

Abstract
Diagnosing ALS can be challenging due to heterogeneity in clinical presentation and overlap with other neurological and neuropsychological conditions. Earlier diagnosis can improve ALS patient outcomes as timely interventions slow disease progression. An evolving awareness of ALS genotypes and phenotypes and new ALS criteria, such as the recent Gold Coast criteria, could expedite diagnosis. Improved prognosis, such as the ENCALS survival model, could inform the patient and their family about disease course and improve end of life planning. Novel staging and scoring systems can help monitor ALS patients’ disease progression and may potentially serve as clinical trial outcomes. Lastly, new tools like biofluid markers, imaging modalities, and neuromuscular electrophysiological measurements may increase diagnostic and prognostic accuracy. We hope that improved diagnostic tools for ALS will shorten time to diagnosis, leading to earlier treatment and potentially better outcomes, whereas improved prognostic tools will help ALS patients understand their likely disease course.
Keywords: Amyotrophic lateral sclerosis, biomarker, brain imaging, clinical overlap, diagnosis, epidemiology, motor neuron disease, phenotype, prognosis, scoring, staging
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease, characterized by progressive, painless muscle weakness due to motor neuron death in the brain, brainstem, and spinal cord. 11 Weakness begins in facial, tongue, and pharyngeal muscles in bulbar onset ALS producing dysarthria and then dysphagia, or in distal upper or lower limb muscles in spinal onset ALS. Most patients with spinal onset ALS present with weakness in one body region that spreads over time to the same region on the contralateral side, as well as regions rostral and caudal to the initial onset region. ALS is now understood as a systems disease and there is significant variation in clinical presentations, including non-motor symptoms, behavioral changes, and cognitive decline, including frontotemporal dementia (FTD). Death from ALS generally occurs within two to four years from diagnosis from respiratory failure, although more slowly progressive forms of the illness occur in a small proportion of patients.
Diagnosing ALS can be challenging, and the process has remained essentially unchanged in clinical practice in the last decade and no test or tool has replaced clinical history and examination for confirming diagnosis, even with the greater adoption of genetic testing. The typical median time between initial symptoms and a definitive diagnosis is 10 to 16 months2 secondary to the rarity and unfamiliarity of disease, incomplete recognition of symptoms, and lack of early and appropriate specialist involvementi.3 Additionally, the prognosis of ALS patients remains suboptimal because the determinants of progression are not fully known.
To facilitate earlier diagnosis and improve prognosis, research is on-going into new ALS criteria and scoring systems, as well as emerging diagnostic and prognostic biofluid markers, imaging modalities, and electrophysiological measurements. This review will highlight these emerging discoveries and focus on the most recent ALS advances in diagnosis and prognosis within the past five years. This review is accompanied by a second more research focused article, which provides an update on complex genetics, pathophysiology, therapeutic development, and exposome science. Both reviews are written for the general neurology community.
ALS epidemiology
ALS incidence and prevalence varies across the globe and estimates are based on different data sources. The availability of registries in some countries enables more accurate calculations of incidence and prevalence, advocating the need for population-based registries worldwide (Panel 1). A recent meta-analysis of 110 incidence and 58 prevalence studies estimates an average global incidence of 1·59 (95% confidence interval [CI] 1·39–1·81) and a prevalence of 4·42 (95%CI 3·92–4·96) per 100,000 individuals.4 Ancestral background and biological sex are linked to ALS rates in an age-dependent manner.5 Despite male predominance, heritability is greater in females, with highest ALS concordance in female-female parent-offspring pairs.6 Male C9orf72 repeat expansion carriers develop ALS at an earlier age by about two years compared to females.7 Thus, an intricate interplay between age, sex, and complex genetics drives ALS risk.5 These sex-dependent differences urge consideration of sex in preclinical and clinical research, to understand the basis of these effects, and in ALS clinical trials for developing therapeutics.
Panel 1. Global incidence and sex in ALS.
Standardized ALS incidence:
Standardized incidence is similar among European populations, which are higher compared to South American and Asian populations.99 Standardized rates are also higher in Oceania and North African populations.99 Data are lacking for sub-Saharan Africa.
| • North Europe, 1·89 per 100,000 | • East Asia, 0·83 per 100,000 |
| • West Europe, 1·71 per 100,000 | • West Asia 0·94 per 100,000 |
| • South Europe 1·75 per 100,000 | • South Asia 0·73 per 100,000 |
| • North America, 1·75 per 100,000 | • Oceania 2·56 per 100,000 |
| • South America, 1·59 per 100,000 | • North Africa 2·03 per 100,000 |
ALS incidence by age:
ALS incidence peaks between the ages of 60 and 75.100 In the United States, the National ALS Registry, which is coordinated by Centers for Disease Control and Prevention, reports a peak ALS prevalence between 60 and 79 years of age.101 Although global ALS burden is anticipated to increase due to an aging population,102 the Irish ALS Register did not observe a rise in incidence between 1995 and 2017.6
ALS incidence by sex:
Sex plays a role in ALS incidence and prevalence. In the South East England ALS Registry, the male-to-female ratio in ALS incidence at younger ages (25–34) was 3·7, which narrows to 1·2 in the 65–74 age group, but then grows slightly to 1·4 above 75 years of age.103 Sex differences in ALS prevalence are present in the US National ALS Registry, which reports that 60% of persons living with ALS are male.101 The Irish ALS Register reports an ALS lifetime risk of 1:347 for males and 1:436 for females.6
ALS clinical presentation
Motor symptoms and phenotypic heterogeneity
ALS was historically considered a relatively uniform disease of progressive painless voluntary muscle weakness.11 Studies over the past decades have redefined ALS as a complex disorder with significant heterogeneity in clinical presentation in site of disease onset and distribution of upper and lower neuron motor signs (Figure 1A, Table 1). Recognizing these multiple heterogeneous ALS phenotypes facilitates earlier diagnosis and informs prognosis.8 The Australian National Motor Neuron Disease (1,677 ALS patients)9 and Italian Piemonte and Valle d’Aosta (2,839 ALS patients)5,10 have documented this heterogeneity in ALS phenotypes, which also correlate with median survival (Figure 1B–C). Uniformly, bulbar onset patients are at a greater risk for FTD than other phenotypes.5 Additionally, less common ALS phenotypes exist, e.g., hemiplegic (Table 1).11 Furthermore, phenotypes correlate with timing of certain treatments. In the Australian registry, feeding tube placement secondary to dysphagia occurs earlier in bulbar versus spinal onset patients,9 as also reported in a European tertiary care ALS cohort.12
Figure 1. ALS phenotypic heterogeneity in initial presentation and staging.
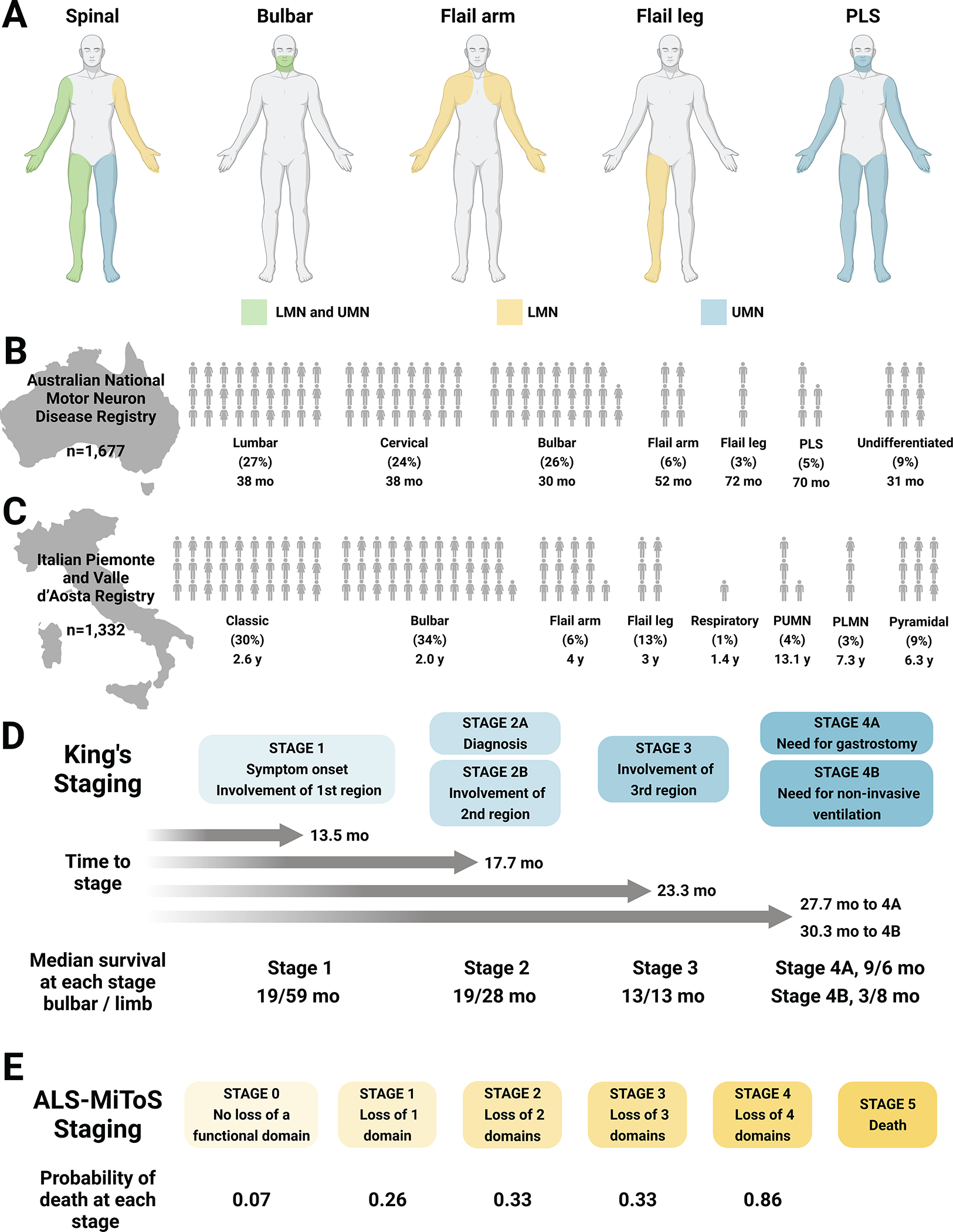
(A) Schematic of involvement of LMN (yellow), UMN (blue), and both LMN and UMN (green) dysfunction at initial presentation in spinal and bulbar onset, flail arm and leg, and PLS phenotypes. Spinal onset ALS involves variable UMN and/or LMN dysfunction in a combination of limbs Bulbar onset ALS involves UMN and/or LMN dysfunction in bulbar muscles (facial, tongue, pharyngeal). Flail arm ALS involves LMN dysfunction in the arms, although mild UMN dysfunction can occur in the legs. Flail leg ALS frequently involves asymmetric LMN dysfunction in the legs. PLS mainly involves UMN dysfunction in the arms and legs or bulbar region, although restricted LMN dysfunction can develop in the later disease stages or become more widespread if it transitions to ALS, often within 4.5 years of symptom onset. (B) Distribution of ALS phenotypes in the Australian National Motor Neuron Disease Registry (n=1,677);9 each human figure represents one percentage point. (C) Distribution of ALS phenotypes in the Italian Piemonte and Valle d’Aosta Registry (n=1,332);5,10 each human figure represents one percentage point; median survival in years is presented under each phenotype. Note that the two registries use slightly different classification systems of ALS phenotypic presentation. (D) King’s staging with four stages indicated (1, 2A/B, 3, 4A/B; blue); time to progress to stages and median survival at each stage (in months) are also annotated. ALS-MiToS staging with six stages indicated (0, 1, 2, 3, 4, 5; orange); staging based on four functional domains from the ALSFRS-R: (i) movement (walking/self-care; ALSFRS-R question 6 or 8); (ii) swallowing (ALSFRS-R question 3); (iii) communicating (ALSFRS-R questions 1 and 4), and (iv) breathing (ALSFRS-R question 10 or 12). Intensifying color indicates progression along stages for both King’s and ALS-MiToS.
LMN, lower motor neuron; PLMN, pure LMN; PLS, primary lateral sclerosis; PUMN, pure UMN; UMN, upper motor neuron; y, year. Created, in part, with BioRender.com.
Table 1.
Clinical spectrum of ALS phenotypes.
| Phenotype | Affected motor neurons | Progression | Additional features |
|---|---|---|---|
| Classical | |||
| Classical bulbar onset | UMN + LMN | Begins with dysarthria then dysphagia then spreads to limbs | |
| Pseudobulbar palsy | UMN | Prominent bulbar features that slowly spread to limbs | Females > males; longer survival; pseudobulbar affect |
| Progressive bulbar palsy | LMN | ||
| Classical cervical onset | UMN + LMN | Typically hand weakness that spreads to bulbar and lumbar regions | |
| Classical lumbar onset | UMN + LMN | Typically foot drop that spreads to cervical and bulbar regions | |
| Flail arm | LMN in UEs; UMN in LEs | Symmetrical proximal > distal upper limb weakness that eventually spreads | Slower progression; males > females |
| Flail leg | LMN in LEs | Symmetrical lower limb weakness | |
| PLS | UMN | May begin in any region and spread over time; if LMN signs develop within 4.5 years, diagnosis is ALS | Normal life expectancy; exclude HSP if involves symmetrical lower limb |
| PMA | LMN | May begin in any region and spread over time; if UMN signs develop within 4.5 years, diagnosis is ALS | |
| Respiratory | UMN + LMN | Limb weakness follows respiratory involvement | Short survival |
| Pseudopolyneuritic | Distal LMN > UMN | ||
| Hemiplegic | Unilateral UMN > LMN | ||
| Cachexia | Weight loss followed by classical ALS | Unexplained weight loss preceding ALS presentation | |
HSP, hereditary spastic paraparesis; LE, lower extremity; LMN, lower motor neuron; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy; UE, upper extremity; UMN, upper motor neuron.
Thus, phenotyping is based on clinical criteria, such as site of disease onset and distribution of upper and lower motor signs.8 Additional relevant clinical variables aid disease classification and can provide prognostic guidance,13 such as age, sex, family history, progression rate, genetic profile and presence of cognitive impairment and other non-motor symptoms (see sections below).
Non-motor ALS symptoms
The concept of ALS as a pure motor disease is now abandoned. In fact, executive dysfunction in 50% and FTD in 15% of ALS patients has been known for decades. Executive dysfunction is evaluated by a suite of neuropsychological tests (Table 2)14 and FTD in ALS is diagnosed by the revised Strong criteria.15 The most characteristic cognitive changes in ALS include impaired language function16 and executive function deficits involving working memory, inhibition, set shifting, and fluency, whereas memory and spatial function are typically spared.17 ALS patients also experience social and cognitive decline, including apathy, disinhibition, irritability, loss of sympathy/empathy, perseveration, reduced concern for hygiene, and changes in eating habits. Similar clinical patterns are present in FTD.17 Additionally, many ALS patients suffer from anxiety, depression, and sleep disorders.18
Table 2. Cognitive changes in ALS.
Domains of cognition function that are impaired in ALS patients, along with the associated symptoms and testing strategies.14,16,17,95,96
| Domain | Symptoms | Neuropsychological test |
|---|---|---|
| Executive function | ||
| Working memory | Unable to “temporarily process, store, and manipulate information in conscious awareness”14 | Digit span subtest (Wechsler Adult Intelligence Scale, Fourth Edition); Corsi Block-Tapping Test or Spatial Span (Wechsler Memory Scale, Third Edition) |
| Inhibition | Inability to ignore stimuli, which can result in impulsive behavior | Flanker task, Continuous Performance Test, antisaccade task (NIH EXAMINER); Stroop test (Delis-Kaplan Executive Function System) |
| Set shifting | Inability to “modify attention and behavior in response to changing circumstances and demands”14 causing rigid thinking and impairments in multitasking | Trail Making Test (Delis-Kaplan Executive Function System); Wisconsin Card Sorting; Set Shifting test (NIH EXAMINER) |
| Fluency | Disorganized thoughts or inability to initiate tasks | Verbal and Design Fluency tests; Category Fluency |
| Language function | Impairment in word naming, spelling, grammatical processing | Psycholinguistic Assessments of Language Processing in Aphasia (PALPA) |
| Social behavior | ||
| Apathy | “Passivity and lack of spontaneity and initiative, loss of interest and motivation for previously rewarding activities, and diminished social interest”96 | Beaumont Behavioural Inventory |
| Disinhibition | “Impulsivity, lack of self-strain, loss of manners, socially inappropriate behaviours, irritability, verbal or physical aggression, disinhibited emotional display, changes in sexual behaviour, and decline in personal hygiene”96 | Beaumont Behavioural Inventory |
| Loss of sympathy/empathy | “Diminished response and understanding of the needs and feelings of others, reduced inter-relatedness and personal warmth, and emotional detachment”96 | Beaumont Behavioural Inventory |
| Perseveration, stereotyped or obsessive-compulsive behaviours | “Simple repetitive movements, more complex ritualistic behaviours, and stereotypy of speech”96 | Beaumont Behavioural Inventory |
| Eating behaviors | “Altered food preferences, increased consumption of cigarettes, binge eating, hyperorality and oral exploration of inedible items”96 | Beaumont Behavioural Inventory |
Executive dysfunction is a negative prognostic indicator, and if present tends to worsen over time.19 Cognitive impairment can later manifest even in patients who appear to be cognitively spared at diagnosis19 and appears to be in part related to the worsening of motor function.17 Thus, there is a growing need to incorporate an evaluation of cognitive function into ALS diagnosis and ongoing management. These behavioral changes can also frustrate family members and caregivers and prevent the patient from accepting medical recommendations, emphasizing the importance of addressing care preferences early in disease.20 These cognitive and behavioral symptoms are accompanied by structural changes to the brain in extra-motor domains (see “Brain and spinal cord imaging” section).
ALS genes influence clinical phenotype
The discovery of mutant SOD1 in a subset of ALS patients in 1993 suggested a potential genetic etiology, which could enhance our understanding of disease risk factors and pathophysiology as well as identify therapeutic targets.21 This possibility was strengthened in 2011 by the discovery of C9orf72 repeat expansions in a larger proportion of individuals, both with and without family history of ALS.22 The genetic architecture of ALS and nuances of familial versus sporadic ALS are fully detailed in the accompanying research-focused review. Over 40 ALS genes are identified to date, which account for approximately 15% of cases. Thus, genetic testing is a growing, albeit non-uniform, component of ALS management. As the cost of genetic profiling drops, we anticipate earlier and broader adoption for ALS patients. First, detecting known pathogenic ALS variants could complement and bolster diagnosis achieved by diagnostic criteria (see “ALS diagnosis” section). Second, though most mutations converge on a typical ALS phenotype, there are important prognostic implications for certain mutant genes linked to unique features (Table 3). For example, ALS2, DCTN1, MATR3, OPTN, and SETX mutations are associated with slower clinical trajectories than classical ALS, information valuable to patients and their families. Furthermore, routine genetic profiling could move past the present and inadequate stratification of patients into sporadic or familial ALS, by uniformly identifying specific gene mutations. Additionally, genetic profiling promotes precision medicine for managing ALS patients23 and clinical trial stratification for targeted therapeutics, e.g., gene therapies (see accompanying review). Therefore, a genetic profile could potentially facilitate diagnosis, prognosis, and treatment for patients harboring known ALS genetic variants.
Table 3. Summary of ALS genotype-phenotype associations and overlap with other diseases.
Adapted, with modifications, from Goutman et al. Handb Clin Neurol, 201897 and Chia et al. Lancet Neurol, 2018.98
| Gene | Genetic effect | %FALS | %SALS | Associated clinical ALS phenotype | Overlap with other diseases |
|---|---|---|---|---|---|
| ALS2 | Autosomal recessive | <1 | <1 | Slowly progressive, infantile and juvenile mainly affecting UMN, PLS | HSP |
| ANG | Autosomal dominant Risk factor |
<1 | <1 | Typical, bulbar-onset tendency, FTD | |
| ANXA11 | Autosomal dominant | ∼1 | ∼1·7 | ND | Autoimmune, sarcoidosis |
| ATXN2 | Autosomal dominant Risk factor |
<1 | <1 | Typical | SCA |
| C9orf72 | Autosomal dominant | 40 | 7 | Typical, FTD | Huntington disease phenocopy, Parkinsonism, essential tremor, myoclonus |
| C21orf2 | ND | <1 | <1 | Typical, FTD | |
| CCNF | Autosomal dominant | ∼1–3·3 | <1 | Typical, FTD, PLS | |
| CHCHD10 | Autosomal dominant | <1 | <1 | Typical, FTD | Cerebellar ataxia, myopathy |
| CHMP2B | Autosomal dominant | <1 | <1 | Typical, PMA | FTD |
| DCTN1 | Autosomal dominant Risk factor |
<1 | <1 | Slowly progressive juvenile | Perry syndrome (Parkinsonism) |
| DNAJC7 | ND | <1 | <1 | ND | |
| ELP3 | Allelic | <1 | <1 | Typical | |
| FUS | Autosomal dominant Autosomal recessive de novo |
4 | 1 | Typical or atypical, FTD, dementia; juvenile, adult onset | Essential tremor* |
| GLT8D1 | Autosomal dominant | <1 | <1 | Typical, shorter and longer survival | Schizophrenia |
| GRN | Autosomal dominant Modifier |
<1 | <1 | Earlier onset, shorter survival | FTD, FTLD, DLB* |
| HNRNPA1 | Autosomal dominant de novo Risk factor |
<1 | <1 | Typical, cognitive impairment | IBM |
| HNRNPA2B1 | Autosomal dominant Risk factor |
<1 | <1 | Typical, cognitive impairment | IBM |
| KIF5A | Autosomal dominant | ∼0·5–3 | <1 | Earlier onset, longer survival | CMT2, PPMS phenocopy*, SPG10 |
| LGALSL | ND | <1 | <1 | Earlier onset, typical | |
| MATR3 | Autosomal dominant | <1 | <1 | Slowly progressive typical or atypical, FTD, myopathy | Distal myopathy |
| NEFH | Autosomal dominant Risk factor |
<1 | <1 | Typical | CMT2* |
| NEK1 | ND | ∼1–2 | <1 | ND | |
| OPTN | Autosomal dominant Autosomal recessive |
<1 | <1 | Slowly progressive atypical | Open-angle glaucoma, Paget’s |
| PFN1 | Autosomal dominant | <1 | <1 | Typical | |
| SETX | Autosomal dominant | <1 | <1 | Slowly progressive juvenile | SCA, progressive motor neuropathy |
| SPG11 | Autosomal recessive | <1 | <1 | Slowly progressive juvenile, mainly affecting UMN | HSP |
| SOD1 | Autosomal dominant Autosomal recessive de novo |
12 | 1–2 | Prominent LMN, cognitive impairment very rare | |
| SQSTM1 | Autosomal dominant | ∼1 | <1 | Typical | Paget’s, FTD, DLB* |
| TARDBP | Autosomal dominant Autosomal recessive de novo |
4 | 1 | Typical, FTD | Supranuclear gaze palsy |
| TBK1 | Autosomal dominant de novo |
∼3 | <1 | Typical, FTD | FTLD, DLB* |
| TIA1 | Autosomal dominant | ∼2·2 | <1 | FTD | DLB* |
| TUBA4A | Autosomal dominant | <1 | <1 | Typical, FTD | |
| UBQLN2 | X-linked autosomal dominant | <1 | <1 | Typical; juvenile, adult onset, FTD | FTD* |
| VAPB | Autosomal dominant | <1 | <1 | Typical or atypical | SMA, essential tremor |
| VCP | Autosomal dominant de novo |
1 | 1 | Typical, FTD | Inclusion body myositis with Paget’s disease, Parkinsonism, SMD, dropped head syndrome |
ALS2, alsin Rho guanine nucleotide exchange factor ALS2; ANG, angiogenin; ANXA11, annexin A11; ATXN2, ataxin 2; C9orf72, chromosome 9 open reading frame 72; C21orf2, chromosome 21 open reading frame 2; CCNF, cyclin F; CHCHD10, coiled-coil-helix-coiled-coil-helix domain containing 10; CHMP2B, charged multivesicular body protein 2B; CMT, Charcot-Marie-Tooth; DCTN1, dynactin subunit 1; DNAJC7, DnaJ homolog subfamily C member 7; DLB, dementia with Lewy bodies; ELP3, elongator acetyltransferase complex subunit 3; FALS, familial ALS; FTD, frontotemporal dementia; FTLD; frontotemporal lobar degeneration; FUS, Fused in Sarcoma; GLT8D1, glycosyltransferase 8 domain containing 1; HNRNPA1, heterogeneous nuclear ribonucleoprotein A1; HNRNPA2B1, heterogeneous nuclear ribonucleoprotein A2/B1; HSP, hereditary spastic paraparesis; IBM, inclusion body myopathy; KIF5A, kinesin family member 5A; LGALSL, galectin-like; LMN, lower motor neuron; MATR3, matrin 3; ND, not determined; NCP, nucleocytoplasmic transport; NEFH, neurofilament heavy chain; NEK1, NIMA (never in mitosis gene a)-related kinase 1; OPTN, optineurin; PFN1, profilin 1; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy; PPMS, primary progressive multiple sclerosis; SCA, spinocerebellar ataxia; SPG10, hereditary spastic paraplegia; SPG11, SPG11 vesicle trafficking associated, spatacsin; SALS, sporadic ALS; SETX, senataxin; SMA, spinal muscular atrophy; SMD, scapuloperoneal muscular dystrophy; SOD1, superoxide dismutase 1; SQSTM1, sequestosome 1; TARDBP, TAR DNA binding protein; TBK1, TANK-binding kinase 1; TIA1, TIA-1 cytotoxic granule-associated RNA binding protein; TUBA4A, tubulin alpha 4a; UBQLN2, ubiquilin 2; UMN, upper motor neuron; VAPB, vesicle-associated membrane protein-associated protein B and C; VCP, valosin-containing protein.
Findings limited to few patients.
ALS diagnosis
Established diagnostic criteria
ALS diagnostic criteria date back to the original El Escorial and later the revised El Escorial (Airlie House) and Awaji criteria. They rate the degree of diagnostic “certainty by clinical assessment alone” from possible to probable to definite ALS, based on the number of affected segments combined with clinical and/or electrophysiological findings.24–26 The El Escorial classification provides prognostic information since, for instance, definite ALS progresses faster.13 Although approaches that score the certainty of diagnosis solely by clinical assessment are reasonable (i.e., possible ALS), they delay diagnosis and confuse patients, their families, and clinicians, who misinterpret these terms as meaning the diagnosis is unlikely or incorrect.27 In reality, nearly all patients diagnosed as possible ALS progress and ultimately die from ALS.
Emerging diagnostic criteria
To address these limitations, an international consensus group reconsidered criteria to improve the diagnostic process for ALS, particularly in the early stages of disease, when clinical symptoms are minimal.28 Recognizing the broad properties of the disease, the Gold Coast criteria define ALS by:
Progressive motor impairment, documented by history or repeated clinical assessment, preceded by normal motor function.
Upper and lower motor neuron dysfunction in at least one body region, or lower motor neuron dysfunction in at least two body regions.
Investigative findings that exclude alternative diseases.
Adopting these simplified criteria for ALS abandons the previous diagnostic categories of possible, probable, and definite. The advent of these new criteria facilitates diagnosing ALS early and definitively. An Australian study found Gold Coast criteria diagnostic sensitivity (92%) was maintained irrespective of functional status, disease duration, or disease onset site and was generally similar to the revised El Escorial (88.6%) and Awaji criteria (90.3%); however, Gold Coast criteria were more sensitive and specific, including for progressive muscular atrophy and for excluding primary lateral sclerosis as a form of ALS, the latter of which meets possible ALS by the revised El Escorial and Awaji criteria.29 This finding was validated in a five-center European study, which similarly found consistent and improved sensitivity of Gold Coast criteria across ALS phenotypes, due to greater sensitivity for identifying progressive muscular atrophy.30 Lastly, a Chinese study corroborated the greater sensitivity of the Gold Coast against the revised El Escorial and Awaji criteria,31 suggesting diagnostic utility would be maintained in racially diverse ALS populations.
Importantly, the Gold Coast criteria were marginally less specific, which clinicians should bear in mind as they monitor their patients’ disease course. However, overall, we anticipate the new Gold Coast criteria will facilitate ALS diagnosis and dispel uncertainty and confusion for patients and their families.
Clinical overlap of ALS with Other Neurodegenerative Disorders
ALS is a multifaceted disease with remarkable phenotypic heterogeneity of motor and non-motor features, as described in previous sections and more fully in our accompanying review. This complexity contributes, in part, to the difficulty of diagnosing ALS, which is rendered more challenging by clinical overlap with other more common neurological and neuromuscular diseases (Table 3). Additionally, C9orf72 repeat expansions, the most common ALS mutations in populations of European descent, are among the strongest determinants of FTD. However, the clinical phenotypes present as a continuum from pure ALS, to ALS-FTD, to pure FTD, sometimes even within the same pedigree. Further complicating the situation, C9orf72 repeat expansions are associated with movement disorders such as parkinsonism, essential tremor, and myoclonus,32 in addition to cognitive impairments. The presence of these conditions in patients may present as atypical ALS, which could contribute to a more difficult and lengthy diagnosis process. Therefore, awareness of additional manifestations of an ALS mutation could facilitate early diagnosis. Additionally, C9orf72 repeat expansions are the most frequent cause of Huntington’s disease (HD) phenocopies, patients with the classical HD phenotype but lacking characteristic huntingtin (HTT) repeat expansions and inclusions.33 Conversely, ALS patients may harbor HTT repeat expansions simultaneously with TDP-43 inclusions,34 underscoring the complexity of genotype-phenotype relationships. Understanding the spectrum of clinical presentations and overlap arising from mutations will expedite ALS diagnosis. Finally, ALS aggregates with neuropsychiatric illnesses, such as psychosis and suicide.35 ALS and schizophrenia share a risk gene, GLT8D1,36 as well as polygenic risk.37 Therefore, one can appreciate the full spectrum of neurodegenerative and neuropsychiatric conditions that may be present an ALS patient’s family history.
ALS prognosis
Established prognostic methods
Nearly every ALS patient asks a series of questions, which usually end with “How much time do I have left?” Access to reliable prognostic methods allows physicians to give patients and their families evidence-based answers. Despite important limitations,38 the ALS community currently relies on the ALS functional rating score-revised (ALSFRS-R),39 a scoring system that monitors the rate of disease progression. ALSFRS-R score changes do not necessarily reflect improvement in disease; for instance, symptom management (e.g., treating sialorrhea) or medical decisions (e.g., discontinuing non-invasive ventilation) impact ALSFRS-R score, even though there is no change in the patient’s underlying disease. The ALSFRS-R multi-dimensionality limits its clinical usefulness, especially in clinical trials,40 as well as its lack of responsiveness during plateau periods, which makes it hard to discern treatment effects in trials.41 Clinicians also derive prognostic value from respiratory tests, such as forced vital capacity (FVC);42 indeed, FVC is a predictive parameter in the ENCALS model (see below).
Emerging prognostic methods
Scoring systems
The self-reported Rasch-Built Overall ALS Disability Scale (ROADS) was developed to overcome ALSFRS-R limitations by ensuring that symptom management or medical decisions do not ameliorate the disease score, which instead reflects true changes in disease progression.43 Versus the ALSFRS-R, the 28-question ROADS better captures functional changes because it accounts for function at the upper and lower ranges of disability. Additionally, the scale has high test-retest reliability and is designed for a 1-point change to represent the same change in function across the whole score spectrum. This is a new scale not in wide clinical use, which will require validation; thus, whether ROADS will supplant or complement the ALSFRS-R requires further study.
Staging systems
A staging system identifies where an individual is in the disease course, thereby improving disease counseling and resource allocation. Staging systems are also useful in clinical trials to determine if an intervention reduces advancement from less to more severe disease stages. The straightforward King’s ALS staging defines four progressive stages linked to survival (Figure 1D).12 King’s staging shows the different progression of patients as well; bulbar onset patients, which require gastrostomy (Stage 4A) before non-invasive ventilation (Stage 4B) versus the opposite in limb-onset patients. ALS Milano-Torino Staging (ALS-MiToS) places patients at one of six stages based on select ALSFRS-R responses in four functional domains.44 In ALS-MiToS, staging depends on the number of functional domains lost (Figure 1E); Stage 0 is no loss, a patient at Stage 1 will have lost one functional domain, a patient at Stage 2 will have lost two functional domains etc., with stage 5 representing death. Patients likely progress from stage to stage, as opposed to skipping stages, with increasing probability of death with each stage. The King’s and ALS-MiToS systems are complementary; the King’s is superior for staging earlier in the disease, whereas ALS-MiToS outperforms later in the disease.45 Although neither is in clinical use, both staging systems describe progression and survival, albeit not without limitations,46 and could be useful in clinical trials.47
ENCALS survival model
The ENCALS survival model is a recent comprehensive approach for predicting ALS patient survival, defined as noninvasive ventilation for over 23 h per day, tracheostomy, or death.13 The model used data from 11,475 ALS patients from 14 ALS centers at several European sites. The model included sixteen clinical predictors, of which only eight reached statistical significance (p<0·001), including age at onset, time to diagnosis, ALSFRS-R progression rate, FVC, bulbar onset, definite ALS by revised El Escorial criteria, FTD, and C9orf72 repeat expansion. These predictors define five survival groups, very short (predicted median survival 17·7 months), short (25·3 months), intermediate (32·2 months), long (43·7 months), and very long (91·0 months). The ENCALS survival model is a new prediction model and unlocks the potential for personalized prognosis for ALS patients, which is essential for a disease of such great heterogeneity. The ENCALS model more accurately modeled the life expectancy of Stephen Hawking,48 in stark contrast to the 2-year expectancy he was given at his diagnosis.
Emerging ALS diagnostic and prognostic biomarkers
Currently, ALS diagnosis relies on an integrative approach, which leverages clinical history (presenting illness, symptom evolution), physical examination (testing strength, reflexes), and/or confirmatory tests, e.g., electromyography (EMG) (see ALS diagnosis section).49 Genetic testing is gaining traction in ALS diagnosis but is not without caveats (Table 3 and accompanying review). EMG and nerve conduction studies are the mainstay of electrodiagnostic tests for ALS, although additional methods are available (Panel 2). While diagnosis remains suboptimal, there is an expanding toolbox of available methods and novel ALS biomarkers. Presently, most of these approaches are only employed in the research setting and have not been validated for clinical use. Herein, we present advances in several recent diagnostic and prognostic ALS biomarkers, including biofluid, imaging (MRI, PET), and electrophysiological measurements.
Panel 2. Prognostic and diagnostic ALS biomarkers.
Diagnostic methods in clinical use
Criteria:
The most frequently used revised El Escorial24 (i.e., Airlie House)26 and Awaji25 criteria; rate the degree of diagnostic certainty (possible to probable to definite), based on number of affected segments and/or electrophysiological findings. El Escorial criteria do offer some prognostic information, e.g., definite ALS progresses faster.13
Electrodiagnostic:
Cornerstone method for clinical practice is needle electromyography (EMG) recordings to confirm the presence and extent of lower motor neuron (LMN) involvement.49 EMG measures fasciculation potentials, fibrillation potentials, and positive sharp waves in resting muscle and recruitment and configuration of motor unit potentials (MUP) with muscle activation.49 Additional techniques include repetitive nerve stimulation and single-fiber EMG to exclude neuromuscular junction disorders in the appropriate clinical context as these can be abnormal in individuals with ALS due and nerve conduction studies to exclude multifocal motor neuropathy.
Ultrasound:
LMN fasciculations; often an early sign.104 Method is not very specific, so differential diagnosis may be needed. Ultrasound can also be used to localize specific muscle groups during needle EMG.
MRI:
To exclude cerebral and spinal ALS mimics.49
Genetic testing:
Around 40 ALS genes are known, and the list is continually growing. Genetic testing is an expanding aspect of clinical ALS care. However, for new variants, there is uncertainty regarding pathogenicity, penetrance, and overlap with other neurological illnesses, which must be considered with care. Please see accompanying review.
Diagnostic methods in the research setting
Criteria:
Gold Coast criteria are simplified criteria to define ALS, particularly in the early stages, by considering:28
Progressive motor impairment, documented by history or repeated clinical assessment, preceded by normal motor function.
Upper and lower motor neuron dysfunction in at least one body region, or lower motor neuron dysfunction in at least two body regions.
Investigative findings that exclude alternative diseases.
Electrodiagnostic:
Motor evoked potential (MEP), motor unit number estimation (MUNE), motor unit number index (MUNIX), and electrical impedance myography, are methods to quantitate the number of functioning LMN motor units.105 Upper motor neuron (UMN) involvement is assessed by cortical hyperexcitability, spectral electroencephalogram (EEG) mapping, and magnetoencephalography. Hyperexcitability measured by transcranial magnetic stimulation (TMS) and has shown some diagnostic utility;84–88 will be useful as an adjunct to existing methods but requires additional research to evaluate how to integrate it into eventual clinical care and to evaluate sensitivity and specificity. EEG and magnetoencephalography are very novel, and their potential as diagnostic tools remains unknown. Previously reviewed.105
MRI and PET:
Advanced brain and spinal cord imaging offer some diagnostic insight;58,61 will be useful as an adjunct to existing methods but require additional research to evaluate how to integrate it into eventual clinical care and better evaluate sensitivity and specificity. Previously reviewed.56
Biosample biomarkers:
Focus is on neurofilaments, but various biosample markers have been previously reviewed.106 Phosphorylated neurofilament heavy chain (pNFH) and neurofilament light chain (NFL) have some diagnostic utility;50,51,55 however, elevated neurofilaments are characteristic of neurodegenerative diseases generally,54 so diagnostic value remains uncertain, though could serve as an adjunct to other methods.
Prognostic methods in clinical use
ALSFRS-R score:
The ALS functional rating score-revised (ALSFRS-R) is an established and clinically used scoring system to monitor the rate of ALS progression.38,39
Spirometry:
Respiratory tests, such as forced vital capacity (FVC), generate prognostic value.42
Prognostic methods in the research setting
Scales and scoring:
The 28-question self-reported Rasch-Built Overall ALS Disability Scale (ROADS) captures functional changes at the upper and lower ranges of disability; 1-point changes reflect the same change in function across the whole score spectrum; has high test-retest reliability;43 still requires validation.
Four-stage King’s ALS staging12 and six-stage ALS Milano-Torino Staging (ALS-MiToS);44 patients progress across stages over the disease course and median survival drops from stage to stage; King’s is superior for staging earlier in the disease, whereas ALS-MiToS outperforms later in the disease;45 neither is in clinical use but could be useful in clinical trials.47
Prediction models:
ENCALS survival model predicts individual ALS patient survival (noninvasive ventilation for over 23 h per day, tracheostomy, or death) by leveraging eight characteristics: age at onset, time to diagnosis, ALSFRS-R progression rate, FVC, bulbar onset, definite ALS by revised El Escorial criteria, FTD, C9orf72 repeat expansion;13 predictors define five survival groups; not in clinical use but could be useful for informing patients and their families.
Electrodiagnostic:
Hyperexcitability by TMS has shown some prognostic utility;84–88 may be useful as an adjunct to existing methods but requires additional research to evaluate how to integrate it into eventual clinical care.89–91
MRI and PET:
Advanced brain and spinal cord imaging offer some prognostic insight;59,60,62 will be useful as an adjunct to existing methods but requires additional research to evaluate how to integrate it into eventual clinical care. Previously reviewed.56
Biosample biomarkers:
Focus is on neurofilaments, but various biosample markers have been previously reviewed.106 pNFH and NFL have some prognostic utility, but it is generally low;50,52 may be useful for lowering ALS trial participants needed by combining with ALSFRS-R.53 Another new biomarker is neutrophil-to-lymphocyte ratio,107 which positively correlates with shorter survival.
Neurofilaments
Neurofilaments are neuronal cytoskeletal proteins that control neuron shape. Two markers exist, phosphorylated neurofilament heavy chain (pNFH) in cerebrospinal fluid (CSF) and neurofilament light chain (NFL) in plasma, serum, or CSF. Elevated pNFH and NFL correlate with ALS versus controls.50 NFL levels also rise in presymptomatic individuals harboring an ALS gene one year before phenoconversion.51 Higher pNFH and NFL levels in ALS patients correlate with more aggressive disease and shorter survival, but are of low prognostic value.50,52 Incorporating baseline NFL into mixed effects models of ALSFRS-R slopes may lower the number of ALS trial participants needed.53 However, elevated neurofilaments are characteristic of neurodegenerative diseases generally54 though they may still be relatively diagnostic of ALS;55 thus, overall, neurofilaments remain of uncertain diagnostic and prognostic use alone, but could add value when combined with other methods.
Brain and spinal cord imaging
Functional and structural brain imaging is a rapidly growing field in ALS56 with considerable recent progress with the advent of multisite ALS imaging protocols,57 studies indicating feasibility for early diagnosis58 and possibility of prognosis,59,60 and for insight into pathogenesis, e.g., quantifying brain atrophy and connectomics (connections between brain regions). Spinal cord MRI is widely used to rule out diagnostic considerations other than ALS,49 but more advanced diagnostic61 and prognostic62 applications are emerging.63
MRI of ALS patients assesses tissue appearance, brain structure volumes, and diffusivity, among other factors (Appendix Table and Figure). Routine MRI does not identify persons with ALS; findings, if present, may be higher corticospinal tract (CST) and corpus callosum intensity in ALS patients.64 A hypo-intensity of the cortical band along the precentral gyrus, called the “motor band sign”, may be characteristic of ALS and can be detected by routine susceptibility-weighted images.65 However, advanced MRI analyses generate deeper insight using post image processing, e.g., assessing brain volumes by mapping brain regions, versus established clinical standards. Advanced MRI of ALS patients indicates, to variable degrees, atrophy in the precentral gyri, posterior cingulate cortex, thalamus, caudate, pallidum, putamen, hippocampus, and amygdala.66 Additional MRI techniques include diffusion tensor imaging (DTI) and diffusion weighted imaging (DWI), which focus on white matter tracts. Studies consistently report changes to CST, corticopontine tract, corticorubral tract, corticostriatal pathway, and corpus callosum.66,67 Diffusion kurtosis, an DTI adjunct, is a newer, more sensitive neuroimaging technique of white matter abnormalities, which may more accurately identify ALS patients.68 White matter changes represent the earliest ALS findings, followed by gray matter changes.69 Spinal cord findings in ALS suggest a drop in CST magnetization transfer ratio and potential DTI changes, although progressive atrophy and cross-sectional area may be the most accurate biomarkers.63
The complexity of ALS pathology advocates multimodal MRI, which combines multiple advanced MRI techniques. Multimodal MRI of both brain volume and white matter integrity has 85·7% sensitivity and 78·4% accuracy for discriminating ALS from control scans.70 A multi-site Italian study evaluated global and lobar connectivity in ALS using DTI, fractional anisotropy (white matter track integrity measure), and resting state functional MRI.71 The results found widespread connectomics dysfunction with early degeneration of brain motor regions followed by breakdown in functional connections, leading to cognitive decline.71 Multimodal longitudinal MRI can monitor spatiotemporal spread in ALS via the brain connectome and potentially serve as a disease biomarker.67 Finally, quantitative susceptibility mapping MRI measures iron accumulation in motor cortex,72 which can be coupled with white matter assessments (DTI, DWI, diffusion kurtosis) to identify early track changes associated with metal toxicity in ALS. Similarly, multimodal MRI of spinal cord in ALS participants leveraged fractional anisotropy, magnetization transfer ratio, and cross-sectional area to build a survival prediction model.62
PET imaging is another modality that may facilitate ALS diagnosis and prognosis (Appendix Table). [18F]-FDG PET to measure glucose metabolism reported hypometabolism in the frontal cortex and hypermetabolism in the temporal cortex, cerebellum, and brainstem in a two-site study of ALS patients.73 PET [11C]-PBR28 brain uptake, a surrogate of microglial activation, is elevated in the bilateral precentral and paracentral gyri of ALS patients versus controls, and colocalizes with cortical thinning, as assessed by integrated MRI imaging,74 but may not correlate with clinical progression.74 Integrating spinal cord with brain in [18F]-FDG PET allows differentiation of ALS from ALS mimics.75
Overall, tremendous progress has been made in advanced brain MRI and PET along with more emerging advanced spinal cord imaging applications, which feasibly could improve diagnosis58,61 and prognosis.59,60,62 Although we anticipate imaging will be useful as an adjunct to existing methods, additional research is required to evaluate how to integrate imaging into eventual clinical care and at the single patient level. Furthermore, most imaging studies focused on imaging in ALS versus controls; however, future studies will need to include patients with ALS mimic disorders to better evaluate sensitivity and specificity.58,75
Spectral electroencephalogram (EEG) mapping and magnetoencephalography
Electrophysiological measures are another technique to assess brain networks. High-density spectral EEG mapping measures the coherence of certain frequency bands between brain regions, generating a functional measure of brain connectivity in ALS.76,77 EEG changes occur to brain connectivity in both motor and non-motor systems, confirming ALS is not a pure motor disease, in agreement with MRI connectomics.77 Magnetoencephalography shows brain networks become increasingly connected during ALS progression, indicating a dysfunctional, modified brain topology.78 These findings are significant because reorganization of brain connections could potentially predict disease spread.67 ALS connectomics studies are needed coupling multimodal MRI, high-density spectral EEG, and magnetoencephalography to further understand how brain structural changes and corresponding connectivity changes associate with ALS symptomatology and disease course. EEG and magnetoencephalography connectomics are very novel techniques not presently in clinical use and their potential as diagnostic and prognostic tools remains unknown.
Hyperexcitability
Excessive cortical excitability, i.e., hyperexcitability, in ALS is increasingly recognised as a pathophysiological mechanism of the neurodegenerative cascade.79 Clinically, hyperexcitability manifests as fasciculations combined with upper motor neuron features of increased tone and hyperreflexia.80 Hyperexcitability is linked to excitotoxicity from excessive glutamate receptor activity at the synaptic cleft, leading to motor neuron death.23,81 Cortical motor neuronal hyperexcitability can be captured by transcranial magnetic stimulation (TMS) techniques.82 A TMS coil is placed over the motor cortex and responses are recorded from the contralateral hand in the abductor pollicis brevis muscle. Short interval intracortical inhibition (SICI) and short interval intracortical facilitation (SICF) are extracted and represent interneuron function.
There is presymptomatic decrease in SICI and increase in SICF in ALS.83 TMS detects cortical hyperexcitability across a range of ALS phenotypes and differentiates ALS from non-ALS disorders with sensitivity (73·21%) and specificity (80·88%) at early disease stages.84 TMS distinguishes ALS, with cortical hyperexcitability predominance, from PLS, with cortical inexcitability predominance.85 TMS can also investigate pathologic spread using hyperexcitability as a surrogate by recording responses to TMS at the tibialis anterior in addition to the abductor pollicis brevis. Analysis of ALS patients demonstrates heterogeneity in cortical dysfunction by body region; cortical hyperexcitability predominates in the upper limbs, whereas cortical inexcitability predominates in the lower limbs versus controls.86 Furthermore, cortical hyperexcitability correlates with the clinically affected body region; ALS patients exhibit focal asymmetry at the onset site early in disease, but widespread hyperexcitability alterations in late stages.87 Cortical motor hyperexcitability may also detect cognitive dysfunction and cortical resting motor threshold distinguishes pure ALS, ALS-FTD, and pure FTD.88
The role of TMS in prognosis is less established compared to diagnosis. A recent longitudinal study of suspected ALS participants found cortical hyperexcitability increases with longer disease duration, indicating a potential link to ALS progression.89 Cortical inexcitability may predict a poorer clinical trajectory in ALS with inexcitability in all four limbs correlating with younger age, lower limb onset, greater extent of functional disability, and more rapid disease progression.90 Thus, cortical hyperexcitability may improve our ability to predict clinical outcomes. It could also serve as a biomarker for drug activity, e.g., in clinical trials of ezogabine, an activator of voltage gated potassium channels.91
Presently, TMS is not in clinical use, although it does appear to offer some diagnostic and prognostic utility and likely will be informative as an adjunct to pre-existing methods. However, future research will determine the full clinical and research potential of TMS in ALS, and determine whether this novel, electrophysiological assessment will become a fully accepted disease biomarker.
Machine learning in ALS
ALS is a highly heterogenous syndrome of various genetic and unknown etiologies converging on a “typical” ALS phenotype with diverse clinical presentations. Machine learning approaches can analyze large datasets (e.g., clinical, demographic, electrophysiological, imaging, morphology) in an agnostic, data-driven manner to develop diagnostic and prognostic models.92 Tang et al. employed unsupervised clustering of clinical data encompassing 8,000 patients, 3 million records, and 200 clinical features over 12 months from the Patient Data Pooled Resource Open-Access ALS Clinical Trials Database archive.93 Unsupervised clustering yielded four consistent computable phenotypes, defined by slope change in ALSFRS-R, with over 95% diagnostic accuracy, based on multivariate features. Deep learning modeling, a form of machine learning, was used for prognosis, and predicted ALS patient survival in this cohort when incorporating TDP-43 aggregation and morphology and MRI connectivity data with clinical characteristics.67 Further research will determine whether machine learning may unlock a way forward for diagnosing and prognosticating for ALS patients at the individual level by integrating multi-domain information into diagnostic and prediction models.
Summary
Overall, most of the recent novel diagnostic and prognostic ALS tests are limited to the research setting. Further studies are needed to determine whether these approaches will be useful in a clinical real-world setting. This will entail studies enrolling participants with diseases mimicking ALS and longitudinal studies against validated prognostic scales to evaluate their potential for improved ALS diagnosis (sensitivity/specificity) and prognosis. Additionally, it will be necessary to determine how to apply findings made from large cohort studies to individual patient diagnosis and prognosis. Until more specific and sensitive tests are developed, ALS diagnosis will remain an integrative and iterative process reliant on clinical history, physical examination, and/or confirmatory clinical electrodiagnostic tests.
Conclusion
Although diagnosis and prognosis procedures in ALS have remained essentially unchanged in the past decade, except for genetic testing, research is on-going into new diagnostic and prognostic criteria, staging and scoring systems, prediction models, and biomarkers, e.g., neurofilament assessment, hyperexcitability, imaging techniques. Even within the realm of genetic testing, questions remain regarding variant pathogenicity, penetrance, and overlap with other neurological disorders, outlined in our accompanying review. It is anticipated and hoped that advances in these areas will expedite ALS diagnosis and prognosis in the future. Faster diagnosis will allow clinicians to initiate care earlier for patients, which may enhance effectiveness or ensure administration within a therapeutic window. Ultimately, insight into the long preclinical phase of ALS will be necessary to truly facilitate early diagnosis.94 Improved prognosis will give ALS patients and their families a better understanding of the anticipated disease course, aiding medical decisions and planning. A major advance is the recognition of ALS as a disease with both motor and non-motor features, which has implications for diagnosis, management, and prognosis. Importantly, cognitive symptoms are not presently considered in clinical criteria and scales, yet integration may improve diagnosis and prognosis. We foresee that our deepening understanding of ALS and widening diagnostic and prognostic toolbox will lead to better care for patients with ALS.
Search strategy and selection criteria
We searched PubMed for English language articles from August 3rd 2021 to August 12th 2021 with the terms, in addition to amyotrophic lateral sclerosis: Epidemiology section: “epidemiology”. ALS phenotypic heterogeneity section: “phenotype”. Established and emerging ALS diagnostic criteria section: “diagnostic”. Non-motor ALS symptoms section: “cognition” and “cognitive”. Clinical overlap: “GWAS” plus each ALS gene in turn. Emerging ALS diagnostic tools section: “neurofilaments”, “Amyotrophic Lateral Sclerosis”[Mesh] and “magnetic[title] or mri[title]”, “Amyotrophic Lateral Sclerosis”[Mesh] and “connectome[title]”, “Amyotrophic Lateral Sclerosis”[Mesh] and “PET[title] or positron[title]”, “EEG”, “hyperexcitability”. ALS prognosis section: “prognosis”. Additional searches during revisions were conducted from November 15th to November 19th with the terms, in addition to amyotrophic lateral sclerosis: Brain and spinal cord imaging section: “spinal cord”, “multimodal MRI”, “PET”. Machine learning in ALS: “machine learning”. Panel 2: “biomarker”, “fluid”, “electrodiagnostic”, “electrophysiological”. In addition, authors used articles from their personal file and references from the identified articles.
Supplementary Material
Acknowledgments
SAG and ELF receive funding from the National ALS Registry/CDC/ATSDR (1R01TS000289; R01TS000327); National ALS Registry/CDC/ATSDR CDCP-DHHS-US (CDC/ATSDR 200–2013-56856); NIEHS K23ES027221; NIEHS R01ES030049; NINDS R01NS127188; NeuroNetwork for Emerging Therapies, the NeuroNetwork Therapeutic Discovery Fund, the Peter R. Clark Fund for ALS Research, the Sinai Medical Staff Foundation, Scott L. Pranger, University of Michigan. OH receives funding from Science Foundation Ireland (13/RC2015, 16/RC/3948), Thierry Latran Foundation, Health Research Board (Ireland). AAC is an NIHR Senior Investigator (NIHR202421). This is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND - www.jpnd.eu (United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1)) and through the Motor Neurone Disease Association. This study represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. AC received funding from the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016–02362405); the Progetti di Rilevante Interesse Nazionale program of the Ministry of Education, University and Research (grant 2017SNW5MB); the European Commission’s Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867); and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. MCK receives funding from the National Health and Medical Research Council of Australia Pro-gram Grant (APP1132524), Partnership Project (APP1153439) and Practitioner Fellowship.
Declaration of interests
Dr. Goutman reports grants from National ALS Registry/CDC/ATSDR, grants from National ALS Registry/CDC/ATSDR, grants from National ALS Registry/CDC/ATSDR CDCP-DHHS-US, grants from NIEHS, grants from NIEHS, grants from NINDS, grants from NINDS, grants from ALS Association, grants from NeuroNetwork for Emerging Therapies, grants from NeuroNetwork Therapeutic Discovery Fund, grants from Peter R. Clark Fund for ALS Research, grants from Sinai Medical Staff Foundation, grants from Scott L. Pranger, University of Michigan, during the conduct of the study; personal fees from Biogen, personal fees from ITF Pharma, personal fees from Watermark, outside the submitted work; In addition, Dr. Goutman has a patent US20200253977A1 issued. Dr. Hardiman reports grants from Science Foundation Ireland, during the conduct of the study; personal fees from Taylor and Francis Publisher, personal fees from Biogen Idec, personal fees from Cytokinetics, grants from Novartis, grants from Merck, grants from Ionis, grants from Cytokinetics, grants from Research Motor Neuron, outside the submitted work. Dr. Al-Chalabi reports grants from Medical Research Council through JPND, grants from Horizon 2020 (European Commission), grants from MND Association, grants from ALS Association, grants from National Institute for Health Research (NIHR), grants from MND Scotland, grants from My Name’5 Doddie Foundation, during the conduct of the study; other from Amylyx, other from Apellis, other from Biogen, other from Brainstorm, other from Cytokinetics, other from GSK, other from Lilly, other from Mitsubishi Tanabe Pharma, other from Novartis, other from OrionPharma, other from Quralis, other from Wave Pharmaceuticals, outside the submitted work; and Scientific lead for the United2EndMND Coalition; Royalties for the following books: OneWorld Publications – The Brain: A Beginner’s Guide, Cold Spring harbor Laboratory Press – The Genetics of Complex Human Disease: A Laboratory Manual. Dr. Chio reports grants from null, during the conduct of the study; personal fees from Mitsubishi Tanabe, personal fees from Biogen, personal fees from Denali Pharmaceutical, personal fees from Cytokinetics, personal fees from Amylyx, personal fees from Lilly, grants from Biogen, outside the submitted work. Dr. Savelieff has nothing to disclose. Dr. Kiernan reports Editor-in-Chief of the Journal of Neurology, Neurosurgery & Psychiatry (BMJ Publishers UK). Dr. Feldman reports grants from National ALS Registry/CDC/ATSDR, grants from National ALS Registry/CDC/ATSDR, grants from National ALS Registry/CDC/ATSDR CDCP-DHHS-US, grants from NIEHS, grants from NIEHS, grants from NINDS, grants from NINDS, grants from ALS Association, grants from NeuroNetwork for Emerging Therapies, grants from NeuroNetwork Therapeutic Discovery Fund, grants from Peter R. Clark Fund for ALS Research, grants from Sinai Medical Staff Foundation, grants from Scott L. Pranger, University of Michigan, during the conduct of the study; In addition, Dr. Feldman has a patent US20200253977A1 issued.
References
- 1.Goutman SA. Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. Continuum (Minneap Minn). Oct 2017;23(5, Peripheral Nerve and Motor Neuron Disorders):1332–1359. doi: 10.1212/con.0000000000000535 [DOI] [PubMed] [Google Scholar]
- 2.Richards D, Morren JA, Pioro EP. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J Neurol Sci. Oct 15 2020;417:117054. doi: 10.1016/j.jns.2020.117054 [DOI] [PubMed] [Google Scholar]
- 3.Galvin M, Ryan P, Maguire S, et al. The path to specialist multidisciplinary care in amyotrophic lateral sclerosis: A population- based study of consultations, interventions and costs. PLoS One. 2017;12(6):e0179796. doi: 10.1371/journal.pone.0179796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu L, Liu T, Liu L, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. Apr 2020;267(4):944–953. doi: 10.1007/s00415-019-09652-y [DOI] [PubMed] [Google Scholar]
- 5.Chiò A, Moglia C, Canosa A, et al. ALS phenotype is influenced by age, sex, and genetics: A population-based study. Neurology. Feb 25 2020;94(8):e802–e810. doi: 10.1212/wnl.0000000000008869 [DOI] [PubMed] [Google Scholar]
- 6.Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. Nov 1 2019;76(11):1367–1374. doi: 10.1001/jamaneurol.2019.2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chio A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. May 18 2017;7(1):2116. doi: 10.1038/s41598-017-02364-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nature reviews Neurology. Nov 2014;10(11):661–70. doi: 10.1038/nrneurol.2014.184 [DOI] [PubMed] [Google Scholar]
- 9.Talman P, Duong T, Vucic S, et al. Identification and outcomes of clinical phenotypes in amyotrophic lateral sclerosis/motor neuron disease: Australian National Motor Neuron Disease observational cohort. BMJ Open. Sep 30 2016;6(9):e012054. doi: 10.1136/bmjopen-2016-012054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiò A, Calvo A, Moglia C, Mazzini L, Mora G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. Jul 2011;82(7):740–6. doi: 10.1136/jnnp.2010.235952 [DOI] [PubMed] [Google Scholar]
- 11.Moglia C, Calvo A, Brunetti M, Chiò A, Grassano M. Broadening the clinical spectrum of FUS mutations: a case with monomelic amyotrophy with a late progression to amyotrophic lateral sclerosis. Neurol Sci. 2021:1207–1209. vol. 3. [DOI] [PubMed] [Google Scholar]
- 12.Roche JC, Rojas-Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain : a journal of neurology. Mar 2012;135(Pt 3):847–52. doi: 10.1093/brain/awr351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. The Lancet Neurology. May 2018;17(5):423–433. doi: 10.1016/S1474-4422(18)30089-9 [DOI] [PubMed] [Google Scholar]
- 14.Rabinovici GD, Stephens ML, Possin KL. Executive dysfunction. Continuum (Minneap Minn). Jun 2015;21(3 Behavioral Neurology and Neuropsychiatry):646–59. doi: 10.1212/01.CON.0000466658.05156.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. May 2017;18(3–4):153–174. doi: 10.1080/21678421.2016.1267768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinto-Grau M, Donohoe B, O’Connor S, et al. Patterns of Language Impairment in Early ALS. Neurology: Clinical Practice. 2020:10.1212/CPJ.0000000000001006. doi: 10.1212/cpj.0000000000001006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. Oct 9 2018;91(15):e1370–e1380. doi: 10.1212/WNL.0000000000006317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholson K, Murphy A, McDonnell E, et al. Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve. Jan 2018;57(1):20–24. doi: 10.1002/mus.25712 [DOI] [PubMed] [Google Scholar]
- 19.Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology. Apr 23 2013;80(17):1590–7. doi: 10.1212/WNL.0b013e31828f18ac [DOI] [PubMed] [Google Scholar]
- 20.Caga J, Hsieh S, Highton-Williamson E, et al. The burden of apathy for caregivers of patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. Nov 2018;19(7–8):599–605. doi: 10.1080/21678421.2018.1497659 [DOI] [PubMed] [Google Scholar]
- 21.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature Mar 4 1993;362(6415):59–62. doi: 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- 22.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron Oct 20 2011;72(2):257–68. doi: 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiernan MC, Vucic S, Talbot K, et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol. Feb 2021;17(2):104–118. doi: 10.1038/s41582-020-00434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. Dec 2000;1(5):293–9. [DOI] [PubMed] [Google Scholar]
- 25.Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. Nov 2012;69(11):1410–6. doi: 10.1001/archneurol.2012.254 [DOI] [PubMed] [Google Scholar]
- 26.van den Berg LH, Sorenson E, Gronseth G, et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology. Apr 2 2019;92(14):e1610–e1623. doi: 10.1212/WNL.0000000000007242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vucic S, Ferguson TA, Cummings C, et al. Gold Coast diagnostic criteria: Implications for ALS diagnosis and clinical trial enrollment. Muscle Nerve. Aug 11 2021;doi: 10.1002/mus.27392 [DOI] [PubMed] [Google Scholar]
- 28.Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. Aug 2020;131(8):1975–1978. doi: 10.1016/j.clinph.2020.04.005 [DOI] [PubMed] [Google Scholar]
- 29.Hannaford A, Pavey N, van den Bos M, et al. Diagnostic Utility of Gold Coast Criteria in Amyotrophic Lateral Sclerosis. Ann Neurol. May 2021;89(5):979–986. doi: 10.1002/ana.26045 [DOI] [PubMed] [Google Scholar]
- 30.Pugdahl K, Camdessanché JP, Cengiz B, et al. Gold Coast diagnostic criteria increase sensitivity in amyotrophic lateral sclerosis. Clin Neurophysiol. Sep 8 2021;doi: 10.1016/j.clinph.2021.08.014 [DOI] [PubMed] [Google Scholar]
- 31.Shen D, Yang X, Wang Y, et al. The Gold Coast criteria increases the diagnostic sensitivity for amyotrophic lateral sclerosis in a Chinese population. Transl Neurodegener. Aug 9 2021;10(1):28. doi: 10.1186/s40035-021-00253-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Estevez-Fraga C, Magrinelli F, Hensman Moss D, et al. Expanding the Spectrum of Movement Disorders Associated With C9orf72 Hexanucleotide Expansions. Neurol Genet. Apr 2021;7(2):e575. doi: 10.1212/NXG.0000000000000575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. Jan 28 2014;82(4):292–9. doi: 10.1212/WNL.0000000000000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewan R, Chia R, Ding J, et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron. Feb 3 2021;109(3):448–460 e4. doi: 10.1016/j.neuron.2020.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devenney EM, Ahmed RM, Halliday G, Piguet O, Kiernan MC, Hodges JR. Psychiatric disorders in C9orf72 kindreds: Study of 1,414 family members. Neurology. Oct 16 2018;91(16):e1498–e1507. doi: 10.1212/WNL.0000000000006344 [DOI] [PubMed] [Google Scholar]
- 36.Yang CP, Li X, Wu Y, et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat Commun. Feb 26 2018;9(1):838. doi: 10.1038/s41467-018-03247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLaughlin RL, Schijven D, van Rheenen W, et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nature Communications. 2017/03/21 2017;8(1):14774. doi: 10.1038/ncomms14774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rooney J, Burke T, Vajda A, Heverin M, Hardiman O. What does the ALSFRS-R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. May 2017;88(5):381–385. doi: 10.1136/jnnp-2016-314661 [DOI] [PubMed] [Google Scholar]
- 39.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). Journal of the neurological sciences. Oct 31 1999;169(1–2):13–21. [DOI] [PubMed] [Google Scholar]
- 40.van Eijk RPA, de Jongh AD, Nikolakopoulos S, et al. An old friend who has overstayed their welcome: the ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. May 2021;22(3–4):300–307. doi: 10.1080/21678421.2021.1879865 [DOI] [PubMed] [Google Scholar]
- 41.Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology. Mar 1 2016;86(9):808–12. doi: 10.1212/wnl.0000000000002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirola A, De Mattia E, Lizio A, et al. The prognostic value of spirometric tests in Amyotrophic Lateral Sclerosis patients. Clin Neurol Neurosurg. Sep 2019;184:105456. doi: 10.1016/j.clineuro.2019.105456 [DOI] [PubMed] [Google Scholar]
- 43.Fournier CN, Bedlack R, Quinn C, et al. Development and Validation of the Rasch-Built Overall Amyotrophic Lateral Sclerosis Disability Scale (ROADS). JAMA neurology. Dec 30 2019;doi: 10.1001/jamaneurol.2019.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry Jan 015;86(1):38–44. doi: 10.1136/jnnp-2013-306589 [DOI] [PubMed] [Google Scholar]
- 45.Fang T, Al Khleifat A, Stahl DR, et al. Comparison of the King’s and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. May 2017;18(3–4):227–232. doi: 10.1080/21678421.2016.1265565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luna J, Couratier P, Lahmadi S, et al. Comparison of the ability of the King’s and MiToS staging systems to predict disease progression and survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. Apr 8 2021:1–9. doi: 10.1080/21678421.2021.1903506 [DOI] [PubMed] [Google Scholar]
- 47.Al-Chalabi A, Chiò A, Merrill C, et al. Clinical staging in amyotrophic lateral sclerosis: analysis of Edaravone Study 19. J Neurol Neurosurg Psychiatry. Feb 2021;92(2):165–171. doi: 10.1136/jnnp-2020-323271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westeneng HJ, Al-Chalabi A, Hardiman O, Debray TP, van den Berg LH. The life expectancy of Stephen Hawking, according to the ENCALS model. Lancet Neurol. Aug 2018;17(8):662–663. doi: 10.1016/s1474-4422(18)30241-2 [DOI] [PubMed] [Google Scholar]
- 49.Lenglet T, Camdessanché JP. Amyotrophic lateral sclerosis or not: Keys for the diagnosis. Rev Neurol (Paris). May 2017;173(5):280–287. doi: 10.1016/j.neurol.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 50.Huang F, Zhu Y, Hsiao-Nakamoto J, et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. Jul 2020;7(7):1103–1116. doi: 10.1002/acn3.51078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. Jul 2018;84(1):130–139. doi: 10.1002/ana.25276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. Jun 13 2017;88(24):2302–2309. doi: 10.1212/WNL.0000000000004029 [DOI] [PubMed] [Google Scholar]
- 53.Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. Jul 7 2020;95(1):e59–e69. doi: 10.1212/wnl.0000000000009559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gafson AR, Barthélemy NR, Bomont P, et al. Neurofilaments: neurobiological foundations for biomarker applications. Brain. Jul 1 2020;143(7):1975–1998. doi: 10.1093/brain/awaa098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halbgebauer S, Steinacker P, Verde F, et al. Comparison of CSF and serum neurofilament light and heavy chain as differential diagnostic biomarkers for ALS. J Neurol Neurosurg Psychiatry. Aug 20 2021;doi: 10.1136/jnnp-2021-327129 [DOI] [PubMed] [Google Scholar]
- 56.Kassubek J, Pagani M. Imaging in amyotrophic lateral sclerosis: MRI and PET. Curr Opin Neurol. Oct 2019;32(5):740–746. doi: 10.1097/wco.0000000000000728 [DOI] [PubMed] [Google Scholar]
- 57.Kalra S, Müller HP, Ishaque A, et al. A prospective harmonized multicenter DTI study of cerebral white matter degeneration in ALS. Neurology. Aug 25 2020;95(8):e943–e952. doi: 10.1212/wnl.0000000000010235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferraro PM, Agosta F, Riva N, et al. Multimodal structural MRI in the diagnosis of motor neuron diseases. Neuroimage Clin. 2017;16:240–247. doi: 10.1016/j.nicl.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agosta F, Spinelli EG, Riva N, et al. Survival prediction models in motor neuron disease. Eur J Neurol. Sep 2019;26(9):1143–1152. doi: 10.1111/ene.13957 [DOI] [PubMed] [Google Scholar]
- 60.Schuster C, Hardiman O, Bede P. Survival prediction in Amyotrophic lateral sclerosis based on MRI measures and clinical characteristics. BMC Neurol. Apr 17 2017;17(1):73. doi: 10.1186/s12883-017-0854-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Querin G, Bede P, El Mendili MM, et al. Presymptomatic spinal cord pathology in c9orf72 mutation carriers: A longitudinal neuroimaging study. Ann Neurol. Aug 2019;86(2):158–167. doi: 10.1002/ana.25520 [DOI] [PubMed] [Google Scholar]
- 62.Querin G, El Mendili MM, Lenglet T, et al. Spinal cord multi-parametric magnetic resonance imaging for survival prediction in amyotrophic lateral sclerosis. Eur J Neurol. Aug 2017;24(8):1040–1046. doi: 10.1111/ene.13329 [DOI] [PubMed] [Google Scholar]
- 63.El Mendili MM, Querin G, Bede P, Pradat PF. Spinal Cord Imaging in Amyotrophic Lateral Sclerosis: Historical Concepts-Novel Techniques. Front Neurol. 2019;10:350. doi: 10.3389/fneur.2019.00350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fabes J, Matthews L, Filippini N, Talbot K, Jenkinson M, Turner MR. Quantitative FLAIR MRI in Amyotrophic Lateral Sclerosis. Acad Radiol. Oct 2017;24(10):1187–1194. doi: 10.1016/j.acra.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roeben B, Wilke C, Bender B, Ziemann U, Synofzik M. The motor band sign in ALS: presentations and frequencies in a consecutive series of ALS patients. J Neurol Sci. Nov 15 2019;406:116440. doi: 10.1016/j.jns.2019.116440 [DOI] [PubMed] [Google Scholar]
- 66.Menke RAL, Proudfoot M, Talbot K, Turner MR. The two-year progression of structural and functional cerebral MRI in amyotrophic lateral sclerosis. Neuroimage Clin. 2018;17:953–961. doi: 10.1016/j.nicl.2017.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meier JM, van der Burgh HK, Nitert AD, et al. Connectome-Based Propagation Model in Amyotrophic Lateral Sclerosis. Ann Neurol. May 2020;87(5):725–738. doi: 10.1002/ana.25706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Welton T, Maller JJ, Lebel RM, Tan ET, Rowe DB, Grieve SM. Diffusion kurtosis and quantitative susceptibility mapping MRI are sensitive to structural abnormalities in amyotrophic lateral sclerosis. Neuroimage Clin. 2019;24:101953. doi: 10.1016/j.nicl.2019.101953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bede P, Hardiman O. Longitudinal structural changes in ALS: a three time-point imaging study of white and gray matter degeneration. Amyotroph Lateral Scler Frontotemporal Degener. May 2018;19(3–4):232–241. doi: 10.1080/21678421.2017.1407795 [DOI] [PubMed] [Google Scholar]
- 70.Schuster C, Hardiman O, Bede P. Development of an Automated MRI-Based Diagnostic Protocol for Amyotrophic Lateral Sclerosis Using Disease-Specific Pathognomonic Features: A Quantitative Disease-State Classification Study. PLoS One. 2016;11(12):e0167331. doi: 10.1371/journal.pone.0167331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Basaia S, Agosta F, Cividini C, et al. Structural and functional brain connectome in motor neuron diseases: A multicenter MRI study. Neurology. Nov 3 2020;95(18):e2552–e2564. doi: 10.1212/WNL.0000000000010731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acosta-Cabronero J, Machts J, Schreiber S, et al. Quantitative Susceptibility MRI to Detect Brain Iron in Amyotrophic Lateral Sclerosis. Radiology. Oct 2018;289(1):195–203. doi: 10.1148/radiol.2018180112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D’Hulst L, Van Weehaeghe D, Chio A, et al. Multicenter validation of [(18)F]-FDG PET and support-vector machine discriminant analysis in automatically classifying patients with amyotrophic lateral sclerosis versus controls. Amyotroph Lateral Scler Frontotemporal Degener. Nov 2018;19(7–8):570–577. doi: 10.1080/21678421.2018.1476548 [DOI] [PubMed] [Google Scholar]
- 74.Alshikho MJ, Zurcher NR, Loggia ML, et al. Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol. Jun 2018;83(6):1186–1197. doi: 10.1002/ana.25251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Weehaeghe D, Devrome M, Schramm G, et al. Combined brain and spinal FDG PET allows differentiation between ALS and ALS mimics. Eur J Nucl Med Mol Imaging. Oct 2020;47(11):2681–2690. doi: 10.1007/s00259-020-04786-y [DOI] [PubMed] [Google Scholar]
- 76.Nasseroleslami B, Dukic S, Broderick M, et al. Characteristic Increases in EEG Connectivity Correlate With Changes of Structural MRI in Amyotrophic Lateral Sclerosis. Cereb Cortex. Jan 1 2019;29(1):27–41. doi: 10.1093/cercor/bhx301 [DOI] [PubMed] [Google Scholar]
- 77.Dukic S, McMackin R, Buxo T, et al. Patterned functional network disruption in amyotrophic lateral sclerosis. Hum Brain Mapp. Nov 1 2019;40(16):4827–4842. doi: 10.1002/hbm.24740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sorrentino P, Rucco R, Jacini F, et al. Brain functional networks become more connected as amyotrophic lateral sclerosis progresses: a source level magnetoencephalographic study. Neuroimage Clin. 2018;20:564–571. doi: 10.1016/j.nicl.2018.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vucic S, Pavey N, Haidar M, Turner BJ, Kiernan MC. Cortical hyperexcitability: Diagnostic and pathogenic biomarker of ALS. Neurosci Lett. Aug 10 2021;759:136039. doi: 10.1016/j.neulet.2021.136039 [DOI] [PubMed] [Google Scholar]
- 80.Swash M, Burke D, Turner MR, et al. Occasional essay: Upper motor neuron syndrome in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. Mar 2020;91(3):227–234. doi: 10.1136/jnnp-2019-321938 [DOI] [PubMed] [Google Scholar]
- 81.Saba L, Viscomi MT, Caioli S, et al. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb Cortex. Apr 2016;26(4):1512–28. doi: 10.1093/cercor/bhu317 [DOI] [PubMed] [Google Scholar]
- 82.Huynh W, Dharmadasa T, Vucic S, Kiernan MC. Functional Biomarkers for Amyotrophic Lateral Sclerosis. Front Neurol. 2018;9:1141. doi: 10.3389/fneur.2018.01141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eisen A, Braak H, Del Tredici K, Lemon R, Ludolph AC, Kiernan MC. Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. Nov 2017;88(11):917–924. doi: 10.1136/jnnp-2017-315573 [DOI] [PubMed] [Google Scholar]
- 84.Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. May 2015;14(5):478–84. doi: 10.1016/s1474-4422(15)00014-9 [DOI] [PubMed] [Google Scholar]
- 85.Geevasinga N, Menon P, Sue CM, et al. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol. May 2015;22(5):826–31, e57–8. doi: 10.1111/ene.12669 [DOI] [PubMed] [Google Scholar]
- 86.Menon P, Yiannikas C, Kiernan MC, Vucic S. Regional motor cortex dysfunction in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. Aug 2019;6(8):1373–1382. doi: 10.1002/acn3.50819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dharmadasa T, Matamala JM, Howells J, Vucic S, Kiernan MC. Early focality and spread of cortical dysfunction in amyotrophic lateral sclerosis: A regional study across the motor cortices. Clin Neurophysiol. Apr 2020;131(4):958–966. doi: 10.1016/j.clinph.2019.11.057 [DOI] [PubMed] [Google Scholar]
- 88.Agarwal S, Highton-Williamson E, Caga J, et al. Motor cortical excitability predicts cognitive phenotypes in amyotrophic lateral sclerosis. Sci Rep. Jan 26 2021;11(1):2172. doi: 10.1038/s41598-021-81612-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Menon P, Higashihara M, van den Bos M, Geevasinga N, Kiernan MC, Vucic S. Cortical hyperexcitability evolves with disease progression in ALS. Ann Clin Transl Neurol. May 2020;7(5):733–741. doi: 10.1002/acn3.51039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dharmadasa T, Howells J, Matamala JM, et al. Cortical inexcitability defines an adverse clinical profile in amyotrophic lateral sclerosis. Eur J Neurol. Jan 2021;28(1):90–97. doi: 10.1111/ene.14515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wainger BJ, Macklin EA, Vucic S, et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. Feb 1 2021;78(2):186–196. doi: 10.1001/jamaneurol.2020.4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grollemund V, Pradat PF, Querin G, et al. Machine Learning in Amyotrophic Lateral Sclerosis: Achievements, Pitfalls, and Future Directions. Front Neurosci. 2019;13:135. doi: 10.3389/fnins.2019.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang M, Gao C, Goutman SA, et al. Model-Based and Model-Free Techniques for Amyotrophic Lateral Sclerosis Diagnostic Prediction and Patient Clustering. Neuroinformatics. Jul 2019;17(3):407–421. doi: 10.1007/s12021-018-9406-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benatar M, Turner MR, Wuu J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. Aug 2019;20(5–6):303–309. doi: 10.1080/21678421.2019.1587634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burke T, Pinto-Grau M, Lonergan K, et al. A Cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: subphenotypes, staging, cognitive predictors, and survival. Ann Clin Transl Neurol. May 2017;4(5):305–317. doi: 10.1002/acn3.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Elamin M, Pinto-Grau M, Burke T, et al. Identifying behavioural changes in ALS: Validation of the Beaumont Behavioural Inventory (BBI). Amyotroph Lateral Scler Frontotemporal Degener. Feb 2017;18(1–2):68–73. doi: 10.1080/21678421.2016.1248976 [DOI] [PubMed] [Google Scholar]
- 97.Goutman SA, Chen KS, Paez-Colasante X, Feldman EL. Emerging understanding of the genotype-phenotype relationship in amyotrophic lateral sclerosis. Handb Clin Neurol. 2018;148:603–623. doi: 10.1016/b978-0-444-64076-5.00039-9 [DOI] [PubMed] [Google Scholar]
- 98.Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. Jan 2018;17(1):94–102. doi: 10.1016/s1474-4422(17)30401-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marin B, Boumediene F, Logroscino G, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. Feb 1 2017;46(1):57–74. doi: 10.1093/ije/dyw061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chio A, Logroscino G, Traynor BJ, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–30. doi: 10.1159/000351153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mehta P, Kaye W, Raymond J, et al. Prevalence of Amyotrophic Lateral Sclerosis - United States, 2015. MMWR Morb Mortal Wkly Rep. Nov 23 2018;67(46):1285–1289. doi: 10.15585/mmwr.mm6746a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. Aug 11 2016;7:12408. doi: 10.1038/ncomms12408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Manjaly ZR, Scott KM, Abhinav K, et al. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. Oct 2010;11(5):439–42. doi: 10.3109/17482961003610853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Carvalho M, Kiernan MC, Swash M. Fasciculation in amyotrophic lateral sclerosis: origin and pathophysiological relevance. J Neurol Neurosurg Psychiatry. Sep 2017;88(9):773–779. doi: 10.1136/jnnp-2017-315574 [DOI] [PubMed] [Google Scholar]
- 105.Vucic S, Rutkove SB. Neurophysiological biomarkers in amyotrophic lateral sclerosis. Curr Opin Neurol. Oct 2018;31(5):640–647. doi: 10.1097/wco.0000000000000593 [DOI] [PubMed] [Google Scholar]
- 106.Verde F, Silani V, Otto M. Neurochemical biomarkers in amyotrophic lateral sclerosis. Curr Opin Neurol. Oct 2019;32(5):747–757. doi: 10.1097/wco.0000000000000744 [DOI] [PubMed] [Google Scholar]
- 107.Choi SJ, Hong YH, Kim SM, Shin JY, Suh YJ, Sung JJ. High neutrophil-to-lymphocyte ratio predicts short survival duration in amyotrophic lateral sclerosis. Sci Rep. Jan 16 2020;10(1):428. doi: 10.1038/s41598-019-57366-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.