

Abstract
Down syndrome (DS) is characterized by chronic neuroinflammation, peripheral inflammation, astrogliosis, imbalanced excitatory/inhibitory neuronal function, and cognitive deficits in both humans and mouse models. Suppression of inflammation has been proposed as a therapeutic approach to treating DS co-morbidities, including intellectual disability (DS/ID). Conversely, we discovered previously that treatment with the innate immune system stimulating cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF), which has both pro- and anti-inflammatory activities, improved cognition and reduced brain pathology in a mouse model of Alzheimer’s disease (AD), another inflammatory disorder, and improved cognition and reduced biomarkers of brain pathology in a phase II trial of humans with mild-to-moderate AD. To investigate the effects of GM-CSF treatment on DS/ID in the absence of AD, we assessed behavior and brain pathology in 12–14 month-old DS mice (Dp[16]1Yey) and their wild-type (WT) littermates, neither of which develop amyloid, and found that subcutaneous GM-CSF treatment (5 µg/day, five days/week, for five weeks) improved performance in the radial arm water maze in both Dp16 and WT mice compared to placebo. Dp16 mice also showed abnormal astrocyte morphology, increased percent area of GFAP staining in the hippocampus, clustering of astrocytes in the hippocampus, and reduced numbers of calretinin-positive interneurons in the entorhinal cortex and subiculum, and all of these brain pathologies were improved by GM-CSF treatment. These findings suggest that stimulating and/or modulating inflammation and the innate immune system with GM-CSF treatment may enhance cognition in both people with DS/ID and in the typical aging population.
Keywords: Down syndrome (DS), granulocyte-macrophage colony-stimulating factor (GM-CSF), intellectual disability (ID), Alzheimer’s disease (AD), cognition, Dp(16)1Yey (Dp16), glial-fibrillary acidic protein (GFAP), astrocyte, calretinin, interneuron
Introduction
Down syndrome (DS) is caused by an extra copy of all or part of human chromosome 21 (Hsa21), (1, 2) and is the most common genetic cause of intellectual disability (ID) (3–5). In addition, every person with DS will develop Alzheimer’s disease (AD) brain pathology by age 40, and most will exhibit AD-like dementia by age 60 (6–12). Lastly, compared to the typical population, people with DS are at increased risk for other nervous system abnormalities, such as seizures and autism spectrum disorder (8, 13). Because there has been a dramatic increase in the life expectancy for people with DS, from an average of approximately 10 years in the 1960s to an average of approximately 60 years today (14), there is a significant need for safe therapeutic treatments to improve the life-long cognitive abilities of people with DS so that they may enjoy expanded educational and employment opportunities, participate more fully in society, and live more independently.
The mechanism by which people with DS develop AD pathology and cognitive dysfunction clearly starts with the presence of the amyloid precursor protein gene (APP) on human chromosome 21 (Hsa21). Specifically, when present in three copies or when mutated, APP causes familial AD (FAD) and is likely to play a key role in the development of AD in people with DS, although other Hsa21-encoded proteins may also contribute (10, 12, 15). In contrast, the cellular and molecular bases of the many other diverse features of DS in both the brain and periphery are not well understood. Reduced brain size and neurological abnormalities, including impaired neurogenesis and oligodendrogenesis, decreased synaptogenesis, imbalanced excitatory/inhibitory neuronal activity, increased gliogenesis, altered astrocyte morphology and signaling, and astrogliosis are common in people with DS and are directly associated with ID (16–22). The long arm of Hsa21 harbors ~165 traditional protein-coding genes, ~50 genes encoding members of the family of keratin-associated proteins, a small number of characterized functional RNA genes, and several hundred genes that may be functional RNAs, protein-coding, or transcriptional noise (23). Although the functional annotation of these genes is far from extensive, several have been shown to play roles in brain development and/or to lead to learning/memory deficits when their gene expression levels are altered (15).
One hypothesis that has received increasing attention and is supported by several lines of evidence is that people with DS are in a state of chronic abnormal inflammation, including features of auto-inflammation. For example, analyses of a selected proteome in both plasma and brain tissue samples from people with DS have revealed dysregulation of inflammatory protein expression, such as striking increases in several pro-inflammatory cytokines and decreases in numerous complement cascade components (24–26). Another study revealed elevated levels of not only pro-inflammatory, but also anti-inflammatory, cytokines in plasma from children with DS (27, 28). These results were recently confirmed and expanded in a comprehensive analysis that showed elevated levels of both pro- and anti-inflammatory markers in the frontal cortex and hippocampi of all ages of people with DS, as well as in primary cultures of DS fetal brain tissue (29). Analyses of protein expression in the brains of mouse models of DS and in lymphoblastoid cell lines from people with DS also show upregulation of inflammation-related genes (15, 30–32).
Relevant to such observations of dysregulated immune responses in DS is the fact that four out of the six genes encoding interferon receptors reside on chromosome 21, thus potentially explaining the finding that the expression of interferon-responsive genes, as well as other downstream inflammatory genes, is upregulated in plasma and/or CSF samples from people with DS (25, 33, 34). The microRNA miR-155, which has been shown to promote inflammatory responses (35), also maps to Hsa21 and is overexpressed in people with DS.
Together, these findings have led investigators to attribute many features of people with DS to their state of chronic abnormal inflammation and to propose that suppressing inflammation may have therapeutic benefits across many features of the disorder (12, 24–26, 29, 36).
However, the role of immune system dysfunction and inflammation in people with DS and even in typical people with AD remains ambiguous. Observations of elevated levels of both pro- and anti-inflammatory biomarkers in DS or AD appear contradictory and led to discussions about which abnormalities are detrimental and which may be compensatory. For example, in contrast to the view that inflammation in AD or DS is the most appropriate target for therapeutic intervention, we proposed and have tested an alternative hypothesis that recruiting the innate immune system with all of its features may successfully ameliorate cognitive and pathological features of many inflammation-associated disorders, including AD and DS. This hypothesis grew out of our previous studies to determine the mechanism by which the inflammatory disease, rheumatoid arthritis, reduces the risk of AD and led to our finding that treatment with the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF), which is upregulated in RA, improves cognition in both a mouse model of AD and in middle-aged wild-type (WT) mice (37).
The finding that GM-CSF was a treatment for AD and normal aging, both of which are associated with inflammation, was surprising and can still be seen as potentially controversial because GM-CSF is usually considered to be pro-inflammatory because it upregulates many classic inflammatory cytokines such as IL-6 and TNFα and has been found in some studies to be upregulated in brains of AD and other dementia patients and in animal models thereof (38–42). Such correlative observations have even led to suggestions that blocking GM-CSF function may be an effective treatment for inflammation-associated diseases, including AD, and, most recently, COVID-19 (39, 43, 44). Nonetheless, our findings that GM-CSF treatment reduced cognitive deficits in mouse models of AD and during normal aging were later replicated by other groups (45, 46). Furthermore, treatment with recombinant human GM-CSF (sargramostim/Leukine®) was associated with improved cognition in leukemia patients with chemotherapy-associated cognitive impairment after chemo-ablation and hematopoietic stem cell transplantation (47), and treatment with GM-CSF/sargramostim was safe, significantly improved Mini-Mental State Exam (MMSE) scores, and partly restored levels of plasma biomarkers of AD brain pathology and neuronal damage (i.e., Aβ40 and Aβ42, total tau, and ubiquitin C-terminal hydrolase L1 [UCH-L1]) toward normal in participants with mild-to-moderate AD after only 15 days of subcutaneous injections in our recently completed Phase 2 clinical trial [NCT01409915], the results of which have been published (48).
Based on all of these findings, we hypothesized that GM-CSF may act as a general cognition enhancer, despite and possibly because of its pro-inflammatory/immune modulatory activity, and thus could potentially ameliorate the cognitive deficits of people of all ages with DS. In the current study, we used a mouse model of DS to test this novel hypothesis, which is in contrast to the commonly held view that inflammation contributes to DS-related, and possibly age-associated, deficits in cognition and should therefore be suppressed to effect therapy.
Methods and Materials
Animals
Animal models are essential for developing and testing therapeutic interventions for DS. It has proven challenging to generate a mouse model that is trisomic for orthologs of all Hsa21 genes because the orthologs map to regions of three mouse chromosomes: Mmu16, Mmu17, and Mmu10. The Mmu16 segment has received the most attention in DS mouse models, in part because of the large number of Hsa21 protein-coding orthologs (i.e., 102) that it harbors. The first viable, and currently the most well-studied, segmental trisomy mouse model of DS, the Ts65Dn mouse, is trisomic for 90 of the 102 Mmu16 orthologs (49). It also exhibits a number of phenotypes relevant to DS, including learning/memory deficits, thus emphasizing the contributions of Mmu16 orthologs to ID in DS. However, the Ts65Dn mouse is also trisomic for ~35 protein-coding genes whose orthologs do not reside on Hsa21 and are not trisomic in people with DS; thus, the presence of those extra genes may confound the interpretation of Ts65Dn phenotypes (23).
To improve on the potential deficiencies of the Ts65Dn mouse as a model of DS, other models have been generated in various ways. For example, the Dp(16)1Yey/+ mouse model of DS (referred to herein as Dp16) was generated using Cre/loxP-mediated chromosomal rearrangement to duplicate a 22.9 Mb segment of Mmu16 spanning the segment syntenic with Hsa21(50). It is not expected that Dp16 mice will show exactly the same deficits that have been reported in Ts65Dn mice because it was designed to better reflect human DS. Thus, these two models have some similarities, but they also have some important differences. Similar to other mouse models of DS, including Ts65Dn, Dp16 mice do not develop AD-related neuropathology, such as amyloid plaques, despite harboring three copies of the mouse APP gene, because the mouse Aβ peptide exhibits little propensity to aggregate into neurotoxic β-sheet oligomers and fibrils (51). One of the major differences is the identities of the triplicated genes they harbor that are related to Hsa21. Dp16 mice contain 100% of the Hsa21 genes that are located on Mmu16 triplicated and no other triplicated genes. In contrast, Ts65Dn mice are trisomic for 90% of the genes that are located on Mmu16, but they are trisomic for only ~65% of the total Hsa21 orthologs. Another significant and more concerning difference is that the Ts65Dn mouse model is also trisomic for ~30 protein coding genes that are located on Mmu17, but are not in the region syntenic with Hsa21. Therefore, it is unknown whether all of the deficits that have been observed in the Ts65Dn mice are attributable to the triplication of the 90% of the Hsa21 genes that are located on Mmu16, or whether they are attributable to the combined effects of the triplication of the 90% of the Hsa21 genes that are located on Mmu16 together with the triplication of the extra 30 genes from mouse Mmu17 that are not orthologs of Hsa21. Thus, although the Ts65Dn mouse is a popular model of DS that has been used widely over the last few decades, it has some limitations when compared to Dp16 mice.
Taking all of these facts into consideration, we decided to employ the Dp16 mouse model to test our hypothesis that GM-CSF may be used to treat cognitive deficits in people with DS. Specifically, we treated Dp16 mice and their WT littermates with GM-CSF or with placebo, assessed their cell and biomarker innate immune system/inflammatory response, their learning/memory performance, and brain pathology in astrocytes and interneurons by indirect immunofluorescence microscopy staining.
Male Dp16 mice on the C57BL/6J background were purchased from Jackson Laboratory (Stock # 013530) and crossed with female C57BL/6J (WT) mice to generate the litters used for these experiments. All animals were bred at the University of Colorado Anschutz Medical Campus and maintained in a room with HEPA-filtered air and a 14:10 light:dark cycle, fed a 6% fat diet, and provided with acidified (pH 2.5–3.0) water ad libitum. All procedures were approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with National Institutes of Health guidelines for the care and use of animals in research. Same-sex littermates were housed in the same cage after weaning. Mice were genotyped by polymerase chain reaction (PCR) using a protocol from the Jackson Laboratory (Bar Harbor, ME). For the behavioral and brain pathology experiments, we started with 19 WT male mice, 11 Dp16 male mice, 13 WT female mice, and 12 Dp16 female mice, but two of the WT male mice did not perform the RAWM task at the baseline assessment (they floated), and only the remaining 17 WT male mice were included in the post-treatment analyses. All physically healthy-looking WT mice were chosen from their littermates. All behavioral and brain pathology experiments were conducted with mice aged 12–14 months. Open Field and Y-maze experiments used both male and female Dp16 mice and their age- and sex-matched WT littermate controls; all other behavioral and brain pathology experiments used only male mice. Open Field and Y-maze tasks were performed between 9:00 AM and 11:00 AM, and the RAWM maze was assessed from 9:00 AM to 1:00 PM because more time is required to complete the task. For studies of plasma biomarkers, we used another set of mice, including 7 WT male mice, 8 Dp16 male mice, 3 WT female mice, and 2 Dp16 female mice, aged 12–15 months.
Drug Administration
All of the mice in the behavioral and brain pathology studies were injected subcutaneously five days/week, Monday-Friday, for five weeks with GM-CSF (5 µg/day) or with an equal volume of saline (200 μl/day) for a total of 24 injections over a period of 32 days including non-treatment days. As discussed, two WT male mice did not perform the RAWM task at the baseline assessment (they merely floated in place), and those two mice were not included in the post-treatment analyses. Mice were divided into four treatment groups: (1) WT mice injected with saline, including 9 males and 6 females; (2) WT mice injected with GM-CSF, including 8 males and 7 females; (3) Dp16 mice injected with saline, including 5 males and 6 females; and (4) Dp16 mice injected with GM-CSF, including 6 males and 6 females (Figure 1 shows the study timeline and assessments). For studies of plasma biomarkers, we used another set of mice, including 7 WT male mice, 8 Dp16 male mice, 3 WT female mice, and 2 Dp16 female mice, aged 12–15 months, that were injected with GM-CSF (5 µg/day) or with an equal volume of saline (200 μl/day) for 10 consecutive days (see Measurements of Plasma Biomarkers below).
Figure 1. Schematic of the Study Timeline.

Injections began on day 1 (D1) and ended on day 24 (D24). After the baseline pre-treatment assessments, the cohorts of Dp16 and WT control mice were injected five days/week with either GM-CSF (5 μg/day) or an equal volume of saline (200 μl/day), Monday-Friday, for five weeks for a total of 24 injections (ending on D24) over a period of 32 days including non-treatment days. There were four treatment groups for both female and male mice: (1) WT mice injected with saline (9 males; 6 females), (2) WT mice injected with GM-CSF (8 males; 7 females), (3) Dp16 mice injected with saline (5 males; 6 females), and (4) Dp16 mice injected with GM-CSF (6 males; 6 females). Both male and female mice were tested in the Open Field task at baseline and in the Y-maze task at baseline and post-treatment. Only male mice were used for the RAWM task at baseline and post-treatment because the female mice have poor swimming abilities at this age.
Behavioral Assessments
For all behavioral/activity assessments, on the day of testing, the mice were brought into the testing room and allowed to habituate for 30 min in their home cage.
Open Field
To evaluate locomotor and exploratory behavior, in naïve mice, each animal was placed in the center of an arena (44 cm x 44 cm) and allowed to explore for 10 min with the room lights on. Activity was recorded by a video tracking system using EthoVision XT software (Noldus). Total distance traveled, total time spent in the center zone, frequency of presence in the center zone, and speed of movement were all analyzed during the 10 min session.
Spontaneous Alternation Y-Maze
The spontaneous alternation Y-maze assesses spatial working memory. The apparatus is composed of three arms set at 120-degree angles from a center area. The arms are 40 cm long, 8 cm wide, and 15 cm high. The mouse was placed in the center area and allowed to explore freely; the session was stopped after the mouse had completed 20 arm entries or had explored for 8 min (if the mouse did not complete 20 entries within 8 min, we counted the total number of entries in 8 min). The EthoVision XT software and video tracking system was used to record the number of arms visited. Spatial working memory was measured as the percentage of the number of successful alternations (sequential entries into all three arms) divided by the total number of arm entries.
Baseline Radial Arm Water Maze (RAWM) Assessments
The RAWM is a hippocampal-based task measuring working memory. Only male mice were used for the RAWM because 12–14 month-old female mice had poor swimming abilities. The RAWM (52) was first performed as a baseline assessment at six days before the first injection of either GM-CSF or saline. The RAWM apparatus consists of six arms and is placed in a circular pool of water. A two-day RAWM task was performed in which a submerged (hidden) platform was placed in arm 4 throughout testing. The maze is located in a room with numerous cues on the walls and surroundings that the mice can use to orient within the maze.
On both Day 1 (learning/training) and Day 2 (testing), each mouse was exposed to 15 trials (60 sec each), run in three sessions: 1st session: 6 trials with 5 min rest between trials; 50 min rest before session 2; 2nd session: 6 trials with 5 min rest between trials; 50 min rest before session 3; and 3rd session: 3 trials with 5 min break between trials. Before the first trial, each mouse was allowed to spend 30 sec on the platform to habituate to the location of the platform with respect to the walls and room surroundings. During each trial, the mouse was started from a different arm using a randomization scheme such that during the 15 trials, the mouse started in each of the five arms without the platform three times. On Day 1 (training day), each mouse was gently placed into one of the five arms (excluding Arm 4, which contained the platform) and was allowed a maximum of 60 sec to find the platform; if the platform was not found during this time, the mouse was gently guided to it. The mouse remained on the platform for 10 sec and was then gently towel-dried and transferred to the home cage located on a heating pad, for a 5 min break between trials. Between sessions, the mouse rested for 50 min in the home cage.
On Day 2 (testing day), spatial memory was tested with the platform still located in arm 4. Similar to Day 1, each mouse was gently placed into one of the five vacant arms and again allowed a maximum of 60 sec to find the platform. As described for Day 1, 15 trials were performed with each mouse, run as three sessions with a 50 min break between sessions. Mouse behavior was recorded with a video camera located directly above the water pool. Both the number of errors (incorrect arm choices) and the escape latency were recorded for each trial. For analysis purposes, both Day 1 and Day 2 data from the RAWM were used separately in baseline assessments. After averaging the data from 15 trials using the time values for each individual mouse, the mean time to reach the platform between Dp16 and WT control mice was compared on both days.
Post-Treatment Alternating Y-maze
After the baseline pre-treatment behavioral testing, the cohorts of Dp16 and WT littermates were injected with either GM-CSF or saline. Post-treatment Y-maze performances of male and female mice were measured starting on treatment day 20 using the same protocol as for baseline measurements.
Post-Treatment RAWM Tasks
Male mice were evaluated post-treatment in the RAWM starting on treatment day 21. The protocol was the same as for baseline measurements, with the exception that on Day 2, the platform was located in arm 6, instead of in arm 4. To evaluate memory function post-treatment, the latency of each saline- or GM-CSF-injected mouse on post-treatment day 1 was normalized with its mean latency at block 5 of pre-treatment day 2 (baseline day 2), and the data were calculated relative to 100% of block 5 pre-treatment day 2. Normalized latencies were analyzed in blocks of three trials (for a total of five blocks from 15 trials), and the GM-CSF-injected group was compared with the saline-injected group. For quantitative analyses, averaging the normalized latency of post-treatment day 1 from blocks 1–5 and combining blocks 1–2 or blocks 4–5 were compared between GM-CSF- and saline-injected mice for each genotype. To assess the effects of GM-CSF treatment over time, the combined normalized latency of blocks 1–2 were compared with the combined normalized latency of blocks 4–5 in GM-CSF-treated Dp16 mice.
On Day 2 of the post-treatment RAWM testing, the platform was moved to a new arm (from arm 4 to arm 6) while other maze cues remained unchanged, and then each mouse performed 15 trials (60 seconds each) to evaluate learning flexibility. To assess learning flexibility, the latency of each saline- or GM-CSF-injected mouse at post-treatment day 2 was normalized with its mean latency at block 5 of post-treatment day 1, and the data were calculated relative to 100% of block 5 post-treatment day 1. Normalized latencies from post-treatment day 2 were analyzed in blocks of three trials (for a total of five blocks from 15 trials), and the GM-CSF-injected group was compared with the saline-injected group. For quantitative analyses, averaging the normalized latencies of post-treatment day 2 from all blocks 1–5 were compared between GM-CSF- and saline-injected mice for each genotype.
Measurements of Plasma Biomarkers
For studies of plasma biomarkers, we used another set of mice (15 males and 5 females, aged 12–15 months) that were injected subcutaneously with GM-CSF (5 µg/day) or with an equal volume of saline (200 μl/day) for 10 consecutive days, with the last injection at 8:00 AM on day 10. The mice were divided into four treatment groups: (1) WT mice injected with saline, including 3 males and 2 females; (2) WT mice injected with GM-CSF, including 4 males and 1 female; (3) Dp16 mice injected with saline, including 4 males and 1 female; and (4) Dp16 mice injected with GM-CSF, including 4 males and 1 female. Blood samples were collected at 4–6 h after the last injection on day 10 in microtubes with EDTA (Sarstedt Inc., Nümbrecht, Germany) from cardiac puncture under anesthesia. The blood samples were centrifuged at 1,500 RCF for 15 min at room temperature to separate the plasma. The plasma was then transferred to a clean microtube and centrifuged a second time at 2,200 RCF for 10 min at 4°C. The resultant plasma was collected and stored at −80°C until use.
We used a custom U-PLEX Metabolic Group 1 (Mouse) Multiplex Assay (Meso Scale Discovery, Rockville, MD, catalog number K152ACL-1) designed to measure 23 different analytes, including cytokines and metabolic proteins, in mouse plasma samples. Results from eight of the analytes were below the limits of detection, but we were able to measure 15 analytes, including IL-6, IL-2, TNFα, IP-10, MCP-1, MDC, IL-1β, IL-5, IL-22, IL-33, IFNγ, VEGF-A, KC/GRO, BDNF, and MMP-9. We used the U-PLEX plate to measure the plasma levels of the analytes according to the manufacturer’s instructions. Briefly, biotinylated capture antibodies were mixed with specific U-PLEX Linkers, added to the appropriate wells, and incubated at room temperature for 30 min. The U-PLEX plates were then washed using an automated plate washer (BioTek ELX5012). Plasma was defrosted, diluted two-fold (Diluent 13 buffer, Meso Scale Discovery), and centrifuged at 2,000 x g for 3 min. Fifty microliters of calibrators or plasma were then added to the wells in triplicate, and the plates were incubated for 2 h at room temperature on a Compact Digital Microplate Shaker (ThermoFisher) at 600 rpm. The plates were washed, and 50 μl of diluted detection antibodies were added and incubated for 1 h at room temperature. After washing, MSD GOLD Read Buffer B (Meso Scale Discovery) was added, the plates were read immediately on a MESO QuickPlex SQ 120 instrument (Meso Scale Discovery), and the data were analyzed using DISCOVERY WORKBENCH software (Meso Scale Discovery). For statistical analyses, the original MSD data were analyzed using ANOVA type models, allowing for different residual variances by group, with Satterthwaite method to determine the denominator degrees of freedom, to compare the plasma cytokine levels between Dp16 and WT mice with GM-CSF treatment versus placebo (saline).
Tissue Processing
Following the post-treatment assessments, on post-treatment day 25, the mice that were assessed in the RAWM (males only) were anesthetized with sodium pentobarbital, perfused intracardially with saline for 5 min, and the brains were then removed rapidly. The left hemisphere was quickly dissected into hippocampus, cerebellum, frontal cortex, and remaining cortex (i.e., total cortex minus frontal cortex), frozen immediately in liquid nitrogen, and then stored at −80°C until use. The right hemisphere was immersed in freshly prepared 4% paraformaldehyde (PFA) in PBS for 24 h at 4°C. After fixing with PFA, 4 μm-thick paraffin-embedded hippocampal brain sections were mounted on glass slides and used for indirect immunofluorescence microscopy staining or stored at 4°C until use.
Immunohistochemistry and Image Analyses
Immunofluorescence microscopy analyses were carried out using 4 μm-thick, paraffin-embedded hippocampal brain sections from male Dp16 and WT mice treated with GM-CSF or saline. Briefly, after standard deparaffination, the slides were incubated for 30 min with just-boiled 10 mM sodium citrate buffer for antigen retrieval. The slides were washed three times (3X) in TBST and incubated in blocking solution (Protein Block Serum-Free, Agilent DAKO, CA, USA) for 30 min at room temperature (RT), followed by incubation with rabbit anti-GFAP primary antibody at a 1:200 dilution (Abcam, MA, USA) in blocking solution, or with a rabbit anti-calretinin primary antibody at a dilution of 1:200 (Swant, Switzerland) in blocking solution overnight at RT. The following day, the slides were washed 3X in TBST with gentle rocking and incubated with Alexa-Flour488-conjugated anti-rabbit secondary antibody (Invitrogen, CA, USA). The slides were mounted using 4,6-diamidino-2-phenylindole (DAPI) Fluoromount-G (SouthernBiotech) and sealed with a coverslip. The preparation and staining of sections from each mouse brain were repeated with two slides, which yielded similar results. Each slide contained three brain sections from each mouse; two sections were incubated with a specific primary antibody, and one section was incubated in blocking solution without antibody as a negative control. There were four immunostained brain sections from each mouse. All sections were imaged using an Olympus IX83 Fluorescence microscope (Center Valley, PA). All images were taken at 20X magnification using the same exposure time. For quantitative analyses, the number of GFAP-positive astrocyte clusters and calretinin-positive interneurons were counted manually within brain regions of interest using cellSens software from Olympus by two different experimenters who were blinded as to genotype and treatment. Before counting, the quality of all images was examined carefully, and only images without broken/folded or smeared tissues were used. At least two sections from each mouse (at least one section from each slide) were usable, for a total of 2–4 brain sections per mouse. After averaging the number of GFAP-positive astrocyte clusters for each mouse, the mean number of clusters was compared between Dp16 and WT mice treated with saline or GM-CSF in the entire hippocampal region. Similarly, after averaging the number of calretinin-positive interneurons from 2–4 repeats for each mouse, the mean numbers of calretinin-positive interneurons were compared between Dp16 and WT mice treated with saline or GM-CSF in the entorhinal cortex and subiculum.
Statistical Analyses
Statistical significance between groups was determined by unpaired Student’s t-test and by ANOVA, followed by Tukey’s and Bonferroni’s multiple comparisons test using GraphPad Prism 7.00. All data are expressed as the mean +/− SEM. A p-value of less than 0.05 was considered statistically significant.
Results
Overview and Study Timeline
For baseline measurements, we first evaluated separate cohorts of untreated male and female Dp16 mice and their WT littermates at 12–14 months of age in the Open Field and the Y-maze tasks, and untreated male mice in the radial arm water maze (RAWM), because female Dp16 mice at this age are poor swimmers. We then investigated the effects of GM-CSF treatment versus placebo (saline) on Y-maze and RAWM performance, and on brain pathology. A timeline showing the experimental design is provided (Figure 1).
Dp16 Male and Female Mice Show Elevated Locomotor Activity in the Open Field Task
Dp16 mice and their WT littermates were first evaluated for genotypic effects on exploratory behavior in the Open Field task. The mice were allowed to explore the Open Field arena for 10 min as a baseline assessment (53). The total distance traveled, total time spent in the center zone, and speed were measured. Both male and female Dp16 mice showed significantly increased distance traveled compared to their WT littermates (p=0.026 for male and p=0.014 for female genotype comparisons) (Figures 2A and 2D). The Dp16 mice also showed a significantly higher speed compared to their WT littermates (p=0.027 for male and p=0.014 for female genotype comparisons) (Figures 2C and 2F). However, the average amount of time spent in the center zone was not significantly different between Dp16 mice and their WT littermates, showing that there are no differences in anxiety behavior between the two groups (Figures 2B and 2E). These results show that both male and female Dp16 mice show hyperactivity, but not increased anxiety, in a novel environment.
Figure 2. Male and Female Dp16 Mice Show Elevated Locomotor Activity in the Open Field Task at Baseline.
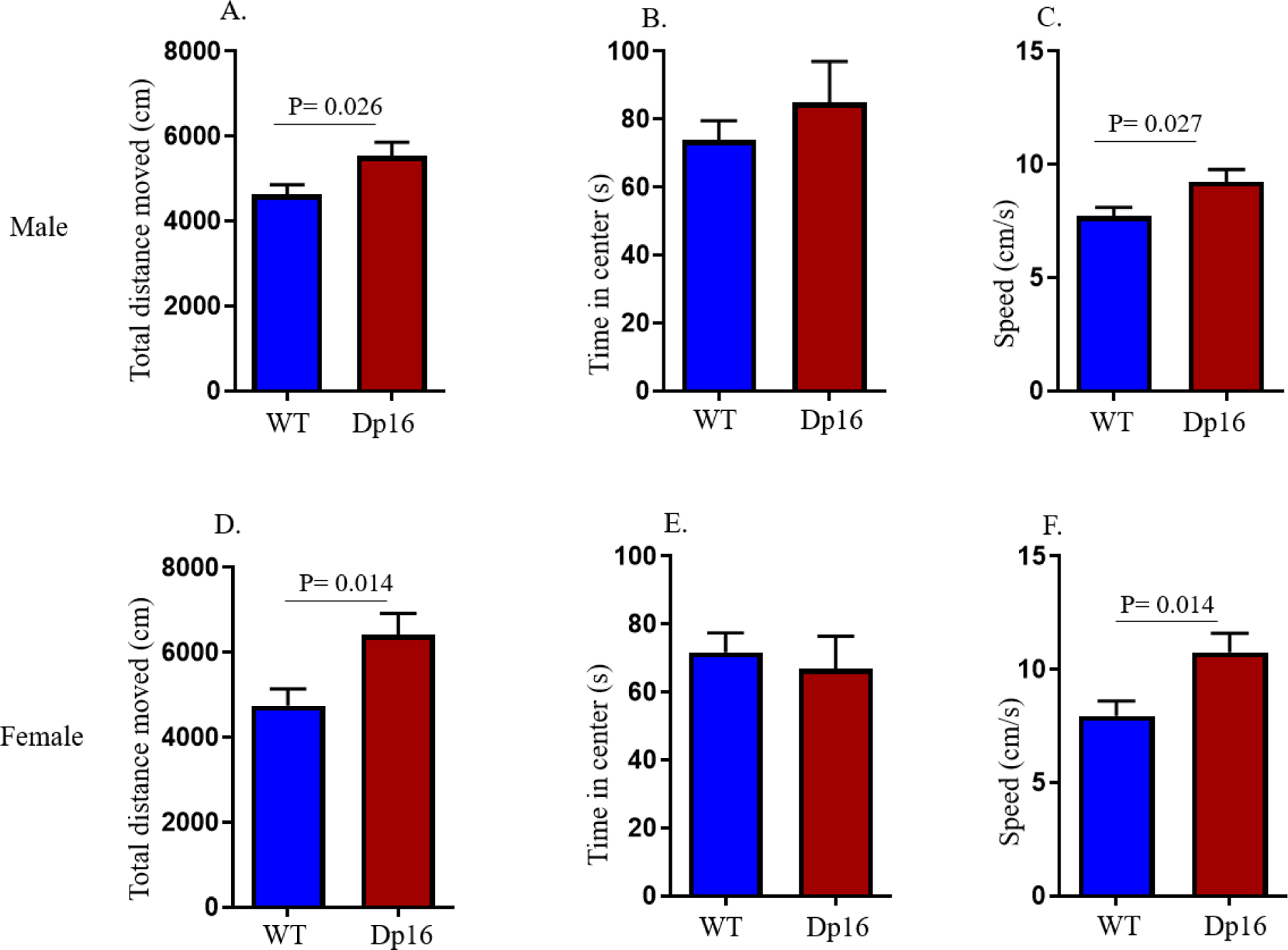
Locomotor and exploratory activity were measured in the Open Field task at baseline. Both male and female Dp16 mice showed a significant increase in total distance traveled (A and D, respectively) and speed of movement (C and F, respectively) compared to male and female WT mice, respectively. No significant differences were observed in the time spent in the center for male and female Dp16 mice (B and E, respectively) compared to male and female WT littermates, respectively. Data are represented as mean ± SEM for separate groups of mice (male WT: n=19; female WT: n=13; male Dp16: n=11; and female Dp16: n=13). Statistical significance was determined by the unpaired Student’s t-test for comparison between Dp16 and WT littermates.
GM-CSF Treatment Does Not Rescue Spatial Working Memory Deficits in Female Dp16 Mice
On the day following the Open Field test, mice were assessed for spatial working memory in the spontaneous alternation version of the Y-maze, where sequential entry of different arms of the maze assumes better working memory. At baseline, female Dp16 mice showed a significantly lower percentage of alternation compared to their female WT littermates (p=0.009) (Figure 3A). Indeed, the 50% alternation rate in female Dp16 mice corresponds to that expected by chance, showing that female Dp16 mice are impaired in working memory. In contrast, Y-maze performance of male Dp16 mice did not differ significantly from that of their male WT littermates (Figure 3B).
Figure 3. Spontaneous Alternation Rates in the Y-maze Task at Baseline and After GM-CSF Treatment: GM-CSF Treatment Does Not Rescue Spatial Working Memory Deficits in Female Dp16 Mice.
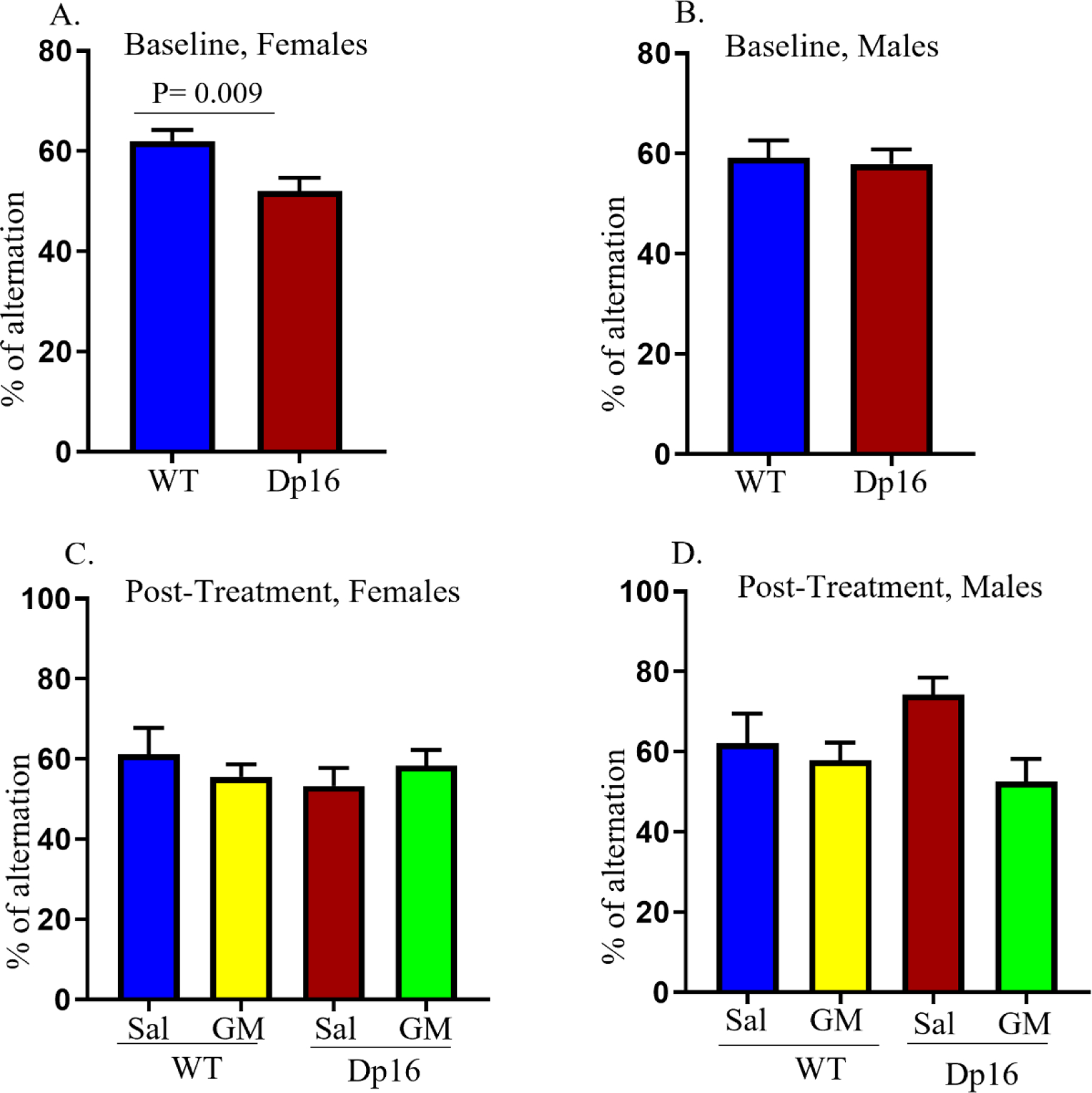
Female Dp16 mice showed a significantly lower alternation rate in the Y-maze compared to female WT littermates at baseline (A). No significant differences in alternation rates in the Y-maze were observed in male Dp16 mice compared to male WT littermates at baseline (B). No significant differences in spontaneous alternation rates in the Y-maze were observed in GM-CSF-treated (GM) female WT or Dp16 mice compared to saline-injected (Sal) female WT or Dp16 mice (F (3,18)=0.49; p=0.69) (C). No significant differences in spontaneous alternation rates in the Y-maze were observed in GM-CSF-treated (GM) male WT or Dp16 mice compared to saline-injected (Sal) male WT or Dp16 mice (p=0.18) (F (3,25)=1.60; p=0.21) (D). Data are represented as mean ± SEM for the separate groups of mice, including WT-Sal (9 males; 6 females), WT-GM (8 males; 7 females), Dp16-Sal (5 males; 6 females), and Dp16-GM (6 males; 6 females). Statistical significance was determined by the unpaired Student’s t-test for baseline comparisons between Dp16 and WT mice (A, B) and by ANOVA followed by Turkey’s multiple comparisons test between groups (C, D). For evaluating the interactions between treatment and genotype, Two-Way ANOVA followed by Bonferroni’s multiple comparisons test were performed. No significant differences in female mice in spontaneous alternation rates of Y-maze were observed between genotype (F (1,18)=0.28; p=0.60), and the interactions between treatment and genotype (F (1.18)=1.23; p=0.28) (C). Similarly, no significant differences in male mice in spontaneous alternation rates of Y-maze were observed between genotype (F (1,25)=0.27; p=0.61), and the interactions between treatment and genotype (F (1,25)=1.75; p=0.20) (D).
Next, the same mice were divided into four treatment groups: (1) WT mice injected with saline, (2) WT mice injected with GM-CSF, (3) Dp16 mice injected with saline, and (4) Dp16 mice injected with GM-CSF (see Figure 1). No significant differences in spontaneous alternation in the Y-maze task were observed in female WT or Dp16 mice treated with GM-CSF compared to female WT or Dp16 mice injected with saline (Figure 3C). Similarly, no significant differences in spontaneous alternation in the Y-maze task were observed in male WT or Dp16 mice treated with GM-CSF compared to male WT or Dp16 mice injected with saline (Figure 3D). Taken together, these findings show that GM-CSF treatment does not rescue working memory deficits in female Dp16 mice or improve working memory in male Dp16 mice as measured by the Y-maze task (Figures 3C and 3D, respectively).
Male Dp16 Mice Show Significant Deficits in Spatial Learning and Memory in the Radial Arm Water Maze (RAWM) Task at Baseline
To evaluate hippocampal-dependent spatial learning and memory, mice were assessed in a two-day RAWM protocol prior to any treatment. Only male mice could be used in these experiments because female Dp16 mice at this age had inadequate swimming abilities. For the baseline assessments, on Day 1, Dp16 mice and their WT littermates were subjected to 15 trials of 60 sec each, and latency to reach the submerged platform was measured. Data were analyzed in blocks of three trials (for a total of five blocks). Latency for both groups decreased over the 15 trials, indicating that they had learned the platform location (Figure 4A). On day two, which served as the memory testing day, both Dp16 and WT mice were again subjected to 15 trials (60 sec each), and the data were analyzed in blocks of three trials. Although Dp16 and WT mice showed no significant differences in memory/latency at the end (block 5) of day 1, they did show a statistically significant difference in the first block of day 2 (p<0.05) (Figures 4A and 4B). The two genotypes showed significant differences in latency between the first four blocks of day 2, but they became indistinguishable by block five (Figure 4B).
Figure 4. Male Dp16 Mice Show Deficits in the RAWM Task at Baseline.
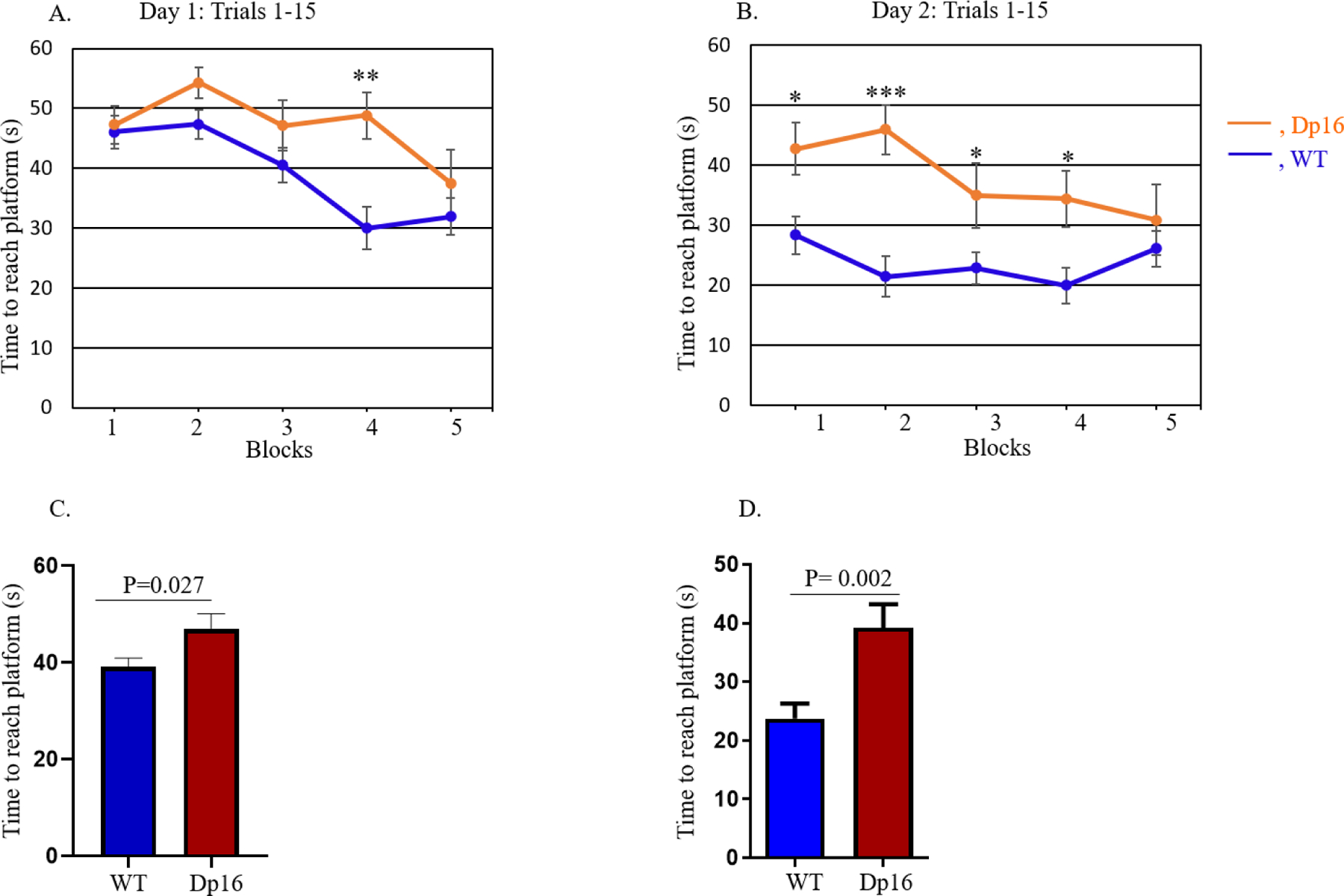
Hippocampal-based spatial learning and memory performance were evaluated in male Dp16 mice for two days using the radial arm water maze (RAWM) task. Data were analyzed considering each group of three out of 15 trials as a block for Day 1, the training day (A), and for Day 2, the testing day (B). On day 2, in blocks 1 and 2, there was a significant difference in memory performance between the Dp16 and WT littermates. After averaging the data from 15 trials using the time values for each individual mouse, the mean time to reach the platform for each day was compared between Dp16 and WT mice for Day 1 (C) and Day 2 (D). Dp16 mice required a significantly longer time to reach the platform than WT mice on both days, although they ultimately learned as well as WT mice, as indicated by non-significant differences in latencies in Block 5. Data are represented as mean ± SEM for separate groups of mice (WT, n=17; Dp16, n=11). Statistical significance was determined by the unpaired Student’s t-test for comparison between Dp16 and WT mice (C, D) and by ANOVA followed by Tukey’s multiple comparisons test (A, B). *p<0.05; ***p<0.001.
Finally, after averaging the data from 15 trials using the time values for each individual mouse, we calculated the mean latency time to reach the platform and compared Dp16 to WT mice on both days. On Day 1, Dp16 mice required a significantly longer time to reach the platform compared to WT controls (p=0.027) (Figures 4C). Similarly, on Day 2, Dp16 mice also required a significantly longer time to reach the platform compared to WT controls (p=0.002) (Figures 4D). These results show that male Dp16 mice exhibit significant cognitive (both learning and memory) deficits overall in the RAWM task. However, by block 5 of both day 1 and day 2, the latencies of the Dp16 and WT mice were not statistically significantly different. Together, these results show that the main learning deficit in the Dp16 mice was in the speed with which they learned the task. With enough practice, the Dp16 mice eventually performed as well as their WT littermates.
GM-CSF Treatment Improves Spatial Memory in Male Dp16 and WT Mice in the RAWM Task
We repeated the RAWM analysis in male Dp16 mice and their male WT littermates after treatment with GM-CSF or saline. The male mice were divided into four treatment groups: (1) WT mice injected with saline, (2) WT mice injected with GM-CSF, (3) Dp16 mice injected with saline, and (4) Dp16 mice injected with GM-CSF (Figure 1). We used the same RAWM protocol for baseline assessments, i.e., each mouse performed 15 trials (60 sec/trial) with the hidden/submerged platform placed in arm 4. Latency for block 5 of pre-treatment day 2 represents maximal learning for baseline assessments; therefore, these values were used for the normalization of post-treatment day 1 latencies. Specifically, we normalized the latencies for each saline- or GM-CSF-injected mouse for each block of post-treatment day 1 with the corresponding latencies from block 5 of pre-treatment day 2 (baseline day 2) and present the values as a percentage of block 5 pre-treatment day 2. Normalized latencies were analyzed in blocks of three trials (for a total of five blocks from 15 trials), and the GM-CSF-treated group was compared with the saline-injected group. Post-treatment latencies in the Dp16 mice treated with GM-CSF showed significant differences from saline-injected Dp16 mice in blocks four and five (Figure 5A). Averaging the normalized latency of post-treatment day 1 from all five blocks (from Figure 5A) showed that the Dp16 mice treated with GM-CSF required significantly less time (shorter latency) to reach the platform compared to the saline-injected Dp16 mice (p=0.009) (Figure 5B). To observe the final learning/memory differences over the course of the day in the Dp16 mice injected with GM-CSF or saline, we combined normalized latency of blocks 4 and 5 and compared the GM-CSF-treated groups with the saline-injected groups. Dp16 mice treated with GM-CSF showed significantly shorter latency compared to the saline-injected Dp16 mice (p=0.008) (Figure 5C). Furthermore, to clarify the longitudinal learning performance of Dp16 mice treated with GM-CSF, we combined data from blocks 1 and 2 and compared it with the combined data from blocks 4 and 5 of Dp16 mice treated with GM-CSF. Figure 5D shows that combined data from the normalized latency of blocks 4 and 5 show significantly smaller latency than the combined latency from blocks 1 and 2 (p=0.03), showing that Dp16 mice treated with GM-CSF are improving their learning and memory over time. WT mice treated with GM-CSF required significantly less time to reach the platform in block 1 compared to saline-injected WT mice (Figure 5E). Because learning is a continuous process and both GM-CSF-treated and saline-injected WT mice continued to learn during the day, there are no significant differences in blocks 2–5, indicating that GM-CSF had little effect on performance at these later times. Combining the normalized latency of blocks 1 and 2, and then comparing GM-CSF-treated WT mice with the saline-injected WT mice revealed that the WT mice treated with GM-CSF showed significantly smaller latency compared to the saline-injected WT mice for the first two blocks of the day (p=0.02) (Figure 5F). Furthermore, even at the very beginning of the task on post-treatment day 1, WT mice treated with GM-CSF showed a significant difference in performance compared to the saline-injected WT mice (Figure 5E, block 1), which remained constant over time, whereas saline-injected WT mice improved in the task and performed similarly to GM-CSF-treated WT mice by the end. To further investigate the learning performance of WT mice injected with saline, we combined data from blocks 1 and 2 and compared them with the combined data from blocks 4 and 5. Figure 5G shows a significantly shorter latency for the results of blocks 4 and 5 compared to that from blocks 1 and 2 (p=0.04), showing that WT mice injected with saline improve their learning and memory performance over time.
Figure 5. GM-CSF Treatment Improves RAWM Performance in Male Dp16 and Wild-type Mice.
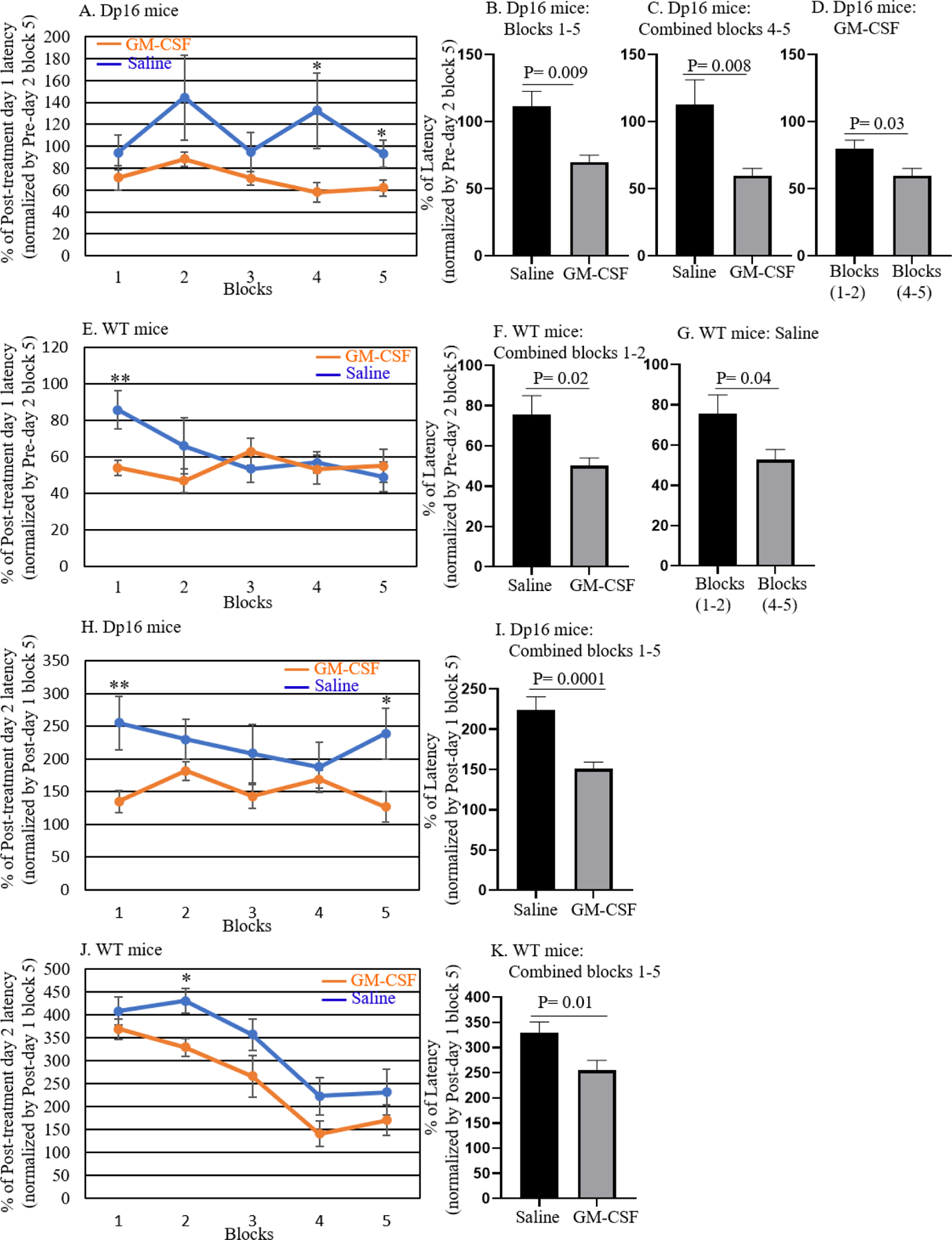
To evaluate the effect of GM-CSF treatment on spatial memory, latencies on post-treatment Day 1 for Dp16 mice and their WT littermates injected with saline or GM-CSF were normalized to their mean latency in block 5 of pre-treatment Day 2, and the data were then compared between saline and GM-CSF injection for each genotype. For quantitative analyses, latencies on post-treatment Day 1 were calculated relative to 100% for block 5 of pre-treatment Day 2 data. Normalized latencies were analyzed in blocks of three trials (with a total of five blocks from 15 trials) and the GM-CSF-treated group was compared with the saline-injected group of Dp16 mice (A; n=5–6) and WT mice (E; n=8–9). After averaging the normalized latencies of post-treatment Day 1 from all blocks, the mean values were compared in Dp16 mice treated with saline or with GM-CSF (B; n=5–6). Combined data from blocks 4–5 of Dp16 mice were compared for saline and GM-CSF injection (C; n=5–6). Combined data from blocks 1–2 were compared with the combined data from blocks 4–5 for Dp16 mice treated with GM-CSF (D; n=5–6). Similarly, combined data from blocks 1–2 of WT mice were compared for saline injection and GM-CSF treatment (F; n=8–9). Combined data from blocks 1–2 were compared with the combined data from blocks 4–5 for WT mice injected with saline (G; n=8–9). The effect of GM-CSF treatment on learning flexibility was evaluated by normalizing the latency of each saline-injected or GM-CSF-treated mouse at post-treatment day 2 with its latency at block 5 of post-treatment day 1 and calculated data relative to 100% of block 5 post-treatment day 1 (H-K). Normalized latencies were calculated in blocks of three trials, and the GM-CSF-injected group was compared with the saline-injected group of Dp16 mice (H; n=5–6) and WT mice (J; n=8–9). Combined data from blocks 1–5 were compared for saline-injected and GM-CSF-treated Dp16 mice (I; n=5–6) and WT mice (K; n=8–9). Data are represented as mean ± SEM for the different groups of mice. Statistical significance was determined by the unpaired Student’s t-test for comparison between groups. *p <0.05; **p <0.01.
To assess flexibility in learning, on testing day 2 of the post-treatment RAWM task, the platform was moved (from arm 4 to arm 6, leaving other maze cues unchanged). We calculated the latency of each GM-CSF-treated or saline-injected mouse on post-treatment day 2 as a percentage of its latency in block 5 of post-treatment day 1. Normalized latencies were analyzed in blocks of three trials (for a total of five blocks from 15 trials), and the GM-CSF-treated group was compared with the saline-injected group. The Dp16 mice treated with GM-CSF showed significantly shorter latencies in blocks 1 and 5 (Figure 5H). Combining the normalized latencies of blocks 1–5 on post-treatment day 2 showed that the Dp16 mice treated with GM-CSF required significantly less time (shorter latencies) to reach the platform compared to the saline-injected Dp16 mice (p=0.0001) (Figure 5I). Similarly, GM-CSF-treated WT mice required significantly less time to reach the platform compared to saline-injected WT mice in block 2 (Figure 5J). Combining the normalized latency of blocks 1–5 in WT mice showed that, overall, GM-CSF-treated WT mice required significantly less time to reach the platform compared to saline-injected WT mice (p=0.01) (Figure 5K).
GM-CSF Treatment Leads to Elevated Cytokine Levels in the Plasma of WT and Dp16 Mice
Although the effects of GM-CSF treatment on cytokine levels have been well-studied in humans and in WT mice (48, 54, 55), the effects of GM-CSF treatment on cytokine levels in Dp16 mice have not been investigated. We assessed the levels of inflammatory cytokines and metabolic proteins in the plasma of WT and Dp16 mice after 10 consecutive days of injection with GM-CSF or with saline using the Meso Scale Discovery (MSD) platform. We measured the levels of 15 analytes (i.e., IL-6, IL-2, TNFα, IP-10, MCP-1, MDC, IL-1β, IL-5, IL-22, IL-33, IFNγ, VEGF-A, KC/GRO, BDNF, and MMP-9) in four treatment groups: (1) WT mice injected with saline, including 3 males and 2 females; (2) WT mice injected with GM-CSF, including 4 males and 1 female; (3) Dp16 mice injected with saline, including 4 males and 1 female; and (4) Dp16 mice injected with GM-CSF, including 4 males and 1 female. Figure 6 shows the plasma levels of six key inflammation-associated cytokines (i.e., IL-6, IL-2, TNFα, IP-10, MCP-1, and MDC) in each of the four treatment groups. Dp16 and WT mice injected with saline did not show significant differences in the levels of these six cytokines (Figure 6A–F), but there was a trend toward increased levels of interleukin-6 (IL-6), interleukin-2 (IL-2), tumor necrosis factor α (TNFα), and macrophage-derived chemokine (MDC) in saline-injected Dp16 mice compared to saline-injected WT mice. In contrast, both WT and Dp16 mice treated with GM-CSF showed significantly higher plasma levels of IL-2 (WT mice, p=0.005; Dp16 mice, p= 0.02; Figure 6B) and MDC (WT mice, p <0.001; Dp16 mice, p= 0.03; Figure 6F) compared to saline-injected WT and Dp16 mice, respectively, whereas the plasma levels of interferon γ-induced protein 10 (IP-10) were significantly higher only in GM-CSF-treated Dp16 mice compared to saline-injected Dp16 mice (p=0.03; Figure 6D). Strong trends toward increased plasma levels of IL-6 (WT mice, p=0.07; Dp16 mice, p= 0.07; Figure 6A) and TNFα (WT mice, p=0.07; Dp16 mice, p=0.06; Figure 6C) were observed in both WT and Dp16 mice after GM-CSF treatment. We did not observe significant differences in the plasma levels of MCP-1 (Figure 6E), or in the plasma levels of the nine other cytokines and metabolic proteins (i.e., IL-1β, IL-5, IL-22, IL-33, IFNγ, VEGF-A, KC/GRO, BDNF, or MMP-9; Supplementary Figure 1) in Dp16 or WT mice injected with GM-CSF or with saline.
Figure 6. GM-CSF Treatment is Associated with Higher Plasma Levels of Cytokines in Both Wild-Type and Dp16 Mice.
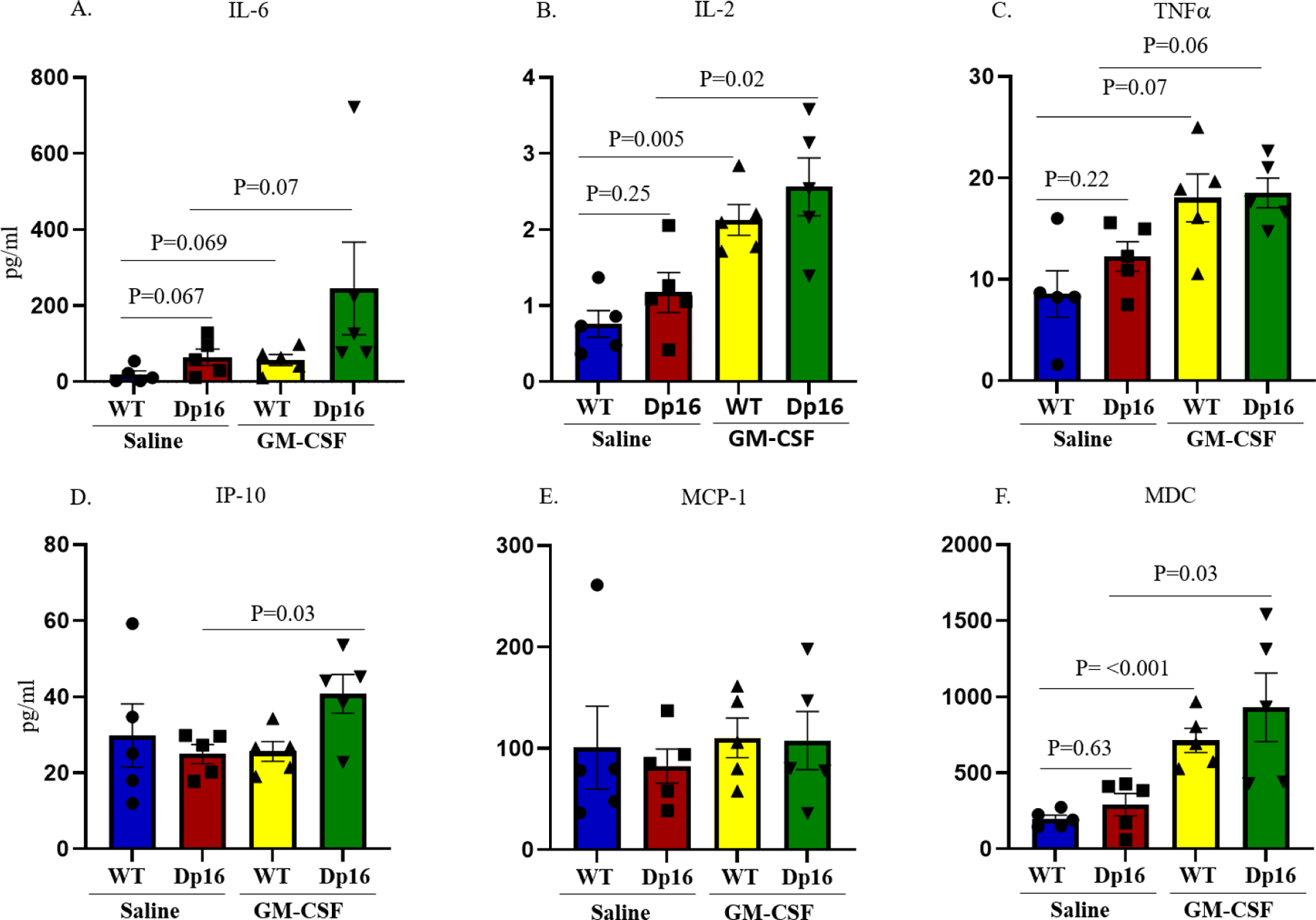
The Meso-Scale Discovery (MSD) platform was used to determine plasma levels of six key inflammation-associated cytokines (i.e., IL-6, IL-2, TNFα, IP-10, MCP-1, and MDC) following GM-CSF treatment or saline injection. No significant differences were observed in the plasma levels of these six cytokines in saline-injected WT mice compared to saline-injected Dp16 mice (A-F). Treatment with GM-CSF led to significantly higher levels of interleukin-2 (IL-2) and macrophage-derived chemokine (MDC) in the plasma of both GM-CSF-treated WT and Dp16 mice compared to saline-injected WT and Dp16 mice (B & F), respectively, whereas the plasma levels of interferon γ-induced protein 10 (IP-10) were significantly higher only in GM-CSF-treated Dp16 mice compared to saline-injected Dp16 mice (D). In addition, there was a strong trend toward higher plasma levels of interleukin-6 (IL-6) and tumor necrosis factor α (TNFα) in the plasma of both GM-CSF-treated WT and Dp16 mice compared to saline-injected WT and Dp16 mice (A & C), respectively. No significant differences were detected in the plasma levels of MCP-1 in GM-CSF-treated or saline-injected WT or Dp16 mice (E).
GM-CSF Treatment Partially Restores the Distribution of Hippocampal Astrocytes and Reverses Astrogliosis in Male Dp16 Mice
Glial cell abnormalities are one of the major cellular phenotypes seen in the brains of people with DS and in mouse models of DS (56, 57). We therefore investigated the expression pattern and distribution of the astrocytic marker protein glial-fibrillary acidic protein (GFAP) in the hippocampi of male Dp16 mice and their male WT littermates. We visually counted the number of GFAP-positive clusters (blinded as to genotype and treatment). WT mice injected with saline or GM-CSF show GFAP-positive astrocytes with similar morphology and distribution (Figures 7A and 7B). In contrast, compared to WT, Dp16 mice treated with saline show GFAP-positive astrocytes with more dense cell bodies that are clustered together with crossed branches, indicating diffuse astrogliosis (Figure 7C). However, Dp16 mice treated with GM-CSF show fewer abnormal astrocyte clusters and a distribution of astrocytes with morphologies more similar to those of WT mice (Figure 7D). This revealed significantly higher numbers of astrocyte clusters in the hippocampi of Dp16 mice injected with saline compared to WT mice (p=0.001) (Figure 7E). In Dp16 mice injected with GM-CSF, the number of astrocyte clusters was significantly reduced compared to saline-injected Dp16 mice (p=0.03) (Figure 7E).
Figure 7. GM-CSF Treatment Partially Restores the Distribution of Hippocampal Astrocytes and Reverses Astrogliosis in Male Dp16 Mice.
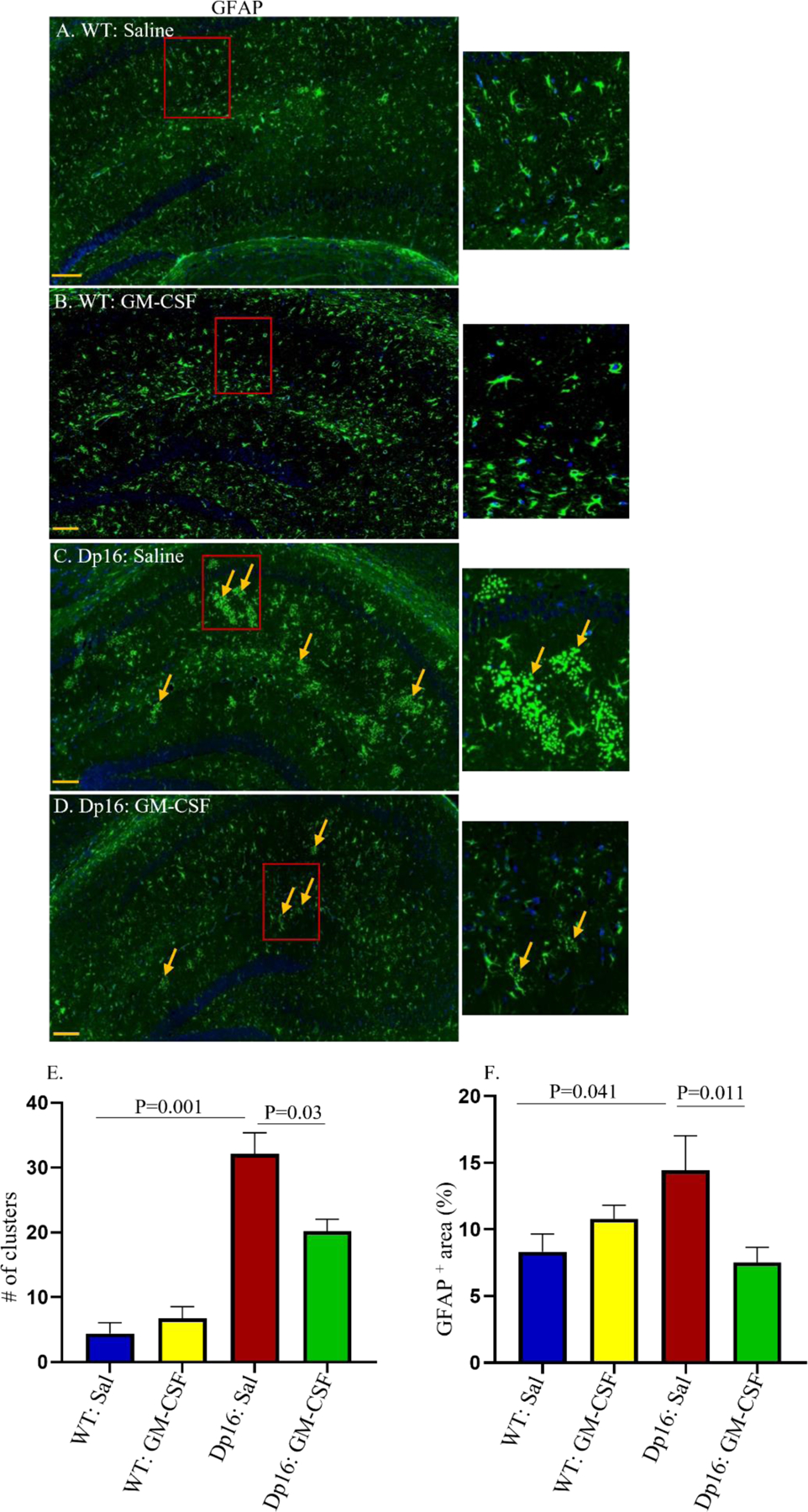
The patterns and distributions of glial fibrillary acidic protein (GFAP)-positive astrocytes (green) were examined in the hippocampi of male WT and Dp16 mice injected with saline or with GM-CSF. Data are shown for WT mice injected with saline (A), WT mice treated with GM-CSF (B), Dp16 mice injected with saline (C), and Dp16 mice treated with GM-CSF (D). Dp16 mice injected with saline (C) showed abnormal clusters of GFAP-positive astrocytes compared to WT mice injected with saline (A). GM-CSF treatment of WT mice had no effect on hippocampal astrocyte distribution (B) compared to WT mice injected with saline. Dp16 mice treated with GM-CSF showed significantly reduced levels of the abnormal clusters of GFAP-positive astrocytes (D) compared to Dp16 mice injected with saline (C). Arrows show abnormal clusters of GFAP-positive astrocytes. Quantitative analyses showed significantly higher numbers of abnormal clusters of GFAP-positive astrocytes in Dp16 mice injected with saline compared to WT mice injected with saline (p=0.001), which were reduced in Dp16 mice treated with GM-CSF (p=0.03) (E). Quantitative analyses of the percent area of GFAP-positive staining showed a significant increase in Dp16 mice injected with saline compared to WT mice injected with saline (p=0.041), which were reduced by GM-CSF treatment (p=0.011) (F). For each bar, the data are represented as mean ± SEM for the separate groups of mice. Statistical significance was determined by the unpaired Student’s t-test for comparison between groups. Scale bar: 200 μm (20X magnification). All experiments were repeated 2–4 times with similar results (n=3 male mice in each of the four groups).
We also measured the percent area of the hippocampi that stained positively for GFAP. Statistical analyses revealed that Dp16 mice injected with saline showed a significantly higher percent area of GFAP-positive astrocyte staining compared to WT mice injected with saline (P=0.041) (Figure 7F). Furthermore, Dp16 mice treated with GM-CSF showed a significantly reduced percent area of GFAP-positive astrocyte staining compared to Dp16 mice injected with saline (p=0.011) (Figure 7F). These results show that treatment with GM-CSF partially reverses the increase in the GFAP-positive staining area in the hippocampi of Dp16 mice. However, no significant difference in the GFAP-positive staining area was observed in WT mice treated with GM-CSF compared to saline-injected WT mice.
Together, these results show that Dp16 mice exhibit astrogliosis/astrocyte abnormalities in their hippocampi, and that treatment with GM-CSF partially reverses the morphological abnormalities of astrocytes and reduces astrocyte clustering and astrogliosis in the hippocampi of Dp16 mice.
To examine the distribution of microglial cells, we assessed expression of Iba1, a marker for reactive microglia, by immune-staining the hippocampi of male Dp16 mice and their WT controls after GM-CSF treatment or saline injection (Supplementary Figure 2A–2E). We measured the percent area of the hippocampi that stained positively with an anti-Iba1 antibody. No significant differences were observed in the Iba1-positive staining area in the hippocampi of Dp16 mice or WT mice treated with GM-CSF or injected with saline (Supplementary Figure 2E). However, our results showed a trend toward a greater area of Iba1-positive staining in the hippocampi of both Dp16 and WT mice following GM-CSF treatment compared to saline-injected Dp16 and WT mice.
GM-CSF Treatment Leads to an Increased Number of Calretinin-Positive Interneurons in Male Dp16 Mice
Previous studies have shown that people with DS exhibit an imbalance between excitatory and inhibitory neuronal activity in the brain, which is also seen in the Ts65Dn mouse model of DS (16, 21). Abnormalities are also observed in interneurons in induced pluripotent stem cells (iPSCs) from people with DS and from Ts65Dn mice (49, 58). We therefore investigated the numbers of calretinin-positive interneurons in the brains of male Dp16 mice and their male WT littermates injected with saline or treated with GM-CSF. Paraffin-embedded tissues from hippocampus and cortex were stained with an anti-calretinin antibody, and the numbers of calretinin-positive interneurons were counted. Compared to saline-injected WT mice, saline-injected Dp16 mice show a trend to a reduction in the number of calretinin-positive interneurons in the entorhinal cortex (p=0.06) (Figures 8A, 8C, and 8E). GM-CSF treatment significantly increased this number (p=0.046) (Figures 8A, 8C, 8D, and 8E), to levels indistinguishable from those of their saline-injected WT littermates. Similarly, the number of calretinin-positive interneurons was significantly reduced (p=0.01) in the subiculum of saline-injected Dp16 mice compared to saline-injected WT mice (Figures 8F, 8H, and 8J). This number was significantly increased in Dp16 mice treated with GM-CSF (p=0.04) (Figures 8H, 8I, and 8J). No significant differences were observed between WT mice injected with saline or treated with GM-CSF in either the entorhinal cortex or the subiculum. Together, these results show that GM-CSF treatment partially reverses the deficit in the number of interneurons in Dp16 male mice.
Figure 8. GM-CSF Treatment Partially Restores the Number of Calretinin-Positive Interneurons in Male Dp16 Mice.
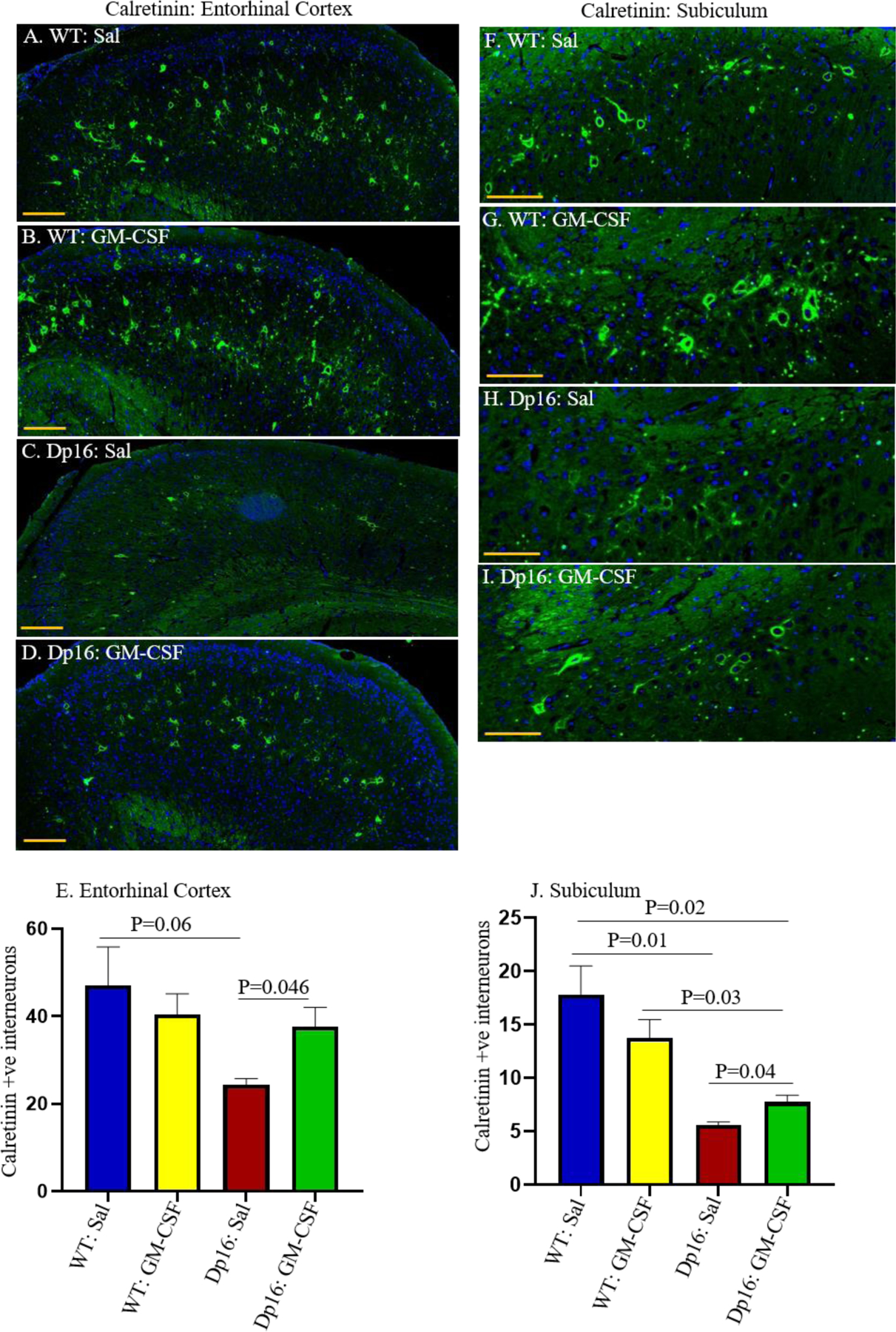
The patterns and distributions of calretinin-positive interneurons (green) were examined in the entorhinal cortex (EC) of male WT mice injected with saline (A), male WT mice treated with GM-CSF (B), male Dp16 mice injected with saline (C), and male Dp16 mice treated with GM-CSF (D). Quantitative analyses showed a trend towards a lower number of calretinin-positive interneurons in the entorhinal cortex of Dp16 mice injected with saline compared to WT mice injected with saline (p=0.06) (E). The number of calretinin-positive interneurons in the entorhinal cortex of the Dp16 mice was significantly increased following GM-CSF treatment compared to Dp16 mice injected with saline (p=0.0406) and was statistically no different than in WT mice injected with saline or treated with GM-CSF (E). The expression patterns and distributions of calretinin-positive interneurons (green) were also examined in the subiculum of male WT mice injected with saline (F), male WT mice treated with GM-CSF (G), male Dp16 mice injected with saline (H), and male Dp16 mice treated with GM-CSF (I). There was a significantly reduced number of calretinin-positive interneurons in the subiculum of Dp16 mice injected with saline compared to WT mice (p=0.01) (J). The number of calretinin-positive interneurons in the subiculum of Dp16 mice was significantly increased following GM-CSF treatment (p=0.04), but it was still significantly lower than in WT mice injected with saline or with GM-CSF (J). For each bar, data are represented as mean ± SEM for separate groups of mice. Statistical significance was determined by the unpaired Student’s t-test for comparison between groups. Scale bar: A-D=100 μm (20X magnification) and F-I=50 μm (20X magnification). All experiments were repeated 2–4 times with similar results (n=3 male mice in each of the four groups).
Discussion
Pro-Inflammatory Effects of GM-CSF Treatment in Dp-16 and WT Mice.
As discussed in the Introduction, GM-CSF has multiple, complex effects on the innate immune system in humans and WT mice, with the majority of downstream responses reported to be pro-inflammatory. GM-CSF treatment has not previously been tested in Dp16 or other mouse models of DS. We found that treatment with GM-CSF increased the levels of many pro-inflammatory cytokines in the plasma of both WT and Dp16 mice, supporting its inflammatory role in both groups of mice.
Sex differences in the Effects of GM-CSF Treatment on Dp-16 Mice.
We analyzed 12–14 month-old male and female Dp16 mice and their age- and sex-matched littermates in the Open Field and alternating Y-maze tasks at baseline, Examining both sexes, when possible, is ideal because sex differences have been noted in the descriptions of cognitive phenotypes observed in people with DS (59, 60), and sex differences are also frequently observed with drug responses in the typical population (61). However, sex differences have not been commonly investigated in DS mouse models, due in part to the fact that the popular Ts65Dn mouse model of DS breeds poorly, and that the male Ts65Dn mice are largely sterile, so that female Ts65Dn mice are generally saved as breeders. However, where investigated, sex differences are evident at the behavioral level (15). Our data show that both male and female Dp16 mice were hyperactive in the Open Field task at baseline, confirming a previous report of increased locomotor activity in male Dp16 mice of a similar age (62). However, this may be an age-dependent phenotype, because younger (2–3 month-old) male Dp16 mice did not show hyperactivity (63). Similarly, increased locomotor activity in the Open Field arena was reported in 9–12 month-old Ts65Dn mice (53), and human studies have shown that children with DS of both sexes are hyperactive compared to typical children of a similar age (64, 65). We also observed sex differences in the Y-maze: at baseline, female Dp16 mice were impaired, performing no better than chance, whereas male Dp16 mice were not impaired.
Choice of Sex for Analysis of Learning Behavior.
Only male mice were analyzed in the RAWM, because the female Dp16 mice at this age are poor swimmers. Male Dp16 mice learned more slowly compared to their male WT littermates, indicating impaired hippocampal function. This finding is consistent with previous reports of impairment of Dp16 mice in other tasks that require hippocampal function, e.g., Morris Water Maze and Contextual Fear Conditioning, and of hippocampal learning deficits in other mouse models of DS, including Ts65Dn and Ts1Cje mice, each of which is trisomic for a subset of the genes that are trisomic in the Dp16 mice (49, 63, 66–69). This finding is also consistent with previous studies that showed that Dp16 mice display abnormal hippocampus-based long-term potentiation (LTP) (62, 63, 69, 70).
In contrast to the commonly held hypotheses that simply blocking or reversing the activated innate immune system and chronic inflammation associated with DS may reverse many of its features, our results show that treatment with the immune modulatory cytokine GM-CSF, which shows substantial pro-inflammatory effects, effectively ameliorated both learning deficits and brain pathology in the Dp16 mouse model of DS. Specifically, daily subcutaneous injection of GM-CSF rescued male Dp16 deficits in the RAWM, improving their performance to a level comparable to that of age-matched, saline-injected male WT littermates. GM-CSF treatment also enhanced RAWM performance in middle-aged WT littermates, as we and others have shown previously (37, 46). Additionally, when the platform was moved to a new arm on testing day 2 of the post-treatment RAWM task, we found that GM-CSF treatment improved learning flexibility in both WT and Dp16 mice. Impaired cognitive flexibility is often observed among individuals with DS (71).
Although studies have shown that GM-CSF treatment improves learning/memory and removes amyloid plaques in mouse models of AD, Dp16 mice do not develop AD amyloid pathology at any age. Therefore, GM-CSF treatment must lead to improved learning/memory in Dp16 mice and WT mice via an amyloid-independent mechanism(s), presumably related to immune-modulatory/stimulatory activity. It is perhaps relevant that GM-CSF has been shown to increase the number of T-regulatory cells (Tregs) in mouse models of Parkinson’s disease (PD), in mouse models of other autoimmune and inflammatory conditions, and in participants in a human PD clinical trial (72–78). Treg levels are deficient in people with DS, and people with DS are susceptible to autoimmune diseases (79, 80), which has been attributed to defective Tregs (80). For example, autoimmune thyroiditis or Hashimoto’s thyroiditis (HT) is prevalent in people with DS (81, 82), and, indeed, GM-CSF-stimulated Treg induction has been shown in several studies to inhibit experimental autoimmune thyroiditis, a common model of HT (77, 79, 83, 84). It is also possible that the increase in proinflammatory cytokines after GM-CSF treatment may also contribute to its learning-enhancing effects.
Effects of GM-CSF on Neuronal Function.
Despite its known immune modulatory and pro-inflammatory properties, GM-CSF also affects multiple CNS processes that are consistent with and may shed light on its unexpected beneficial effects on learning/memory in the Dp16 mouse model of DS. For example, we found that the number of cells that stain positively for calretinin, a marker for interneurons, is decreased in the entorhinal cortex and in the subiculum of male Dp16 mice, and that GM-CSF treatment leads to a partial restoration of these numbers. Reduced density of calretinin-positive interneurons has also been seen in iPSCs from people with DS (58). Because inhibitory interneurons contribute to neuronal activity, information processing for neuronal networks, synaptic plasticity, and learning/memory (85–87), their impaired function might impact cognitive processing in the brains of people with DS. However, reduced interneuron numbers cannot fully explain the learning/memory deficits in Dp16 mice because, in contrast to our observations in 12–14 month-old mice, in 4 month-old Dp16 mice, the density of calretinin-positive interneurons was normal in the striatum oriens of the hippocampus (63, 68). Notably, 2–4 month-old Dp16 mice show impairment in some hippocampal-based tasks, but we did not detect defects in the RAWM at this age, suggesting that other cellular abnormalities must underlie some of the learning/memory deficits at this age. Furthermore, because we did not find that GM-CSF treatment increases the number of interneurons in 12–14 month-old WT mice, the improved learning/memory function observed in WT mice must occur via a different mechanism(s).
Other studies support a beneficial effect of GM-CSF on recovery from neuronal damage or dysfunction, modulation of apoptosis-related genes and prevention of programmed neuronal cell death, restoration of cerebral blood flow, and promotion of axonal growth, regeneration, and survival in animal models of several CNS-related disorders (88–96). (97–99). Additionally, GM-CSF is a growth factor for neural stem cells (NSCs) and it prevents neuronal apoptosis (100–105), with effects only on cell survival at low concentrations and with effects on cell survival, proliferation, differentiation, and functional activation at higher concentrations (106). GM-CSF can cross the blood-brain barrier (107), and is also produced within the brain, where numerous cell types express the GM-CSF receptor, including neurons, oligodendrocytes, microglia, astroglia, and endothelial cells (108, 109). Indeed, GM-CSF has also been shown to play a major role in neuronal plasticity that is critical for learning/memory (110). In addition to its effects on NSCs, GM-CSF has been shown to mobilize mesenchymal stem cells (MSCs) from the bone marrow and to enhance their differentiation and migration in response to injury (111, 112). MSCs have been reported to have beneficial cognitive and regenerative effects and are being investigated as potential positive effectors in several neurological disorders, including AD, PD, and spinal cord injury (113–119). Whether the beneficial effects of GM-CSF on the nervous system are related to its immune stimulatory and pro-inflammatory activity or due to a different response to activation of the GM-CSF receptor on cells of the nervous system is unknown.
We previously showed neurological benefits of GM-CSF treatment in a mouse model of AD that was also unexpected, because the well-documented existence of inflammation and its contribution to AD led many to predict that, in general, non-steroidal anti-inflammatory drugs (NSAIDs) would be protective against AD, and that a pro-inflammatory molecule such as GM-CSF would be expected to exacerbate AD (44, 120–123). However, the NSAID clinical trials in AD and mild cognitive impairment (MCI) participants failed to show any reversal or slowing of disease symptoms and may have led to accelerated cognitive decline (124, 125). Furthermore, treatment of a mouse model of AD with GM-CSF reversed cognitive deficits in the RAWM, reduced amyloidosis, and increased synaptophysin expression and microglial density (37). Similar to the current study, cognitive improvement was also reported in age-matched WT mice treated with GM-CSF (37). These results showing improved cognition in WT and AD mice treated with GM-CSF and reversal of neuropathology in AD mice have been replicated by other groups (45, 46). Our recently completed clinical trial in which mild-to-moderate AD participants were treated with recombinant human GM-CSF (sargramostim/Leukine®) for 15 days showed that sargramostim treatment was safe, led to significantly improved MMSE scores, and partly restored levels of plasma biomarkers of AD brain pathology and neuronal damage (i.e., Aβ40 and Aβ42, total tau, and ubiquitin C-terminal hydrolase L1 [UCH-L1]) towards normal (48, 126). Before the onset of AD pathology, people with DS also exhibit increased brain levels of Tau and low levels of UCH-L1(127). We are collaborating to measure these proteins in the plasma of people with DS throughout the lifespan, including at ages before AD-related brain pathology is present, compared to typical age-matched controls.
Role of Astrocytes in Learning/Memory.
Astrocytes play an essential supportive role with regard to neuronal function, both directly and indirectly, by regulating glucose metabolism, controlling cerebral blood flow, and maintaining homeostasis of ion and neurotransmitter levels in the CNS (reviewed in (128)), as well as by playing pivotal roles in information processing, brain wiring, and cellular programming (129–131). Astrocytes modulate synaptic activity, regulate synapse formation, synaptic transmission, and synaptic plasticity, and are required for the maintenance of LTP via their interactions with neurons and/or with other astrocytes (132). Aberrations in the generation, number, morphology, and proliferation of astrocytes are observed in many neurodegenerative and neuropsychiatric disorders, including AD, schizophrenia, autism, Rett’s syndrome, and epileptic and non-epileptic seizures (133–135). Recent studies also demonstrated that region-specific changes in the morphological complexity of astrocytes are associated with brain aging in mice (136). Numerous studies have also described abnormalities in the numbers and morphologies of astrocytes in people with DS (17, 18, 56, 137, 138). Our results showed that GFAP-expressing astrocytes in the hippocampi of male Dp16 mice are morphologically abnormal and that GM-CSF treatment partially reversed these abnormalities. GM-CSF treatment also reverses a defect in the percent area of GFAP-positive astrocytes in Dp16 mice, which correlates with improvements in the RAWM task. In agreement with this finding, studies of spinal cord injury showed that GM-CSF treatment reduced GFAP astrogliosis, inhibited glial scar formation, prevented neuronal cell death, preserved axon and myelin structure and facilitated axonal regeneration, and promoted functional recovery (139, 140). WT littermates in our studies also showed improved RAWM performance with GM-CSF treatment without changes in astrocyte morphology or in the percent area of GFAP staining, suggesting that the GM-CSF-induced improvements occur via alternative mechanisms in WT mice.
In summary, these data show that treatment with GM-CSF, a cytokine with immune system modulatory, pro-inflammatory, and anti-inflammatory effects, improves learning/memory and neuropathology in the Dp16 mouse model of DS and improves learning/memory in WT mice. However, as discussed, multiple studies have shown that people with DS and typical aged people (141–144) exhibit an auto-inflammatory or ‘inflammaging’ syndrome that would seem to preclude any benefit from GM-CSF treatment. However, it is more likely that this apparent dichotomy may be explained by the fact that GM-CSF is not merely a pro-inflammatory molecule. A more accurate description would be that GM-CSF modulates the innate immune system, especially in the setting of immune system dysregulation and in the brain (37, 40, 42, 79). Indeed, GM-CSF treatment not only increases the levels of many cellular and cytokine biomarkers of inflammation in the blood of mild-to-moderate AD patients (e.g., neutrophils, monocytes, lymphocytes, IL-2, IL-6, and TNFα), but also reduces the level of the inflammatory cytokine IL-8 and increases the level of the typically anti-inflammatory cytokine IL-10 (48). Thus, GM-CSF has a much more complex physiological effect than simply being pro-inflammatory, and as mentioned above has direct beneficial effects on neurons, their precursors, and other cells of the nervous system. Furthermore, suppressing the inflammation associated with DS in the periphery may be beneficial in the setting of certain acute disorders (36). Thus, the results reported here, although somewhat unexpected, contribute to our growing understanding of the complexity of the innate immune system in the context of inflammation in people with DS and in typical aging populations.
The main limitation of the study is that the precise mechanism by which GM-CSF improves pathology and behavior in Dp16 and WT mice was not and probably cannot be precisely determined, given the multiple and complex effects of GM-CSF on the immune and nervous systems, any or all of which may be beneficial.
Conclusions
The population of people with DS continues to grow, and the average lifespan of people with DS continues to increase. There is, therefore, a critical need to develop therapeutics to improve cognitive function in people with DS and to help them to live more independently. In contrast to previous expectations that inhibiting inflammation and the innate immune system would be the most effective therapy for co-morbidities of DS, we report that treatment with GM-CSF, which has pro-inflammatory, anti-inflammatory and immune regulatory activities, which we have also confirmed in the Dp16 mice, reverses learning/memory deficits in a hippocampal-based task, ameliorates astrocyte abnormalities, and partially normalizes the numbers of interneurons that are reduced in 12–14 month-old male Dp16 mice, while also improving cognitive function in their WT littermates. These findings were unexpected in view of the extensive body of literature describing abnormal and chronic inflammatory perturbations across the lifespan in people with DS and in the typical aging population. Collectively, our results should highlight the possibility that the field’s current approach to cognitive therapy in people with DS, as well as in the typical aging population, should be reconsidered, reversed, or at least modified. Furthermore, these new results, taken together with previous findings of the benefits of GM-CSF treatment in AD mice and other models of neurological disease, and of our results showing GM-CSF/sargramostim efficacy with regard to cognition in clinical trial participants with mild-to-moderate AD as well as in cancer patients, combine to support the hypothesis that GM-CSF may promote neuronal recovery from injury or neurological disease through multiple mechanisms, some of which may also enhance cognitive function. The beneficial effects of GM-CSF on learning and memory may reflect its pro-inflammatory activity, its other physiological effects, or both.
These results indicate that with an almost 30-year history of safety in numerous patient populations across the lifespan, including in vulnerable populations, sargramostim should be tested for safety, tolerability, and efficacy for improving cognition in people with DS. Indeed, a grant to carry out such a clinical trial has recently been awarded to us by the NIH/National Institute on Aging, “GM-CSF/sargramostim treatment to improve cognition in Down syndrome,” which aims to determine the safety and tolerability of sargramostim treatment in young adults with Down syndrome, and to investigate its potential efficacy with regard to cognitive function, quality of life measures, and biomarkers associated with neuronal damage.
Supplementary Material
NBD highlights.
Dp16 mouse model of Down syndrome exhibits Deficits in Spatial Learning and Memory.
GM-CSF improves spatial memory in Dp16 and WT mice.
GM-CSF reverses abnormal clustering of hippocampal astrocytes in Dp16 mice.
GM-CSF partially reverses loss of calretinin-positive interneurons in Dp16 mice.
Acknowledgements
Research support was provided by NIH grant R01 AG037942-01A1, the State of Colorado, the University of Colorado School of Medicine, the Linda Crnic Institute for Down Syndrome, Don and Sue Fisher, the Hewit Family Foundation, Marcy and Bruce Benson, the Global Down Syndrome Foundation, and other generous philanthropists. In addition, we would like to thank the Animal Behavior Core, which is supported by NIH grant 5P30NS048154, Dr. Kimberly Jordan, and the Human Immunology & Immunotherapy Initiative at the University of Colorado Anschutz Medical Campus.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests. Dr. Boyd was recently hired by Partner Therapeutics, the manufacturer of human recombinant GM-CSF.
References
- 1.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet 2004;5(10):725–38. Epub 2004/10/29. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 2.Opitz JM, Gilbert-Barness EF. Reflections on the pathogenesis of Down syndrome. Am J Med Genet Suppl 1990;7:38–51. [DOI] [PubMed] [Google Scholar]
- 3.Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet Suppl 1990;7:31–7. Epub 1990/01/01. doi: 10.1002/ajmg.1320370706. [DOI] [PubMed] [Google Scholar]
- 4.Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev 2000;6(2):84–95. Epub 2000/07/19. doi: . [DOI] [PubMed] [Google Scholar]
- 5.Silverman W Down syndrome: cognitive phenotype. Ment Retard Dev Disabil Res Rev 2007;13(3):228–36. Epub 2007/10/03. doi: 10.1002/mrdd.20156. [DOI] [PubMed] [Google Scholar]
- 6.Head E, Lott IT, Wilcock DM, Lemere CA. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr Alzheimer Res 2016;13(1):18–29. Epub 2015/12/15. doi: 10.2174/1567205012666151020114607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strydom A, Coppus A, Blesa R, Danek A, Fortea J, Hardy J, Levin J, Nuebling G, Rebillat AS, Ritchie C, van Duijn C, Zaman S, Zetterberg H. Alzheimer’s disease in Down syndrome: An overlooked population for prevention trials. Alzheimers Dement (N Y) 2018;4:703–13. Epub 2018/12/26. doi: 10.1016/j.trci.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol 2010;9(6):623–33. Epub 2010/05/25. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 9.Teipel SJ, Hampel H. Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer’s disease. Behav Genet 2006;36(3):405–15. Epub 2006/02/18. doi: 10.1007/s10519-006-9047-x. [DOI] [PubMed] [Google Scholar]
- 10.Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, Granholm AC, Iqbal K, Krams M, Lemere C, Lott I, Mobley W, Ness S, Nixon R, Potter H, Reeves R, Sabbagh M, Silverman W, Tycko B, Whitten M, Wisniewski T. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement 2015;11(6):700–9. Epub 2014/12/17. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging 2005;26(3):383–9. Epub 2005/01/11. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Snyder HM, Bain LJ, Brickman AM, Carrillo MC, Esbensen AJ, Espinosa JM, Fernandez F, Fortea J, Hartley SL, Head E, Hendrix J, Kishnani PS, Lai F, Lao P, Lemere C, Mobley W, Mufson EJ, Potter H, Zaman SH, Granholm AC, Rosas HD, Strydom A, Whitten MS, Rafii MS. Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement 2020;16(7):1065–77. Epub 2020/06/17. doi: 10.1002/alz.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis MAC, Spriggs A, Rodgers A, Campbell J. The Effects of a Peer-Delivered Social Skills Intervention for Adults with Comorbid Down Syndrome and Autism Spectrum Disorder. J Autism Dev Disord 2018;48(6):1869–85. Epub 2017/12/24. doi: 10.1007/s10803-017-3437-1. [DOI] [PubMed] [Google Scholar]
- 14.Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur J Public Health 2007;17(2):221–5. Epub 2006/07/22. doi: 10.1093/eurpub/ckl103. [DOI] [PubMed] [Google Scholar]
- 15.Block A, Ahmed MM, Dhanasekaran AR, Tong S, Gardiner KJ. Sex differences in protein expression in the mouse brain and their perturbations in a model of Down syndrome. Biol Sex Differ 2015;6:24. Epub 2015/11/12. doi: 10.1186/s13293-015-0043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci 2012;35(2):81–91. Epub 2011/12/16. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colombo JA, Reisin HD, Jones M, Bentham C. Development of interlaminar astroglial processes in the cerebral cortex of control and Down’s syndrome human cases. Exp Neurol 2005;193(1):207–17. Epub 2005/04/09. doi: 10.1016/j.expneurol.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Dossi E, Vasile F, Rouach N. Human astrocytes in the diseased brain. Brain Res Bull 2018;136:139–56. Epub 2017/02/19. doi: 10.1016/j.brainresbull.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zorrilla de San Martin J, Delabar JM, Bacci A, Potier MC. GABAergic over-inhibition, a promising hypothesis for cognitive deficits in Down syndrome. Free Radic Biol Med 2018;114:33–9. Epub 2017/10/11. doi: 10.1016/j.freeradbiomed.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Reiche L, Kury P, Gottle P. Aberrant Oligodendrogenesis in Down Syndrome: Shift in Gliogenesis? Cells 2019;8(12). Epub 2019/12/11. doi: 10.3390/cells8121591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rissman RA, Mobley WC. Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer’s disease. J Neurochem 2011;117(4):613–22. Epub 2011/03/11. doi: 10.1111/j.1471-4159.2011.07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds GP, Warner CE. Amino acid neurotransmitter deficits in adult Down’s syndrome brain tissue. Neurosci Lett 1988;94(1–2):224–7. Epub 1988/11/22. doi: 10.1016/0304-3940(88)90299-6. [DOI] [PubMed] [Google Scholar]
- 23.Gupta M, Dhanasekaran AR, Gardiner KJ. Mouse models of Down syndrome: gene content and consequences. Mamm Genome 2016;27(11–12):538–55. Epub 2016/08/20. doi: 10.1007/s00335-016-9661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Che M, Yuan J, Yu Y, Cao C, Qin XY, Cheng Y. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget 2017;8(48):84489–96. Epub 2017/11/16. doi: 10.18632/oncotarget.21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, DeGregori J, Espinosa JM. Trisomy 21 consistently activates the interferon response. Elife 2016;5. Epub 2016/07/30. doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, Rachubinski AL, Wolter-Warmerdam K, Hickey F, Espinosa JM, Blumenthal T. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci Rep 2017;7(1):14818. Epub 2017/11/03. doi: 10.1038/s41598-017-13858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huggard D, Doherty DG, Molloy EJ. Immune Dysregulation in Children With Down Syndrome. Front Pediatr 2020;8:73. Epub 2020/03/17. doi: 10.3389/fped.2020.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huggard D, Kelly L, Ryan E, McGrane F, Lagan N, Roche E, Balfe J, Leahy TR, Franklin O, Doherty DG, Molloy EJ. Increased systemic inflammation in children with Down syndrome. Cytokine 2020;127:154938. Epub 2019/12/01. doi: 10.1016/j.cyto.2019.154938. [DOI] [PubMed] [Google Scholar]
- 29.Flores-Aguilar L, Iulita MF, Kovecses O, Torres MD, Levi SM, Zhang Y, Askenazi M, Wisniewski T, Busciglio J, Cuello AC. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 2020. Epub 2020/11/19. doi: 10.1093/brain/awaa326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed MM, Sturgeon X, Ellison M, Davisson MT, Gardiner KJ. Loss of correlations among proteins in brains of the Ts65Dn mouse model of down syndrome. J Proteome Res 2012;11(2):1251–63. Epub 2012/01/05. doi: 10.1021/pr2011582. [DOI] [PubMed] [Google Scholar]
- 31.Spellman C, Ahmed MM, Dubach D, Gardiner KJ. Expression of trisomic proteins in Down syndrome model systems. Gene 2013;512(2):219–25. Epub 2012/10/30. doi: 10.1016/j.gene.2012.10.051. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed MM, Dhanasekaran AR, Tong S, Wiseman FK, Fisher EM, Tybulewicz VL, Gardiner KJ. Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum Mol Genet 2013;22(9):1709–24. Epub 2013/01/26. doi: 10.1093/hmg/ddt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araya P, Waugh KA, Sullivan KD, Núñez NG, Roselli E, Smith KP, Granrath RE, Rachubinski AL, Enriquez Estrada B, Butcher ET, Minter R, Tuttle KD, Bruno TC, Maccioni M, Espinosa JM. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci U S A 2019;116(48):24231–41. Epub 2019/11/09. doi: 10.1073/pnas.1908129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powers RK, Culp-Hill R, Ludwig MP, Smith KP, Waugh KA, Minter R, Tuttle KD, Lewis HC, Rachubinski AL, Granrath RE, Carmona-Iragui M, Wilkerson RB, Kahn DE, Joshi M, Lleó A, Blesa R, Fortea J, D’Alessandro A, Costello JC, Sullivan KD, Espinosa JM. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun 2019;10(1):4766. Epub 2019/10/20. doi: 10.1038/s41467-019-12739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y, Hong W, Wang X, Zhang P, Körner H, Tu J, Wei W. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma? Front Mol Neurosci 2019;12:125. Epub 2019/05/28. doi: 10.3389/fnmol.2019.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, Espinosa JM. Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep 2019;5(4):365–7. Epub 2019/04/23. doi: 10.1016/j.jdcr.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M, Padmanabhan J, Neame P, Wefes I, Sanchez-Ramos J, Arendash GW, Potter H. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. Journal of Alzheimer’s disease : JAD 2010;21(2):507–18. doi: 10.3233/JAD-2010-091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varga-Medveczky Z, Kovács N, Tóth ME, Sántha M, Horváth I, Bors LA, Fónagy K, Imre T, Szabó P, Máthé D, Erdő F. Age-Related Inflammatory Balance Shift, Nasal Barrier Function, and Cerebro-Morphological Status in Healthy and Diseased Rodents. Front Neurosci 2021;15:700729. Epub 2021/08/10. doi: 10.3389/fnins.2021.700729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitz M, Hermann P, Oikonomou P, Stoeck K, Ebert E, Poliakova T, Schmidt C, Llorens F, Zafar S, Zerr I. Cytokine profiles and the role of cellular prion protein in patients with vascular dementia and vascular encephalopathy. Neurobiol Aging 2015;36(9):2597–606. Epub 2015/07/15. doi: 10.1016/j.neurobiolaging.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharya P, Budnick I, Singh M, Thiruppathi M, Alharshawi K, Elshabrawy H, Holterman MJ, Prabhakar BS. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J Interferon Cytokine Res 2015;35(8):585–99. Epub 2015/03/25. doi: 10.1089/jir.2014.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridwan S, Bauer H, Frauenknecht K, von Pein H, Sommer CJ. Distribution of granulocyte-monocyte colony-stimulating factor and its receptor α-subunit in the adult human brain with specific reference to Alzheimer’s disease. J Neural Transm (Vienna) 2012;119(11):1389–406. Epub 2012/03/21. doi: 10.1007/s00702-012-0794-y. [DOI] [PubMed] [Google Scholar]
- 42.Borriello F, Galdiero MR, Varricchi G, Loffredo S, Spadaro G, Marone G. Innate Immune Modulation by GM-CSF and IL-3 in Health and Disease. Int J Mol Sci 2019;20(4). Epub 2019/02/17. doi: 10.3390/ijms20040834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonaventura A, Vecchié A, Wang TS, Lee E, Cremer PC, Carey B, Rajendram P, Hudock KM, Korbee L, Van Tassell BW, Dagna L, Abbate A. Targeting GM-CSF in COVID-19 Pneumonia: Rationale and Strategies. Front Immunol 2020;11:1625. Epub 2020/07/29. doi: 10.3389/fimmu.2020.01625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manczak M, Mao P, Nakamura K, Bebbington C, Park B, Reddy PH. Neutralization of granulocyte macrophage colony-stimulating factor decreases amyloid beta 1–42 and suppresses microglial activity in a transgenic mouse model of Alzheimer’s disease. Hum Mol Genet 2009;18(20):3876–93. Epub 2009/07/21. doi: 10.1093/hmg/ddp331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Yokoyama I, Mosley RL, Gendelman HE. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J Neuroimmunol 2018;319:80–92. Epub 2018/03/27. doi: 10.1016/j.jneuroim.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, Shen JC, Zou B, Xie XS, Tingle M, Hinkson IV, Angst MS, Wyss-Coray T. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 2017;544(7651):488–92. Epub 2017/04/21. doi: 10.1038/nature22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jim HS, Boyd TD, Booth-Jones M, Pidala J, Potter H. Granulocyte Macrophage Colony Stimulating Factor Treatment is Associated with Improved Cognition in Cancer Patients. Brain disorders & therapy 2012;1(1). doi: 10.4172/bdt.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Potter H, Woodcock JH, Boyd TD, Coughlan CM, O’Shaughnessy JR, Borges MT, Thaker AA, Raj BA, Adamszuk K, Scott D, Adame V, Anton P, Chial HJ, Gray H, Daniels J, Stocker ME, Sillau SH. Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimers Dement (N Y) 2021;7(1):e12158. Epub 2021/03/30. doi: 10.1002/trc2.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 1995;11(2):177–84. Epub 1995/10/01. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet 2007;16(11):1359–66. Epub 2007/04/07. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- 51.Jankowsky JL, Younkin LH, Gonzales V, Fadale DJ, Slunt HH, Lester HA, Younkin SG, Borchelt DR. Rodent A beta modulates the solubility and distribution of amyloid deposits in transgenic mice. J Biol Chem 2007;282(31):22707–20. Epub 2007/06/09. doi: 10.1074/jbc.M611050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc 2006;1(4):1671–9. Epub 2007/05/10. doi: 10.1038/nprot.2006.275. [DOI] [PubMed] [Google Scholar]
- 53.Faizi M, Bader PL, Tun C, Encarnacion A, Kleschevnikov A, Belichenko P, Saw N, Priestley M, Tsien RW, Mobley WC, Shamloo M. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down syndrome: activation of beta1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol Dis 2011;43(2):397–413. Epub 2011/04/30. doi: 10.1016/j.nbd.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owen JL, Torroella-Kouri M, Handel-Fernandez ME, Iragavarapu-Charyulu V. GM-CSF up-regulates the expression of CCL2 by T lymphocytes in mammary tumor-bearing mice. Int J Mol Med 2007;20(1):129–36. Epub 2007/06/06. [PubMed] [Google Scholar]
- 55.Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 2007;178(8):5245–52. Epub 2007/04/04. doi: 10.4049/jimmunol.178.8.5245. [DOI] [PubMed] [Google Scholar]
- 56.Perry JC, Pakkenberg B, Vann SD. Striking reduction in neurons and glial cells in anterior thalamic nuclei of older patients with Down syndrome. Neurobiol Aging 2019;75:54–61. Epub 2018/12/15. doi: 10.1016/j.neurobiolaging.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lockrow JP, Fortress AM, Granholm AC. Age-related neurodegeneration and memory loss in down syndrome. Curr Gerontol Geriatr Res 2012;2012:463909. Epub 2012/05/01. doi: 10.1155/2012/463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huo HQ, Qu ZY, Yuan F, Ma L, Yao L, Xu M, Hu Y, Ji J, Bhattacharyya A, Zhang SC, Liu Y. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Reports 2018;10(4):1251–66. Epub 2018/03/13. doi: 10.1016/j.stemcr.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kittler P, Krinsky-McHale SJ, Devenny DA. Sex differences in performance over 7 years on the Wechsler Intelligence Scale for Children--Revised among adults with intellectual disability. J Intellect Disabil Res 2004;48(Pt 2):114–22. Epub 2004/01/16. doi: 10.1111/j.1365-2788.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 60.de Sola S, de la Torre R, Sánchez-Benavides G, Benejam B, Cuenca-Royo A, Del Hoyo L, Rodríguez J, Catuara-Solarz S, Sanchez-Gutierrez J, Dueñas-Espin I, Hernandez G, Peña-Casanova J, Langohr K, Videla S, Blehaut H, Farre M, Dierssen M. A new cognitive evaluation battery for Down syndrome and its relevance for clinical trials. Front Psychol 2015;6:708. Epub 2015/06/20. doi: 10.3389/fpsyg.2015.00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franconi F, Campesi I. Pharmacogenomics, pharmacokinetics and pharmacodynamics: interaction with biological differences between men and women. Br J Pharmacol 2014;171(3):580–94. Epub 2013/08/29. doi: 10.1111/bph.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levenga J, Peterson DJ, Cain P, Hoeffer CA. Sleep Behavior and EEG Oscillations in Aged Dp(16)1Yey/+ Mice: A Down Syndrome Model. Neuroscience 2018;376:117–26. Epub 2018/02/20. doi: 10.1016/j.neuroscience.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goodliffe JW, Olmos-Serrano JL, Aziz NM, Pennings JL, Guedj F, Bianchi DW, Haydar TF. Absence of Prenatal Forebrain Defects in the Dp(16)1Yey/+ Mouse Model of Down Syndrome. J Neurosci 2016;36(10):2926–44. Epub 2016/03/11. doi: 10.1523/jneurosci.2513-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Capone G, Goyal P, Ares W, Lannigan E. Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. Am J Med Genet C Semin Med Genet 2006;142c(3):158–72. Epub 2006/07/14. doi: 10.1002/ajmg.c.30097. [DOI] [PubMed] [Google Scholar]
- 65.Godfrey M, Lee NR. Memory profiles in Down syndrome across development: a review of memory abilities through the lifespan. J Neurodev Disord 2018;10(1):5. Epub 2018/01/31. doi: 10.1186/s11689-017-9220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herault Y, Duchon A, Velot E, Marechal D, Brault V. The in vivo Down syndrome genomic library in mouse. Prog Brain Res 2012;197:169–97. Epub 2012/05/01. doi: 10.1016/b978-0-444-54299-1.00009-1. [DOI] [PubMed] [Google Scholar]
- 67.Stasko MR, Costa AC. Experimental parameters affecting the Morris water maze performance of a mouse model of Down syndrome. Behav Brain Res 2004;154(1):1–17. Epub 2004/08/11. doi: 10.1016/j.bbr.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 68.Raveau M, Polygalov D, Boehringer R, Amano K, Yamakawa K, McHugh TJ. Alterations of in vivo CA1 network activity in Dp(16)1Yey Down syndrome model mice. Elife 2018;7. Epub 2018/02/28. doi: 10.7554/eLife.31543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aziz NM, Guedj F, Pennings JLA, Olmos-Serrano JL, Siegel A, Haydar TF, Bianchi DW. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Dis Model Mech 2018;11(6). Epub 2018/05/03. doi: 10.1242/dmm.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu T, Liu C, Belichenko P, Clapcote SJ, Li S, Pao A, Kleschevnikov A, Bechard AR, Asrar S, Chen R, Fan N, Zhou Z, Jia Z, Chen C, Roder JC, Liu B, Baldini A, Mobley WC, Yu YE. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res 2010;1366:162–71. Epub 2010/10/12. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campbell C, Landry O, Russo N, Flores H, Jacques S, Burack JA. Cognitive flexibility among individuals with Down syndrome: assessing the influence of verbal and nonverbal abilities. Am J Intellect Dev Disabil 2013;118(3):193–200. Epub 2013/06/06. doi: 10.1352/1944-7558-118.3.193. [DOI] [PubMed] [Google Scholar]
- 72.Zou T, Satake A, Ojha P, Kambayashi T. Cellular therapies supplement: the role of granulocyte macrophage colony-stimulating factor and dendritic cells in regulatory T-cell homeostasis and expansion. Transfusion 2011;51 Suppl 4:160s–8s. Epub 2011/12/07. doi: 10.1111/j.1537-2995.2011.03379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rowin J, Thiruppathi M, Arhebamen E, Sheng J, Prabhakar BS, Meriggioli MN. Granulocyte macrophage colony-stimulating factor treatment of a patient in myasthenic crisis: effects on regulatory T cells. Muscle Nerve 2012;46(3):449–53. Epub 2012/08/22. doi: 10.1002/mus.23488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weinhage T, Dabritz J, Brockhausen A, Wirth T, Bruckner M, Belz M, Foell D, Varga G. Granulocyte Macrophage Colony-Stimulating Factor-Activated CD39(+)/CD73(+) Murine Monocytes Modulate Intestinal Inflammation via Induction of Regulatory T Cells. Cell Mol Gastroenterol Hepatol 2015;1(4):433–49.e1. Epub 2015/05/06. doi: 10.1016/j.jcmgh.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hotta M, Yoshimura H, Satake A, Tsubokura Y, Ito T, Nomura S. GM-CSF therapy inhibits chronic graft-versus-host disease via expansion of regulatory T cells. Eur J Immunol 2019;49(1):179–91. Epub 2018/11/21. doi: 10.1002/eji.201847684. [DOI] [PubMed] [Google Scholar]
- 76.Sheng JR, Muthusamy T, Prabhakar BS, Meriggioli MN. GM-CSF-induced regulatory T cells selectively inhibit anti-acetylcholine receptor-specific immune responses in experimental myasthenia gravis. J Neuroimmunol 2011;240–241:65–73. Epub 2011/11/22. doi: 10.1016/j.jneuroim.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c+CD8a--dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol 2009;21(3):269–82. Epub 2009/01/29. doi: 10.1093/intimm/dxn147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dabritz J, Weinhage T, Varga G, Wirth T, Walscheid K, Brockhausen A, Schwarzmaier D, Bruckner M, Ross M, Bettenworth D, Roth J, Ehrchen JM, Foell D. Reprogramming of monocytes by GM-CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol 2015;194(5):2424–38. Epub 2015/02/06. doi: 10.4049/jimmunol.1401482. [DOI] [PubMed] [Google Scholar]
- 79.Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BS. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015;75(2):261–71. Epub 2015/06/27. doi: 10.1016/j.cyto.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pellegrini FP, Marinoni M, Frangione V, Tedeschi A, Gandini V, Ciglia F, Mortara L, Accolla RS, Nespoli L. Down syndrome, autoimmunity and T regulatory cells. Clin Exp Immunol 2012;169(3):238–43. Epub 2012/08/07. doi: 10.1111/j.1365-2249.2012.04610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amr NH. Thyroid Disorders in Subjects with Down Syndrome: An Update. Acta Biomed 2018;89(1):132–9. Epub 2018/04/11. doi: 10.23750/abm.v89i1.7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aversa T, Crisafulli G, Zirilli G, De Luca F, Gallizzi R, Valenzise M. Epidemiological and clinical aspects of autoimmune thyroid diseases in children with Down’s syndrome. Ital J Pediatr 2018;44(1):39. Epub 2018/03/23. doi: 10.1186/s13052-018-0478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vasu C, Dogan RN, Holterman MJ, Prabhakar BS. Selective induction of dendritic cells using granulocyte macrophage-colony stimulating factor, but not fms-like tyrosine kinase receptor 3-ligand, activates thyroglobulin-specific CD4+/CD25+ T cells and suppresses experimental autoimmune thyroiditis. J Immunol 2003;170(11):5511–22. Epub 2003/05/22. doi: 10.4049/jimmunol.170.11.5511. [DOI] [PubMed] [Google Scholar]
- 84.Gangi E, Vasu C, Cheatem D, Prabhakar BS. IL-10-producing CD4+CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J Immunol 2005;174(11):7006–13. Epub 2005/05/21. doi: 10.4049/jimmunol.174.11.7006. [DOI] [PubMed] [Google Scholar]
- 85.Antonucci F, Alpar A, Kacza J, Caleo M, Verderio C, Giani A, Martens H, Chaudhry FA, Allegra M, Grosche J, Michalski D, Erck C, Hoffmann A, Harkany T, Matteoli M, Hartig W. Cracking down on inhibition: selective removal of GABAergic interneurons from hippocampal networks. J Neurosci 2012;32(6):1989–2001. Epub 2012/02/11. doi: 10.1523/jneurosci.2720-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tremblay R, Lee S, Rudy B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016;91(2):260–92. Epub 2016/08/02. doi: 10.1016/j.neuron.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Deng X, Gu L, Sui N, Guo J, Liang J. Parvalbumin interneuron in the ventral hippocampus functions as a discriminator in social memory. Proc Natl Acad Sci U S A 2019;116(33):16583–92. Epub 2019/07/31. doi: 10.1073/pnas.1819133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buschmann IR, Busch HJ, Mies G, Hossmann KA. Therapeutic induction of arteriogenesis in hypoperfused rat brain via granulocyte-macrophage colony-stimulating factor. Circulation 2003;108(5):610–5. Epub 2003/07/02. doi: 10.1161/01.Cir.0000074209.17561.99. [DOI] [PubMed] [Google Scholar]
- 89.Hossmann KA, Buschmann IR. Granulocyte-macrophage colony-stimulating factor as an arteriogenic factor in the treatment of ischaemic stroke. Expert Opin Biol Ther 2005;5(12):1547–56. Epub 2005/12/02. doi: 10.1517/14712598.5.12.1547. [DOI] [PubMed] [Google Scholar]
- 90.Schneeloch E, Mies G, Busch HJ, Buschmann IR, Hossmann KA. Granulocyte-macrophage colony-stimulating factor-induced arteriogenesis reduces energy failure in hemodynamic stroke. Proc Natl Acad Sci U S A 2004;101(34):12730–5. Epub 2004/08/13. doi: 10.1073/pnas.0404880101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res 2006;1089(1):179–85. Epub 2006/05/09. doi: 10.1016/j.brainres.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 92.Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, Gassler N, Aronowski J, Mier W, Kirsch F, Dittgen T, Bach A, Sommer C, Schneider A. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood Flow Metab 2008;28(1):29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- 93.Schneider UC, Schilling L, Schroeck H, Nebe CT, Vajkoczy P, Woitzik J. Granulocyte-macrophage colony-stimulating factor-induced vessel growth restores cerebral blood supply after bilateral carotid artery occlusion. Stroke 2007;38(4):1320–8. doi: 10.1161/01.STR.0000259707.43496.71. [DOI] [PubMed] [Google Scholar]
- 94.Todo K, Kitagawa K, Sasaki T, Omura-Matsuoka E, Terasaki Y, Oyama N, Yagita Y, Hori M. Granulocyte-macrophage colony-stimulating factor enhances leptomeningeal collateral growth induced by common carotid artery occlusion. Stroke 2008;39(6):1875–82. Epub 2008/04/05. doi: 10.1161/strokeaha.107.503433. [DOI] [PubMed] [Google Scholar]
- 95.Kong T, Choi JK, Park H, Choi BH, Snyder BJ, Bukhari S, Kim NK, Huang X, Park SR, Park HC, Ha Y. Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats. Laboratory investigation. J Neurosurg 2009;111(1):155–63. doi: 10.3171/2008.12.JNS08172. [DOI] [PubMed] [Google Scholar]
- 96.Theoret JK, Jadavji NM, Zhang M, Smith PD. Granulocyte macrophage colony-stimulating factor treatment results in recovery of motor function after white matter damage in mice. Eur J Neurosci 2016;43(1):17–24. Epub 2015/10/17. doi: 10.1111/ejn.13105. [DOI] [PubMed] [Google Scholar]
- 97.Schallenberg M, Charalambous P, Thanos S. GM-CSF regulates the ERK1/2 pathways and protects injured retinal ganglion cells from induced death. Exp Eye Res 2009;89(5):665–77. Epub 2009/06/30. doi: 10.1016/j.exer.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 98.Schallenberg M, Charalambous P, Thanos S. GM-CSF protects rat photoreceptors from death by activating the SRC-dependent signalling and elevating anti-apoptotic factors and neurotrophins. Graefes Arch Clin Exp Ophthalmol 2012;250(5):699–712. Epub 2012/02/03. doi: 10.1007/s00417-012-1932-9. [DOI] [PubMed] [Google Scholar]
- 99.Legacy J, Hanea S, Theoret J, Smith PD. Granulocyte macrophage colony-stimulating factor promotes regeneration of retinal ganglion cells in vitro through a mammalian target of rapamycin-dependent mechanism. J Neurosci Res 2013;91(6):771–9. Epub 2013/03/05. doi: 10.1002/jnr.23205. [DOI] [PubMed] [Google Scholar]
- 100.Kim JK, Choi BH, Park HC, Park SR, Kim YS, Yoon SH, Park HS, Kim EY, Ha Y. Effects of GM-CSF on the neural progenitor cells. Neuroreport 2004;15(14):2161–5. [DOI] [PubMed] [Google Scholar]
- 101.Huang X, Choi JK, Park SR, Ha Y, Park H, Yoon SH, Park HC, Park JO, Choi BH. GM-CSF inhibits apoptosis of neural cells via regulating the expression of apoptosis-related proteins. Neurosci Res 2007;58(1):50–7. Epub 2007/03/03. doi: 10.1016/j.neures.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 102.Choi JK, Choi BH, Ha Y, Park H, Yoon SH, Park HC, Park SR. Signal transduction pathways of GM-CSF in neural cell lines. Neurosci Lett 2007;420(3):217–22. Epub 2007/06/09. doi: 10.1016/j.neulet.2007.03.065. [DOI] [PubMed] [Google Scholar]
- 103.Choi JK, Kim KH, Park H, Park SR, Choi BH. Granulocyte macrophage-colony stimulating factor shows anti-apoptotic activity in neural progenitor cells via JAK/STAT5-Bcl-2 pathway. Apoptosis 2011;16(2):127–34. Epub 2010/11/06. doi: 10.1007/s10495-010-0552-2. [DOI] [PubMed] [Google Scholar]
- 104.Choi JK, Kim KH, Park SR, Choi BH. Granulocyte macrophage colony-stimulating factor shows anti-apoptotic activity via the PI3K-NF-kappaB-HIF-1alpha-survivin pathway in mouse neural progenitor cells. Mol Neurobiol 2014;49(2):724–33. Epub 2013/09/12. doi: 10.1007/s12035-013-8550-3. [DOI] [PubMed] [Google Scholar]
- 105.Kruger C, Laage R, Pitzer C, Schabitz WR, Schneider A. The hematopoietic factor GM-CSF (granulocyte-macrophage colony-stimulating factor) promotes neuronal differentiation of adult neural stem cells in vitro. BMC Neurosci 2007;8:88. doi: 10.1186/1471-2202-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guthridge MA, Powell JA, Barry EF, Stomski FC, McClure BJ, Ramshaw H, Felquer FA, Dottore M, Thomas DT, To B, Begley CG, Lopez AF. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. Embo j 2006;25(3):479–89. Epub 2006/01/27. doi: 10.1038/sj.emboj.7600948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McLay RN, Kimura M, Banks WA, Kastin AJ. Granulocyte-macrophage colony-stimulating factor crosses the blood--brain and blood--spinal cord barriers. Brain 1997;120 (Pt 11):2083–91. Epub 1997/12/16. doi: 10.1093/brain/120.11.2083. [DOI] [PubMed] [Google Scholar]
- 108.Sawada M, Itoh Y, Suzumura A, Marunouchi T. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci Lett 1993;160(2):131–4. Epub 1993/10/01. doi: 10.1016/0304-3940(93)90396-3. [DOI] [PubMed] [Google Scholar]
- 109.Baldwin GC, Benveniste EN, Chung GY, Gasson JC, Golde DW. Identification and characterization of a high-affinity granulocyte-macrophage colony-stimulating factor receptor on primary rat oligodendrocytes. Blood 1993;82(11):3279–82. Epub 1993/12/01. [PubMed] [Google Scholar]
- 110.Krieger M, Both M, Kranig SA, Pitzer C, Klugmann M, Vogt G, Draguhn A, Schneider A. The hematopoietic cytokine granulocyte-macrophage colony stimulating factor is important for cognitive functions. Sci Rep 2012;2:697. doi: 10.1038/srep00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim J, Kim NK, Park SR, Choi BH. GM-CSF Enhances Mobilization of Bone Marrow Mesenchymal Stem Cells via a CXCR4-Medicated Mechanism. Tissue Eng Regen Med 2019;16(1):59–68. Epub 2019/03/01. doi: 10.1007/s13770-018-0163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park SR, Cho A, Kim JW, Lee HY, Hong IS. A Novel Endogenous Damage Signal, CSF-2, Activates Multiple Beneficial Functions of Adipose Tissue-Derived Mesenchymal Stem Cells. Mol Ther 2019;27(6):1087–100. Epub 2019/04/10. doi: 10.1016/j.ymthe.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chakari-Khiavi F, Dolati S, Chakari-Khiavi A, Abbaszadeh H, Aghebati-Maleki L, Pourlak T, Mehdizadeh A, Yousefi M. Prospects for the application of mesenchymal stem cells in Alzheimer’s disease treatment. Life Sci 2019;231:116564. Epub 2019/06/17. doi: 10.1016/j.lfs.2019.116564. [DOI] [PubMed] [Google Scholar]
- 114.Ge M, Zhang Y, Hao Q, Zhao Y, Dong B. Effects of mesenchymal stem cells transplantation on cognitive deficits in animal models of Alzheimer’s disease: A systematic review and meta-analysis. Brain Behav 2018;8(7):e00982. Epub 2018/06/08. doi: 10.1002/brb3.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jiang Y, Gao H, Yuan H, Xu H, Tian M, Du G, Xie W. Amelioration of postoperative cognitive dysfunction in mice by mesenchymal stem cell-conditioned medium treatments is associated with reduced inflammation, oxidative stress and increased BDNF expression in brain tissues. Neurosci Lett 2019;709:134372. Epub 2019/07/12. doi: 10.1016/j.neulet.2019.134372. [DOI] [PubMed] [Google Scholar]
- 116.Maldonado-Lasunción I, Verhaagen J, Oudega M. Mesenchymal Stem Cell-Macrophage Choreography Supporting Spinal Cord Repair. Neurotherapeutics 2018;15(3):578–87. Epub 2018/05/08. doi: 10.1007/s13311-018-0629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mendes Filho D, Ribeiro PDC, Oliveira LF, de Paula DRM, Capuano V, de Assunção TSF, da Silva VJD. Therapy With Mesenchymal Stem Cells in Parkinson Disease: History and Perspectives. Neurologist 2018;23(4):141–7. Epub 2018/06/29. doi: 10.1097/nrl.0000000000000188. [DOI] [PubMed] [Google Scholar]
- 118.Nakazaki M, Sasaki M, Kataoka-Sasaki Y, Oka S, Suzuki J, Sasaki Y, Nagahama H, Hashi K, Kocsis JD, Honmou O. Intravenous infusion of mesenchymal stem cells improves impaired cognitive function in a cerebral small vessel disease model. Neuroscience 2019;408:361–77. Epub 2019/04/19. doi: 10.1016/j.neuroscience.2019.04.018. [DOI] [PubMed] [Google Scholar]
- 119.Uccelli A, Laroni A, Brundin L, Clanet M, Fernandez O, Nabavi SM, Muraro PA, Oliveri RS, Radue EW, Sellner J, Soelberg Sorensen P, Sormani MP, Wuerfel JT, Battaglia MA, Freedman MS. MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): a randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 2019;20(1):263. Epub 2019/05/11. doi: 10.1186/s13063-019-3346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996;47(2):425–32. Epub 1996/08/01. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 121.Tarkowski E, Wallin A, Regland B, Blennow K, Tarkowski A. Local and systemic GM-CSF increase in Alzheimer’s disease and vascular dementia. Acta Neurol Scand 2001;103(3):166–74. Epub 2001/03/10. doi: 10.1034/j.1600-0404.2001.103003166.x. [DOI] [PubMed] [Google Scholar]
- 122.Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J Neuroinflammation 2005;2(1):9. Epub 2005/03/15. doi: 10.1186/1742-2094-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Volmar CH, Ait-Ghezala G, Frieling J, Paris D, Mullan MJ. The granulocyte macrophage colony stimulating factor (GM-CSF) regulates amyloid beta (Abeta) production. Cytokine 2008;42(3):336–44. Epub 2008/04/25. doi: 10.1016/j.cyto.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 124.Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 2008;65(7):896–905. Epub 2008/05/14. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.ADAPT_FS_Research_Group. Follow-up evaluation of cognitive function in the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial and its Follow-up Study. Alzheimers Dement 2015;11(2):216–25.e1. Epub 2014/07/16. doi: 10.1016/j.jalz.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ahmed MM, Johnson NR, Boyd TD, Coughlan CM, Chial HJ, Potter H. Innate Immune System Activation and Neuroinflammation in Down Syndrome and Neurodegeneration: Therapeutic Targets or Partners? Front Aging Neurosci 2021. Epub 2021/09/16. doi: 10.3389/fnagi.2021.718426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta 2013;1832(8):1249–59. Epub 2013/04/23. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 2010;119(1):7–35. Epub 2009/12/17. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011;144(5):810–23. Epub 2011/03/08. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bezzi P, Volterra A. Astrocytes: powering memory. Cell 2011;144(5):644–5. Epub 2011/03/08. doi: 10.1016/j.cell.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 131.Diniz LP, Tortelli V, Garcia MN, Araujo AP, Melo HM, Silva GS, Felice FG, Alves-Leon SV, Souza JM, Romao LF, Castro NG, Gomes FC. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia 2014;62(12):1917–31. Epub 2014/07/22. doi: 10.1002/glia.22713. [DOI] [PubMed] [Google Scholar]
- 132.Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, Eroglu C. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 2017;551(7679):192–7. Epub 2017/11/10. doi: 10.1038/nature24638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci 2012;32(46):16129–40. Epub 2012/11/16. doi: 10.1523/jneurosci.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett’s syndrome. Nature 2011;475(7357):497–500. Epub 2011/07/01. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tarasov VV, Svistunov AA, Chubarev VN, Sologova SS, Mukhortova P, Levushkin D, Somasundaram SG, Kirkland CE, Bachurin SO, Aliev G. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front Pharmacol 2019;10:1612. Epub 2020/03/03. doi: 10.3389/fphar.2019.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bondi H, Bortolotto V, Canonico PL, Grilli M. Complex and regional-specific changes in the morphological complexity of GFAP(+) astrocytes in middle-aged mice. Neurobiol Aging 2021;100:59–71. Epub 2021/01/26. doi: 10.1016/j.neurobiolaging.2020.12.018. [DOI] [PubMed] [Google Scholar]
- 137.Kanaumi T, Milenkovic I, Adle-Biassette H, Aronica E, Kovacs GG. Non-neuronal cell responses differ between normal and Down syndrome developing brains. Int J Dev Neurosci 2013;31(8):796–803. Epub 2013/10/12. doi: 10.1016/j.ijdevneu.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 138.Goodison KL, Parhad IM, White CL III, Sima AAF, Clark AW. Neuronal and Glial Gene Expression in Neocortex of Down’s Syndrome and Alzheimer’s Disease. Journal of Neuropathology & Experimental Neurology 1993;52(3):192–8. doi: 10.1097/00005072-199305000-00002. [DOI] [PubMed] [Google Scholar]
- 139.Huang X, Kim JM, Kong TH, Park SR, Ha Y, Kim MH, Park H, Yoon SH, Park HC, Park JO, Min BH, Choi BH. GM-CSF inhibits glial scar formation and shows long-term protective effect after spinal cord injury. J Neurol Sci 2009;277(1–2):87–97. Epub 2008/11/27. doi: 10.1016/j.jns.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 140.Chung J, Kim MH, Yoon YJ, Kim KH, Park SR, Choi BH. Effects of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor on glial scar formation after spinal cord injury in rats. J Neurosurg Spine 2014;21(6):966–73. Epub 2014/10/04. doi: 10.3171/2014.8.Spine131090. [DOI] [PubMed] [Google Scholar]
- 141.Serre-Miranda C, Roque S, Santos NC, Costa P, Sousa N, Palha JA, Correia-Neves M. Cognition Is Associated With Peripheral Immune Molecules in Healthy Older Adults: A Cross-Sectional Study. Front Immunol 2020;11:2045. Epub 2020/09/29. doi: 10.3389/fimmu.2020.02045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Frasca D, Blomberg BB. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016;17(1):7–19. Epub 2015/04/30. doi: 10.1007/s10522-015-9578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ashraf-Ganjouei A, Moradi K, Bagheri S, Aarabi MH. The association between systemic inflammation and cognitive performance in healthy adults. J Neuroimmunol 2020;345:577272. Epub 2020/06/14. doi: 10.1016/j.jneuroim.2020.577272. [DOI] [PubMed] [Google Scholar]
- 144.Trollor JN, Smith E, Agars E, Kuan SA, Baune BT, Campbell L, Samaras K, Crawford J, Lux O, Kochan NA, Brodaty H, Sachdev P. The association between systemic inflammation and cognitive performance in the elderly: the Sydney Memory and Ageing Study. Age (Dordr) 2012;34(5):1295–308. Epub 2011/08/20. doi: 10.1007/s11357-011-9301-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.