

Abstract
Background and Objectives
Little is known of the functional potential of the gut microbiome in pediatric-onset multiple sclerosis (MS). We performed metagenomic analyses using stool samples from individuals with pediatric-onset MS and unaffected controls.
Methods
Persons ≤21 years old enrolled in the Canadian Pediatric Demyelinating Disease Network providing a stool sample were eligible. Twenty patients with MS (McDonald criteria) with symptom onset <18 years were matched to 20 controls by sex, age (±3 years), stool consistency, and race. Microbial taxonomy and functional potentials were estimated from stool sample–derived metagenomic reads and compared by disease status (MS vs controls) and disease-modifying drug (DMD) exposure using alpha diversity, relative abundance, and prevalence using Wilcoxon rank sum, ALDEx2, and Fisher exact tests, respectively.
Results
Individuals with MS were aged 13.6 years (mean) at symptom onset and 8 were DMD-naive. Mean ages at stool sample were 16.1 and 15.4 years for MS and control participants, respectively; 80% were girls. Alpha diversity of enzymes and proteins did not differ by disease or DMD status (p > 0.20), but metabolic pathways, gene annotations, and microbial taxonomy did. Individuals with MS (vs controls) exhibited higher methanogenesis prevalence (odds ratio 10, p = 0.044) and Methanobrevibacter abundance (log2 fold change [LFC] 1.7, p = 0.0014), but lower homolactic fermentation abundance (LFC −0.48, p = 0.039). Differences by DMD status included lower phosphate butyryl transferase for DMD-naive vs exposed patients with MS (LFC −1.0, p = 0.033).
Discussion
The gut microbiome's functional potential and taxonomy differed between individuals with pediatric-onset MS vs controls, including higher prevalence of a methane-producing pathway from Archaea and depletion of the lactate fermentation pathway. DMD exposure was associated with butyrate-producing enzyme enrichment. Together these findings indicate that the gut microbiome of individuals with MS may have a disturbed functional potential.
Although the causes of multiple sclerosis (MS), a neuroinflammatory and neurodegenerative disease, are unknown, environmental and lifestyle factors are considered to have a greater role than genetics in the susceptibility to MS, and there is a major need to identify these factors.1 One of the potential environmental factors that has received recent attention is the gut microbiota. The gut microbiota plays a critical role in host immune development and response and can affect the function of the CNS, as well as mediating or modifying the effect of other environmental exposures on health, such as diet-associated weight changes or the clinical effect of drugs.2-4
Pediatric-onset MS (POMS), although relatively rare (affecting <5% of all patients with MS), represents a unique opportunity to examine exposures, such as the gut microbiome, very close to the actual biological onset of MS. As such, evaluation of the gut microbiota in pediatric patients with MS may advance the search for potential early triggers of MS. Information derived from POMS populations likely extends to the adult-onset relapsing-remitting MS population as environmental risk factors and host genetic variants associated with MS are considered similar.5-7 Furthermore, epidemiologic studies in adult-onset MS indicate that the risk is largely determined during a window of opportunity occurring prior to adulthood.4 Studying POMS allows for an exploration of the microbiome from persons who have had fewer lifetime exposures, such as different medications or varying diets, all of which may affect the gut microbiome.
The functional potential of the MS microbiome is not understood, in part because of the lack of studies utilizing metagenomic sequencing.8-11 With the use of metagenomics, it is possible to survey all the genes present in the gut microbiome and infer the abundance of microbial functions. A higher relative abundance of a specific gene also implies a higher potential of the gut microbiome to produce a related product. Whereas other -omics, such as metabolomics and proteomics, provide a survey of functional products, they alone do not provide information on the functional potential because gene products are not always expressed or detected.12 The functional potential of the MS microbiome requires further exploration and has not been investigated in people with POMS. We examined the functional potential of the MS gut microbiome and compared the gut microbiome's metabolic pathways, gene functions, and microbial taxonomy between individuals with and without POMS using metagenomic analysis of stool samples.
Methods
Participant Selection and Data Sources
Persons ≤21 years old enrolled from 1 of 23 sites across Canada and 1 in the United States (Children's Hospital of Philadelphia) involved in the Canadian Pediatric Demyelinating Disease Network (CPDDS) and who provided a stool sample between 2015 and 2019 and had not taken an antibiotic or corticosteroids within the previous 30 days were potentially eligible.13 All patients with MS met McDonald criteria (2017), had symptom onset <18 years of age, and had either no previous disease-modifying drug (DMD) exposure or were exposed to a platform DMD (interferon-β or glatiramer acetate). Healthy youth (controls) were enrolled in the CPDDS by community advertisement. For this current metagenomics analysis, participants were matched (20 patients with MS to 20 healthy controls) based on sex, age (±3 years), stool consistency (hard, medium, or loose stool [types 1–2, 3–5, or 6–7, respectively]), and, when possible, by race (White or non-White). Stool consistency was ascertained using the Bristol Stool Scale, which is a 7-point ordinal scale completed by the parent/caregiver or participant at the time of stool sample procurement.14 Dietary intake was assessed using the Block Kids Food Screener (NutritionQuest), a validated food frequency questionnaire,15 with the questionnaire completed closest to the date of stool sample procurement selected (excluding those with implausibly low or high energy intake; i.e. <500 or >3,500 kcal/d).16
Basic demographics, race, and comorbidity-related information were collected using a questionnaire, typically completed by the parent/caregiver and reviewed with the clinical research coordinator. Height and weight were captured using standardized case report forms at study visits or from routine medical records. Other detailed clinical data were also obtained using standardized forms, completed by site neurologists and facilitated by a study coordinator.
Stool Handling, Data Processing, Gene Annotation, and Phylogenetic Profiling
Stool was collected and shipped on ice, then stored at −80°C before DNA was extracted from stool samples using Zymo Quick-DNA Fecal/Soil Microbe Miniprep Kit (D6010). Shotgun metagenomic paired-end reads (2 × 250 base pairs) were generated using the Illumina NextSeq platform (using the NextSeq500/550 High v2.5 sequencing reagent kits with 300 cycles). All read quality control steps were performed using KneadData (v. 0.7.4) software. Specifically, human contaminant reads were removed based on the 1,000 Genomes Project Consortium's Decoy Genome and extensive sequences provided in relevant published work.17 Trimmomatic (v. 0.33) was used to trim adapter contents and to discard reads in which the quality of the sliding window average of 4 base pairs fell below 20, or a read had a length less than 75 base pairs. Repeated sequences were removed using the tandem repeats finder program (v. 4.09).18
The Human Microbiome Project Unified Metabolic Analysis Network (HUMAnN v. 3.0.0 alpha) was then used to construct metabolic pathways from the MetaCyc database and to identify the microbial species that contributed to the abundance of each pathway. Gene functions were annotated using the Kyoto Encyclopedia of Genes and Genomes Orthologs (KO), Enzyme Commissions (EC, level 4), and the Gene Ontology knowledgebase.19-21
Microbial taxonomic classifications and their relative abundance were generated from the metagenomic reads using exact k-mer matches (Kraken2 v. 2.0.9 beta) and the relative abundances were re-estimated using Bayesian Reestimation of Abundance after Classification with KrakEN (BRACKEN v. 2.6.0).22,23
Standard Protocol Approvals, Registrations, and Patient Consents
Informed consent or assent was obtained from individual participants and their parent or legal guardian. The relevant research ethics boards at each institution approved the study.
Statistics
Cohort characteristics were summarized quantitatively by mean, SD, and range for continuous variables and count and percent prevalence for categorical variables. The relative abundances of microbiome functions and taxonomy from individuals with MS were compared to controls. Only those reaching nominal statistical significance (p < 0.05) were subsequently compared between patients with MS by DMD status to controls (i.e., pairwise comparisons of DMD-exposed patients with MS, DMD-naive patients with MS, and controls).
The KOs and ECs total relative abundances of each sample were rarefied to the relative abundance of the smallest sample, and assessed using alpha diversity metrics (observed richness and inverse Simpson). Rarefication enables comparability of the diversity measures across samples. Metabolic pathways and gene functional annotations were assigned using the Gene Ontology knowledgebase and assessed as relative abundance and prevalence (being present if their relative abundance was >0).
The relative abundance of metabolic pathways, gene function annotations, and microbial taxonomy were compared between individuals with and without MS following a compositional data analysis approach.24 Briefly, the relative abundances were modeled by the Dirichlet distribution from which 1,000 Monte Carlo instances were drawn. For each instance, the additive-log ratio (alr) transformation was applied. The alr-transformed values of each feature were then compared between the MS and control participants (groups) using the Wilcoxon rank sum test. The median of the uncorrected and corrected p values (q values) of all instances were reported. All were performed using the ALDEx2 R package (v. 1.21.1).24 The log2 fold change (LFC) of the relative abundances for each group comparison was also reported.
For the alr transformation, the microbiome function or microbial taxonomy with the lowest variance of the centered-log transformed abundances was selected as the standard reference denominator (the pathway reference corresponded to the bacterial superpathway of hexitol degradation, selected based on the benchmarking simulations available at GitHub).25 A distinct singular log ratio reference was selected for gene function annotations within each of the 3 Gene Ontology domains that corresponded with gluconeogenesis (GO 0006094) for biological processes, kinase activity (GO 0016301) for molecular function, and plasma membrane (GO 0005886) for the cellular component. Neisseria and Desulfosporosinus youngiae were selected for the microbial genera and species log ratio reference, respectively.
The alpha diversity was compared between the MS and control participants using the Wilcoxon rank sum test and pairwise comparisons between patients with MS with and without prior DMD exposure and controls were performed using the nonparametric Dunn Kruskal-Wallis test.
Differential prevalence of pathways, gene function annotations, and microbial taxonomy were performed using the Fisher exact test and results were reported as odds ratios (ORs), 95% confidence intervals, and p and q values. All p values were Benjamini-Hochberg corrected using the “p.adjust” function from the R package “stats” (v.4.04).
Microbiome functions and taxonomy that were present—that is, had a non-zero raw count—in ≤5% (≤2/40) of the samples (an arbitrary cutoff) were excluded from all analyses (except for diversity). Those present in all samples were excluded from the prevalence analyses. Therefore, as differential abundance analysis and differential prevalence analysis have different filtering criteria, the final number of microbiome functions and taxonomy compared in each analysis may differ. KOs and ECs were excluded from the diversity analyses if there was zero abundance after rarefaction. All statistical analyses were performed using R.26
Data Availability
Code measures used for data processing and statistical analysis can be found at GitHub.25
Results
Participant Characteristics
Twenty individuals with MS were successfully matched to 20 controls by sex (80% were female), age (mean age, 16.1 years for patients with MS, 15.4 years for controls), and stool type (median Bristol Stool Scale score of 3), with 11 patients with MS and 9 controls self-identifying as White. None of the individuals with MS or matched controls had loose stool. Individuals with MS were similar to the controls for all other characteristics (Table 1; e.g., country of residence, birth country, body mass index, and energy intake; all p > 0.1 not shown).
Table 1.
Characteristics of the Individuals With Pediatric-Onset Multiple Sclerosis and Unaffected Controls

All participants with MS had a relapsing-remitting disease course and the mean disease duration at stool sample collection (from MS symptom onset) was 2.5 years. Twelve (60%) were ever exposed to a DMD for MS within 90 days prior to stool sample procurement (Table 1).
Gut Microbiome Enzyme and Protein Diversity
Participants with POMS did not differ compared to the controls in terms of the richness and diversity of gut microbiome enzymes and proteins (p > 0.30; Figure 1, A and C). Similarly, we did not observe differences by DMD exposure (p > 0.20; Figure 1, B and D).
Figure 1. Alpha Diversity of the Gut Microbiota Enzymes and Proteins for the Individuals With Pediatric-Onset Multiple Sclerosis and Controls, Including by Disease-Modifying Drug Exposure Status.

Differences in alpha diversity of Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologs (KOs), enzyme commissions (ECs), and microbial species relative abundance were assessed by disease status (multiple sclerosis [MS] and controls) and by disease-modifying drug (DMD) exposure. Diversity measures includes observed number (richness) and inverse Simpson (true diversity). None of the comparisons was statistically significant (p > 0.05, Wilcoxon rank sum test and Dunn test). (A, B) ECs alpha diversity comparing individuals with MS and controls and by DMD exposure, respectively. (C, D) KO alpha diversity comparing individuals with MS and controls and by DMD exposure, respectively.
Metabolic Pathways
Out of 381 gut microbiome metabolic pathways identified, 5 differed (p < 0.050) in relative abundance between MS and control participants. Relative to controls, participants with MS exhibited a 4-log2 fold higher relative abundance for the pathway pyrimidine deoxyribonucleotides biosynthesis (from cytidine triphosphate; p = 0.015, q = 0.77). Lower relative abundances were observed for peptidoglycan maturation (meso-diaminopimelate containing), homolactic fermentation, superpathway of pyrimidine ribonucleosides salvage, and glycolysis II (from fructose 6-phosphate), with a LFC ranging from −0.46 to −0.54 and all p < 0.046, but q > 0.77 (Figure 2A and eTable 1, links.lww.com/WNL/B727).
Figure 2. Gut Microbiome Metabolic Pathway Relative Abundance and Prevalence for the Individuals With Pediatric-Onset Multiple Sclerosis and Controls, Including by Disease-Modifying Drug Exposure Status.

Relative abundance of metabolic pathways (pathways from the MetaCyc database) were estimated for 40 participants: 20 controls and 20 individuals with multiple sclerosis (MS) (of whom 8 were disease-modifying drug [DMD]–naive and 12 DMD-exposed MS). All groups were compared pairwise by differences in relative abundance (A) and prevalence of the pathways (B). Pathways that were significantly different between individuals with MS and controls were assessed further for differences by DMD exposure. (A) Differences in the median log ratio transformed abundances between each pairwise group comparison. In total, 1 pathway was significantly higher and 4 significantly lower in individuals with MS compared to controls. (B) Prevalence of significantly different pathways (3 total) among participants within each group and subgroup, represented as bars. The odds of MS onset given presence of the pathway compared to absence of the pathway is reported under the asterisk. *p < 0.05; **p < 0.01; Inf = infinite because this genus was not found among controls.
Out of 245 metabolic pathways identified, 3 differed and were all more prevalent among individuals with MS compared to controls: l-glutamate degradation VIII (to propanoate), 2 archaeal pathways, methanogenesis from H2 and CO2, and flavin biosynthesis II, with OR >9.0, p < 0.047, but q = 1.0 (Figure 2B and eTable 2, links.lww.com/WNL/B727).
The pathways methanogenesis from H2 and CO2 and flavin biosynthesis II were exclusively detected from the Archaea Methanobrevibacter smithii genome. Fifty-four microbes present across all participants contained the peptidoglycan maturation pathway, with the largest percent originating from Escherichia coli (25%). Most of the microbial origin of the other pathways that differed significantly between MS and control participants were unknown (eTable 3, links.lww.com/WNL/B727).
Gene Function Annotations
Out of a total of 4,503 gene function annotations identified, 34 were enriched and 34 were depleted in individuals with MS compared to controls. The gene function annotations with the largest LFC in relative abundance between MS and control participants identified were methanogenesis (methane production from Archaea), viral (bacteriophage) genome packaging, archaeal flavin biosynthesis, and enterobactin syntheses (bacterial secreted compound to acquire iron), all of which were higher for individuals with MS, with LFCs ranging from 1.9 to 4.7 (p and q values ranged from 0.0031 to 0.030 and 0.81 to 1.0, respectively; Figure 3, eTable 4, links.lww.com/WNL/B727). As bacteria synthesize enterobactin in low iron environments, we also performed a post hoc assessment of participants’ dietary iron intake (from food). Iron intake was lower for individuals with MS relative to controls (median [interquartile range] = 5.7 [3.8] and 9.0 [4.9] mg/d, respectively); however, this did not reach nominal significance (p = 0.09).
Figure 3. Gene Functional Annotations Relative Abundance for the Individuals With Pediatric-Onset Multiple Sclerosis and Controls, Including by Disease-Modifying Drug Exposure Status.

Abundance of gene function annotations among 3 independent domains of the Gene Ontology knowledgebase—biological process (BP), cellular component (CC), and molecular function (MF)—were estimated for all participants. Gene annotations were grouped into higher order biological functions. All groups were compared pairwise by differences in relative abundance of the gene annotations. Gene annotations that were significantly different between individuals with multiple sclerosis (MS) and controls were further assessed for differences by disease-modifying drug (DMD) exposure. Differences in the median log ratio transformed abundances between each pairwise comparison. The relative abundance of 68 total gene annotations were significantly different between individuals with MS and controls, of which 60 informative gene annotations are displayed in the heatmap. *p < 0.05; **p < 0.01; ***p < 0.005.
Genes involved in several amino acids metabolism were lower in relative abundance for individuals with MS relative to controls, among which threonine metabolism was depleted the most (i.e., d-threonine aldolase activity, LFC −2.5, p = 0.041, q = 1.0). Genes involved in alanine, aspartate, and cysteine metabolism were modestly depleted in individuals with MS relative to controls, with LFCs ranging from −0.13 to −0.55, all p < 0.044, but q = 1.0. In contrast, genes involved in the adenine metabolism were modestly enriched for individuals with MS relative to controls (LFC 0.35, p = 0.012, q = 1.0). Modest depletions for individuals with MS were also observed for genes involved in various carbohydrate degrading enzymes, such as pullulanase (degrades pullulan and starch), sialate O-acetylesterase (degrades carbohydrate from intestinal mucosal glycoproteins), and α-amylase (degrades a wide variety of carbohydrates), with LFCs ranging from −0.22 to −0.70, all p < 0.022, but q = 1.0 (Figure 3 and eTable 4, links.lww.com/WNL/B727). Because depletion of gut microbiome carbohydrate-degrading enzymes can result from a diet low in complex carbohydrates, we performed a post hoc assessment of dietary fiber, vegetables (excluding potatoes and legumes), and whole grain intake. Compared to controls, we observed that individuals with MS consumed less than half the quantity of whole grains (median ounce equivalence/day [interquartile range] = 0.54 [0.80] and 0.24 [0.35], p = 0.061, respectively) and less fiber (median grams/d [interquartile range] = 11.0 [6.9] and 8.7 [6.5], p = 0.085, respectively), but neither reached nominal significance; vegetable intake was similar for MS and control participants (data not shown, p = 0.65).
We observed depletion in a gene that encodes an enzyme involved in the final steps of the butyrate pathway (i.e., phosphate butyryl transferase, p = 0.021, LFC −0.82; Figure 3 and eTable 4, links.lww.com/WNL/B727). Genes that encode an enzyme involved in a step in the acetyl-CoA pathway to butyrate were modestly enriched in individuals with MS compared to controls (i.e., 3-hydroxybutyryl-CoA dehydrogenase, LFC 0.26, p = 0.034, q = 1.0; Figure 3 and eTable 4).
Other modest differences (i.e., absolute LFCs ranging from 0 to 1) included a depletion of genes involved in long-chain fatty acid biosynthesis, lipopolysaccharide biosynthesis, and folate metabolism (vitamin B9), and an enrichment of genes involved in heavy metal activity (i.e., arsenic transportation), hydrolysis of phenyl acetate, vitamin B5 transport, and molybdopterin biosynthesis, in participants with MS relative to controls (p values ranged from 0.0057 to 0.032, but q ≥ 0.81; Figure 3 and eTable 4, links.lww.com/WNL/B727).27
Out of 2,835 gene function annotations identified, 57 differed in prevalence by disease status: 56 were more and 1 was less prevalent among individuals with MS relative to controls. A total of 14 gene function annotations related to methanogenesis or flavin metabolism were more prevalent in individuals with MS, which were similarly observed at the pathway level (OR ≥10, p values ranged from 0.031 to 0.047, but all q = 1.0). Compared to controls, individuals with MS also exhibited a higher prevalence of gene function annotations related to viral activity, metabolism of various amino acids (such as lysine, arginine, and the enzyme dehydroquinate synthase), diphthamide synthesis (a nonsecreted peptide that is the target of diphtheria toxin), transport of bactericidal polyketides (i.e., daunorubicin and doxorubicin), and response to the heavy metal cadmium, with ORs ranging from 7 to 12 (p values ranged from 0.0011 to 0.047, but all q = 1.0). The only gene function annotation less prevalent in individuals with MS compared to control participants was acetate-CoA ligase (involved in acetate production and also known as acetyl-CoA synthetase; OR <0.1, p = 0.047, but q = 1.0; eFigure 1 and eTable 5, links.lww.com/WNL/B727).
Microbial Taxonomy
From 1,580 genera and 5,908 species identified, 32 genera (27 bacteria, 4 archaea, and 1 virus) and 44 species (40 bacteria, 3 archaea, and 1 virus) differed between individuals with MS and controls. Compared to controls, 28 genera were higher and 4 were lower, and 27 species were higher and 17 were lower, in relative abundance for individuals with MS. Specifically, methanogens were more abundant for individuals with MS, including the genus Methanobrevibacter and its species, M. smithii (LFC 2.69, p = 0.0076 q = 1.0). Because constipation and hard stool have been associated with higher M. smithii abundance or higher gut-associated methanogenesis, we performed a post hoc assessment of the association of individual (raw) or grouped Bristol Stool Scale scores with Methanobrevibacter and M. smithii.28 We did not find any significant associations (generalized linear regression and Wilcoxon rank sum test, p > 0.23). Individuals with MS also exhibited a 3-log2-fold lower relative abundance of a Faecalibacterium virus (bacteriophage) from the genus Taranis virus relative to controls (p = 0.022, q = 1.0). Because Faecalibacterium taranis virus can infect Faecalibacterium prausnitzii strains and may modify its abundance, we were interested to know if there was a correlation between the phage and F. prausnitzii in our cohort.29 We did not find an association (post hoc analyses, Kendall rank correlation, tau = 0.18, p = 0.096). Furthermore, the relative abundance of F prausnitzii and its genus did not differ between individuals with MS and controls (p > 0.67, data not shown). The relative abundance of 10 species and their corresponding genera differed significantly between individuals with MS and controls, including a higher relative abundance of Methanobrevibacter millerae and Methanohalophilus sp for individuals with MS (LFC 1.48 and 1.14, p = 0.0054 and 0.041, q = 1.0 and 1.0, respectively; Figures 4 and 5 and eTable 6, links.lww.com/WNL/B727).
Figure 4. Genus-Level Gut Microbial Taxonomy Relative Abundance for the Individuals With Pediatric-Onset Multiple Sclerosis and Controls, Including by Disease-Modifying Drug Exposure Status.
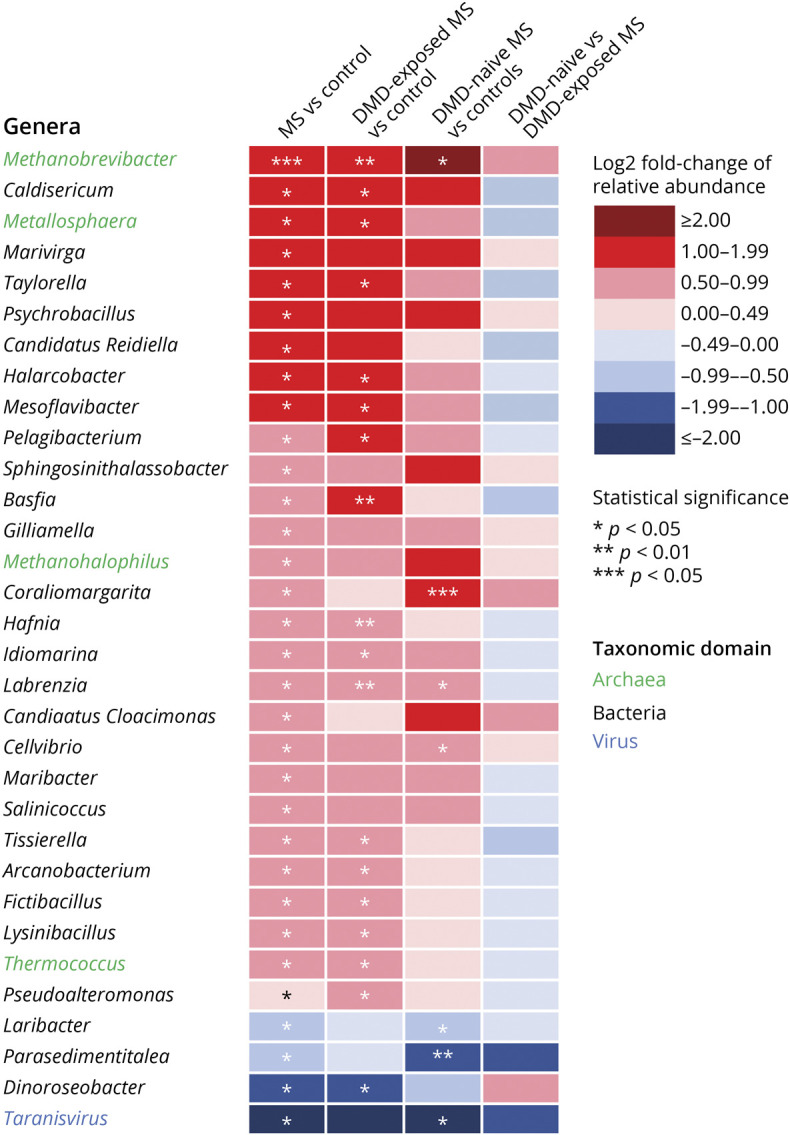
Genera relative abundance of bacteria, archaea, and virus were estimated for all 40 participants. All groups were compared pairwise by differences in relative abundance of genera. Differences in the median log-ratio transformed abundances between each pairwise comparison. In total, 27 bacterial (black text), 4 archaeal (green text), and 1 viral (blue font) genera significantly different between individuals with multiple sclerosis (MS) and controls are displayed in the heatmap. Genera were indicated for higher (red shading) or lower (blue shading) median relative abundance of the 1st group compared to the 2nd group, respectively. *p < 0.05; **p < 0.01; ***p < 0.005. DMD = disease-modifying drug.
Figure 5. Microbial Species Relative Abundance for the Individuals With Pediatric-Onset Multiple Sclerosis and Controls, Including by Disease-Modifying Drug Exposure Status.

Species relative abundance of bacteria, archaea, and a virus species were estimated for all 40 participants. All groups were compared pairwise by differences in relative abundance. In total, 40 bacterial (black font) and 3 archaeal (green text) and 1 viral (blue text) species differed significantly between individuals with MS and controls are displayed in the heatmap. Red shading indicates a higher and blue a lower median relative abundance of the 1st group (e.g., multiple sclerosis [MS]) compared to the 2nd group (e.g., controls). *p < 0.05; **p < 0.01; ***p < 0.005. DMD = disease-modifying drug.
From 1,107 genera identified, 3 were more and 1 was less prevalent among individuals with MS relative to controls: Polymorphum, Thermomonas, and Metallosphaera (an archaeon) and Candidatus vesicomyosocius, respectively. Of 5,409 species identified, 31 were more and 9 less prevalent among individuals with MS relative to controls. Methanobacterium sp, the only Archaeon species that differed, was more prevalent in individuals with MS relative to controls. Two unclassified Bacillus species were exclusively present among individuals with MS (prevalent in ≥25% of patients with MS) while Enterobacter cloacae complex sp was exclusively present among controls (prevalent in 25% of controls; eFigures 2 and 3 and eTable 7, links.lww.com/WNL/B727).
Because our results indicated a lower potential in the abundance in butyrate or lactate in individuals with MS, we performed post hoc analyses to investigate if there were any corresponding differences in the relative abundance of the bacterial species known to produce these metabolites, but none were found (data not shown, p > 0.05).27
Associations With DMD Exposure
The direction of findings was similar regardless of DMD exposure in virtually all analyses, with 3 exceptions: (1) the peptidoglycan maturation pathway was higher for the DMD-naive vs exposed patients with MS (LFC 0.63, p = 0.016, q = 0.080; Figure 2A and eTable 1, links.lww.com/WNL/B727); (2) genes involved in phosphate butyryl transferase synthesis, a precursor enzyme for butyrate production, were lower in the DMD-naive vs exposed patients with MS (LFC −1.0, p = 0.033, q = 0.63; Figure 3 and eTable 4); and (3) Bacillus sp JS was present in 50% of the DMD-exposed patients with MS and none of the DMD-naive patients with MS (p = 0.042, q = 1.0; eFigure 3 and eTable 7).
Discussion
We observed differences in the functional potential and taxonomy of the gut microbiome of individuals with POMS relative to community control participants. While overall functional alpha diversity of the gut microbiome did not differ, specific functions and taxonomy relative abundance and prevalence did. Key findings included a higher prevalence of a methane producing pathway from Archaea, as well as enrichment of several methanogens, such as M smithii, and depletion of the lactate fermentation pathway for individuals with MS relative to controls. Prior use of a DMD was also associated with differences, with, for example, the gut microbiome of DMD-exposed (vs naive) patients with MS being enriched for the genes encoding the butyrate-producing enzyme phosphate butyryl transferase.
We were able to find just 3 other published studies comparing the gut microbiome's functional profiles between individuals with MS and unaffected controls via metagenomic sequencing, although none involved individuals with POMS. We were unable to identify a microbiome function associated with MS that was similarly observed across all 3 studies. A German study of 34 monozygotic twins discordant for MS, in which the affected twin had longstanding disease (averaging 13.2 years since clinical onset of MS), observed no significant differences in metabolic pathways or microbial taxa between individuals with MS and controls (but actual results were not shown).30 A US study compared 24 treatment-naive patients with MS, nearly 85% of whom were within 1 year of symptom onset, and 25 controls, and similar to our study they reported differences in species relative abundance and metabolic pathways. However, the direction of findings or significance of the specific species and pathways they identified (e.g., inosine metabolism) did not completely concur with ours.9 A Japanese study compared 62 patients with relapsing-remitting MS with 55 controls for differences in gene groups and pathways. Although they did not quantify microbial taxa from metagenomic reads, similar to our findings, the authors reported a depletion in gene groups involved in propionate, butyrate, and pyruvate metabolism for individuals with MS relative to controls.10
Genes involved in methanogenesis, the production of methane from Archaea (a domain of life that, in the human gut, consists mostly of methanogens), and related methanogenesis pathway enzymes were overrepresented in the gut microbiome of our participants with POMS.31 Furthermore, specific methanogen species were more abundant, including M smithii. Our findings broadly concur with others, including in adults with longstanding MS who had elevated breath methane compared to controls.32 However, higher levels of intestinal methane can directly delay intestinal transit time and have been associated with constipation. This is a common gastrointestinal complaint in individuals with MS, particularly in participants with longstanding disease who are often enrolled in studies of the MS microbiome.28,33 While our ability to match participants with MS and controls according to stool consistency was a study strength, this may not entirely account for the presence of constipation. Understanding the potential link between methanogenesis, the microbiome, and the potential relationship with constipation in individuals with MS is warranted and may facilitate a more targeted approach to managing this common, understudied, but quality of life reducing symptom.
Gut microbiome metabolites, such as short-chain fatty acids and lactate, can affect host physiology by regulating immune cell development and suppressing inflammation.27,34 Our results suggest that lactate and several short-chain fatty acids (butyrate, propionate, and acetate) producing genes were disrupted for our participants with MS relative to controls. For example, genes that express the enzyme phosphate butyryl transferase involved in the late phases of the butyrate synthesis pathway were depleted for our participants with MS relative to controls. Homolactic fermentation, a pathway that produces lactate, a precursor metabolite for short-chain fatty acid production, was also depleted in our participants with MS.34 In line with our observations, others have reported lower levels of short-chain fatty acids in stool samples from individuals with MS relative to controls.10,35,36 There were no short-chain fatty acid or lactate-producing species identified that explained the corresponding lower relative abundance in these functions, indicating that the gut microbiome's community genetic composition, not individual species abundance, may be more important. Both short-chain fatty acids and lactate are products of microbiome carbohydrate digestion and our results suggest that the MS gut microbiome might be associated with a perturbed (lower) potential for carbohydrate digestion. For example, participants with MS were depleted in genes that express carbohydrate-degrading enzymes, specifically α-amylases, pullulanase, and sialate O-acetylesterase.37,38 Consistent with a lower ability for carbohydrate degradation, we observed lower relative abundance of the glycolysis (from fructose 6-phosphate) metabolic pathway for our participants with MS relative to controls, as well as lower dietary intake of fiber and whole grains (from our post hoc analyses; neither reached nominal significance). Thus, if individuals with MS have a suboptimal diet and fewer carbohydrate-degrading enzymes in their gut, these individuals may be depleted of various potentially beneficial anti-inflammatory metabolites synthesized by the microbiome.37
Abnormal iron distribution has been measured using brain MRI of individuals with MS, but the cause and mechanisms surrounding this are unknown.39 Our findings raise the possibility that the gut microbiome may be involved. Compared to controls, stool samples from participants with MS were enriched in genes involved in the synthesis of enterobactin (a siderophore), which enables the scavenging of iron when its bioavailability is low. Gut bacteria may also upregulate enterobactin synthesis when the import of the enterobactin–iron complex is perturbed, for example, when other competing bacteria or host mammalian cells intercept the complex.40,41 While enterobactin enrichment can occur independent of dietary iron intake, lower intake has been found in individuals with POMS relative to unaffected controls.42 We also observed a lower dietary iron intake for our participants with MS, but this did not reach statistical significance. Nonetheless, intestinal innate cells sequester, import, and transfer the enterobactin-iron complex from the intestines to the host cells.40,43 Thus, there is a potentially complex relationship between the gut microbiome and the host surrounding the acquisition of dietary iron.40,44 Considering that iron may play a role in MS progression, the potential relationship of MS and the influence of the gut microbiome in modulating host iron levels warrants further investigation.39
Microbial genes involved in responding to the heavy metals cadmium ion and arsenite were overrepresented in our participants with MS relative to controls. The redox-inactive metals cadmium and arsenic can lead to neurotoxicity in the brain and warrant further investigation into whether, for example, individuals with MS have been overexposed to heavy metals.45,46
This study provides insight into the functional potential of the POMS gut microbiome. Other study strengths included our ability to examine the gut microbiome relatively close to MS onset. We also matched participants with MS and controls for multiple characteristics, including stool consistency, which is thought to be a major confounder in gut microbiome research and is seldom considered in MS studies.47,48 In addition to relative abundance, we were also able to quantify the prevalence of gut microbiome functions and examine taxonomic profiles. We also observed results that concurred across metabolic pathways, functional annotation of genes, and microbial taxonomy, including methanogenesis-related pathways. Our observed depletion in genes involved in carbohydrate fermentation, lactate, and short-chain fatty acids synthesis in participants with MS (compared to controls) may explain the corresponding depletion of stool short-chain fatty acids in previous studies on MS.10,35,36 Together, these strengthen the biological plausibility and confidence in the associations observed. POMS is rare, such that our sample size was relatively modest, limiting our ability to formally assess the effect of potentially important cohort characteristics, such as diet. Furthermore, after correcting our p values for multiple comparisons, few reached significance. Nonetheless, our findings can be considered as hypotheses-generating, worthy of validation and further investigation, preferably in large longitudinal studies capable of establishing the potential causal relationship of the gut microbiome with MS, and the contribution of other factors, such as dietary patterns.49 The relative rarity of POMS is such that multisite collaborative efforts will be required, such as enrollment of participants and accrual of samples through established networks of pediatric clinics. Ultimately, this study advances understanding surrounding the potential association of the microbiome functions in individuals with MS.
Acknowledgment
The authors thank the children and youth with MS and their parents; the investigators and their institutions; the Tremlett team (University of British Columbia); Thomas Duggan for facilitating study setup, coordination, and data collection; Bonnie Leung for additional study coordination; Michael Sargent (Department of Internal Medicine and the University of Manitoba IBD Clinical and Research Centre laboratory, Winnipeg, Canada) for managing the biobank; Christine Bonner (National Microbiology Laboratory, Winnipeg, Canada) for processing samples and generating the sequence read data; and all the investigators and study teams at each site involved in the Canadian Pediatric Demyelinating Disease Network study.
Glossary
- alr
additive-log ratio
- CPDDS
Canadian Pediatric Demyelinating Disease Network
- DMD
disease-modifying drug
- EC
Enzyme Commissions
- KO
Kyoto Encyclopedia of Genes and Genomes orthologs
- LFC
log2 fold change
- MS
multiple sclerosis
- OR
odds ratio
- POMS
pediatric-onset multiple sclerosis
Appendix. Authors

Footnotes
CME Course: NPub.org/cmelist
Study Funding
MS Society of Canada endMS Doctoral Studentship (EGID: 3246) and The Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636).
Disclosure
A.I. Mirza is funded through the MS Society of Canada endMS Doctoral Studentship (EGID: 3246) and was funded through the Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636). G. Van Domselaar is the Chief Bioinformatics Scientist with the National Microbiology Laboratory–Public Health Agency of Canada and has received research support in the past 3 years from the National MS Society, the Canadian Institute of Health Research, and Genome Canada. C.N. Bernstein has served on advisory boards for AbbVie Canada, Roche Canada, Janssen Canada, Takeda Canada, and Pfizer Canada; consulted to Mylan Pharmaceuticals; has received educational grants from AbbVie Canada, Pfizer Canada, Takeda Canada, and Janssen Canada; and has been on the speaker's panel for Medtronic Canada, Janssen Canada, Takeda Canada, and AbbVie Canada. E. Ann Yeh has received research support in the past 3 years from the National MS Society, Canadian Institutes of Health Research, NIH, Ontario Institute of Regenerative Medicine, Stem Cell Network, SickKids Foundation, Peterson Foundation, MS Society of Canada, and MS Scientific Research Foundation; has received funding for investigator-initiated research from Biogen; and has served on scientific advisory boards for Biogen, Alexion, and Hoffman-LaRoche. A. Bar-Or is funded by the NIH, ITN, NMSS, and MSSOC; and has participated as a speaker in meetings sponsored by and received consulting fees and/or grant support from Janssen/Actelion, Atara Biotherapeutics, Biogen Idec, Celgene/Receptos, Roche/Genentech, Medimmune, Merck/EMD Serono, Novartis, and Sanofi-Genzyme. B. Banwell serves as a consultant to Novartis, UCB, and Roche; provides non-remunerated advice on clinical trial design to Novartis, Biogen, and Teva Neuroscience; and is funded by the NMSS, NIH, and Canadian MS Society. E. Waubant is funded by the NMSS, the NIH, PCORI, and the Race to Erase MS; has received consulting honoraria from Jazz Pharma, Emerald, and DBV; and volunteers on a clinical trial committee for Novartis. H. Tremlett is the Canada Research Chair for Neuroepidemiology and Multiple Sclerosis; current research support is received from the National Multiple Sclerosis Society, the Canadian Institutes of Health Research, the Multiple Sclerosis Society of Canada, and the Multiple Sclerosis Scientific Research Foundation; and in the past 5 years, has received research support from the UK MS Trust, travel expenses to present at CME conferences from the Consortium of MS Centres (2018), the National MS Society (2016, 2018), ECTRIMS/ACTRIMS (2015, 2016, 2017, 2018, 2019, 2020), and American Academy of Neurology (2015, 2016, 2019); speaker honoraria are either declined or donated to an MS charity or to an unrestricted grant for use by H.T.'s research group. D.L. Arnold reports consulting fees or grant support from Alexion, Biogen, Celgene, Geneuro, Genentech, Merck, Novartis, Roche, and Sanofi; and grant support from the International Progressive MS Alliance, MS Society of Canada, CIHR, as well as an equity interest in NeuroRx Research. Y. Zhao and F. Zhu were funded through research grants held by H.T., including The Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636). J.D. Forbes was partly funded through research grants held by H.T., including The Multiple Sclerosis Scientific and Research Foundation (PI: Tremlett, EGID: 2636). M. Graham, N. Knox, R.A. Marrie, J. Hart, J. O'Mahony, and W. Hsiao report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. [DOI] [PubMed] [Google Scholar]
- 2.Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19(1):55-71. [DOI] [PubMed] [Google Scholar]
- 3.Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease: a review. Ann Neurol. 2017;81(3):369-382. [DOI] [PubMed] [Google Scholar]
- 4.Sun L, Xie C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waubant E, Lucas R, Mowry E, et al. Environmental and genetic risk factors for MS: an integrated review. Ann Clin Transl Neurol. 2019;6(9):1905-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waubant E, Ponsonby AL, Pugliatti M, Hanwell H, Mowry EM, Hintzen RQ. Environmental and genetic factors in pediatric inflammatory demyelinating diseases. Neurology. 2016;87(9 suppl 2):S20-S27. [DOI] [PubMed] [Google Scholar]
- 7.Gianfrancesco MA, Stridh P, Shao X, et al. Genetic risk factors for pediatric-onset multiple sclerosis. Mult Scler 2018;24(14):1825-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirza A, Forbes JD, Zhu F, et al. The multiple sclerosis gut microbiota: a systematic review. Mult Scler Relat Disord. 2020;37:101427. [DOI] [PubMed] [Google Scholar]
- 9.Ventura RE, Iizumi T, Battaglia T, et al. Gut microbiome of treatment-naïve MS patients of different ethnicities early in disease course. Sci Rep. 2019;9(1):16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takewaki D, Suda W, Sato W, et al. Alterations of the gut ecological and functional microenvironment in different stages of multiple sclerosis. Proc Natl Acad Sci USA. 2020;117(36):22402-22412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boullerne AI, Adami GR, Schwartz JL, et al. Deep DNA metagenomic sequencing reveals oral microbiome divergence between monozygotic twins discordant for multiple sclerosis severity. J Neuroimmunol. 2020;343:577237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7(1):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banwell B, Bar-Or A, Arnold DL, et al. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol. 2011;10(5):436-445. [DOI] [PubMed] [Google Scholar]
- 14.O'Donnell LJ, Virjee J, Heaton KW. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ. 1990;300(6722):439-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunsberger M, O'Malley J, Block T, Norris JC. Relative validation of Block Kids Food Screener for dietary assessment in children and adolescents. Matern Child Nutr. 2015;11(2):260-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willett W. Nutritional Epidemiology, 3rd ed. Oxford University Press; 2013. [Google Scholar]
- 17.Breitwieser FP, Pertea M, Zimin AV, Salzberg SL. Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res. 2019;29(6):954-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franzosa EA, McIver LJ, Rahnavard G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15(11):962-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caspi R, Billington R, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46(D1):D633-D639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457-D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Computer Sci. 2017;3:e104. [Google Scholar]
- 24.Fernandes AD, Reid JN, Macklaim JM, McMurrough TA, Edgell DR, Gloor GB. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome. 2014;2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Analysis Workflow and Supplementary Information. 2021. Accessed April 21, 2021. github.com/aimirza/PedsMSMetagenomics2020. [Google Scholar]
- 26.R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2021. [Google Scholar]
- 27.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29-41. [DOI] [PubMed] [Google Scholar]
- 28.Triantafyllou K, Chang C, Pimentel M. Methanogens, methane and gastrointestinal motility. J Neurogastroenterol Motil. 2014;20(1):31-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornuault JK, Petit MA, Mariadassou M, et al. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome. 2018;6(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berer K, Gerdes LA, Cekanaviciute E, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. 2017;114(40):10719-10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JY, Whon TW, Lim MY, et al. The human gut archaeome: identification of diverse haloarchaea in Korean subjects. Microbiome. 2020;8(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munteis E, Andreu M, Téllez MJ, Mon D, Ois A, Roquer J. Anorectal dysfunction in multiple sclerosis. Mult Scler. 2006;12(2):215-218. [DOI] [PubMed] [Google Scholar]
- 34.Pessione E. Lactic acid bacteria contribution to gut microbiota complexity: lights and shadows. Front Cell Infect Microbiol. 2012;2:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng Q, Junli Gong G, Liu X, et al. Gut dysbiosis and lack of short chain fatty acids in a Chinese cohort of patients with multiple sclerosis. Neurochem Int. 2019;129:104468. [DOI] [PubMed] [Google Scholar]
- 36.Duscha A, Gisevius B, Hirschberg S, et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell. 2020;180(6):1067–e16. [DOI] [PubMed] [Google Scholar]
- 37.Cerqueira FM, Photenhauer AL, Pollet RM, Brown HA, Koropatkin NM. Starch digestion by gut bacteria: crowdsourcing for carbs. Trends Microbiol. 2020;28(2):95-108. [DOI] [PubMed] [Google Scholar]
- 38.Baxter NT, Schmidt AW, Venkataraman A, Kim KS, Waldron C, Schmidt TM. Dynamics of human gut microbiota and short-chain fatty acids in response to dietary interventions with three fermentable fibers. mBio. 2019;10(1):e02566-e02518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zivadinov R, Tavazzi E, Bergsland N, et al. Brain iron at quantitative MRI is associated with disability in multiple sclerosis. Radiology. 2018;289(2):487-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi B, Han M. Microbial siderophore enterobactin promotes mitochondrial iron uptake and development of the host via interaction with ATP synthase. Cell. 2018;175(2):571–e11. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Li X, Olmedo M, et al. A delicate balance between bacterial iron and reactive oxygen species supports optimal C elegans development. Cell Host Microbe. 2019;26(3):400–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bagur MJ, Murcia MA, Jiménez-Monreal AM, et al. Influence of diet in multiple sclerosis: a systematic review. Adv Nutr. 2017;8(3):463-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao X, Yeoh BS, Vijay-Kumar M. Lipocalin 2: an emerging player in iron homeostasis and inflammation. Annu Rev Nutr. 2017;37:103-130. [DOI] [PubMed] [Google Scholar]
- 44.Das NK, Schwartz AJ, Barthel G, et al. Microbial metabolite signaling is required for systemic iron homeostasis. Cell Metab. 2020;31(1):115–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheykhansari S, Kozielski K, Bill J, et al. Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: a review. Cell Death Dis. 2018;9(3):348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh V, Gera R, Kushwaha R, Sharma AK, Patnaik S, Ghosh D. Hijacking microglial glutathione by inorganic arsenic impels bystander death of immature neurons through extracellular cystine/glutamate imbalance. Sci Rep. 2016;6:30601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352(6285):560-564. [DOI] [PubMed] [Google Scholar]
- 48.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65(1):57-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulz CA, Oluwagbemigun K, Nothlings U. Advances in dietary pattern analysis in nutritional epidemiology. Eur J Nutr. 2021;60(8):4115-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Code measures used for data processing and statistical analysis can be found at GitHub.25