

Abstract
The nuclear factor kappa B (NFκB) family of transcription factors is a key regulator of immune development, immune responses, inflammation, and cancer. The NFκB signaling system (defined by the interactions between NFκB dimers, IκB regulators, and IKK complexes) is responsive to a number of stimuli, and upon ligand–receptor engagement, distinct cellular outcomes, appropriate to the specific signal received, are set into motion. After almost three decades of study, many signaling mechanisms are well understood, rendering them amenable to mathematical modeling, which can reveal deeper insights about the regulatory design principles. While other reviews have focused on upstream, receptor proximal signaling (Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev 2004, 18:2195–2224; Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-κB: variations on a common theme. Cell Mol Life Sci 2008, 65:2964–2978), and advances through computational modeling (Basak S, Behar M, Hoffmann A. Lessons from mathematically modeling the NF-κB pathway. Immunol Rev 2012, 246:221–238; Williams R, Timmis J, Qwarnstrom E. Computational models of the NF-KB signalling pathway. Computation 2014, 2:131), in this review we aim to summarize the current understanding of the NFκB signaling system itself, the molecular mechanisms, and systems properties that are key to its diverse biological functions, and we discuss remaining questions in the field.
INTRODUCTION
Nuclear factor kappa B (NFκB) is a family of dimeric transcription factors central to coordinating inflammatory responses; innate and adaptive immunity; and cellular differentiation, proliferation, and survival in almost all multicellular organisms.1–4 The NFκB system is tightly regulated, and misregulation of NFκB has been implicated in a wide range of diseases ranging from cancers to inflammatory and immune disorders. As a result, the NFκB regulatory network and its dynamics offer a multitude of promising therapeutic targets that remain to be fully explored and translated into clinical use.5–7 However, there continues to be an untapped potential for finer grained therapeutic targeting of the NFκB signaling system that requires a quantitative understanding of dynamical control and the integration of various physiological and pathological signals and stimuli.8–10
In Mammalia, the NFκB network consists of five family member protein monomers (p65/RelA, RelB, cRel, p50, and p52) that form homodimers or heterodimers that bind DNA differentially11–14 and are regulated by two pathways: the canonical, NFκB essential modulator (NEMO)-dependent pathway and the noncanonical, NEMO-independent pathway. These pathways tightly control the levels and dynamics of the transcriptionally active NFκB dimer repertoire constitutively and in response to stimuli, and thus control broad gene expression programs15,16 via the recruitment of co-activators17 or interplay with other transcription factors.18,19 The activation pathways control NFκB activity through multiple mechanisms: degradation of IκB inhibitor proteins, processing of NFκB precursor proteins, and expression of NFκB monomer proteins.10,20 Signals from tumor necrosis factor receptor (TNFR), toll-like receptor (TLR) superfamilies, interleukin receptor (IL-1R) and metabolic genotoxic, and shear stresses are integrated by the IκB/NFκB signaling network to produce signal-specific, context-specific, and cell-type-specific transcriptional responses.21–23
CANONICAL SIGNALING
Signaling via the NEMO-Associated IKK Complex
The canonical NFκB signaling pathway (a.k.a. NEMO-dependent pathway) is mediated by kinase complexes consisting of the scaffold/adaptor protein NEMO (a.k.a. IKKγ) and two IκB kinases (IKK1/2, a.k.a. IKKα and IKKβ) (Figure 1(a)). This IKK complex is activated by mechanisms that are NEMO dependent. The IKK kinases are activated by phosphorylation of serines in the activation T-loop, characteristic of the MAPK superfamily, and three mechanisms for IKK activation have emerged: (1) NEMO multimerizes IKK subunits27 to allow for activation via trans-autophosphorylation, (2) and/or brings them in proximity to upstream kinases such as TAK1,28 which may also lead to mutual activation forming positive feedback and digital dose-response characteristics.29,30 These mechanisms are mediated by NEMO’s ubiquitin-binding domain that allows for IKK’s recruitment to nondegradative K63-linked ubiquitin chains, which are a hallmark of inflammatory signaling. Finally, (3) NEMO itself is a substrate of ubiquitination, particularly linear ubiquitin chains produced by the LUBAC enzyme,31 which also facilitates the formation of transient signalsomes.
FIGURE 1 |.
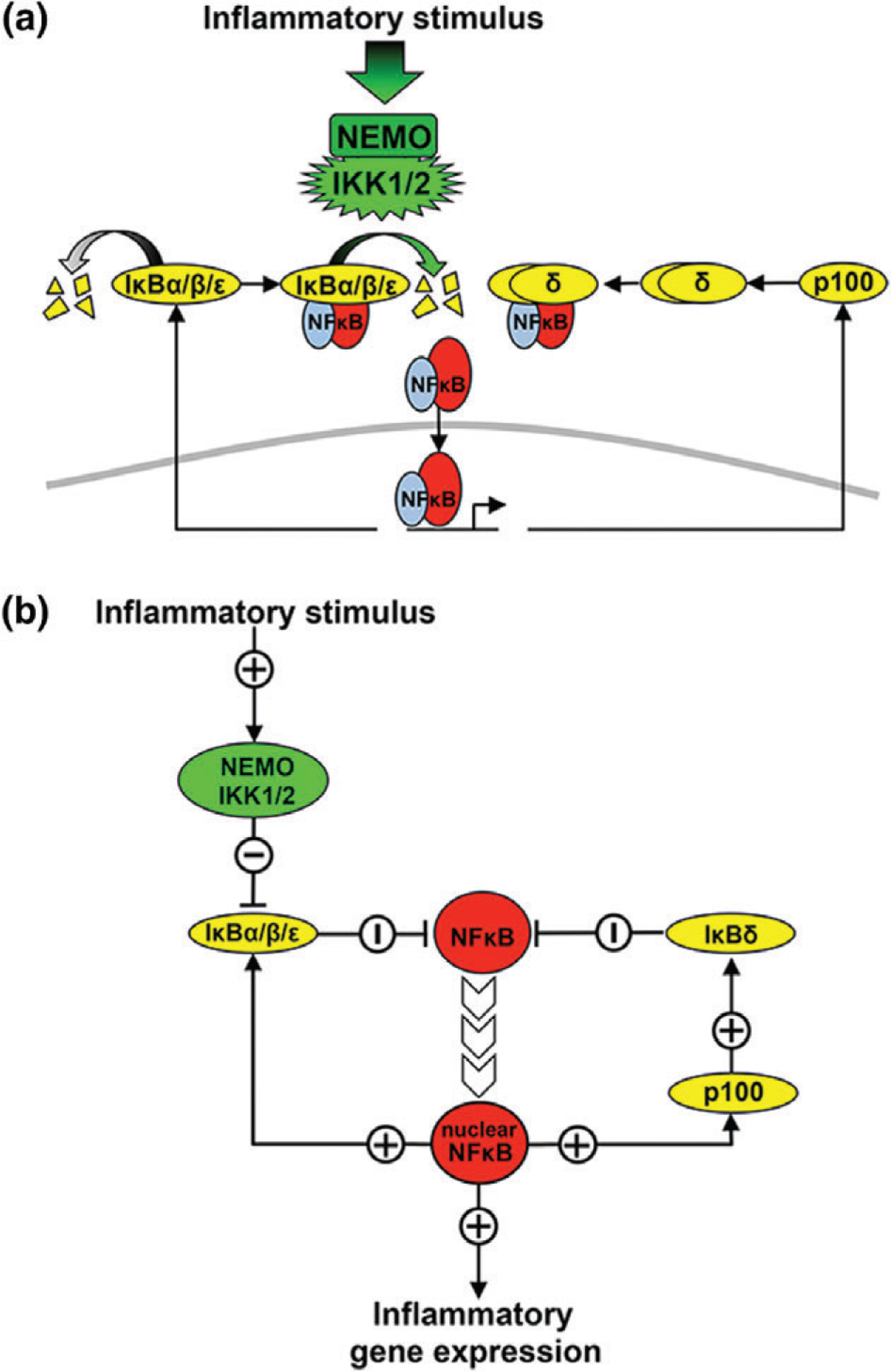
The canonical nuclear factor κ B (NFκB) activation pathway. (a) Schematic depiction of the canonical NFκB signaling pathway. Multiple inflammatory signals activate the complex containing NEMO and IKK1/2. IKK1/2 phosphorylates NFκB-bound IκBs, targeting them for ubiquitination and proteasomal degradation.24,25 Free IκBs also undergo constitutive degradation via a ubiquitin-independent proteasomal degradation pathway. As IκBs are degraded, free NFκB is then able to translocate to the nucleus where it binds to κB sites on DNA and activates gene expression.21,22 IκBα, β, and ε are themselves NFκB target genes, along with p100 that can form higher-molecular-weight complexes that inhibit NFκB.25,26 (b) Diagram of the regulatory logic of the canonical NFκB signaling network. Canonical signals activate NEMO/IKK, downregulating IκBs and reducing inhibition of NFκB. Free NFκB then translocates to the nucleus where it upregulates IκBs and p100 and in turn IκBδ.
A wide variety of inflammatory cytokines (such as TNF and IL-1), a wide variety of pathogen-associated molecular patterns (PAMPs), or antigen/immune stimulatory signals result in IKK phosphorylation-dependent activation of the NEMO-containing complex, by one or a subset of these mechanisms,24 resulting in complex dynamical control.32 Once activated, the complex binds to and phosphorylates IκB proteins on specific serines in the N-terminal, leading to ubiquitination and subsequent proteasomal degradation.25 The degradation of inhibitors releases NFκB dimers associated with them, freeing NFκB dimers and allowing them to bind κB site-containing DNA and thus rapidly accumulate in the nucleus. Following IκB release the NFκB subunits are subject to a variety of posttranslational modification that fine-tune gene-expression control.33
Further, NEMO was shown to function as a scaffold between IKK and IκBα, thereby directing IKK activity to IκBα.34 This mechanism ensures that the activation of NFκB dimers associated with IκBα, which is RelA:p50 in most cells and conditions, is directly linked to signals propagating through the NEMO hub. Hence, canonical signaling is often thought to be synonymous with RelA activation, but this is not always the case. In inflammatory dendritic cells, IκBα was also shown to be associated with RelB:p50 dimers, thus rendering RelB a key transcriptional effector of the canonical NFκB pathway in that cell type.35
While the activation mechanism of IKK is beginning to be elucidated with the aid of recent structural and biophysical characterizations36,37 and, for example, combined single-cell computational studies that have identified distinct pathways for robust digital responses and noisy sustained responses,38 how IKK is inactivated remains unclear. One attractive proposal is that IKK is regulated in an autocatalytic cycle of at least three states in which activation occurs from a poised state and is followed by an inactive state. While the dynamic properties of such a kinase control cycle have been studied,39 the biophysical evidence remains scant, but could involve trans-autophosphorylation of an inhibitory C-terminal domain in IKKβ40 and/or conformational changes of the complex.41
IκBα Negative Feedback
Among the target genes regulated by κB sites are the IκBs that, upon transcriptional induction and resynthesis, are able to translocate to the nucleus, bind to and inhibit NFκB activity, trafficking it back to the cytosol. This constitutes the primary component of the self-regulating negative feedback loop25 (Figure 1(b)). This feedback loop not only prevents run-away NFκB activity in response to transient inflammatory signals but also poises the system for reactivation when IKK activity is longer lasting. IκBα negative feedback has been studied in some detail with a combined experimental and mathematical modeling approach, and interesting properties have emerged: (1) Given the delay intrinsic to IκBα resynthesis, even very transient cytokine exposure (1 min TNF) results in almost a full hour of NFκB activity.42 That 1 h of NFκB activity is invariant to the duration of the signal unless the incoming signal lasts longer than approximately 45 min. (2) Transcriptional induction of IκBα is necessary but not sufficient for mediating this effective negative feedback control of NFκB activity.43 IκBβ, when controlled by an NFκB-inducible promoter, is unable to provide normal physiologically observed negative feedback44; nuclear import, export, and protein degradation mechanisms specific to IκBα are critically important to recapitulating proper negative feedback and the NFκB dynamic responses characteristic of normal activity.45–49 In addition, IκBα has been shown to be able to strip NFκB off DNA (or associate with chromatin50), a function that IκBβ does not possess.51 (3) Given effective IκBα negative feedback, stimulation conditions that produce longer lasting IKK activities allow for repeated cycles of reactivation, leading to oscillatory NFκB activity observed in biochemical bulk population assays25 or single-cell assays.38,52–54 Intrinsic variability in the kinetic mechanisms that govern IκBα feedback is thought to render the oscillatory behavior more robust to variations in dynamic signals.55,56 However, the potential function of NFκB oscillations remains unclear, and no satisfying answer has been presented as to how such oscillations are interpreted by downstream gene regulatory networks.57
IκBδ Negative Feedback
Another transcriptional target gene that is induced by nuclear NFκB activity is the nfkb2 gene, which produces the p100 protein.4 p100 was first known as the precursor for the p52 protein, a dimerization partner of RelB and potentially other Rel proteins. However, p100 that is not processed to p52 is able to form higher-molecular-weight complexes that are capable of binding to NFκB, acting as an IκB, termed IκBδ.26 As such, NFκB control of nfkb2 forms another negative feedback loop that may terminate NFκB signaling.58 However, transcriptional induction and protein synthesis are slow, in part due to the length and half-life of the mRNA and protein, and the subsequent required oligomerization step also takes several hours.59 Thus, in contrast to IκBα that functions rapidly, IκBδ’s role is primarily in attenuating persistent signals.58 Indeed, while IκBα negative feedback is reversible as it is a sensitive substrate for continued canonical IKK activity, IκBδ is insensitive to canonical signals and thus attenuates the canonical pathway regardless of whether incoming signals persist. Further, because IκBδ has a longer half-life than other IκBs, it contributes to a signaling memory in which sequential stimulation events are dampened.58 However, as a substrate for noncanonical signaling (see below), IκBδ is a signaling crosstalk node that integrates canonical and noncanonical signals that may result in noncanonical signals emanating from LTβR or BAFF to activate (or prolong the activation of) NFκB RelA or cRel dimers.26,60,61
Other Feedback Mechanisms
There are several other negative and positive feedback mechanisms that contribute to the complex and potentially oscillatory dynamics of nuclear NFκB.58,62 IκBε was shown to provide delayed negative feedback (due to a transcriptional delay) that may partially compensate for the loss of IκBα,63 and was suggested to dampen IκBα-mediated oscillations by forming a dual, antiphase negative feedback system.62 There is also evidence that IκBα and ε preferentially inhibit distinct NFκB family members.64 In B cells, IκBε has been shown to play a key role in regulating cRel containing NFκB dimers, with loss of IκBε resulting in increased B-cell survival and proliferation.21
Within the TNF pathway, both negative and positive feedback has been reported. Expression of the de-ubiquitinase A20 is strongly NFκB inducible. However, owing to a long protein half-life and enzymatic effector function, its negative feedback effects do not shape NFκB dynamics acutely, but rather integrate the history of prior exposure to render the NFκB pathway less sensitive to subsequent stimuli.42 TNF itself is NFκB inducible, but for full activation, additional signaling mechanisms controlling splicing, mRNA half-life, pro-TNF processing, and secretion must be activated.65,66 Hence, TNF functions more like a feed-forward loop in response to PAMPs rather than a positive feedback loop per se.
NONCANONICAL NFκB SIGNALING
Signaling via NIK and IKKα
Noncanonical signals are NFκB activating signals that are transduced in a NEMO-independent, but NIK and IKKα-dependent manner. Noncanonical pathways activating signals are primarily developmental signals that activate TNF receptors (BAFFR, CD40, LTβR, RANK, TNFR2, Fn14, etc.), some of which also activate the canonical NFκB pathway.67–74 Noncanonical NFκB signaling is known to control a wide variety of developmental phenotypes including B-cell survival and maturation, dendritic cell activation, and bone metabolism.75 Several chemokines that regulate lymphoid organogenesis are induced specifically by noncanonical NFκB activation.69 Base pair differences in κB sites may contribute to noncanonical pathway-specific gene expression.76,77
While canonical signals transduced by NEMO require phosphorylation-dependent activation of the IKK kinase complex, noncanonical signals transduced by NIK require stabilization and accumulation of the kinase, which, in the absence of signal, is rapidly degraded by a TRAF–cIAP complex.78 This ubiquitination-dependent degradation ensures very low basal NIK levels.79 Noncanonical stimuli trigger TRAF2-dependent cIAP1–cIAP2 activation, which in turn leads to the proteasomal degradation of TRAF3; this disrupts the cIAP–TRAF complex reducing NIK degradation and leads to accumulation of NIK.79–81 NIK activity is dependent on IKK1, but independent of IKK2. Once activated, the NFκB-inducing-kinase (NIK) complex has dual roles within the noncanonical pathway (Figure 2(a)). Originally identified as a MAP kinase kinase kinase (encoded by Map3k14), NIK activates IKKα by phosphorylation of Ser and Thr residues within an activation loop between subdomains VII and VIII of the kinase domain.84,85 Although NIK overexpression results in canonical NFκB activation,85,86 NIK knockouts are not defective in the canonical activation of IKK and NFκB in response to inflammatory cytokines.82
FIGURE 2 |.
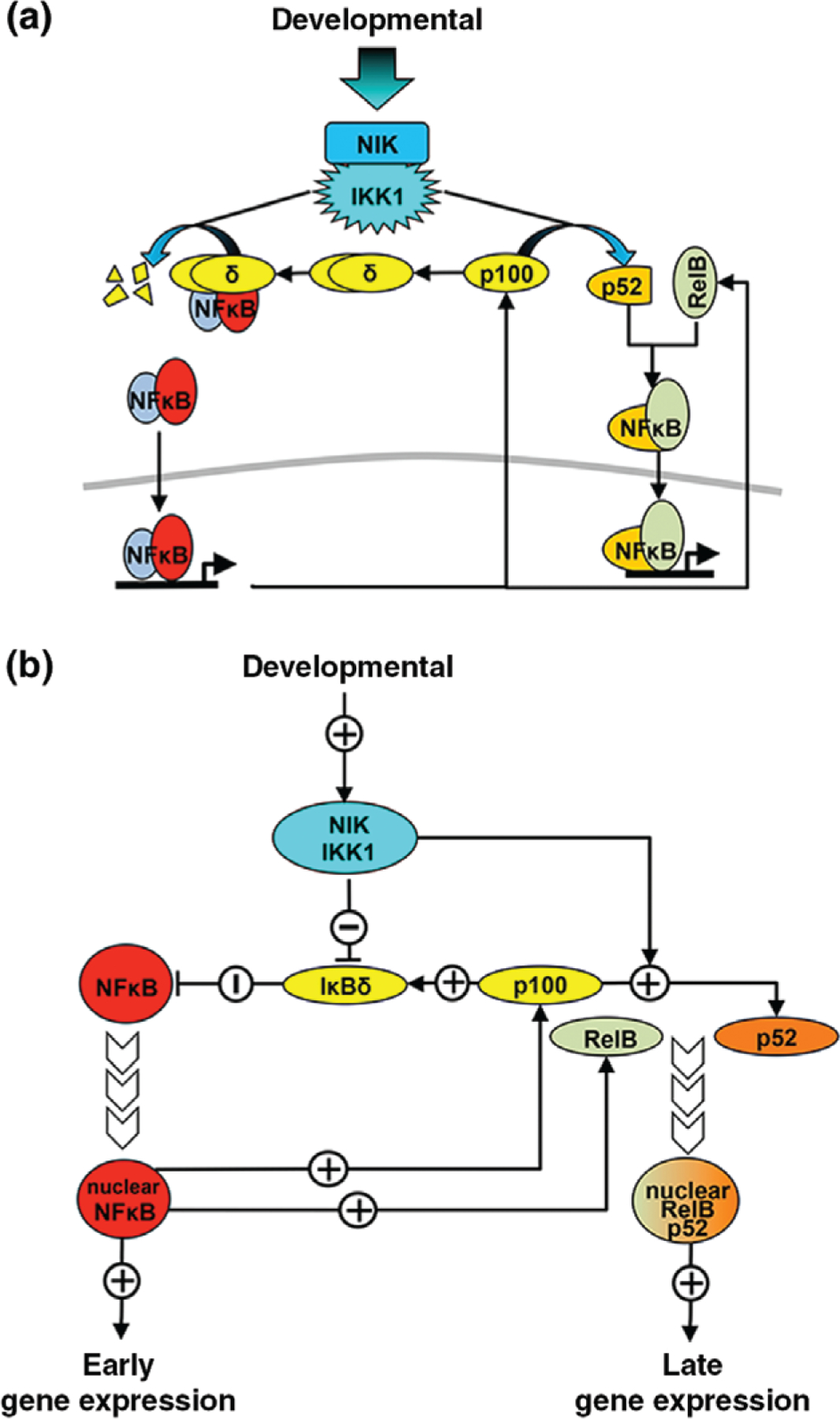
The noncanonical nuclear factor κ B (NFκB) activation pathway. (a) Schematic depiction of the noncanonical NFκB signaling pathway. Developmental signals activate the NIK/IKK1 complex that phosphorylates p100. Most p100 is found in a higher-molecular-weight inhibitory complex (IκBδ). Upon phosphorylation, p100 is processed into p52 and is then available to bind RelB, creating a dimer that localizes to the nucleus and binds DNA to activate transcription.82,83 Active NIK/IKK1 complex also phosphorylates the p100 within IκBδ, resulting in its partial degradation and releasing bound NFκB dimers for nuclear localization and gene activation.26 (b) Diagram of the regulatory logic of the noncanonical NFκB signaling network. Noncanonical signals activate NIK/IKK1 that suppresses IκBδ and activates processing of p100 into p52. The suppression of IκBδ that was sequestering preexisting NFκB dimers results in nuclear localization of NFκB and early-phase gene expression. NIK-dependent p100 processing results in p52 production and the formation of new RelB:p52 dimers that can activate a late-phase gene expression response.
IκBδ Degradation to Release Preformed Dimers
NIK’s first role is in targeting the oligomeric IκB complex, causing IκBδ degradation and release of preexisting NFκB dimers for nuclear localization.26 This initial response relies on phosphorylation events and occurs quickly, as IκBδ bound to preexisting dimers can release NFκB to localize to the nucleus as soon as it is modified by NIK (Figure 2(b)). While NIK is primarily considered the transducer of noncanonical NFκB signaling and IκBδ differs from other IκBs in its ability to inhibit RelB-containing dimers and respond to noncanonical stimuli, IκBδ also inhibits RelA-containing dimers. Therefore, NIK-induced IκBδ degradation may induce an inflammatory and/or developmental response depending on the existing NFκB dimer repertoire poised within the cell prior to stimulation. Both RelB and RelA induction in response to LTβR (noncanonical only) are abrogated in NIK knockout.26
P100 Processing to p52 to Generate New RelB:p52
The second role of NIK is in initiating the proteasome-mediated processing of p100 into p52. Phosphorylation of C-terminal serine residues leads to p100 being recognized by SCF/βTRCP ubiquitin ligase and subsequent partial degradation of the ARD by the 26S proteasome.82,83 This processing produces a mature p52 monomer that is then able to dimerize to form transcriptionally active RelB:p52 and other NFκB dimers.87 It appears that only newly translated p100 is able to be processed into p52,88 probably because p100 oliomerizies into a complex that renders it unavailable for processing.26 The multidomain interactions between RelB and p52 (and also p100) co-stabilize both proteins, as RelB protein levels are decreased in Nfkb2−/− cells and p100 protein is decreased in RelB−/− cells.89,90 While p52 is only produced at very low levels in most mammalian cells, certain cell types, including B cells, show active generation of p52.91 Processing is tightly controlled by a processing-suppressive region in the C-terminal portion of the p100 protein and disruption of this domain leads to constitutive p100 processing.82,92 p100 that does not form p52 is free to form higher-molecular-weight inhibitors of NFκB (IκBδ), and therefore tight regulation is important to ensure production of IκBδ in the absence of noncanonical stimuli to regulate late-phase NFκB activity. NIK-induced p100 processing, and subsequent dimerization with RelB, is a relatively slow process compared to the release of preexisting NFκB dimer from inhibitor, and therefore this results in a late-phase and sustained gene-expression response.
During B-cell maturation, BAFF activates non-canonical NFκB signaling and the consequence of this is dependent on the state of p100 synthesis within the NFκB network: at moderate p100 synthesis rates BAFF-induced p52 production fully depletes p100 and prevents the formation of IκBδ, whereas in the context of high p100 synthesis and resultant IκBδ formation BAFF causes IκBδ degradation altering cRel activity and affecting B-cell expansion.61 Constitutive P100 degradation also contributes to p100 homeostasis and hence is important for a variety of cellular functions.93
ALTERNATIVE NFκB ACTIVATION MECHANISMS
Ribotoxic Stress
Ribotoxic stress, as induced by ultraviolet light (UV) exposure or by unfolded protein response (UPR) inducers, has been found to activate NFκB by inhibiting translation of IκBα.94,95 Thus, these stimuli engage in crosstalk with inflammatory signaling and amplify NFκB activity (Figure 3(a)). Indeed, UV-induced activation of NFκB was shown to occur in enucleated cells, eliminating UV-induced DNA damage as the primary transducer of this pathway. Instead, in response to UV, eukaryotic initiation factor 2 α (eIF2α) is phosphorylated through stress response kinases GCN2/PERK. Phosphorylation of eIF2α broadly inhibits transcription initiation, resulting in reduced synthesis of IκBα.94 As free IκBα is constantly degraded and synthesized, the UV-induced reduction in synthesis results in decreased IκBα and amplified NFκB responses (Figure 3(b)). Interestingly, in response to chronic reactive oxygen species exposure, NFκB may repress prosurvival genes and induce prodeath genes.97
FIGURE 3 |.
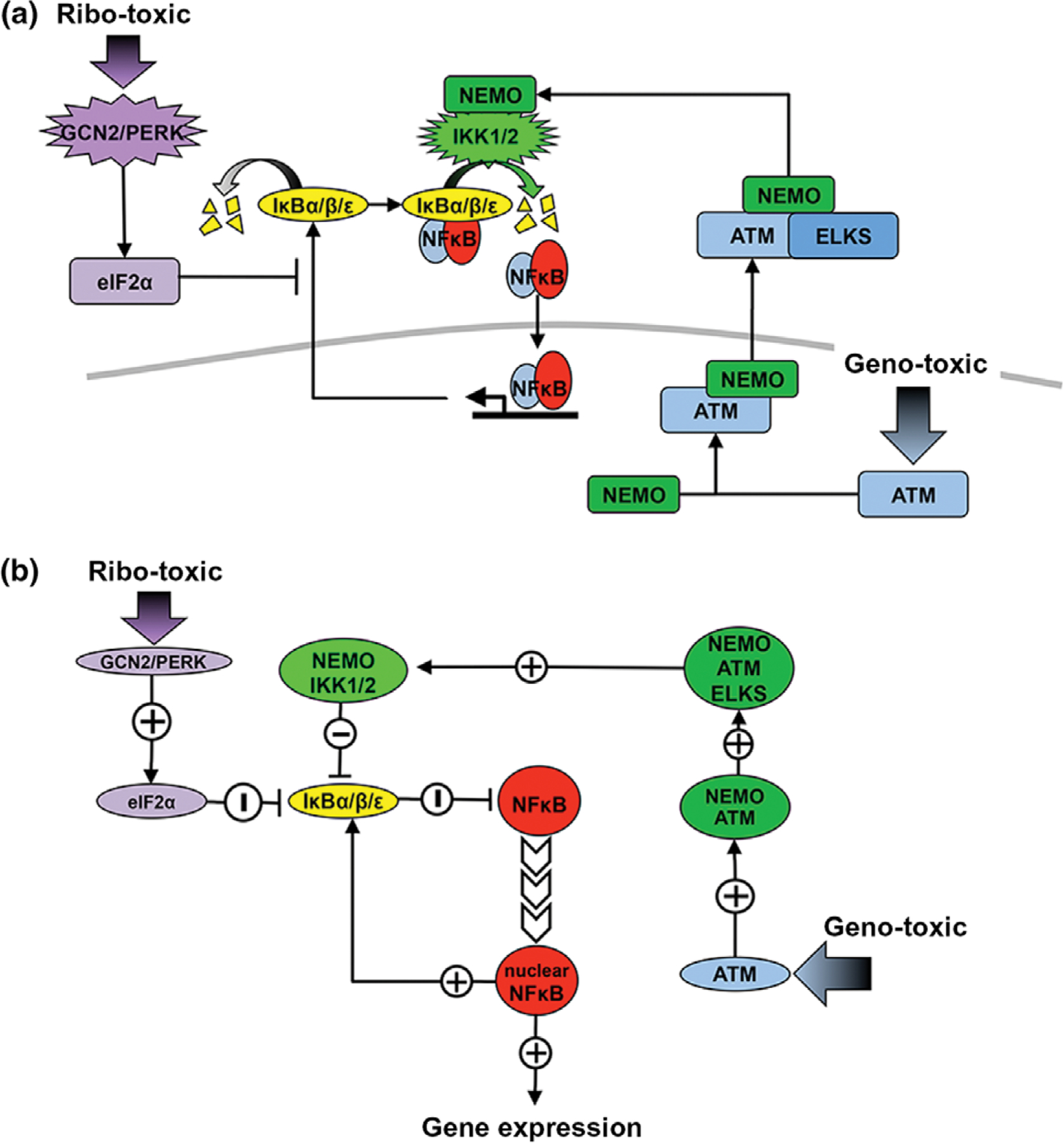
Nuclear factor κ B (NFκB) activation by ribotoxic and genotoxic stresses. (a) Schematic depiction of alternative methods of NFκB activation. Ribotoxic stress inducers lead to the phosphorylation of initiation factor eIF2α through the action of kinases GCN2 and PERK. Once active, eIF2α inhibits translation initiation, thereby reducing synthesis of IκBs.94 The reduction in IκB leads to more free NFκB that localizes to the nucleus and binds DNA to promote target gene expression. Genotoxic stress inducers lead to the phosphorylation of ATM and induce complex formation with NEMO in the nucleus.96 NEMO is phosphorylated and exported into the cytoplasm where it associates with ELKS and stimulates IKK2-containing complexes. Activation of NEMO/IKK2 complexes results in increased IκB degradation and localization of NFκB to the nucleus. (b) Diagram of the regulatory logic of alternative methods of NFκB activation. UV stress response through GCN2/PERK upregulates eIF2α which in turn suppresses IκBs. The reduced IκB synthesis reduces the inhibition of NFκB and increases nuclear NFκB and target gene expression. In response to DNA damage, ATM is upregulated and activates NEMO through a complex with ELKS. Increased NEMO/IKK2 activation results in suppression of IκBs.
Genotoxic Stress
Genotoxic stress also activates NFκB, although through distinct mechanisms.96 In this context, the initiation signal for NFκB responses originates from within the nucleus and is propagated to the cytosol via mechanisms that involve the nuclear export of upstream signaling molecules. In response to DNA damage NEMO is localized to the nucleus as a result of SUMO-1 attachment.98 DNA damage-activated ATM (ataxia telangiectasia mutated) then phosphorylates NEMO in the nucleus and triggers mono-ubiquitination of NEMO. ATM then binds to this modified NEMO, which is exported to the cytoplasm and activates IKK to result in degradation of IκBs. NFκB activation upon genotoxic stress is markedly slower and lower in amplitude than that observed for immune receptor signaling, and its physiological role remains unclear.
Shear Stress
Mechanical forces exerted on cells can also activate NFκB. Shear stress (mechanotransduction) activates NFκB in osteoblasts through phospholipase pathways that release intracellular Ca2+. Activated NFκB in response to shear stress upregulates COX-2, which plays an important role in the response of bone to mechanical loading.99 Hemodynamic forces also exert shear stresses on vascular cells during development of atherosclerosis that result in activation of NFκB through a mechanism independent from canonical IκBα degradation.100
NFκB GENERATION MECHANISMS
NFκB dimeric transcription factors are formed by five monomers (Figure 4). Of the 15 possible dimers, 12 are thought to bind the DNA κB element, and 3 (RelB:RelB, RelB:RelA, and RelB:cRel) form low-affinity intertwined dimers that are unable to bind DNA.101 Of the 12 DNA-binding dimers, 9 contain at least one of the activator proteins, RelA, cRel, or RelB (RelA being the most potent and RelB the least), and generally function as transcriptional activators. The remaining three (the abundant p50:p50 homodimer, and the lesser p52:p52 and p50:p52 homodimers and heterodimers, respectively) may function as activators in conjunction with co-activators, including Bcl3 and IκBζ.
FIGURE 4 |.
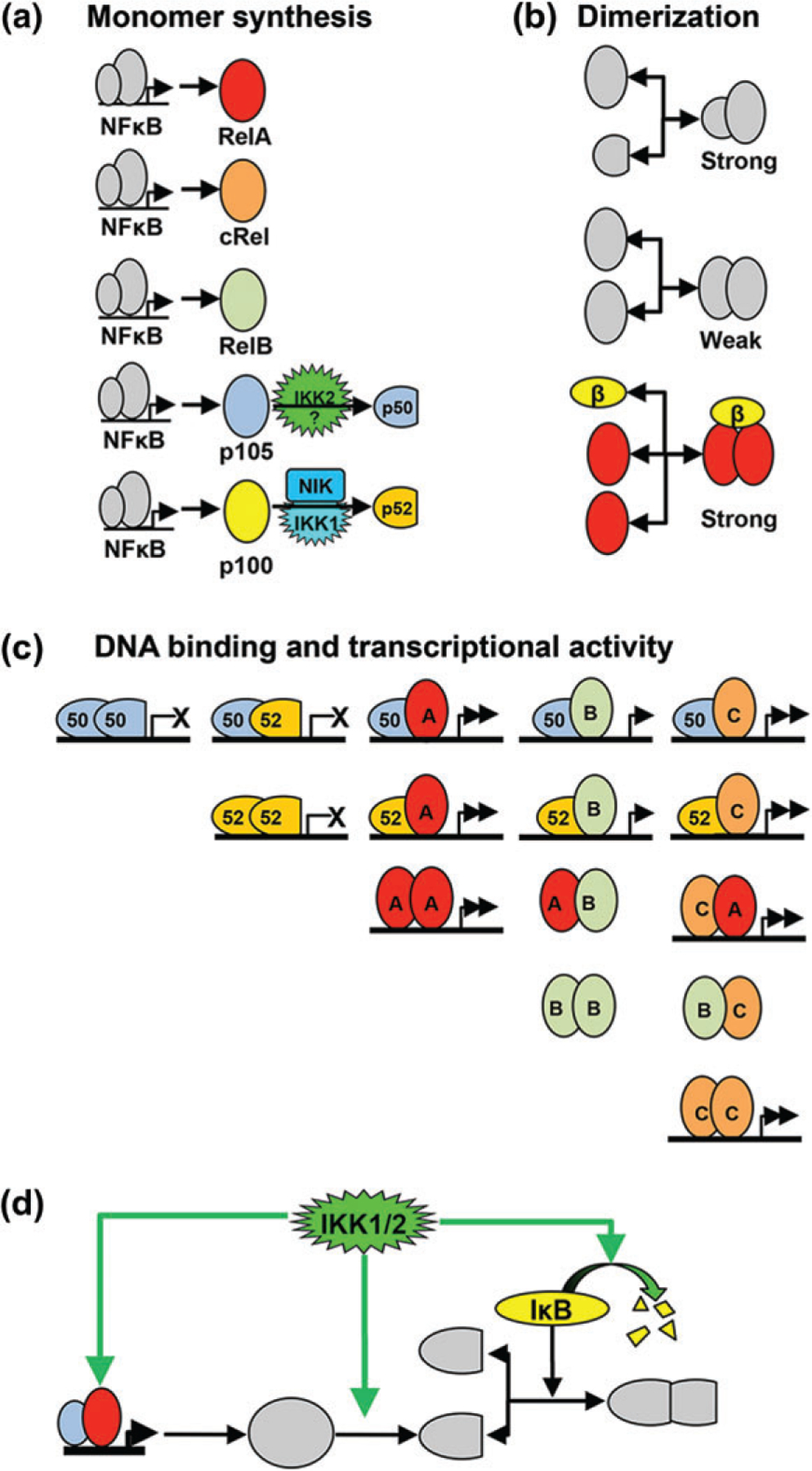
Mechanisms regulating nuclear factor κ B (NFκB) dimer generation. (a) Diagram of NFκB monomer synthesis and processing.10,20 All NFκB monomers and precursors are NFκB target genes and induced, to varying extents, by NFκB. RelA/RelB/cRel polypeptides are synthesized in a complete form, ready to dimerize into functional NFκB dimers.61,104 p105 is a precursor to p50 that must be cleaved in a process thought to be dependent on IKK2.2 p100 must be processed via a NIK/IKK1-dependent pathway into mature p52 before it can dimerize into NFκB.82 (b) Schematic of the NFκB dimerization process. Monomers must dimerize before they are transcriptionally active. The affinity of binding between monomers varies with two large, activation domain proteins having low affinity. IκBβ can act as a chaperone, enhancing the effective binding affinity of RelA to form homodimer by stabilizing this normally weak affinity dimer. (c) Table of the combinatorial composition of potential NFκB dimers, indicating their capacities to bind DNA (indicated by horizontal line) and to activate transcription (indicated by arrows). (d) Diagram of IKK’s multiple points of control over NFκB dimer formation. (1) The IKK kinases upregulates monomer expression by activating NFκB-responsive promoters. (2) IKK1 and IKK2 activities promote processing of p100 to p52 and p105 to p50. (3) IKKs lead to the degradation of IκBs that may function as dimerization chaperones (as for example IκBβ for RelA homodimer) as well as inhibitors.
The mechanisms underlying NFκB dimer generation (Figure 4) have only recently received attention, but are critical to understanding how different cell types produce different NFκB dimer repertoires during cell differentiation and development. In some cases, a shift in the dimer repertoire has been documented: for example, B cells shift from a RelA:p50 predominant state at the pre-B stage to a cRel:p50 dominant state in mature B-lymphoid cells and then show strong upregulation of RelB and p52 in terminally differentiated B cells.102 Also, while most monocyte lineages rely on RelA:p50, GM-CSF-derived inflammatory dendritic cells show high levels of the unusual RelB:p50 dimer, whose generation was shown to be dependent on high constitutive RelB expression and NIK activity.35 These examples show that the NFκB dimer repertoire is responsive to stimuli, albeit at longer timescales (>8 h) than the activation of NFκB from the latent dimer repertoire.
NFκB family members are obligate dimers; as monomers they are unstable and are thought to be quickly degraded. Thus, the NFκB dimer generation system is highly dynamic and homeostatic. Below, we summarize the key regulatory mechanisms that contribute to the NFκB dimer repertoire. How these function together to determine the specific NFκB signaling system of specific cell types ought to be a focus of future increasingly quantitative studies.
Dimerization Affinities
Key determinants of the NFκB dimer repertoire are the interaction rate constants between the five monomers. Indirect evidence suggests that the 15 NFκB dimers have dramatically different dimerization affinities, yet remarkably little quantitative information has been published. Recently, analytical ultracentrifugation determined the affinities of the RelA:p50, p50:p50, and RelA:RelA dimers to be in the range of 1–5 nM, 20–50 nM, and 0.8–1.5 μM, respectively.103 It is not unreasonable to speculate that other dimers fall into these orders of magnitude, with large dimers (RelA:cRel, cRel:cRel and the DNA-binding incompetent dimers RelB:RelA, RelB; RelB, and RelB:cRel) having low affinities close to the μM range, small dimers (p50:p52, p52:p52) in the high nM range, and dimers composed of one large and one small subunit (RelA:p52, RelB:p50, RelB:p52, cRel:p50, and cRel:p52) having the tightest affinities. However, even if these broad rules prove correct, differences between them are likely to be important in determining the dimer repertoire. Further, it is likely that association and dissociation rate constants, rather than steady-state affinities derived from ratios of these rates, may also be important, as a slow kon rates will lead to a kinetic disadvantage within this potentially competitive dynamical system. If experimental pipelines are established, we expect that posttranslational modifications will likely be found that modulate these important parameters.
Expression of NFκB Monomer Genes
In order to produce NFκB dimers, a monomer must be expressed, yet given the interdependence of combinatorial dimerization, expression of all monomers must be considered in order to predict the abundance of a single dimer. NFκB monomer genes are known to be expressed differentially. RelA is thought to be ubiquitously expressed at high levels, whereas cRel is largely restricted to lymphoid lineages (as well as inflammatory dendritic cells), and RelB is also known to be expressed at high levels in specific cell types. All NFκB genes are to some degree NFκB inducible, with cRel, RelB, Nfkb1, and Nfkb2 being long-recognized targets of RelA,61,104 and RelA recently reported to be a RelA target as well.105 However, it is unlikely that autoregulation alone accounts for the cell-type-specific expression patterns and little information is available about other transcription factors that control the expression of NFκB genes.
Processing of NFκB Monomer Precursors
Expression of Nfkb1 and Nfkb2 leads to the precursor proteins p105 and p100 that must be processed before the p50 and p52 dimerization partners are available. p105 is endoproteolytically cleaved into a mature protein, p50, that is able to dimerize with other NFκB proteins (predominantly RelA).2 p105 processing is generally a constitutive process that occurs in unstimulated cells.106 It is unclear whether IKK2 may process p105 in a signal-responsive manner.107
Nfkb2/p100 must be processed into p52 before it can dimerize, predominantly with RelB, to form an NFκB dimer. In response to developmental signals p52 processing is increased as NIK is activated.82 p100 to p52 processing is not only significant as it generates p52 but also as it depletes p100 which would otherwise complex into the inhibitory complex IκBδ. Competition for binding to RelA and RelB between p50 and p52 also contributes to p100 processing rate as RelB inhibits p100 to p52 processing.90 The level of precursor protein controls the maximal signal-responsive induction of protein processing that can be achieved. When elevated, p100 processing depletes the cellular pool of p100 and noncanonical signaling is unable to strongly induce further p52 production or RelB:p52 formation. Similarly, when all RelB is able to bind to p50 (for example, in nfkb2−/− where no p52 is produced), the pool of precursor p105 is depleted.90
Monomer Competition During Dimerization
The combinatorial nature of NFκB dimerization implies the potential for competition between monomers for the generation of specific dimers. This competition was first reported with regard to the formation of RelA:p50 versus RelB:p52, where a slightly higher affinity of p105/p50 for RelA reduces the opportunity for p100/p52 from complexing with RelA.108 Conversely, a slightly higher affinity of p100/p52 for RelB diminishes the potential formation of RelA:p52 dimers. This model explains the appearance of such dimers in the respective knockouts.90
More recently, a quantitative analysis of RelA homodimerization and heterodimerization (with p50) revealed the degree to which monomer competition reduces the abundance of low-affinity dimers103: the high-affinity RelA:p50 dimer dramatically reduces the abundance of the low-affinity RelA homodimer, whose formation hence becomes entirely dependent on a chaperone. Only when p50 is genetically diminished does RelA homodimer formation become chaperone independent.103
Dimer Stabilization and Chaperones
In addition to the combinatorial dimerization affinities and physiochemical properties of the monomers, dimer abundances may be enhanced by ‘third-party’ stabilizers, and dimer generation may be enhanced by dimerization chaperones. To date, one such example has been reported103,109: IκBβ was identified as increasing the effective binding affinity of RelA homodimer.103 As mentioned, RelA homodimer levels suffer not only from a poor affinity but also from competition from RelA:p50 dimerization. This effect is counteracted by IκBβ, which is able to increase the binding of the low-affinity RelA homodimer. A quantitative analysis arrives at such a high effective affinity that a two-step model for IκBβ function is plausible, i.e. that IκBβ binds one monomer first and then a second, enhancing the effective association rate constant and rendering it a bona fide ‘chaperone.’ However, direct evidence is currently outstanding.
But regardless of whether IκBs function as chaperones or merely stabilizers of specific dimers, their potential role adds a more dynamic and complex component to the control of the cell-type-specific NFκB dimer repertoire, as well as a redefinition of IκBs from being merely inhibitors to also being ‘licensing factors’ of NFκB activity.
Dimer Degradation
NFκB dimers are thought to be stable when associated with IκBs. They neither come apart nor are they degraded. However, while DNA interactions may also stabilize dimerizing interactions, NFκB dimers bound to DNA are thought to be subject to regulated degradation. Though an attractive hypothesis, the mechanisms remain less than clear. RelA was shown to be removed from specific chromatin sites even in the absence of new IκBα feedback synthesis110; RelA removal from chromatin was impaired in IKKα-deficient macrophages111; and the peptidyl-prolyl isomerase Pin1 and the E3 ligases SOCS1112 and COMMD1/Cul2113 have been implicated in RelA degradation.
SIGNALING CROSSTALK MECHANISMS
While the canonical and noncanonical pathways are mediated by distinct kinases and immediate substrates, given the large number of shared components within the NFκB system, there is a great potential for crosstalk between the two pathways.
Noncanonical Control of Canonical Signaling
Nfkb2/p100 is the primary signaling node at which canonical and noncanonical signals interact (Figure 5 (a)). That is because (1) Nfkb2 expression is inducible by RelA, (2) any p100 that is not processed into p52 forms a higher-molecular-weight inhibitor of NFκB, the IκBδ-containing IκBsome, that may trap NFκB dimers and thus diminish their association with canonical IκBs,26,114 and (3) p100 processing to p52 and IκBδ degradation is triggered by noncanonical NIK activity. As a result, noncanonical signaling may extend the duration of canonical NFκB activation,61 or in its absence may diminish canonical NFκB activation.58 Thus, noncanonical pathway activity tunes the potency of the canonical pathway in activating NFκB (Figure 5(b)). Interestingly, in conditions with chronically elevated noncanonical activity, as in inflammatory dendritic cells, canonical pathway signaling is not only altered quantitatively but also qualitatively: in GM-CSF-derived dendritic cells, high noncanonical pathway activity diminishes p100 to such a degree that the RelB:p50 dimer forms, which then associates with IκBα (and to some degree IκBε), rendering it responsive to noncanonical signals.35 Hence, TLR-triggered maturation of these dendritic cells involves activation of RelB:p50, in addition to the expected RelA:p50 and cRel:p50 dimers.
FIGURE 5 |.
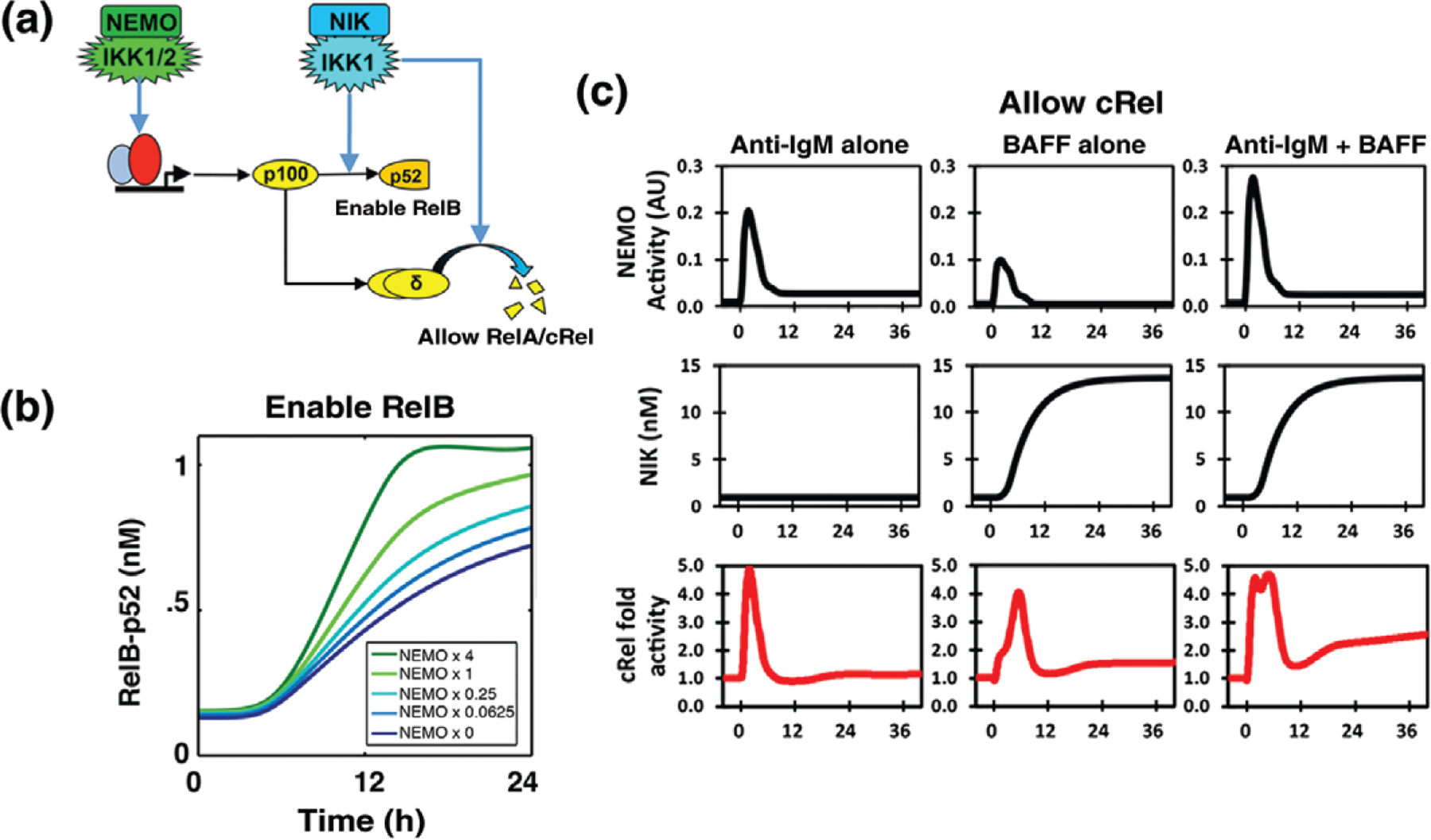
Signaling crosstalk between canonical and noncanonical pathways. (a) Diagram of dual roles of Nfkb2/p100 and NIK within the noncanonical pathway that together with the inducible expression of Nfkb2/p100 mediate two crosstalk functions. (1) NIK/IKK1 processes p100 into p52, enabling the activity of RelB.82 (2) NIK degrades IκBδ, allowing for sustained RelA activity.61 (b) Canonical pathway activity may boost noncanonical pathway activation of RelB:p52.90 Novel model simulations that illustrate how noncanonical pathway activation of RelB:p52 may be boosted by increasing constitutive canonical pathway activities. (c) A noncanonical pathway stimulus may prolong canonical pathway-induced NFκB activity. In B cells, BAFF may potentiate late IgM-induced cRel61 activity.
Canonical Control of Noncanonical Signaling
In addition to Nfkb2, the Relb gene is also induced by NFκB dimers in response to canonical pathway activity.104,115 Therefore, canonical pathway activity is essential for noncanonical activation of the RelB: p52 dimer.90 The dependency of noncanonical signaling on canonical activity impacts lymph node development in RelA-deficient mice.87
This potential for crosstalk may in principle also allow elevated canonical pathway activity to amplify noncanonical activation of RelB:p52 (Figure 5(b)), with potentially pathologic consequences.108 However, this does not appear to be the case—yet the mechanism that insulates canonical signaling from high canonical activity remains to be elucidated.
Posttranslational Modifications
Several posttranslational modifications of RelA have been identified, including phosphorylation of serines and threonines in the DNA binding and activation domains, methylation, acetylation, and glycosylation.116,117 Mutational analysis of the modification acceptor residues has in many cases been shown to have detrimental effects on proper NFκB control and target gene expression, and it appears that a variety of regulatory steps (e.g., nuclear localization, DNA binding, and co-activator recruitment) may be affected. However, in many cases, it remains unclear whether the posttranslational modification occurs constitutively, is induced by the same stimulus as IκB degradation, or may in fact be induced by a different stimulus or cell-type specifically. Evidence for the latter scenario would support the attractive hypothesis that NFκB proteins function as integrators of distinct signals (to control nuclear localization and posttranslational modifications) to fine-tune NFκB-responsive transcriptional programs. Such a scenario would suggest a signaling crosstalk that ought to be examined with a range of biochemical, cell biological, and computational research tools.
SUMMARY
NFκB has been a key nexus of scientific interest over the past several decades. The breadth and depth of investigation, and the compiled knowledge of this key family of transcription factors are unparalleled. From early insights into its function as a transcription factor and immune response regulator to later studies on cellular signaling, dynamical responses, gene regulatory networks, and feedback mechanisms, it is clear that NFκB has become a standard bearer for research into the inner workings of cellular communication and signal response paradigms. New work on signaling crosstalk and remaining questions and challenges in the field, an amenability to signaling and information transmission studies informed by computational modeling, as well as NFκB’s continued interest as a therapeutic target ensure a future of extensive interest and publication on the subject. Ongoing work to expand the scope of computational models and extend their applicability from the cellular scale to the tissue scale will require agent-based modeling techniques that may also leverage minimal model formulations.118–120 Identification and construction of appropriate minimal models for this purpose depend on identifying the physiologically relevant features within NFκB’s intricate and varied dynamic responses.
Footnotes
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1.Gerondakis S, Grumont R, Gugasyan R, Wong L, Isomura I, Ho W, Banerjee A. Unravelling the complexities of the NF-κB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25:6781–6799. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol 2009, 27:693–733. [DOI] [PubMed] [Google Scholar]
- 3.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012, 26:203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev 2006, 210:171–186. [DOI] [PubMed] [Google Scholar]
- 5.Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 2009, 8:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheong R, Hoffmann A, Levchenko A. Understanding NF-κB signaling via mathematical modeling. Mol Syst Biol 2008, 4:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kearns JD, Hoffmann A. Integrating computational and biochemical studies to explore mechanisms in NF-κB signaling. J Biol Chem 2009, 284:5439–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Basak S, Behar M, Hoffmann A. Lessons from mathematically modeling the NF-κB pathway. Immunol Rev 2012, 246:221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Dea E, Hoffmann A. NF-κB signaling. WIREs Syst Biol Med 2009, 1:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Dea E, Hoffmann A. The regulatory logic of the NF-κB signaling system. Cold Spring Harb Perspect Biol 2010, 2:a000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J 2003, 22:5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 2004, 118:453–464. [DOI] [PubMed] [Google Scholar]
- 13.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci USA 2000, 97:12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanjabi S, Williams KJ, Saccani S, Zhou L, Hoffmann A, Ghosh G, Gerondakis S, Natoli G, Smale ST. A c-Rel subdomain responsible for enhanced DNA-binding affinity and selective gene activation. Genes Dev 2005, 19:2138–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25:6706–6716. [DOI] [PubMed] [Google Scholar]
- 16.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res 2011, 21:86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol 2013, 11:e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 2009, 138:114–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, Chang M, Ponomarenko JV, Sun SC, Benedict CA, et al. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci Signal 2011, 4:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huxford T, Hoffmann A, Ghosh G. Understanding the logic of IκB: NF-κB regulation in structural terms. In: NF-kB in Health and Disease. Berlin Heidelberg: Springer; 2011, 1–24. [DOI] [PubMed] [Google Scholar]
- 21.Alves BN, Tsui R, Almaden J, Shokhirev MN, Davis-Turak J, Fujimoto J, Birnbaum H, Ponomarenko J, Hoffmann A. IκBε is a key regulator of B cell expansion by providing negative feedback on cRel and RelA in a stimulus-specific manner. J Immunol 2014, 192:3121–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Escoubet-Lozach L, Benner C, Kaikkonen MU, Lozach J, Heinz S, Spann NJ, Crotti A, Stender J, Ghisletti S, Reichart D. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet 2011, 7:e1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent Iκ B α phosphorylation marks the NF-κB inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA 1995, 92:10599–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev 2012, 246:239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 2002, 298:1241–1245. [DOI] [PubMed] [Google Scholar]
- 26.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, et al. A fourth IκB protein within the NF-κB signaling module. Cell 2007, 128:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwai K Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol 2012, 22:355–364. [DOI] [PubMed] [Google Scholar]
- 28.Adhikari A, Xu M, Chen Z. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26:3214–3226. [DOI] [PubMed] [Google Scholar]
- 29.Cheong R, Bergmann A, Werner SL, Regal J, Hoffmann A, Levchenko A. Transient IκB kinase activity mediates temporal NF-κB dynamics in response to a wide range of tumor necrosis factor-α doses. J Biol Chem 2006, 281:2945–2950. [DOI] [PubMed] [Google Scholar]
- 30.Shinohara H, Behar M, Inoue K, Hiroshima M, Yasuda T, Nagashima T, Kimura S, Sanjo H, Maeda S, Yumoto N, et al. Positive feedback within a kinase signaling complex functions as a switch mechanism for NF-κB activation. Science 2014, 344:760–764. [DOI] [PubMed] [Google Scholar]
- 31.Niu J, Shi Y, Iwai K, Wu ZH. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J 2011, 30:3741–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 2005, 309:1857–1861. [DOI] [PubMed] [Google Scholar]
- 33.Chen L-F, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol 2004, 5:392–401. [DOI] [PubMed] [Google Scholar]
- 34.Schröfelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A. NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol Cell 2012, 47:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, Yu T, Fagerlund R, Asagiri M, Zuniga EI. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat Immunol 2012, 13:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu G, Lo Y-C, Li Q, Napolitano G, Wu X, Jiang X, Dreano M, Karin M, Wu H. Crystal structure of inhibitor of κB kinase β. Nature 2011, 472:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polley S, Huang D-B, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schröfelbauer B, Kim Y, Hoffmann A, Verma IM. A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol 2013, 11: e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng Z, Taylor B, Ourthiague DR, Hoffmann A. Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci Signal 2015, 8:ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behar M, Hoffmann A. Tunable signal processing through a kinase control cycle: the IKK signaling node. Biophys J 2013, 105:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 1999, 284:309–313. [DOI] [PubMed] [Google Scholar]
- 41.Schröfelbauer B, Hoffmann A. How do pleiotropic kinase hubs mediate specific signaling by TNFR superfamily members? Immunol Rev 2011, 244:29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O’Dea E, Boldin MP, Ma A, Baltimore D, Hoffmann A. Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes Dev 2008, 22:2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell S, Tsui R, Hoffmann A. Studying NF-κB signaling with mathematical models. Methods Mol Biol 2015, 1280:647–661. [DOI] [PubMed] [Google Scholar]
- 44.Fagerlund R, Behar M, Fortmann KT, Lin YE, Vargas JD, Hoffmann A. Anatomy of a negative feedback loop: the case of IκBα. J R Soc Interface 2015, 12:0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachdev S, Hoffmann A, Hannink M. Nuclear localization of IκB α is mediated by the second ankyrin repeat: the IκB α ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol Cell Biol 1998, 18:2524–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathes E, O’Dea EL, Hoffmann A, Ghosh G. NF-κB dictates the degradation pathway of IκBα. EMBO J 2008, 27:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, Levchenko A, Hoffmann A. A homeostatic model of IκB metabolism to control constitutive NF-κB activity. Mol Syst Biol 2007, 3:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fortmann KT, Lewis RD, Ngo KA, Fagerlund R, Hoffmann A. A regulated, ubiquitin-independent degron in IκBα. J Mol Biol 2015, 427:2748–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wuerzberger-Davis SM, Chen Y, Yang DT, Kearns JD, Bates PW, Lynch C, Ladell NC, Yu M, Podd A, Zeng H. Nuclear export of the NF-κB inhibitor IκBα is required for proper B cell and secondary lymphoid tissue formation. Immunity 2011, 34:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulero MC, Ferres-Marco D, Islam A, Margalef P, Pecoraro M, Toll A, Drechsel N, Charneco C, Davis S, Bellora N. Chromatin-bound IκBα regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 2013, 24:151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-κB[C1]DNA dissociation by IκBα. Proc Natl Acad Sci USA 2009, 106:19328–19333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson D, Ihekwaba A, Elliott M, Johnson J, Gibney C, Foreman B, Nelson G, See V, Horton C, Spiller D. Oscillations in NF-κB signaling control the dynamics of gene expression. Science 2004, 306:704–708. [DOI] [PubMed] [Google Scholar]
- 53.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157:1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sung M-H, Bagain L, Chen Z, Karpova T, Yang X, Silvin C, Voss TC, McNally JG, Van Waes C, Hager GL. Dynamic effect of bortezomib on nuclear factor-κB activity and gene expression in tumor cells. Mol Pharmacol 2008, 74:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kellogg RA, Tay S. Noise facilitates transcriptional control under dynamic inputs. Cell 2015, 160:381–392. [DOI] [PubMed] [Google Scholar]
- 56.Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, Hoffmann A, Tsimring L, Wollman R. Accurate information transmission through dynamic biochemical signaling networks. Science 2014, 346:1370–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Behar M, Hoffmann A. Understanding the temporal codes of intra-cellular signals. Curr Opin Genet Dev 2010, 20:684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shih VF-S, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A. Kinetic control of negative feedback regulators of NF-κB/RelA determines their pathogen-and cytokine-receptor signaling specificity. Proc Natl Acad Sci USA 2009, 106:9619–9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savinova OV, Hoffmann A, Ghosh G. The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell 2009, 34:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banoth B, Chatterjee B, Vijayaragavan B, Prasad MV, Roy P, Basak S. Stimulus-selective crosstalk via the NF-κB signaling system reinforces innate immune response to alleviate gut infection. Elife 2015, 4: eLife.05648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almaden JV, Tsui R, Liu YC, Birnbaum H, Shokhirev MN, Ngo KA, Davis-Turak JC, Otero D, Basak S, Rickert RC. A pathway switch directs BAFF signaling to distinct NFκB transcription factors in maturing and proliferating B cells. Cell Rep 2014, 9:2098–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Longo DM, Selimkhanov J, Kearns JD, Hasty J, Hoffmann A, Tsimring LS. Dual delayed feedback provides sensitivity and robustness to the NF-κB signaling module. PLoS Comput Biol 2013, 9:e1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keams J, Basak S, Wemer S, Huang C, Hoffmann A. IκBε provides negative feedback to control NF-κB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol 2006, 173:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malek S, Chen Y, Huxford T, Ghosh G. IκBβ, but not IκBα, functions as a classical cytoplasmic inhibitor of NF-κB dimers by masking both NF-κB nuclear localization sequences in resting cells. J Biol Chem 2001, 276:45225–45235. [DOI] [PubMed] [Google Scholar]
- 65.Caldwell AB, Cheng Z, Vargas JD, Birnbaum HA, Hoffmann A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes Dev 2014, 28:2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis-Turak JC, Allison K, Shokhirev MN, Ponomarenko P, Tsimring LS, Glass CK, Johnson TL, Hoffmann A. Considering the kinetics of mRNA synthesis in the analysis of the genome and epigenome reveals determinants of co-transcriptional splicing. Nucleic Acids Res 2015, 43:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coope H, Atkinson P, Huhse B, Belich M, Janzen J, Holman M, Klaus G, Johnston L, Ley S. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J 2002, 21:5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-κ B2 in maturing B cells. Nat Immunol 2002, 3:958–965. [DOI] [PubMed] [Google Scholar]
- 69.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li Z-W, Karin M, Ware CF, Green DR. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002, 17:525–535. [DOI] [PubMed] [Google Scholar]
- 70.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity 2002, 17:515–524. [DOI] [PubMed] [Google Scholar]
- 71.Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL. The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J Exp Med 2003, 198:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Munroe ME, Bishop GA. Role of tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) in distinct and overlapping CD40 and TNF receptor 2/CD120b-mediated B lymphocyte activation. J Biol Chem 2004, 279:53222–53231. [DOI] [PubMed] [Google Scholar]
- 73.Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-κB2 p100 processing and long lasting NF-κB activation. J Biol Chem 2003, 278:36005–36012. [DOI] [PubMed] [Google Scholar]
- 74.Wicovsky A, Salzmann S, Roos C, Ehrenschwender M, Rosenthal T, Siegmund D, Henkler F, Gohlke F, Kneitz C, Wajant H. TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ 2009, 16:1445–1459. [DOI] [PubMed] [Google Scholar]
- 75.Dejardin E The alternative NF-κB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 2006, 72:1161–1179. [DOI] [PubMed] [Google Scholar]
- 76.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J 2004, 23:4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang VY-F, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, Ghosh G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep 2012, 2:824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun S-C. Non-canonical NF-κB signaling pathway. Cell Res 2011, 21:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao G, Zhang M, Harhaj EW, Sun S-C. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 2004, 279:26243–26250. [DOI] [PubMed] [Google Scholar]
- 80.He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G. Rescue of TRAF3-null mice by p100 NF-κB deficiency. J Exp Med 2006, 203:2413–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng P-H, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol 2008, 9:1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao G, Harhaj EW, Sun S-C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell 2001, 7:401–409. [DOI] [PubMed] [Google Scholar]
- 83.Senftleben U, Li Z-W, Baud V, Karin M. IKKβ is essential for protecting T cells from TNFα-induced apoptosis. Immunity 2001, 14:217–230. [DOI] [PubMed] [Google Scholar]
- 84.Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T. Alympho-plasia is caused by a point mutation in the mouse gene encoding NF-κB-inducing kinase. Nat Genet 1999, 22:74–77. [DOI] [PubMed] [Google Scholar]
- 85.Ling L, Cao Z, Goeddel DV. NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc Natl Acad Sci USA 1998, 95:3792–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lallena M-J, Diaz-Meco MT, Bren G, Payá CV, Moscat J. Activation of IκB kinase β by protein kinase C isoforms. Mol Cell Biol 1999, 19:2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX. Coordination between NF-κB family members p50 and p52 is essential for mediating LTβR signals in the development and organization of secondary lymphoid tissues. Blood 2006, 107:1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mordmüller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF-κB-p52 generation by a co-translational mechanism. EMBO Rep 2003, 4:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fusco AJ, Savinova OV, Talwar R, Kearns JD, Hoffmann A, Ghosh G. Stabilization of RelB requires multidomain interactions with p100/p52. J Biol Chem 2008, 283:12324–12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Basak S, Shih VF, Hoffmann A. Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system. Mol Cell Biol 2008, 28:3139–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV. RelB cellular regulation and transcriptional activity are regulated by p100. J Biol Chem 2002, 277:1405–1418. [DOI] [PubMed] [Google Scholar]
- 92.Liao G, Sun S-C. Regulation of NF-κB2/p100 processing by its nuclear shuttling. Oncogene 2003, 22:4868–4874. [DOI] [PubMed] [Google Scholar]
- 93.Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O’Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7α- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol 2012, 14:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Dea EL, Kearns JD, Hoffmann A. UV as an amplifier rather than inducer of NF-κB activity. Mol Cell 2008, 30:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tam AB, Mercado EL, Hoffmann A, Niwa M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS One 2012, 7: e45078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat Cell Biol 2006, 8:986–993. [DOI] [PubMed] [Google Scholar]
- 97.Ho JQ, Asagiri M, Hoffmann A, Ghosh G. NF-κB potentiates caspase independent hydrogen peroxide induced cell death. PLoS One 2011, 6:e16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang TT, Wuerzberger-Davis SM, Wu Z-H, Miyamoto S. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 2003, 115:565–576. [DOI] [PubMed] [Google Scholar]
- 99.Chen NX, Geist DJ, Genetos DC, Pavalko FM, Duncan RL. Fluid shear-induced NFκB translocation in osteoblasts is mediated by intracellular calcium release. Bone 2003, 33:399–410. [DOI] [PubMed] [Google Scholar]
- 100.Ganguli A, Persson L, Palmer IR, Evans I, Yang L, Smallwood R, Black R, Qwarnstrom EE. Distinct NF-κB regulation by shear stress through Ras-dependent IκBα oscillations: real-time analysis of flow-mediated activation in live cells. Circ Res 2005, 96:626–634. [DOI] [PubMed] [Google Scholar]
- 101.Huang D-B, Vu D, Ghosh G. NF-κB RelB forms an intertwined homodimer. Structure 2005, 13:1365–1373. [DOI] [PubMed] [Google Scholar]
- 102.Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta 2011, 1815:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsui R, Kearns JD, Lynch C, Vu D, Ngo KA, Basak S, Ghosh G, Hoffmann A. IκBβ enhances the generation of the low-affinity NFκB/RelA homodimer. Nat Commun 2015, 6:7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-kB. Oncogene 2001, 20:7722–7733. [DOI] [PubMed] [Google Scholar]
- 105.Sung M-H, Li N, Lao Q, Gottschalk RA, Hager GL, Fraser ID. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-κB signaling. Sci Signal 2014, 7:ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beinke S, Ley S. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J 2004, 382:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orian A, Gonen H, Bercovich B, Fajerman I, Eytan E, Israël A, Mercurio F, Iwai K, Schwartz AL, Ciechanover A. SCFβ-TrCP ubiquitin ligase-mediated processing of NF-κB p105 requires phosphorylation of its C-terminus by IκB kinase. EMBO J 2000, 19:2580–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Basak S, Hoffmann A. Crosstalk via the NF-κB signaling system. Cytokine Growth Factor Rev 2008, 19:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rao P, Hayden MS, Long M, Scott ML, West AP, Zhang D, Oeckinghaus A, Lynch C, Hoffmann A, Baltimore D, et al. IκBβ acts to inhibit and activate gene expression during the inflammatory response. Nature 2010, 466:1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med 2004, 200:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 2005, 434:1138–1143. [DOI] [PubMed] [Google Scholar]
- 112.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 2003, 12:1413–1426. [DOI] [PubMed] [Google Scholar]
- 113.Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, Repaka A, Mayo MW, Burstein E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev 2009, 23:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mercurio F, DiDonato J, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev 1993, 7:705–718. [DOI] [PubMed] [Google Scholar]
- 115.Liptay S, Schmid RM, Nabel EG, Nabel GJ. Transcriptional regulation of NF-κ B2: evidence for κ B-mediated positive and negative autoregulation. Mol Cell Biol 1994, 14:7695–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang B, Yang XD, Lamb A, Chen LF. Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell Signal 2010, 22:1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu T, Stark GR. NF-κB: regulation by methylation. Cancer Res 2015, 75:3692–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williams R, Timmis J, Qwarnstrom E. Computational models of the NF-KB signalling pathway. Computation 2014, 2:131. [Google Scholar]
- 119.Shokhirev MN, Almaden J, Davis-Turak J, Birnbaum HA, Russell TM, Vargas JA, Hoffmann A. A multi-scale approach reveals that NF-κB cRel enforces a B-cell decision to divide. Mol Syst Biol 2015, 11:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fallahi-Sichani M, Kirschner DE, Linderman JJ. NF-κB signaling dynamics play a key role in infection control in tuberculosis. Front Physiol 2012, 3:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
FURTHER READING
- Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev 2004, 18:2195–2224. [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-κB: variations on a common theme. Cell Mol Life Sci 2008, 65:2964–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]