytes were distributed in multiple (10 to 40) aliquots, each of which contained sufficiently few lymphocytes so that there was not more than one infected cell in each aliquot. Typically for asymptomatic individuals, each aliquot contained 400 to 1000 cells, whereas for AIDS patients each aliquot contained 50 to 400 cells. Total cellular DNA was extracted from each aliquot, mixed with purified carrier salmon sperm DNA (1 μg) and resolved on 0.8% low gelling temperature agarose–tris borate gels at 2.5 V/cm for 10 to 14 hours. After electrophoresis, DNA was visualized by ethidium bromide staining. Gel regions containing DNA of 4 to 15 kb (low molecular weight fraction) and over 15 kb (high molecular weight fraction) were excised. Each gel fragment was melted at 68°C, and DNA was extracted and purified as described elsewhere (6). Purified DNA from each high and low molecular weight fraction was pelleted, dried, and resuspended in 10 μl H2O for analysis by PCR.
- 13.Folks TM et al. , J. Exp. Med 164, 280 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevenson M et al. , J. Virol 64, 2421 (1990b). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho DD, Moudgil T, Alam M, N. Engl. J. Med 321, 1621 (1989). [DOI] [PubMed] [Google Scholar]
- 16.Panganiban AT and Fiore D, Science 241, 1064 (1988); [DOI] [PubMed] [Google Scholar]; Hu W-S and Temin HM, ibid 250, 1227 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Sommerfelt MA and Weiss RA, Virology 176, 58 (1990); [DOI] [PubMed] [Google Scholar]; Steck FT and Rubin H, ibid 29, 642 (1966); [DOI] [PubMed] [Google Scholar]; Dorner AJ and Coffin JM, Cell 45, 365 (1986). [DOI] [PubMed] [Google Scholar]
- 18.Reinherz EL, et al. , J. Exp. Med 150, 1472 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bukrinsky MI et al. , unpublished data. [Google Scholar]
- 20.Deen KC, et al. , Nature 331, 82 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Mueller PR and Wold B, Science 246, 780 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Grandgenett DP and Munn SR, Cell 60, 3 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Shank PR et al. , ibid 15, 1383 (1978). [Google Scholar]
- 24.Shaw GM et al. , Science 226, 1165 (1984); [DOI] [PubMed] [Google Scholar]; Pang SY et al. , Nature 343, 85 (1990). [DOI] [PubMed] [Google Scholar]
- 25.PCR was performed essentially as described (6), with the following modifications. PCR was performed in a reaction volume of 25 μl, and each cycle of amplification comprised a 30-s denaturation step (95°C), a 30-s annealing step (56°C), and a 1-min extension step (72°C). In addition, there was one 5-min step at 72°C at the end of each series of cycles to ensure complete extension of amplified DNA. The sequences of all primers used in this study are available from M. Stevenson on request.
- 26.Ratner L et al. , Nature 313, 277 (1985). [DOI] [PubMed] [Google Scholar]
- 27.Padmanabhan R et al. , Anal. Biochem 170, 341 (1988). [DOI] [PubMed] [Google Scholar]