

Abstract
Parp inhibitors (Parpi) are commonly used as single agents for the management of tumors with homologous recombination repair (HRR) deficiencies, but combination with radiotherapy (RT) is not widely considered due to the modest radiosensitization typically observed. BMN673 is one of the most recently developed Parpi and has been shown to mediate strong cell sensitization to methylating agents. Here, we explore the mechanisms of BMN673 radiosensitization to killing, aiming to combine it with RT. We demonstrate markedly stronger radiosensitization by BMN673 at concentrations substantially lower (50 nmol/L) than olaparib (3 μmol/L) or AG14361 (0.4 μmol/L) and dramatically lower as compared with second-generation inhibitors such as PJ34 (5 μmol/L). Notably, BMN673 radiosensitization peaks after surprisingly short contact times (~1 hour) and at pharmacologically achievable concentrations in vivo. BMN673 exerts a complex set of effects on DNA double-strand break (DSB) processing, including inhibition of classic nonhomologous end-joining (cNHEJ) and alternative end-joining (altEJ) pathway at high doses of ionizing radiation (IR). BMN673 enhances resection at DSB and favors HRR and altEJ at low clinically relevant IR doses. The combined outcome of these effects is an abrogation in the inherent balance of DSB processing culminating in the formation of chromosomal translocations that underpin radiosensitization. Our observations pave the way to clinical trials exploring inherent benefits in combining BMN673 with RT for the treatment of various forms of cancer.
Introduction
Poly (ADP-ribose) polymerase-1 (Parp1) is the founding member of a family of enzymes consisting of 17 members that catalyze the addition of ADP-ribose units to a wide range of proteins, including proteins involved in DNA repair (1). Parpi as single agents have shown great promise in breast cancer treatment through synthetic lethality with defects in BRCA1/2 and other components of homologous recombination repair (HRR; refs. 2-5). This recognition prompted the development of highly promising third-generation Parpi that are presently tested in clinical trials—including AG14361, olaparib, niraparib, veliparib, and talazoparib (BMN673; ref. 6).
The mechanism of action of Parpi is not completely understood (7) but is often attributed to secondary production of DSB. Parp1 is involved in single-strand break (SSB) repair (8, 9), as well as in base excision repair (BER; ref. 10). The prevailing model is that Parpi-mediated inhibition of these repair pathways leads to the accumulation of SSB converting to DSB during DNA replication. Ineffective processing of these DSB in HRR-deficient cells was originally proposed as a mechanism of Parpi-induced cell lethality (7). However, SSB often fail to accumulate during Parp inhibition when Parp expression is downregulated (9) and some SSB repair-deficient mutants fail to show synthetic lethality with BRCA deficiency (9, 11). Moreover, Parp1 is dispensable for BER (9). It is thus likely that additional mechanisms underpin synthetic lethality between Parp1 inhibition and HRR defects.
Indeed, a role of Parp1 in DSB processing is increasingly considered (7). Among DNA lesions, the DSB is the most deleterious and is processed by various repair pathways. Classic nonhomologous end-joining (cNHEJ) and HRR are the two main DSB repair pathways, which if they fail to function or engage properly are backed-up by an error-prone alternative end-joining (altEJ; refs. 12, 13).
Parp1 has been implicated in altEJ together with Lig3 and Xrcc1 and increase in Parp1 mediated altEJ is evident in the absence of functional HRR (14, 15). Parp1 may contribute to the survival of BRCA1/2-deficient cells by promoting altEJ of accidentally induced DSB and Parp1 inhibition may cause synthetic lethality by suppressing altEJ. Indeed, Parp1 is hyperactivated in HRR deficient cells (16) and cNHEJ activity is elevated in BRCA deficient cells treated with Parpi (11).
RT is a central pillar of cancer treatment. It is used in the management of the majority of cancers and is responsible for about 40% of the cures achieved (17). Most of radiotherapy (RT) effects are thought to derive from the induction of DSB. Whereas DSB repair proficiency is beneficial for the genomic integrity of normal cells, it helps tumor cells to develop radioresistance. Thus, targeting DSB repair using inhibitors specifically in tumor cells offers means for improving RT.
The possibility of using Parpi with RT has been discussed but not extensively considered, mainly due to the relatively modest radiosensitization achieved with tested compounds (18-20). Even olaparib showed modest radiosensitization restricted to proliferating cells (19), although a recent report shows marked radiosensitization in selected tumor cell lines (21). On the other hand, Parp1-dependent altEJ robustly catalyzes IR-induced translocations, when cNHEJ or HRR are compromised. Indeed, the mechanisms of Parpi radiosensitization by altEJ inhibition may resemble those invoked to explain synthetic lethality with HRR. These parallels hint to potentially unexplored benefits from combinations with Parpi of RT or other DNA-damaging agents.
Talazoparib (BMN673) is one of the most recently developed Parpi (22) and is shown to mediate strong sensitization of cells to methylating agents (23, 24). Here, we explore the mechanisms of BMN673 radiosensitization to killing aiming toward a possible combination with RT.
Materials and Methods
Cell culture
All cell lines were maintained at 37°C in an atmosphere with 5% CO2 and 95% air. Hamster cells CHO10B4 wild-type (WT), V3 (DNA-PKcsm), a gift from Dr. D. Chen (UT Southwestern Medical Center, Dallas, TX), human cells HCT116 wt, A549, U2OS, and U2OS DR-GFP (a gift from Dr. J. Stark [Beckman Research Institute of the City of Hope]; ref. 25) were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum (FBS) and antibiotics. The CHO mutant irs1SF (Xrcc3m), a gift from Dr. L. Thompson (Lawrence Livermore National Labora-tory), and 82-6 hTert (human fibroblast cells), a gift from Dr. M. Loebrich (Darmstadt University of Technology, Germany), were grown in minimum essential medium (MEM) supplemen-ted with 10% FBS and antibiotics. Mouse embryonic fibroblasts (MEF), WT, Parp1−/− (26), a gift from Dr. Z.Q. Wang (Leibniz Institute on Aging, Jena) and human RPE-1 cells, a gift from Dr. K.W. Caldecott (University of Sussex, UK), were grown in Dulbecco's modified MEM (DMEM) supplemented with 10% FBS and antibiotics. BT-12 (human, atypical teratoid/rhabdoid tumor) cells, a gift from Dr. P. Houghton, were grown in RPMI medium supplemented with 15% FBS and antibiotics. CHLA-9 (human, Ewing sarcoma) cells (a gift from Dr. P. Houghton [Nationwide Children's Hospital, Columbus, Ohio]) were grown in Iscove's Modified Dulbecco's Medium supplemented with 20%FBS, 1% insulin–transferrin–selenium (Invitrogen) and antibio-tics. Cell lines were passaged thrice a week. A549, HCT116, 82-6 hTert, and U2OS-282C were authenticated using Multiplex Cell Authentication by Multiplexion as described (27). Human hTert RPE-1 cells were directly traced to ATCC (28). BT12 and CHLA-9 cells were obtained directly from the original source; the cell lines are not widely distributed. Cells were tested for mycoplasma before freezing using MycoAlert Plus Mycoplasma detection kit from Lonza (LT07-705).
Inhibitors
BMN673 was obtained from Medivation, a collaborator of NCI. The Parp1/2 inhibitor PJ34 (ref. 29; Calbiochem) was used at 5 μmol/L final concentration. 8-(4-Dibenzothienyl)-2-(4-morpholinyl)-4H-1-benzopyran-4-one (NU7441, Tocris), a highly specific DNA-PKcs inhibitor (30), was used at 5 μmol/L final concentration. Rad51 inhibitor B-02 (ref. 31; Merck-Millipore) was used at 25 μmol/L final concentration. AG14361 (specific Parp1 inhibitor; ref. 32), olaparib (Parp1/2 inhibitor), ME0328 (Parp3 inhibitor; ref. 33), and UPF1069 (Parp2 inhibitor; ref. 34) were purchased from Selleckchem and used at 400 nmol/L (AG14361), 1 μmol/L (UPF1069) or 3 μmol/L (all others).
Radiation exposure
Irradiations were carried out with an X-ray machine (GE Healthcare) operated at 320 kV, 10 mA with a 1.65 mm Al filter (effective photon energy approximately 90 kV), at a distance of 50 cm and a dose rate of approximately 1.3 Gy/minute. Cells were returned to the incubator immediately after exposure to IR.
Clonogenic survival assay
Protocol 1:
Exponentially growing cells were treated with inhibitor for 1 hour and exposed to 0, 2, 4, 6, or 8Gy. Cells were subsequently trypsinized, diluted, and plated for colony formation. Parpi was again added in this protocol after seeding cells for colony formation, i.e., inhibitors were present during colony formation. Feeder cells were included in appropriate numbers when they improved plating efficiency. Colonies were stained with 1% crystal violet and counted under a stereomicroscope. Plating efficiency (PE) of untreated cells was calculated as ratio between colonies counted to cells seeded. Surviving fractions (SF) in irradiated cells were calculated also as ratios between colonies counted to cells seeded after correcting for PE. For drug-treated samples, SF was calculated using the PE of unirradiated and untreated cells as a denominator (18) to allow visualization of drug toxicity.
Protocol 2:
In this protocol, inhibitors were added 1 hour before IR and were allowed to act for the indicated times after IR. Subsequently, cells were plated for colony formation in regular growth medium, i.e., inhibitors were not present during colony formation.
Quantitative estimates of radiosensitization achieved by an inhibitor are given as dose modification factor (DMF10) calculated at 10% survival as follows: DMF10 = IR dose for 10% survival without inhibitor/IR dose for 10% survival with inhibitor. DMF10 values higher than 1 reflect radiosensitization. DMF10 values for different inhibitors and cell lines are summarized in Supplementary Table S1. The cytotoxicity of different inhibitors in CHO cells (at 0Gy) is summarized in Supplementary Table S3.
Analysis of chromosomal translocations
Cytogenetic analysis was done in irradiated G2 phase cells as described before (14, 35). Exponentially growing cells were exposed to 1 Gy X-rays and were incubated at 37°C for 4 hours before adding 0.1 μg/mL Colcemid (L-6221, Biochrom AG) for 1 hour. Metaphase-enriched cultures were harvested and processed. Bright field microscopy (Olympus, Vanox-T) and a Meta-Systems station (Altlussheim) with a microscope (AxioImager.Z2, Zeiss) and automated image capture and analysis capabilities were used for scoring chromosome aberrations. Standard criteria were used for scoring chromatid translocations.
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) was used to assess induction and repair of DSB. The methodology has been described previously (36). PFGE gels were scanned in a fluorescence scanner and the fraction of DNA released (FDR) from the plug into the lane quantified by ImageQuant 5.2 software (GE Healthcare). Dose response curves are plotted as FDR versus radiation dose. These curves are used to calculate the equivalent Gy dose values (DEQ) for each FDR measured at a given repair time point; repair kinetics are given as plots of DEQ versus time.
Indirect immunofluorescence
Cells were grown on coverslips and treated with the indicated inhibitors for 1 hour prior to IR. Coverslips were rinsed in phosphate-buffered saline (PBS) and fixed with 2% paraformaldehyde (PFA) solution followed by permeabilization with P-solution (100 mmol/L Tris pH 7.4, 50 mmol/L EDTA, 0.5% Triton X-100) at different times after IR. Fixed cells were blocked in PBG solution (0.2% gelatin, 0.5% BSA fraction V in PBS) overnight at 4°C. Cells were incubated with appropriately diluted antibodies as indicated for 1.5 hour at room temperature. After washing with PBS, cells were incubated with AlexaFluor488 (AF488)-, AF568-, or AF647-conjugated secondary antibodies, as required, for 1 hour at room temperature. Cells were finally counterstained with DAPI (50 ng/mL in distilled water) for 10 minutes at room temperature and mounted in Prolong-Antifade mounting media. Samples were scanned on a Leica TCS-SP5 confocal microscope and foci were counted using Imaris software (Bitplane). Sources and dilutions of antibodies used are listed in Supplementary Table S2.
Poly (ADP-ribose) PAR staining
Cells were grown on coverslips for ~48 hours and treated with the indicated inhibitors or DMSO for 1 hour at 37°C. H2O2 was diluted in PBS and added at a final concentration of 10 mmol/L. Cells were then processed for immunofluorescence as described above.
DSB repair analysis using genomically integrated reporter constructs
U2OS cells (2 × 106) containing an HRR reporter construct (25) were transfected with 1 μg I-SceI expression plasmid and allowed to attach for 90 minutes. Then, cells were treated with indicated inhibitors. Seventy-two hours later, cells were analyzed for GFP expression by flow cytometry.
Statistical analysis
Graphs were created in SigmaPlot 11.0. Statistical significance was determined using Student t test available in SigmaPlot 11.0. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
Among Parpi, BMN673 exerts the strongest radiosensitization with a short, time and sequence flexible exposure
We began our investigations with CHO and mouse cells to take advantage of the large repertoire of available DSB repair mutants that enable analysis of radiosensitization mechanisms. CHO cells preexposed to a variety of Parpi for 1 hour, irradiated and plated immediately thereafter in growth medium, also supplemented with inhibitors, show surprisingly variable degrees of radiosensitization (Fig. 1A). Strikingly, BMN673 is by far the strongest radiosensitizer leading to radiosensitivities only known from cNHEJ or HRR mutants (Fig. 1B). All inhibitors are used at concentrations sufficiently high to reduce H2O2 induced parylation below detection (Supplementary Fig. S1A). We conclude that differences in radiosensitization derive from mechanisms operating beyond simple Parp inhibition.
Figure 1.
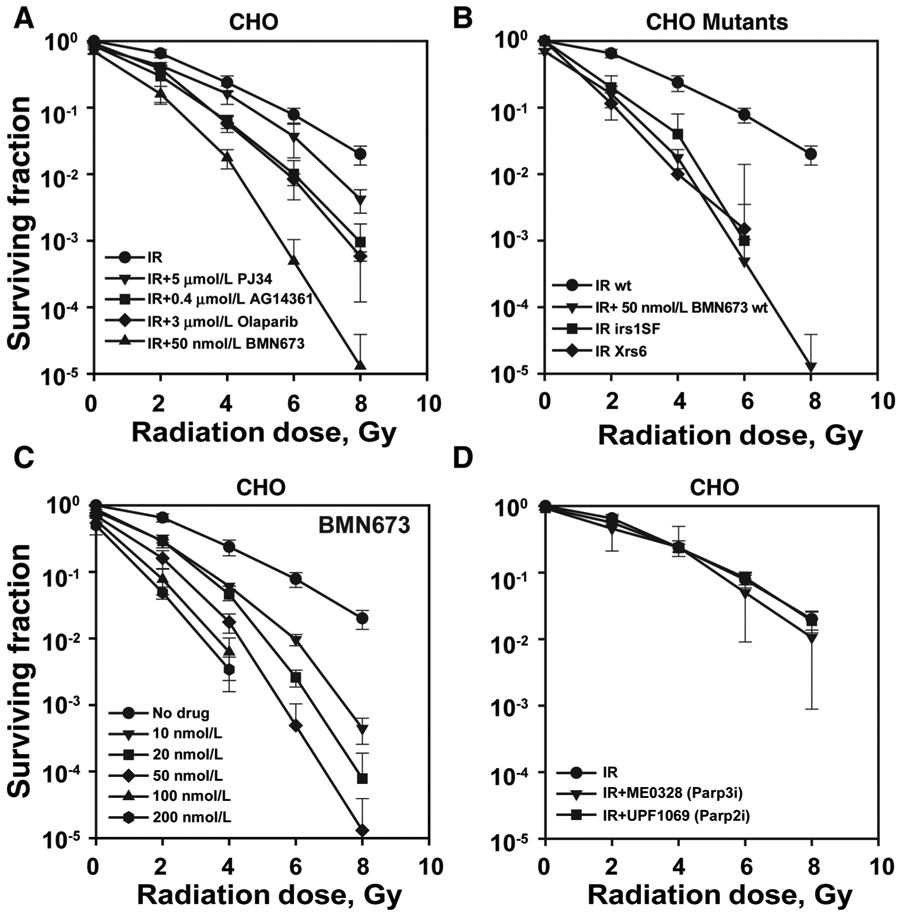
Among Parpi, BMN673 exerts the strongest radiosensitization. Exponentially growing cells were treated with indicated inhibitors for 1 hour, irradiated, trypsinized, and seeded in appropriate numbers in the presence of inhibitors to form colonies for 7 to 8 days. Drug toxicity is indicated by reduced surviving fraction at 0 Gy. A, Impact of various Parpi on radiosensitivity in CHO cells. B, Comparison of clonogenic survival of BMN673-treated CHO cells with Xrs6 (Ku80m) or irs1SF (Xrcc3m) cells. C, Effect of various concentrations of BMN673 on CHO radiosensitization. D, Impact of Parp2 and Parp3 inhibition on CHO radiosensitization. Data, mean ± SD calculated from three to four independent experiments.
BMN673 is an effective radiosensitizer with 10 nmol/L generating radiosensitization comparable to that of 3 μmol/L olaparib (compare Figs. 1A and C). Notably, the radiosensitizing effect of BMN673 plateaus at about 50 nmol/L, a concentration that is clinically achievable (37). The cytotoxicity data for CHO cells after treatment with various concentrations of BMN673 are given in Supplementary Table S3, where it can be seen that the IC50 is about 200 nmol/L. BMN673-induced radiosensitization derives from inhibition of Parp1, as specific inhibition of Parp2 with UPF1069 or of Parp3 with ME0328 fails to generate statistically significant levels of radiosensitization (Fig. 1D, see Supplementary Table S1 for DMF10 values). In addition, Parp1−/− MEFs show no radiosensitization by BMN673 (Supplementary Fig. S1B).
When repair inhibitors are combined with RT, it is important to ensure that their concentrations in the blood will be sufficiently high at the time of patient irradiation and that they will be maintained high for several hours after irradiation—in order to efficiently interfere with DSB processing. The precise administration schedule will depend on their behavior as repair inhibitors and their pharmacokinetics. To begin understanding the properties of BMN673 as radiosensitizer, we tested the drug exposure times required for maximum effect in CHO cells. Strikingly, we discovered that treatment with 50 nmol/L for only 1 hour prior to IR is sufficient to generate nearly maximum radiosensitization (Fig. 2A; see Supplementary Table S1 for DMF10 values). An experiment in which cells were first plated for colony formation, treated with BMN673 for 1 hour, irradiated and incubated for up to 72 hours before transferring to BMN673 free growth medium, allows similar conclusions (Supplementary Fig. S1C).
Figure 2.
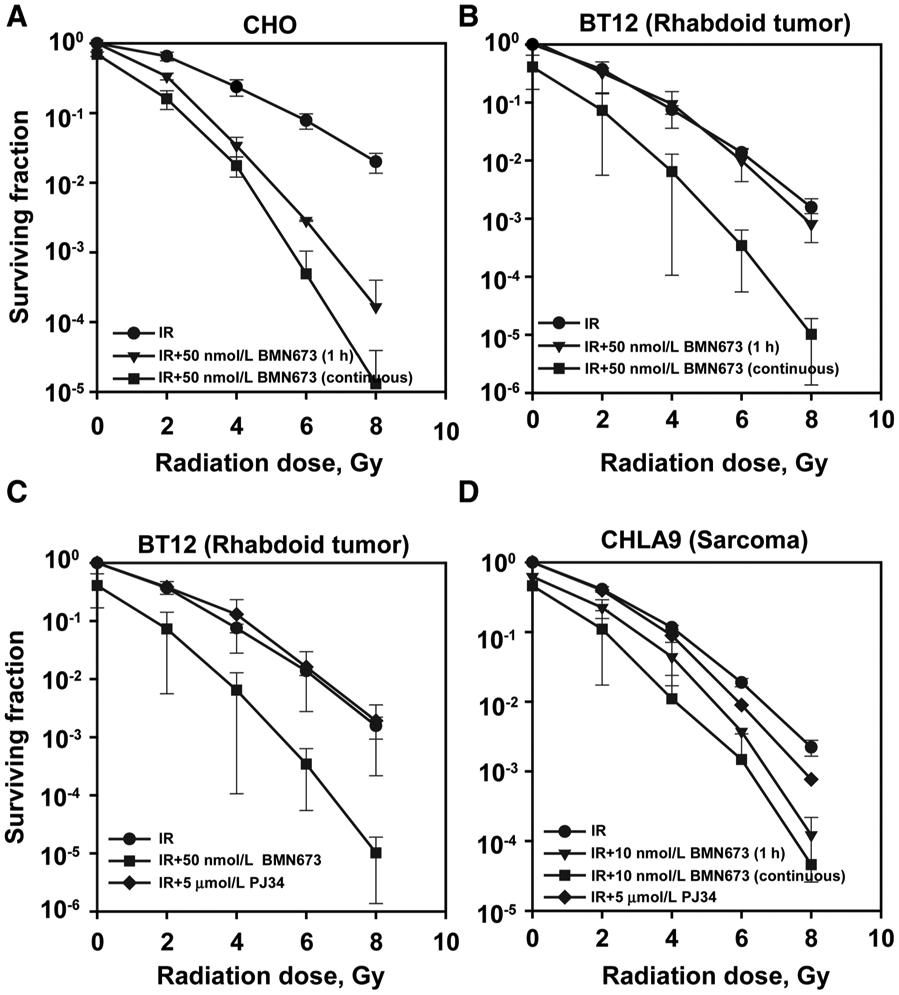
BMN673 efficiently radiosensitizes human rhabdoid and sarcoma cell lines. Exponentially growing cells were treated with indicated inhibitors for 1 hour, irradiated, trypsinized, and seeded to form colonies either under drug-free conditions or under continuous exposure to inhibitors. Drug toxicity is indicated by the surviving fraction at 0 Gy. A, Impact of short (1 hour) vs. continuous treatment with BMN673 on CHO radiosensitization. B, Impact of short (1 hour) vs. continuous treatment with 50 nmol/L BMN673 on radiosensitization in BT12 rhabdoid human cells. C, Effect of PJ34 vs. BMN673 continuous treatment on radiosensitization in BT12 rhabdoid human cells. D, Impact of short (1 hour) vs. continuous pretreatment with 10 nmol/L BMN673 on radiosensitization in CHLA9 Ewing sarcoma cells. Data, mean ± SD calculated from three to four independent experiments.
We inquired whether radiosensitization established in CHO cells also holds for human tumor cell lines. Because our team participates in the NRG sarcoma group, which focuses on the development of novel treatments for tumors of mesenchymal origin, we tested BMN673 radiosensitization in tumor cell lines of such origin. Preexposure for 1 hour with BMN673 of human rhabdoid BT12 cells fails to generate detectable radiosensitization, but continuous treatment generates marked radiosensitization (Fig 2B; see Supplementary Table S1 for DMF10 values). We conclude that exposure time for maximum radiosensitization will be cell line dependent and that biomarkers of response are required for optimal administration in the clinical setting (see below). Notably, BMN673 is a much stronger radiosensitizer than PJ34, which is completely ineffective in BT12 cells (Fig. 2C; Supplementary Table S1 for DMF10 values).
The human Ewing sarcoma CHLA9 cells also show considerable radiosensitization after 1 hour, and even more after continuous, treatment with low concentrations (10 nmol/L) of BMN673 (higher concentrations were toxic), although PJ34 is relatively ineffective (Fig. 2D; Supplementary Table S1 for DMF10 values). Considering that BMN673, as single agent, shows limited activity against xenografts grown from these cells (38), the results suggest benefits from combining BMN673 with IR in these childhood cancers.
We also examined BMN673 radiosensitization in human tumor cell lines widely used in mechanistic studies of the DNA damage response (Fig. 3). We observed marked and practically maximal radiosensitization in U2OS, HCT116, and A549 cells treated with 25 nmol/L BMN673 for 1 hour before irradiation and then immediately plated for colony formation in medium without BMN673 (Fig. 3A-C; Supplementary Fig. S2 for results on inhibition of parylation and Supplementary Table S1 for DMF10 values). These results extend the validity of the results presented above with CHO cells.
Figure 3.
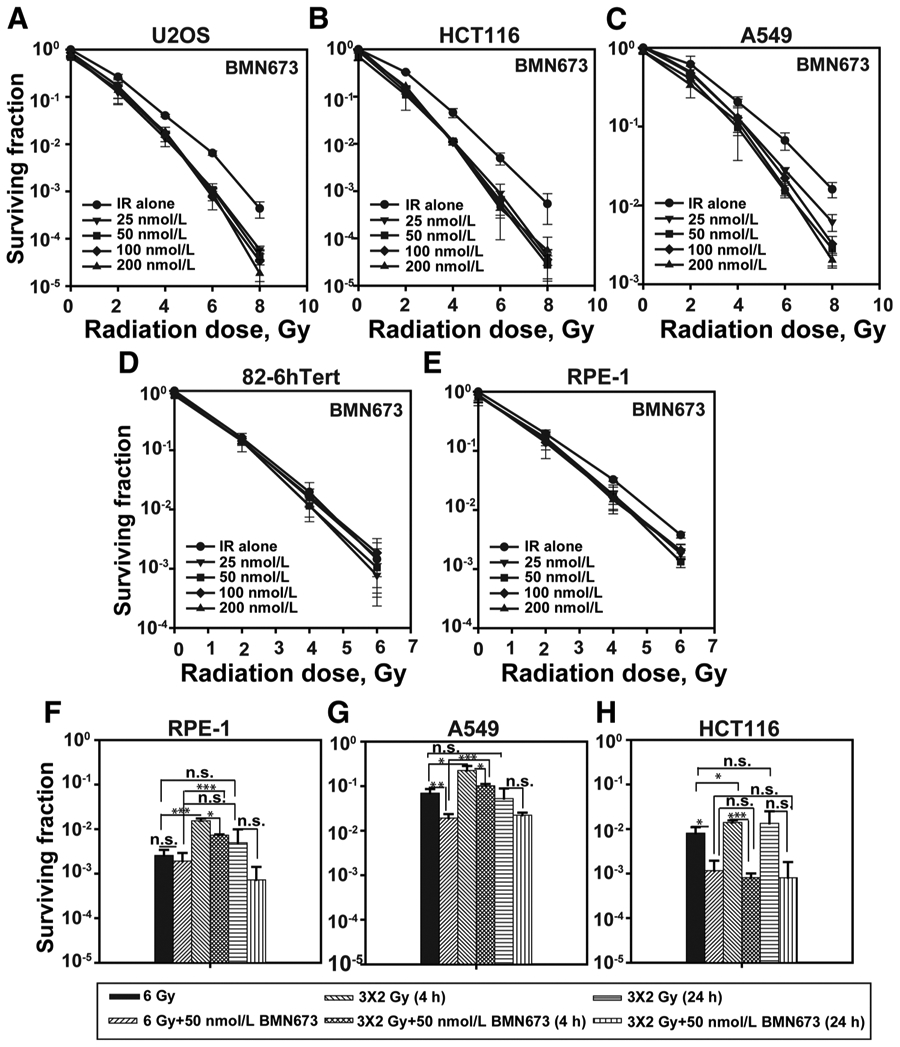
BMN673 radiosensitization is specific for tumor cells. Exponentially growing cells were treated with indicated inhibitors for 1 hour prior to IR (continuous treatment was toxic in these cell lines), immediately trypsinized, seeded at appropriate numbers in the absence of BMN673, and allowed to form colonies for 7 to 8 days in drug-free medium. A, Impact of short (1 hour) pretreatment with BMN673 on radiosensitization in U2OS cells. B, As in A for HCT116 cells. C, As in A for A549 cells. D, As in A for normal human fibroblast 82-6 hTert cells. E, As in A for normal RPE-1 cells. F, BMN673 radiosensitization after fractionated irradiation (3 fractions of 2 Gy separated by 4 or 24 hours) in RPE-1 cells. G, As in F for A549 cells. H, As in F for HCT116 cells. The significance of differences between individual measurements is indicated by connecting lines between bars; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., nonsignificant. Data, mean ± SD calculated from three independent experiments.
An important consideration for the clinical application of BMN673 is the radiosensitization of normal cells that will lead to normal tissue toxicity. We therefore tested BMN673 radiosensitization in two normal human cell lines. Normal human fibroblasts 82-6 hTert and normal retinal epithelial cells RPE-1 show no signs of radiosensitization by BMN673 (Fig. 3D and E). In these cells, BMN673 strongly inhibits H2O2-induced parylation, confirming inhibition of Parp1 (Supplementary Fig. S2). We conclude that BMN673 may act specifically on tumor cells, fulfilling a key requirement for a clinically relevant, tumor-specific radiosensitizer.
IR is applied in daily fractions in the clinical setting. We examined, therefore, the effect of IR dose fractionation on BMN673-mediated radiosensitization. The results summarized in Fig. 3F, G, and H again show cell line-dependent reduction in BMN673-mediated radiosensitization following the application of 6 Gy in 3 fractions of 2 Gy separated by 4 or 24 hours.
Collectively, the low and clinically achievable concentrations of BMN673 for maximum radiosensitization, the specificity for tumor cells, the mere 1 hour or so of required drug exposure, and the embedded flexibility in the timing of IR and drug administration uncover properties never before reported for a radiosensitizer. Together, they define the "perfect" scenario for the combined application of BMN673 with IR in cancer treatment and make elucidation of the underpinning molecular mechanisms a high priority task. Work along these lines is described next.
BMN673 suppresses DSB repair at low IR doses
We used γ-H2AX immunofluorescence staining to assess DSB processing at low doses of IR. CHO cells exposed to 2 Gy show pronounced increase in γ-H2AX foci formation, as compared with nonirradiated controls (Supplementary Fig. S3A and S3B), which reaches a maximum at 1 hour after IR. The number of foci decreases and reaches values only slightly above background at 8 hours (Supplementary Fig. S3A), suggesting efficient processing of DSBs. A similar response is also observed in cells incubated with AG14361 for 1 hour before IR and 8 hour thereafter, indicating no detectable effect of the inhibitor on DSB processing (Supplementary Fig. S3A and S3B). Notably, treatment with BMN673 increases the number of γ-H2AX foci scored at 1 hour and strongly inhibits their resolution in the ensuing 8 hours, documenting profound global inhibition of DSB processing. Also, the intensity of the γ-H2AX foci is increased after treatment with BMN673, an effect also observed with AG14361. Similar results are also obtained with A549 cells (Supplementary Fig. S3C). Although our results clearly implicate inhibition of DSB processing in BMN673 radiosensitization, they do not provide information as to whether the effect relies on inhibition of a specific DSB repair pathway (cNHEJ, HRR, or altEJ). Pathway specificity of BMN673-induced DSB repair inhibition was therefore studied next.
BMN673 favors HRR by promoting hyper-resection and suppressing recruitment of 53BP1 at DSBs
To study BMN673 effects on HRR, we measured DNA end resection (referred to as "resection" here), the first step in HRR, by means of RPA foci formation—a widely accepted marker, detected here as RPA70 immunofluorescence.
Although a relatively small number of RPA70 foci forms in cells exposed to 2Gy, a dramatic increase occurs after incubation with BMN673, particularly at 3 and 6 hours after IR (Fig. 4A-C). Treatment of nonirradiated cells with BMN673 generates a modest increase in RPA70 foci; therefore, Fig. 4C shows the results of Fig. 4B after subtraction of this background. The dramatic increase in RPA foci formation suggests a shift in the fraction of DSB shunted for resection, i.e., a shift from cNHEJ to either HRR or altEJ.
Figure 4.
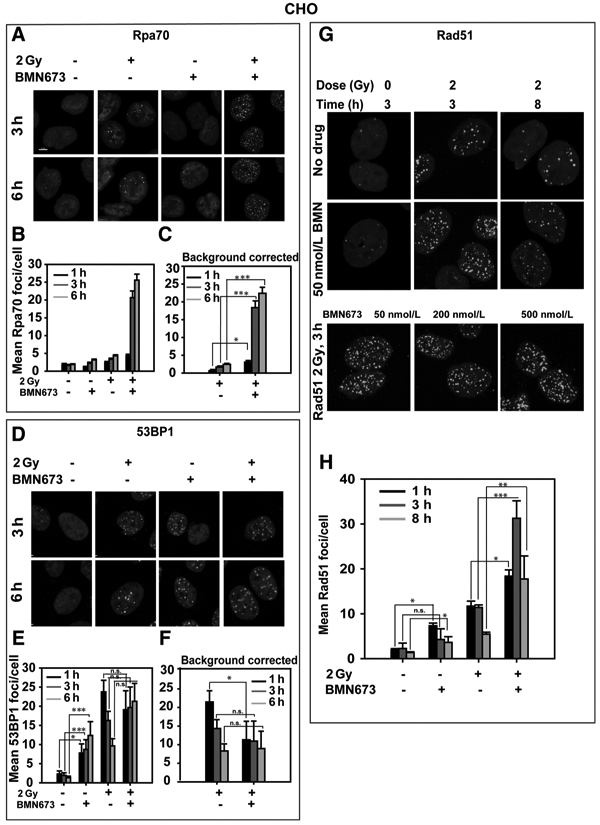
BMN673 promotes resection and increases Rad51 foci formation. Cells were exposed to 2 Gy IR in the presence or absence of BMN673 and kinetics of RPA70, 53BP1, and Rad51 foci formation, and decay was measured by immunofluorescence. A, Representative images showing formation and resolution of RPA70 foci after 2 Gy in the absence or presence of 50 nmol/L BMN673. B, Graphical representation of RPA70 foci formation and resolution without background (0 Gy) correction. C, Graphical representation of RPA70 foci formation and resolution after background correction. D, Representative images showing formation and resolution of 53BP1 foci after 2 Gy in the absence or presence of 50 nmol/L BMN673. E, Graphical representation of 53BP1 foci formation and resolution without background correction. F, Graphical representation of 53BP1 foci formation and resolution after background correction. G, Representative images showing formation and resolution of Rad51 foci after 2 Gy in the absence or presence of 50 nmol/L BMN673. H, Graphical representation of Rad51 foci formation and resolution at 1, 3, and 8 hours after IR. Data, mean ± SD calculated from two to three independent experiments. The significance of differences between individual measurements is indicated by connecting lines between bars; *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., nonsignificant.
53BP1 is thought to suppress resection at DSB (39), and this prompted us to inquire whether the increase in resection observed is accompanied by a decrease in 53BP1 foci. In the absence of IR, CHO cells display low numbers of 53BP1 foci that increase abruptly 1 hour after exposure to 2 Gy (Fig. 4D-F). 53BP1 foci numbers gradually decrease in irradiated cells at 3 and 6 hours, indicating completion in DSB processing. Notably, treatment with BMN673 causes a large increase in 53BP1 foci even in nonirradiated cells, which becomes apparent at 1 hour and reaches values over 5-fold above background after 3 and 6 hours of incubation (Fig. 4E). IR causes only a small additional increase in 53BP1 foci in this setting (Fig. 4F), an effect clearly evident after subtraction of the background in nonirradiated cells. We conclude that BMN673 suppresses 53BP1 foci formation at early times after IR.
EdU (5-ethynyl-2′-deoxyuridine) labeling of S-phase cells (Supplementary Fig. S4A) shows that 53BP1 foci developing in nonirradiated cells after treatment with BMN673 are specific for S-phase cells and reflect therefore effects of Parpi on DNA replication. IR exposure in S-phase causes only a modest further increase in 53BP1 foci formation. In EdU-negative cells (Supplementary Fig. S4B), BMN673 has no effect on 53BP1 foci formation in the absence of IR and only a modest effect after exposure to IR. Thus, suppression of IR-dependent 53BP1 foci formation by BMN673 is specific for S-phase cells.
For direct analysis of the effect of BMN673 on HRR, we used immunofluorescence to score formation and resolution of foci formed by Rad51, the central mitotic recombinase in eukaryotic cells. CHO cells exposed to 2 Gy develop Rad51 foci 3 hours later demonstrating engagement of HRR(Fig. 4G). Rad51 foci decrease at 8 hours signifying the gradual completion of this form of processing. Notably, treatment with BMN673 causes initially a 3-fold increase in Rad51 foci (Fig. 4G and H), suggesting increased engagement of HRR. However, at 8 hours, although a clear reduction in Rad51 foci is measured, residual numbers remain high, suggesting that processing by HRR is incomplete in BMN673-treated cells. Similar results are obtained at higher concentration of BMN673 (Fig. 4G). Rad51 foci colocalize with γ-H2AX foci as would be expected from DSB processing by HRR (Supplementary Fig. S5A and S5B).
We introduced a functional reporter assay to study the effect of Parpi on HRR. U2OS-DRGFP cells have integrated a construct in their genome that carries two nonfunctional copies of the GFP gene (ref. 25; Supplementary Fig. S6A). Introduction of a DSB within this construct by transient expression of I-Sce I, a restriction endonuclease recognizing a DNA sequence normally not present in the human genome, and processing by HRR results in expression of GFP that is detected by flow cytometry. Supplementary Fig. S6A shows that BMN673, added 1.5 hours after transfection and kept for the duration of the experiment (72 hours), has no effect on HRR when tested at 50 nmol/L and even 10 μmol/L fails to generate any effect. HRR remains unaffected also by olaparib, AG14361, or PJ34. In contrast to Parpi, a Rad51 inhibitor (B02) abrogates HRR and DNA-PK inhibitor (NU7441) exerts the expected increase in the frequency of HRR events.
How could BMN673, on the one hand, increase resection and Rad51 foci formation and, on the other hand, leave HRR as measured by functional assays unchanged? We considered the possibility that inhibition of cNHEJ by BMN673 allows resection at DSB that feeds futile HRR. Futile HRR events, in turn, may be rescued by altEJ causing translocations (40). We therefore examined the effect of BMN673 on translocations and cNHEJ.
Contrary to other Parpi, BMN673 increases IR-induced translocations
Our previous studies show that IR-induced translocations, particularly those generated under conditions of HRR or cNHEJ deficiency, require Parp1-mediated altEJ (14, 35) and are reduced after treatment with PJ34 or olaparib (14, 35, 41). We inquired whether BMN673 exerts similar effects. CHO cells irradiated in G2 phase and treated with NU7441 to inhibit cNHEJ show a marked increase in the incidence of chromosomal translocations as they reach metaphase 4 hours after IR (Fig. 5A and B). Treatment of these cells with PJ34, olaparib, or AG14361 causes the expected decrease in translocations. Strikingly, BMN673 treatment in combination with NU7441 has opposite effects of increasing the incidence of translocations by nearly a factor of two (Fig. 5B). BMN673 also increases translocation in HCT116 cells exposed to 1 Gy (Fig. 5C). Notably, RPE-1 cells show no increase in translocation formation after BMN673 treatment (Fig. 5C), in line with the lack of radiosensitization shown in Fig. 3E. We conclude that, compared with other Parpi, BMN673 has a distinct spectrum of activities on IR-induced lesions.
Figure 5.

BMN673 treatment causes translocations in irradiated cells. Cells were exposed to 1 Gy X-rays in the presence or absence of the indicated inhibitors and analyzed for chromosome damage at metaphase 4 hours after IR to limit analysis to cells irradiated in G2 phase of the cell cycle. The protocol is a modification of one previously used for this type of analysis. Chromatid translocations were scored. No translocations were detected in unirradiated cells. A, Representative image of IR-induced chromatid translocation (indicated by arrow). B, Translocations forming in CHO cells after exposure to 1 Gy IR and treatment with NU7441 and various Parpi as indicated. C, Translocations in HCT116 WT and RPE-1 cells after exposure to 1 Gy, alone or in combination with 50 nmol/L BMN673. Data, mean ± SD calculated from two to three independent experiments. The significance of differences between individual measurements is indicated by connecting lines between bars; *, P < 0.05; n.s., nonsignificant.
BMN673 compromises cNHEJ at high doses of IR
DSB generated in CHO cells after exposure to 20 Gy are processed with fast kinetics in untreated cells (Fig. 6A) with nearly 90% being removed within 1 hour. Remaining DSBs are removed within 8 hours with slower kinetics. It is thought that DSB processing detected under these conditions by PFGE mainly reflects the function of cNHEJ (42).
Figure 6.

At high doses of IR, BMN673 compromises cNHEJ and altEJ. Exponentially growing cells were irradiated with 20 Gy in the presence or absence of indicated inhibitors. A total of 500 nmol/L BMN673, 3 μmol/L olaparib, 5 μmol/L PJ34, and 5 μmol/L NU7441 were added 1 hour prior to IR and maintained during the experiment. Repair kinetics were measured using PFGE. A, DSB repair kinetics in CHO cells incubated in the presence or absence of indicated inhibitors. B, DSB repair kinetics in MEFs incubated with the indicated Parpi. C, DSB repair kinetics measured in Parp1−/− MEFs incubated with the indicated Parpi. D, DSB repair kinetics in V3 (DNA-PKcsm) cells incubated with the indicated Parpi. Data above represent the mean ± SD calculated from four determinations in two independent experiments. E, At low IR doses, BMN673 enhances DSB end resection, increases Rad51 foci formation and possibly futile HRR. Resected ends exclude cNHEJ and promote error-prone altEJ, causing translocations and radiosensitization. F, At high IR doses, BMN673 suppresses both cNHEJ and altEJ. Resection and HRR are reduced after exposure to high IR doses (45, 46).
Notably, although PJ34 has no detectable effect and olaparib only inhibits the slow component, BMN673 has a pronounced effect on DSB processing (Fig. 6A). Indeed, the inhibition achieved by BMN673 is indistinguishable from that of NU7441, a specific inhibitor of DNA-PKcs (Fig. 6A). Because combined treatment with NU7441 and BMN673 generates additional inhibition, we conclude that beyond cNHEJ, BMN673 also inhibits altEJ, which is also assessed in this type of experiments (36).
BMN673 inhibits DSB processing to a greater degree than PJ34 and olaparib in MEFs as well (Fig. 6B), an effect that is dependent on Parp1, as it is absent in Parp1−/− mutants (Fig. 6C). BMN673 inhibits DSB processing stronger than olaparib or PJ34 in V3 as well, a CHO mutant defective in DNA-PKcs (Fig. 6D). Finally, BMN673 exerts marked inhibition of cNHEJ in CHLA9 cells and a modest inhibition in BT12 cells (Supplementary Fig. S6B).
Discussion
We report a novel spectrum of activities for BMN673 on IR-induced DSB repair causing marked radiosensitization specifically in tumor cells. Notably, nontransformed cells remain largely unaffected. BMN673 radiosensitization peaks after surprisingly short and flexible contact times (~1 h) and at pharmacologically achievable concentrations in vivo. Collectively, these observations suggest clear benefits from a combination of BMN673 with RT.
The majority of Parpi tested thus far show only modest-to-intermediate, cell line–dependent radiosensitization (18, 19, 21, 43), explaining why Parpi have not been considered extensively hitherto as clinical radiosensitizers. The effects we report here for BMN673 suggest a paradigm change. Because tumor resistance to Parpi, and thus possibly also to BMN673, remains a problem, when used as single agents, combination with RT offers a means to overcome this limitation.
BMN673 exerts a complex set of effects on DSB processing, the complete elucidation of which will require further work. The present study shows that at low, clinically relevant IR doses, BMN673 strongly enhances resection and shifts DSB processing toward resection-dependent pathways, such as HRR and altEJ (Fig. 6E). Indeed, the strong increase observed in translocation formation supports an increase in the engagement of altEJ. Translocations are one of the key drivers of oncogenesis and the culprits of IR-induced cell killing (44). Strikingly, other Parpi cause 40% to 80% suppression of translocation formation; refs. 13, 14, 35, 41), indicating that the effects of BMN673 on resection and subsequently on altEJ are unique, with mechanistic underpinnings that will require further investigations.
Another resection-dependent pathway of DSB processing is HRR, and indeed BMN673 causes a large increase in IR-induced Rad51 foci formation. Because reporter assays fail to detect an increase in the overall function of HRR, we propose that increased resection in BMN673-treated cells increases initiation of HRR events at DSB not predestined for HRR processing, which aborts later feeding, altEJ.
A different picture develops at high doses of IR (Fig. 6F), where resection is limited through mechanisms (45, 46) that are presently under investigation. In this setting, BMN673 cannot increase resection because the required apparatus is inherently compromised and acts, instead, as a regular Parpi suppressing altEJ. This is indeed observed by PFGE (Fig. 6A-D).
In the high dose range, BMN673 also inhibits cNHEJ. Whether BMN673 commensurately inhibits cNHEJ at low IR doses cannot be assessed from existing data, as the strong inhibition observed in the resolution of γ-H2AX foci reflects effects on all DSB repair pathways. It remains to be investigated whether BMN673 inhibits cNHEJ by the same mechanism at high and low IR doses and how the effect changes throughout the cell cycle.
Collectively, our results show that BMN673 abrogates the inherent balance of DSB processing culminating in the formation of chromosomal abnormalities that underpin radiosensitization. Dysregulation or imbalance of DNA repair pathways is found in many human malignancies (47) and opens windows of opportunity for the combination of IR with repair inhibitors, including BMN673, which already shows promising results as a single agent in preclinical and clinical trials (48, 49). Inhibition of Chk1 was shown to potentiate the efficacy of BMN673 (50), which suggests benefits from combinations of BMN673 with checkpoint inhibitors. Testing the radiosensitizing effect of BMN673 in animal models to pave the way for clinical trials is now a priority. Finally, Parpi are primarily indicated for treatment of breast cancer. Therefore, testing BMN673 radiosensitization in breast cancer cell lines will be highly instructive and relevant.
Supplementary Material
Acknowledgments
This work was supported by grants from the German Federal Ministry of Education and Research (BMBF-02NUK037B and 02NUK043B) and the German Research Foundation (DFG-GRK1739) to G.E. Iliakis. We would like to thank NCI Collaborator Medivation and NCI for providing talazoparib (BMN673). We are also thankful to Drs. K.W. Caldecott, D. Chen, P.J. Houghton, M. Loebrich, J. Stark, L.H. Thompson, and Z.Q. Wang for providing cell lines.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther 2015;8:519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 3.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 4.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123–34. [DOI] [PubMed] [Google Scholar]
- 5.Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 2017;18:610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer 2016;114:713–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helleday T The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011;5:387–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol 2007;27: 5597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ström CE, Johansson F, Uhlen M, Al-Khalili Szigyarto C, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res 2011;39:3166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, et al. Involvement of poly (ADP-ribose) polymerase in base excision repair. Biochimie 1999;81:69–75. [DOI] [PubMed] [Google Scholar]
- 11.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA 2011; 108:3406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res 2011;711:61–72. [DOI] [PubMed] [Google Scholar]
- 13.Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res /Genet Toxicol Envir Mutag 2015; 793:166–75. [DOI] [PubMed] [Google Scholar]
- 14.Soni A, Siemann M, Grabos M, Murmann T, Pantelias GE, Iliakis G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res 2014;42:6380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter AC, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res 2016;44: 5743–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res 2010;70:5389–98. [DOI] [PubMed] [Google Scholar]
- 17.Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci 2012;9:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalmers A, Johnston PG, Woodcock M, Joiner M, Marples B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int J Radiat Oncol Biol Phys 2004;58:410–9. [DOI] [PubMed] [Google Scholar]
- 19.Dungey FA, Löser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys 2008;72:1188–97. [DOI] [PubMed] [Google Scholar]
- 20.Löser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther 2010;9:1775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kötter A, Cornils K, Borgmann K, Dahm-Daphi J, Petersen C, Dikomey E, et al. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol Oncol 2014; 8:1616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Chu D, Feng Y, Shen Y, Aoyagi-Scharber M, Post LE. Discovery and characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a novel, highly potent, and orally efficacious poly(ADP-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J Med Chem 2016;59:335–57. [DOI] [PubMed] [Google Scholar]
- 23.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 2014;13:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Aoyagi-Scharber M, Wang B. Trapping Poly(ADP-ribose) polymerase. J Pharmacol Exp Ther 2015;353:446–57. [DOI] [PubMed] [Google Scholar]
- 25.Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand-breaks. Methods Mol Biol 2012;920:379–91. [DOI] [PubMed] [Google Scholar]
- 26.Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004;23: 3872–82. [DOI] [PubMed] [Google Scholar]
- 27.Castro F, Dirks WG, Fahnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer 2013;132:308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanzlikova H, Gittens W, Krejcikova K, Zeng Z, Caldecott KW. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res 2017;45: 2546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, et al. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med 2001;7:255–60. [PubMed] [Google Scholar]
- 30.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 2004;14:6083–7. [DOI] [PubMed] [Google Scholar]
- 31.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 2011;6:628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst 2004;96:56–67. [DOI] [PubMed] [Google Scholar]
- 33.Lindgren AE, Karlberg T, Thorsell AG, Hesse M, Spjut S, Ekblad T, et al. PARP inhibitor with selectivity toward ADP-ribosyltransferase ARTD3/PARP3. ACS Chem Biol 2013;8:1698–1703. [DOI] [PubMed] [Google Scholar]
- 34.Moroni F, Formentini L, Gerace E, Camaioni E, Pellegrini-Giampietro DE, Chiarugi A, et al. Selective PARP-2 inhibitors increase apoptosis in hippocampal slices but protect cortical cells in models of post-ischaemic brain damage. Br J Pharmacol 2009;157:854–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soni A, Siemann M, Pantelias GE, Iliakis G. Marked cell cycle-dependent contribution of alternative end joining to formation of chromosome translocations by stochastically induced DNA double strand breaks in human cells. Mutat Res 2015;793:2–8. [DOI] [PubMed] [Google Scholar]
- 36.Wu W, Wang M, Wu W, Singh SK, Mussfeldt T, Iliakis G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair (Amst) 2008;7:329–38. [DOI] [PubMed] [Google Scholar]
- 37.de Bono JS, Mina LA, Gonzalez M, Curtin NJ, Wang E, Henshaw JW, et al. First-in-human trial of novel oral PARP inhibtior BMN673 in patients with solid tumors. J Clin Oncol 2013; 31:(suppl; abstr 2580). [Google Scholar]
- 38.Smith MA, Hampton OA, Reynolds CP, Kang MH, Maris JM, Gorlick R, et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer 2015; 62: 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 2010;141:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004;23:546–53. [DOI] [PubMed] [Google Scholar]
- 41.Wray J, Williamson EA, Singh SB, Wu Y, Cogle CR, Weinstock DM, et al. PARP1 is required for chromosomal translocations. Blood 2013;121: 4359–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iliakis G Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother Oncol 2009;92:310–5. [DOI] [PubMed] [Google Scholar]
- 43.Wurster S, Hennes F, Parplys AC, Seelbach JI, Mansour WY, Zielinski A, et al. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016;7:9732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer 2013;13:443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grøfte M, et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 2012;150:697–709. [DOI] [PubMed] [Google Scholar]
- 46.Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol 2016;23: 714–21. [DOI] [PubMed] [Google Scholar]
- 47.Jackson SP, Helleday T. DNA REPAIR. Drugging DNA repair. Science 2016; 352:1178–9. [DOI] [PubMed] [Google Scholar]
- 48.de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Disc 2017;7:620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res 2013;19:5003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin Y, Shen Q, Zhang P, Tao R, Chang W, Li R, et al. Chk1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer. Am J Cancer Res 2017; 7: 473–83. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.