

Significance Statement
When podocytes are injured, foot processes efface and detach, leading to severe proteinuria. Endocytic trafficking maintains the integrity of this crucial glomerular interface. Epsins, a family of membrane proteins, assist in the endocytosis and also take part in cell signaling. Mice that have lost podocyte-associated epsins develop proteinuria and kidney failure, due to diminished activity of the transcription factor serum response factor (SRF), which reduces cell division control protein 42 homolog activation and β1 integrin expression. Podocyte-specific Srf knockout mice also demonstrate proteinuria and kidney failure. These findings suggest that, in podocytes, epsins are required to coordinate a proper signaling platform, beyond their known endocytic properties.
Keywords: cell adhesion, glomerulosclerosis, podocyte
Visual Abstract

Abstract
Background
Epsins, a family of evolutionarily conserved membrane proteins, play an essential role in endocytosis and signaling in podocytes.
Methods
Podocyte-specific Epn1, Epn2, Epn3 triple-knockout mice were generated to examine downstream regulation of serum response factor (SRF) by cell division control protein 42 homolog (Cdc42).
Results
Podocyte-specific loss of epsins resulted in increased albuminuria and foot process effacement. Primary podocytes isolated from these knockout mice exhibited abnormalities in cell adhesion and spreading, which may be attributed to reduced activation of cell division control protein Cdc42 and SRF, resulting in diminished β1 integrin expression. In addition, podocyte-specific loss of Srf resulted in severe albuminuria and foot process effacement, and defects in cell adhesion and spreading, along with decreased β1 integrin expression.
Conclusions
Epsins play an indispensable role in maintaining properly functioning podocytes through the regulation of Cdc42 and SRF-dependent β1 integrin expression.
A normally functioning glomerular filtration barrier acts as a semipermeable membrane, preventing large mol wt proteins from seeping into the urine. Podocytes play a crucial role in maintaining the integrity and stability of the glomerular filtration barrier. When these cells are injured, foot process effacement and detachment from the glomerular basement membrane (GBM) ensue, resulting in proteinuria and glomerulosclerosis.1 Recently, continuing evidence supports the importance of podocyte endocytic trafficking, not only in murine models,2–5 but also in humans.6 It has been shown previously that proteins within the clathrin-mediated endocytic pathway, such as dynamin, synaptojanin1, and endophilin, are critical to normal kidney function, because mice lacking these genes developed massive albuminuria.7 Furthermore, we also identified that endocytic proteins in podocytes mediate proper actin cytoskeletal dynamics, slit diaphragm turnover, and membrane receptor internalization.5,7,8
Epsins, acting as either endocytic accessory proteins or alternate adaptors (for protein receptors like EGFR and VEGFR2), play a critical physiologic role in both clathrin-mediated endocytosis and clathrin-independent cargo internalization.9–14 Complementary to their endocytic role, epsins also have signaling functions and are involved in a variety of cellular activities, including signal transduction, ion-channel activity, and miscellaneous nuclear functions.15–18 Epsins contain a conserved N-terminal homology (ENTH) domain, which has been implicated in sensing and generating cellular membrane curvature upon interaction with the phospholipid bilayer.19–23 Additionally, the ENTH domain of epsin has been shown to interact with GTPase-activating proteins (GAPs), which, in turn, regulate the activation of a small Rho GTPase, cell division control protein 42 homolog (Cdc42).24–26 As a small GTPase of the Rho family, Cdc42 has been shown to regulate filopodia formation and cell migration.27–29
Epsins are encoded by different genes in mammals (Epn1, Epn2, and Epn3), and are thought to have redundant functions, because mice lacking only Epn1, Epn2, or Epn3 are viable without an overt phenotype. However, global loss of both Epn1 and Epn2 simultaneously resulted in embryonic lethality.16 Many studies have shown that epsin 1 and epsin 2 are ubiquitously expressed,16,30 whereas epsin 3 is enriched in keratinocytes of the epidermis and in parietal cells of the stomach.31,32 In this study, we demonstrate that epsin 1, 2, and 3 are expressed in kidney podocytes. To further determine the roles of these three genes, mice with global Epn2 and Epn3 deletions, along with a podocyte-selective deletion of Epn1, were generated using the Cre-LoxP crossed with Rosa-Dtrfl system. Mice harboring these genetic defects did not develop an overt phenotype during embryonic development, but demonstrated severe albuminuria, foot process effacement, and kidney failure after 6 months of age. We further determined that podocytes devoid of Epn1, Epn2, and Epn3 exhibited a reduction in Cdc42 activity and β1 integrin expression, through reduced binding of a transcription factor, serum response factor (SRF), to the Itgb1 promoter. Furthermore, podocyte-specific knockout (KO) of Srf in mice resulted in massive proteinuria and kidney failure. Our results indicate that the endocytic protein, epsin, is critical in maintaining the integrity of the kidney filtration barrier, due to its ability to regulate proper podocyte adhesion to the GBM in a Cdc42-SRF-β1 integrin–dependent manner.
Methods
Generation of Mice
Epn1fl/fl mice were interbred with Epn2−/− and Epn3 −/− mice, as previously described,13,16,32,33 kindly gifted by Pietro De Camilli (Yale University, New Haven, CT). These mice were bred with podocyte-specific Podocin-Cre and Rosa- Diphtheria toxin receptor (Dtr)fl/fl mice,34 gifted by Lloyd Cantley (Yale University) to generate podocyte-specific Epn triple KO; Dtr fl/fl mice (Pod-Epn TKO). All mice were on a C57BL/6J congenic background. Srffl/fl mice were gifted from Stephanie Halene (Yale University)35 and bred to podocyte-specific podocin-Cre and Rosa-Dtrfl/fl mice. These mice were backcrossed ten generations on a C57BL/6J congenic background to develop Pod-Srf−/− Dtrfl/fl mice (Pod-Srf KO).
Antibodies and Reagents
Antibodies (Abs) used in this study were as follows: mouse anti–Wilms tumor, clone 6F-H2 (anti-WT1; catalog MAB4234; Millipore Sigma); rabbit anti–Wilms tumor (CAN-R9[IHC]-56-2; anti-WT1; ab89901; Abcam); guinea pig anti-nephrin (GP-N2; Progen); mouse anti-Rac1, clone 23A8 (catalog 05-389; Millipore Sigma); mouse anti-RhoA (catalog ARH04; Cytoskeleton); mouse anti-Cdc42, clone 28-10 (catalog MABN 2485-25UL; Millipore sigma); rat anti–integrin β1 clone MB1.2 (catalog MAB1997-25UG; Millipore Sigma); rabbit anti-SRF (catalog 5147; Cell Signaling Technology); rabbit anti–glyceraldehyde-3-phosphate dehydrogenase (catalog 5174; Cell Signaling Technology); mouse anti-Fascin1(D-10; catalog sc-46675; Santa Cruz Biotechnology); Alexa Fluor 488 goat anti-rabbit IgG Ab (catalog A-11008; Invitrogen); Alexa Fluor 488 goat anti-mouse IgG Ab (catalog A-11001; Invitrogen); Alexa Fluor 488 goat anti-rat IgG Ab (catalog A-11006; Invitrogen); Alexa Fluor 594 goat anti–guinea pig IgG Ab (catalogA-11076; Invitrogen); Alexa Fluor 594 goat anti-rabbit IgG Ab (catalog A-11012; Invitrogen); goat anti-rat IgG secondary Ab, horseradish peroxidase (HRP) (catalog AP136P; EMD Millipore); goat anti-mouse IgG secondary Ab, HRP (catalog AP160P; EMD Millipore); and goat anti-rabbit IgG secondary Ab, HRP (catalog AP187P; EMD Millipore). The following Abs were gifts: mouse anti–epsin 1 (Pier-Paolo Di Fiore, IFOM, Milan, Italy); and rabbit anti–epsin 2, rabbit anti–clathrin light chain (anti-CLC), and mouse anti–adaptin α subunit (Pietro De Camilli, Yale University). The reagents used in this study were as follows: Collagen I, bovine, type 1 (catalog 354231; Corning). Natural mouse laminin (catalog 23017-015; Invitrogen), Cignal Lenti SRE Reporter (luc; catalog 226851, CLS-10L-1; Qiagen), SureENTRY Transduction Reagent (catalog 336921), Dual-Luciferase Assay System (catalog E1910; Promega), and ML 141 (SML0407; Sigma-Aldrich).
Cell Culture
Isolation of primary podocytes from postnatal day 1 (P1) to P3 control, Pod-Srf KO, and Pod-Epn TKO pups were performed as described previously.34,36 Glomerular cells were seeded on type-1 collagen (catalog 354231)–coated cell-culture dishes and cultured in RPMI 1640 medium (catalog 11875-093; Gibco) with 10% FBS (catalog 10082147; Gibco), 100 U/ml penicillin, 100 µg/ml streptomycin, 100 mM HEPES (American Bio), 1 mM sodium bicarbonate, and 1 mM sodium pyruvate (catalog 11360070; Gibco) in a humidified 5% carbon dioxide (CO2) incubator. Two days after the cells were seeded, medium including 0.1 μg/ml diphtheria toxin (D0564; Sigma-Aldrich) was changed every other day. Subculture was performed by detaching podocytes with 0.25% trypsin-EDTA (Invitrogen) and passing through a 40-μm cell strainer (Falcon; BD Biosciences) and cultured on collagen type 1–coated dishes. All experiments were performed with P1 podocytes.
Adhesion, Spreading, and Wound-Healing Assay
For the podocyte-adhesion assay, primary Pod-Epn TKO, Pod-Srf KO, and control podocytes were trypsinized and counted using a hemocytometer, with equal amounts of cells (6×103 per well) from each group seeded on 96-well plates coated with collagen type 1 (40 μg/ml) and laminin (10 μg/ml) for 2 hours. After the removal of nonadherent cells by gentle washing with PBS three times, the adherent cells were fixed by 95% ethanol. Podocytes were stained with 0.1% crystal violet (Sigma-Aldrich) for 15 minutes at room temperature (RT), washed with water, and then lysed with 1% SDS while shaking for 5 minutes, or until a uniform color was obtained. The absorbance was measured at 595 nm by a Microplate Reader (model 550; Bio-Rad). For the cell-spreading assay, primary podocytes isolated from Pod-Epn TKO, Pod-Srf KO, and control mice were seeded on a collagen type 1–coated tissue culture dish and kept in a stage incubator at 37°C with 5% CO2. Image acquisition started 5 minutes after podocytes attached, and, 2 hours afterward, by phase-contrast microscopy with a Nikon Eclipse TE200 equipped with a 20× objective. Images were captured with Hoffman modulation and a Spot RT camera (Diagnostic Instruments). After collecting images, cell area change over time was analyzed by National Institutes of Health (NIH) ImageJ software, in a blinded manner, by randomly examining 20 cells per genotype for each experiment. For the wound-healing assay, the confluent monolayer of primary podocytes isolated from Pod-Epn TKO, Pod-Srf KO, and control mice were scraped with a 20-μl pipette and visualized immediately (T0) and 24 hours afterward (T24 hours) using Hoffman modulation (×10) and a Spot RT camera. Results were presented as percentage of wound closure, calculated using NIH ImageJ software as follows: ([area T24 hours−area T0)/area T0).
Quantitative PCR Analysis
Total RNA was extracted from the primary podocytes from control and Pod-Epn TKO mice by using Trizol (Thermo Fisher Scientific). The RNA concentration was measured by spectrophotometry (Nanodrop Technologies), and 2 μg of total RNA was used for reverse transcription by a High-Capacity cDNA Reverse Transcription Kit (catalog 4368814; Applied Biosystems) according to the manufacturer’s instructions. The quantitative PCR (qPCR) amplifications were performed using Power SYBR Green PCR Master Mix (Applied Biosystems) with a 7300 Real-Time PCR machine (Applied Biosystems).
Immunoblotting
Primary podocytes were lysed in lysis buffer containing 50 mM Tris-hydrochloride (pH 7.6), 500 mM sodium chloride, 0.1% SDS, 0.5% deoxycholate, 1% Triton X-100, 0.5 mM magnesium chloride, phosphatase inhibitor cocktail (Sigma), 1 mM PMSF, 1 mM sodium orthovanadate, 50 mM sodium fluoride, and protease inhibitor cocktail (Roche Diagnostics). Protein concentrations were quantified using the Bradford Assay (Bio-Rad Protein Assay Kits). Equal amounts of podocyte lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (EMD Millipore). The membrane was blocked with 5% nonfat milk (AmericanBio) or 5% BSA (Sigma-Aldrich) in Tris-buffered saline and Tween 20 and incubated with the appropriate primary Ab at 4°C overnight. After three washes with Tris-buffered saline and Tween 20, the appropriate peroxidase-labeled anti-IgG secondary Ab (EMD Millipore) was added, and signals were detected using enhanced chemiluminescence reagents (Bio-Rad) and exposed with Odyssey (LI-COR Biosciences). For quantification, densitometry was performed using ImageJ software.
Immunofluorescence Staining
Primary podocytes on collagen type 1–coated coverslips were washed by PBS and fixed with 4% paraformaldehyde for 20 minutes. The cells were permeabilized with 0.1% Triton X-100 in PBS for 20 minutes. Kidney cryosections were subjected to antigen retrieval at 95°C for 10 minutes in Retrievagen A solution (pH 6.0; BD Biosciences). Primary podocytes or kidney cryosections were then blocked with 5% BSA in 1× PBS for 1 hour at RT. Immunostaining was performed with the appropriate primary Abs overnight at 4°C, followed by Alexa Fluor 488– and/or 594–conjugated secondary Abs at RT for 1 hour, and samples were mounted with SlowFade containing 4′,6-diamidino-2-phenylindole (Invitrogen). Images were taken by an Andor CSU-WDi spinning disk confocal microscope equipped with a Nikon Ti-E CFI Plan Apochromat Lambda 60× oil immersion objective for immunofluorescence analysis, and images were processed using ImageJ software (version 1.51H) or Adobe Photoshop CS 2018.
Cdc42, Rac 1, and RhoA Activation Assay
The CDC42, Rac1, and RhoA activity assays were performed in control and Pod-Epn TKO primary podocytes according to manufacturer’s protocol (catalog 17-441, Rac1/Cdc42 Activation Assay Kit; EMD Millipore; catalog BK036, RhoA Pull-down Activation Assay Biochem Kit; Cytoskeleton). The lysates were incubated with PAK-1 PBD-bound agarose beads (for Cdc42 and Rac1) or GST-tagged Rhotekin-RBD protein-bound agarose beads (for RhoA), followed by SDS-PAGE and immunoblotting with anti-Cdc42, Rac1, or RhoA Abs.
Chromatin Immunoprecipitation Assay
Primary podocytes were crosslinked using 1% formaldehyde, and a chromatin immunoprecipitation (ChIP) assay was performed using SRF Ab with the SimpleChIP Enzymatic Chromatin IP Kit, according to the manufacturer’s protocol (catalog 9004; Cell Signaling Technology). Coprecipitated DNA and input DNA were quantified by RT-PCR calculations as the ratio of the PCR signal from coprecipitated DNA to that of input DNA. Because the mouse Itgb1 gene promoter contains putative serum response elements, approximately 1770 bp proximal to the translation start site, we generated primers in mouse Itgb1 promoter as follows: forward primer, 5′-ATCATAGTTTGTCTCTCTGACTTGT-3′; reverse primer, 5′-GGATATACTCCTTCAGGCTACCTT-3′.
Luciferase Reporter Assay
Primary podocytes (1000 per well) were seeded on collagen type 1–coated, 96-well tissue culture plates with fresh media and incubated 20 hours at 37°C in a humidified incubator with an atmosphere of 5% CO2. Medium was removed from the well, and then lentiviral particles (50 MOI) containing SRF transcription response element and firefly luciferase reporter gene (Qiagen) were added and incubated for 20 hours. Media containing lentiviral particles were removed from wells and changed to fresh media for another 72 hours. For Cdc42 inhibitor studies, primary podocytes were pretreated with 20 μM of ML 141 for 1 hour before performing the assay. The expression of the reporter gene was measured by performing dual-luciferase reporter assays following the manufacturer’s protocol (Promega) using a luminometer (Promega).
Biochemical Measurements, Urine Albumin, Plasma Creatinine, and BUN
Urine and plasma samples were collected from control, Pod-Epn TKO mice, and Pod-Srf KO mice at different time points. Albuminuria was qualitatively assessed by a 10% SDS-PAGE, followed by Coomassie blue staining. Urine albumin was quantified in duplicates using an Albumin ELISA Quantitation kit (Bethyl Laboratories Inc), according to the manufacturer’s protocol. Plasma and urine creatinine were measured in duplicate for each sample, at the time points indicated in the figures, using an ELISA quantification kit (BioAssay Systems, Hayward, CA) at an absorbance of 490 nm (Microplate Reader). The urine albumin concentration of each sample was normalized to its urine creatinine concentration, expressed as the albumin-creatinine ratio (ACR). BUN was measured by the Yale O’Brien Kidney Center using blood collected at the indicated times.
Kidney Tissue for Histology, Immunofluorescence, and Electron Microscopy Quantification
Mice were anesthetized via an intraperitoneal injection of ketamine and xylazine, followed by perfusion fixation with 30 ml of 4% paraformaldehyde with or without 2% glutaraldehyde through the left ventricle for histology and immunofluorescence or electron microscopy (EM), respectively. Kidney tissues were postfixed with osmium in 0.1 M sodium cacodylate and 0.1 M sucrose, pH 7.4. For histology, kidneys were sent to the Yale Pathology Core Tissue Services for hematoxylin and eosin, Periodic acid–Schiff (PAS), and Masson trichrome (TRI) staining. For EM, kidneys were sent to the Cellular and Molecular Physiology Core Services at Yale University. To evaluate glomerulosclerosis and interstitial fibrosis, kidney sections on TRI and PAS staining were assessed as previously described.37,38 Briefly, at least 50 glomeruli from each specimen were assessed on PAS-stained sections, and the severity of glomerulosclerosis in each glomerulus was semiquantitatively scored in a blinded manner as follows: 0, no sclerosis; 1, sclerosis of <10% of the glomeruli; 2, sclerosis of 10%–25% of the glomeruli; 3, sclerosis of 25%–50% of the glomeruli; and 4, sclerosis of >50% of glomeruli. To evaluate interstitial fibrosis, 20 fields of each section were examined on TRI-stained sections. Semiquantitative analysis of each field was assessed as follows: 0, no fibrosis; 1, fibrosis of <10% of areas; 2, fibrosis of 10%–25% of areas; 3, fibrosis of 25%–50% of areas; and 4, fibrosis of >50% of areas. The averages of the glomerulosclerosis and interstitial fibrosis scores were evaluated and calculated from the total glomeruli or interstitial lesions in each section from each group. For quantitative ultrastructural analysis of the glomerulus by transmission EM (TEM), the number of podocyte foot processes present in each micrograph was divided by the total length of GBM; this was done to calculate the mean density of podocyte foot processes. The thickness in each image was measured by ImageJ software. For quantification of podocyte number, 20 glomeruli were evaluated blindly for each section by counting the WT1-positive nuclei in each glomerulus.38,39
Statistical Analyses
All data are represented as means±SEM. The number of replicates for each experiment is shown in the figure legends. Statistical analysis was performed by GraphPad Prism 8 software using two-tailed t test or one-way ANOVA alone with Dunnett multiple-comparisons test. Statistical significance was determined with P<0.05.
Study Approval
The Committee on the Use and Care of Animals Institutional Review Board at Yale University approved all animal experiments (2019-11196). All work was carried out in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals (Revised 2011).40
Results
Loss of Epn 1, Epn 2, and Epn3 Results in Albuminuria, Interstitial Fibrosis, and Kidney Failure
To examine the importance of epsins in podocyte biology, we generated the podocyte-specific Epn1 KO mice, on the background of the Epn2 and Epn3 global KO (Figure 1A), by mating Epn1fl/fl Epn2−/− Epn3−/− mice with Podocin-Cre and Rosa-Dtrfl/fl mice. The mutant mice were verified via PCR-based tail genotyping of the Epn1 alleles, Podocin-Cre, and the Rosa-Dtrfl/fl transgene (Figure 1B). Epn2 and Epn3 global KO mice were used as a parental strain to mediate concerns of Cre recombinase completely deleting all three Epn genes along with the Dtr. To ensure a pure population of primary podocytes, we leveraged our Rosa-Dtrfl/fl mice that have DTR selectively deleted from the podocytes upon Cre activation, making all other non-Cre-expressing cells susceptible to diphtheria toxin exposure while sparing the podocytes.34 We confirmed, via immunofluorescence, the loss of both epsin 1 and epsin 2 immunoreactivity in the podocytes colabeled with nephrin, a podocyte-specific marker (Figure 1C). By Western blot, we also observed that enriched Pod-Epn TKO primary podocytes lacked epsin 1 and 2 expression (Figure 1D), whereas there was an absence of the punctate appearance of epsin 1 after immunostaining (Figure 1E). Due to a lack of a reliable epsin 3–specific Ab, qPCR was performed and it showed a loss of Epn3 expression when compared with control (Epn1 +/+, Pod- Cre, Epn 2, and Epn 3 +/+, Dtrfl/fl), confirming deletion of Epn3 in podocytes (Figure 1F).
Figure 1.
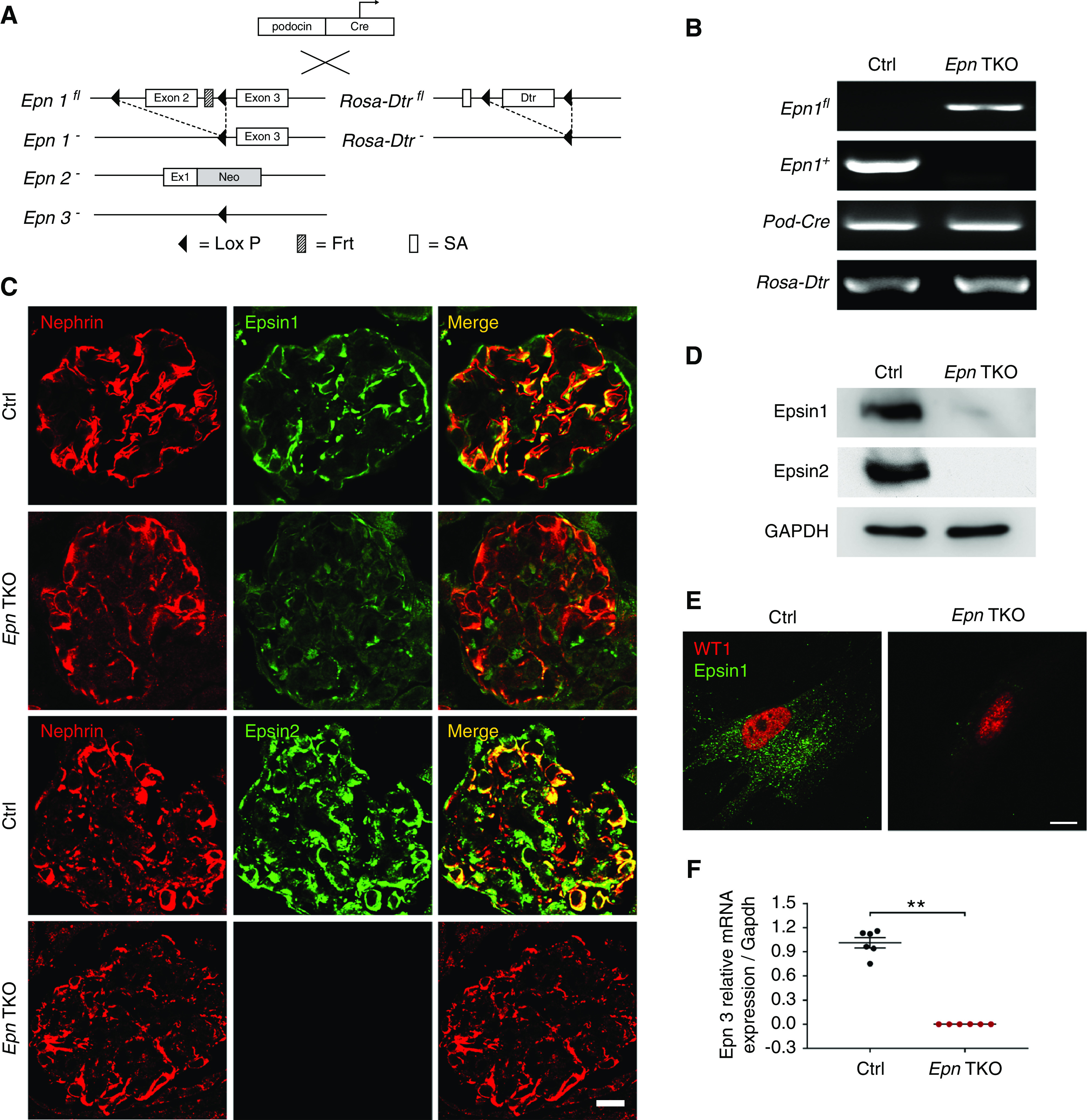
Generation of Podocin-Cre, Rosa-Dtrfl/fl, Epn1fl/fl, Epn2−/−, Epn3−/− (Pod-Epn TKO) mice. (A) Schematic representation of the mating scheme in Podocin-Cre, Rosa-Dtrfl/fl, Epn1fl/fl, Epn2−/−, Epn3−/− (Pod-Epn TKO) mice. (B) Identification of Epn1flox, Epn1+, Podocin-Cre, and Rosa-Dtrfl/fl by tail genotyping. (C) Immunofluorescence images of epsin 1 (green), epsin 2 (green), and nephrin (red) in control (Ctrl) and Pod-Epn TKO mice glomeruli. (D) Immunoblot images of epsin 1, epsin 2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in Ctrl and Pod-Epn TKO mice primary podocytes. (E) Immunofluorescence images of epsin 1 (green) and WT1 (red) from enriched primary podocytes in control and Pod-Epn TKO mice. (F) Epn3 mRNA expression levels in control (black) and Pod-Epn TKO mice primary podocytes (red). n=6. **P<0.01. Scale bar, 10 μm.
The Epn2; Epn3; Pod-Epn1 TKO (Pod-Epn TKO) mice exhibited Mendelian frequencies of inheritance and had no overt phenotype at birth. As the mice reached 6 months of age, they presented with severe albuminuria, which further progressed at 12 months of age, as examined qualitatively by Coomassie blue staining (Figure 2A). Quantitative analysis of urine ACR by ELISA validated that modest albuminuria was observed as early as a month after birth, and it progressively increased by 12 months of age (Figure 2B). The Pod-Epn TKO mice also displayed kidney failure, demonstrated by elevated plasma creatinine by 6 months of age, which further progressed by 12 months of age (Figure 2E), leading to death due to ESKD in 20% of mutant mice (data not shown). Histologic examinations of Pod-Epn TKO mice kidneys by hematoxylin and eosin, PAS, and TRI demonstrated normal features at 1 month of age. However, increased mesangial expansion, along with modest to severe glomerulosclerosis, were observed at both 6 and 12 months of age (Figure 2C and quantified in Figure 2F). Moreover, modest interstitial fibrosis was observed in 6-month-old Pod-Epn TKO mice kidneys by TRI staining, which progressed to severe interstitial scarring, tubular dilation, and glomerular sclerosis at 12 months of age (Figure 2D and quantified in Figure 2G).
Figure 2.
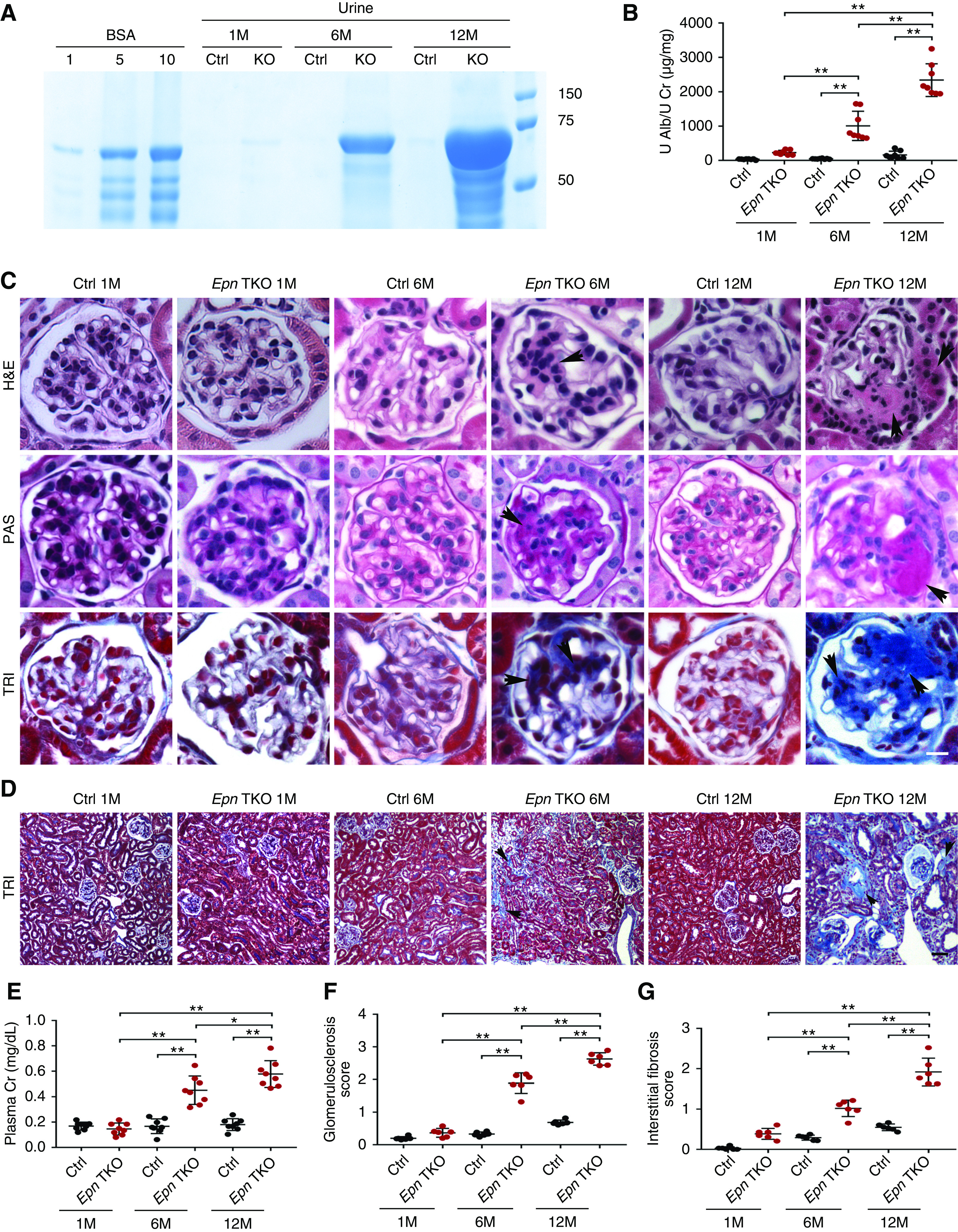
Pod-Epn TKO mice demonstrate albuminuria, glomerulosclerosis, and kidney failure. (A) SDS-PAGE (Coomassie blue staining) of standard BSA (numbers indicate μg) and urine from control and Pod-Epn TKO mice at 1, 6, and 12 months of age. (B) Quantification of urine albumin normalized to creatinine in control (black) and Pod-Epn TKO mice (red) at 1, 6, and 12 months of age. n=8. **P<0.01. (C) Representative light microscopic images (hematoxylin and eosin [H&E], PAS, and TRI) of a single glomerulus from control and Pod-Epn TKO mice at 1, 6, and 12 months of age. Arrowheads show mesangial matrix accumulation in H&E and PAS stain, and glomerulosclerosis in TRI stain. Scale bar, 10 μm. (D) Representative TRI-staining images of kidney interstitium from control and Pod-Epn TKO mice at 1, 6, and 12 months of age. Arrowheads display interstitial fibrosis. Scale bar, 50 μm. (E) Plasma creatinine in control (black) and Pod-Epn TKO (red) mice at 1, 6, and 12 months of age. n=8. (F) Quantification of glomerulosclerosis in (C). n=6 different mice. (G) Quantification of interstitial fibrosis in (D). n=6 different mice. **P<0.01. Alb, albumin; Cr, urinary creatinine; Ctrl, control; U, urinary.
Electron Microscopic Examination Reveals Podocyte Foot Process Effacement and Thickened GBM in Pod-Epn TKO Mice
After detection of severe albuminuria and glomerulosclerosis in the Pod-Epn TKO mice, we analyzed the effect of these genetic KOs on the podocyte ultrastructure by TEM. At 1 month of age, the Pod-Epn TKO mice showed relatively normal podocyte foot process architecture. But, at 6 months of age, in comparison with age-matched controls, the Pod-Epn TKO mice exhibited podocyte foot process effacement, which progressed to generalized podocyte foot process effacement by 12 months of age. (Figure 3A and quantified in Figure 3B). The thickening of the GBM was also observed by 6 months of age in the Pod-Epn TKO mice, which became more striking at 12 months of age (Figure 3A and quantified in Figure 3C).
Figure 3.

Pod-Epn TKO mice kidneys demonstrate podocyte foot process effacement and thickened GBM. (A) Transmission electron micrographs in control and Pod-Epn mice foot process at 1, 6, and 12 months of age. The white and black asterisks depict foot process effacement and thickened GBM, respectively. Scale bar, 2 μm. (B) Quantification of the number of foot processes per micrometer of GBM in control (black) and Pod-Epn TKO mice (red) at 1, 6, and 12 months of age. n=3 different mice. (C) Quantification of GBM thickness in control (black) and Pod-Epn TKO mice (red) at 1, 6, and 12 months of age. n=3 different mice. *P<0.05; **P<0.01.
Pod-Epn TKO Podocytes Show Reduced Cdc42 Activity
Deletion of Epn1, -2, and -3 in primary mouse embryonic fibroblasts has been shown to impair endocytosis and result in increased clathrin density.13 To determine whether podocytes that lack all three Epns also demonstrate accumulation of clathrin-coated pits, primary podocytes were immunostained with CLC and α-adaptin. Surprisingly, when stained with CLC and α-adaptin, Pod-Epn TKO podocytes did not reveal an obvious accumulation of these endocytic proteins (Supplemental Figure 1, A and C, quantified in Supplemental Figure 1, B and D, respectively).
We next turned our attention to the Epsin’s conserved N-terminal phosphatidylinositol 4,5-bisphosphate–binding ENTH domain that has been shown to regulate Cdc42/Rac1 activity through its interactions with a GAP termed RalBP1/RLIP76.24–26 The regulatory role of epsin’s interactions with GAP on Cdc42/Rac1 activity is thought to be conserved in higher eukaryotes, but has not been confirmed yet in podocytes. Thereafter, we investigated whether loss of epsins would alter the activities of the three well-studied Rho GTPases (Cdc42, Rac1, and RhoA). Primary podocytes isolated from Pod-Epn TKO mice exhibited a striking reduction in Cdc42 activity, as measured by pull-down assay with p21-activated protein kinase coupled to agarose beads (Figure 4A and quantified in Figure 4D). Interestingly, no overt differences in Rac1 (Figure 4B and quantified in Figure 4E) or RhoA activity (Figure 4C and quantified in Figure 4F) was observed. It is worth mentioning that podocyte-specific loss of Cdc42 has been shown to result in severe proteinuria.41–43 In addition, Cdc42 critically regulates cell morphology, specifically filopodia formation, via the actin cytoskeleton.29,44–46 Thus, we next explored if the reduction in Cdc42 activity in Pod-Epn TKO podocytes results in reduced filopodial formation. To assess changes in filopodial formation, primary podocytes isolated from control and Pod-Epn TKO mice were immunostained with fascin1, the major actin-binding protein highly expressed in filopodia.47,48 Unexpectedly, quantitative analysis of fascin1 staining in Pod-Epn TKO podocytes did not show significant reduction in filopodia formation when compared with control conditions (Figure 4G and quantified in Figure 4H).
Figure 4.
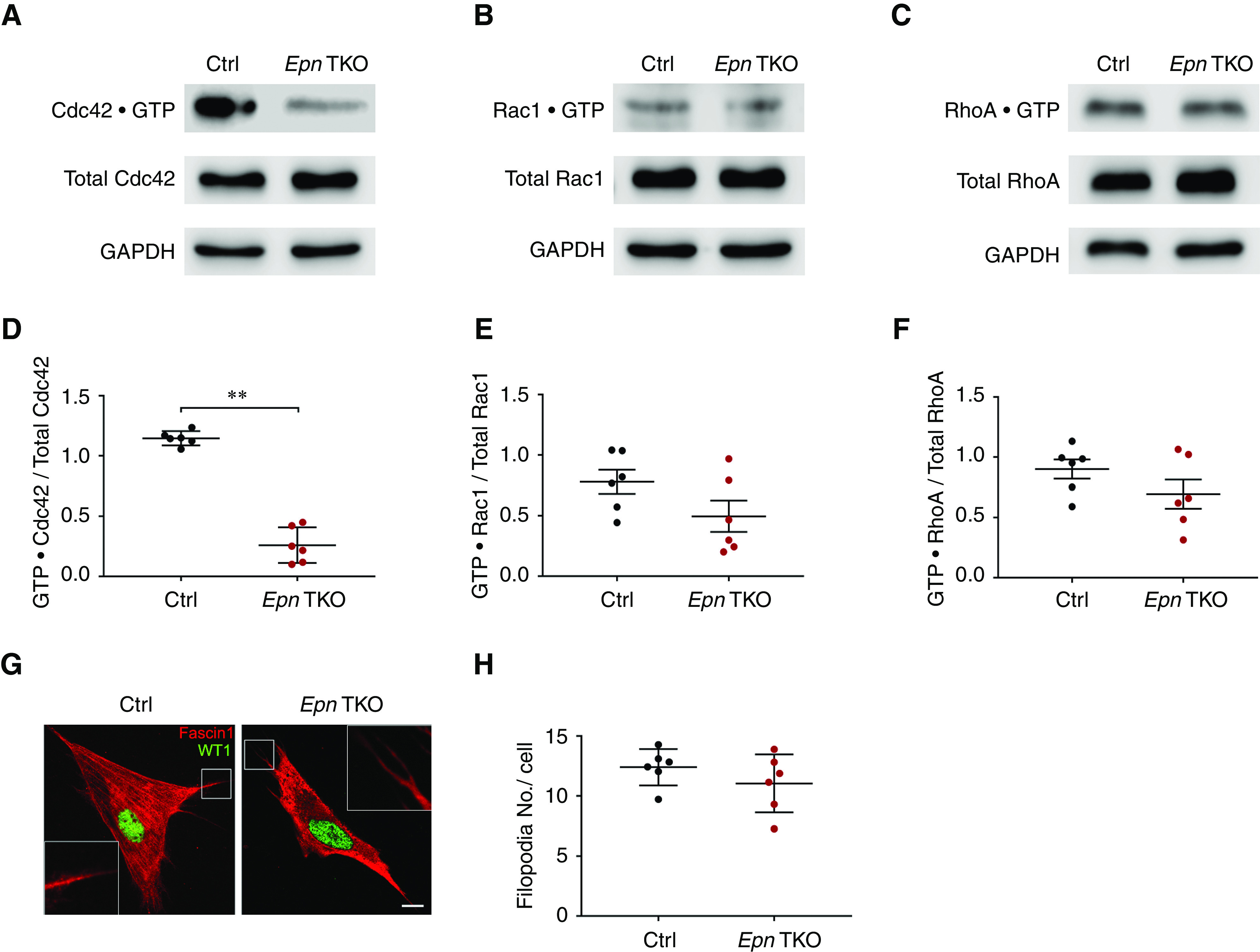
Pod-Epn TKO podocytes demonstrate reduced Cdc42 activity. (A) Representative immunoblot images of GTP·Cdc42 and total Cdc42 in control (Ctrl) and Pod-Epn TKO primary podocytes. (B) Representative immunoblot images of GTP·Rac1 and total Rac1 in Ctrl and Pod-Epn TKO primary podocytes. (C) Representative immunoblot images of GTP·RhoA and total RhoA in Ctrl and Pod-Epn TKO primary podocytes. (D) Quantification of immunoblots in (A). n=6. **P<0.01. (E) Quantification of immunoblots in (B). n=6. (F) Quantification of immunoblots in (C). n=6. (G) Representative immunofluorescence images of fascin1 (red) and WT1 (green) in Ctrl and Pod-Epn TKO primary podocytes. Insets show the boxed regions at high magnification. Scale bar, 10 μm. (H) Quantification of the number of filopodia in (G). n=6. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Pod-Epn TKO Podocytes Have Reduced Cell Adhesion and Cell Spreading, Accompanied with Diminished β1 Integrin Expression and SRF Activity
The Rho family of GTPases play a central role in regulating the organization of cytoskeletal filaments, and cooperate to promote cell movement.49,50 Because Cdc42 has also been shown to regulate cell–extracellular matrix (ECM) adhesion and migration,29,51 we next examined how the loss of epsins in podocytes would affect cell adhesion and spreading by seeding podocytes on both laminin and type-1 collagen–coated cell-culture plates. We observed impairment in Pod-Epn TKO podocyte adhesion to both laminin and collagen type 1 substrates, as compared with control conditions, assessed by crystal violet (Figure 5A). To confirm the reduced adhesion of Pod-Epn TKO podocytes resulted in their detachment from the GBM, we counted podocyte numbers on kidney sections from control and Pod-Epn TKO mice. Immunofluorescence staining with the podocyte-specific transcription factor WT1 revealed fewer podocytes on Pod-Epn TKO mice kidney sections compared with the control condition (Figure 5B and quantified in Figure 5C). In addition, cell migration, as measured by a wound-healing assay, was also impaired in Pod-Epn TKO–enriched podocytes as compared with control conditions (Figure 5D). Similarly, a reduction in cell spreading was observed in Pod-Epn TKO podocytes (Figure 5E and quantified in Figure 5F). To gain further insight into the molecular basis of the defects seen in Pod-Epn TKO podocytes, we next examined β1 integrin expression levels in Pod-Epn TKO podocytes. As a subunit of a large family of transmembrane receptors, β1 integrin plays a critical role in the interaction between cells and the underlying ECM.52 Primary podocytes isolated from Pod-Epn TKO mice demonstrated a remarkable reduction in β1 integrin expression compared with control, as analyzed by Western blotting (Figure 5G and quantified in Figure 5H). Because protein expression of β1 integrin was dramatically decreased, we investigated whether Cdc42 was regulating the expression of β1 integrin at a transcriptional level in the Pod-Epn TKO primary podocytes. As expected, the level of Itgb1 mRNA isolated from Pod-Epn TKO podocytes was markedly reduced compared with control podocytes (Figure 5I). It has been reported that the SRF/megakaryoblastic leukemia 1 transcription factor complex is capable of regulating β1 integrin transcription.51,53,54 To test this hypothesis, SRF protein expression level was measured by Western blot, whereas SRF activity was quantified via a luciferase reporter assay after transient expression in both control and Pod-Epn TKO primary podocytes. Compared with controls, Pod-Epn TKO podocytes displayed normal SRF protein expression (Figure 5J and quantified in Figure 5K), but a striking reduction in SRF activity (Figure 5L). To further elucidate whether SRF regulates β1 integrin expression by binding to Itgb1 promoter, we next performed a ChIP–qPCR assay using an Ab for SRF and a primer set for the Itgb1 promoter. We observed reduced binding of SRF to the Itgb1 promoter in the Pod-Epn TKO podocytes compared with control conditions (Figure 5M), suggesting the diminished β1 integrin expression in Pod-Epn TKO podocytes is partially due to reduced SRF activity. To next determine whether Cdc42 activation can regulate SRF activity, we pharmacologically inhibited Cdc42 by using ML 141. After validation that ML 141 blocked Cdc42 activity in enriched primary podocytes (Figure 5N), we also observed reduced SRF activity, suggesting Cdc42 is acting upstream of SRF (Figure 5O).
Figure 5.
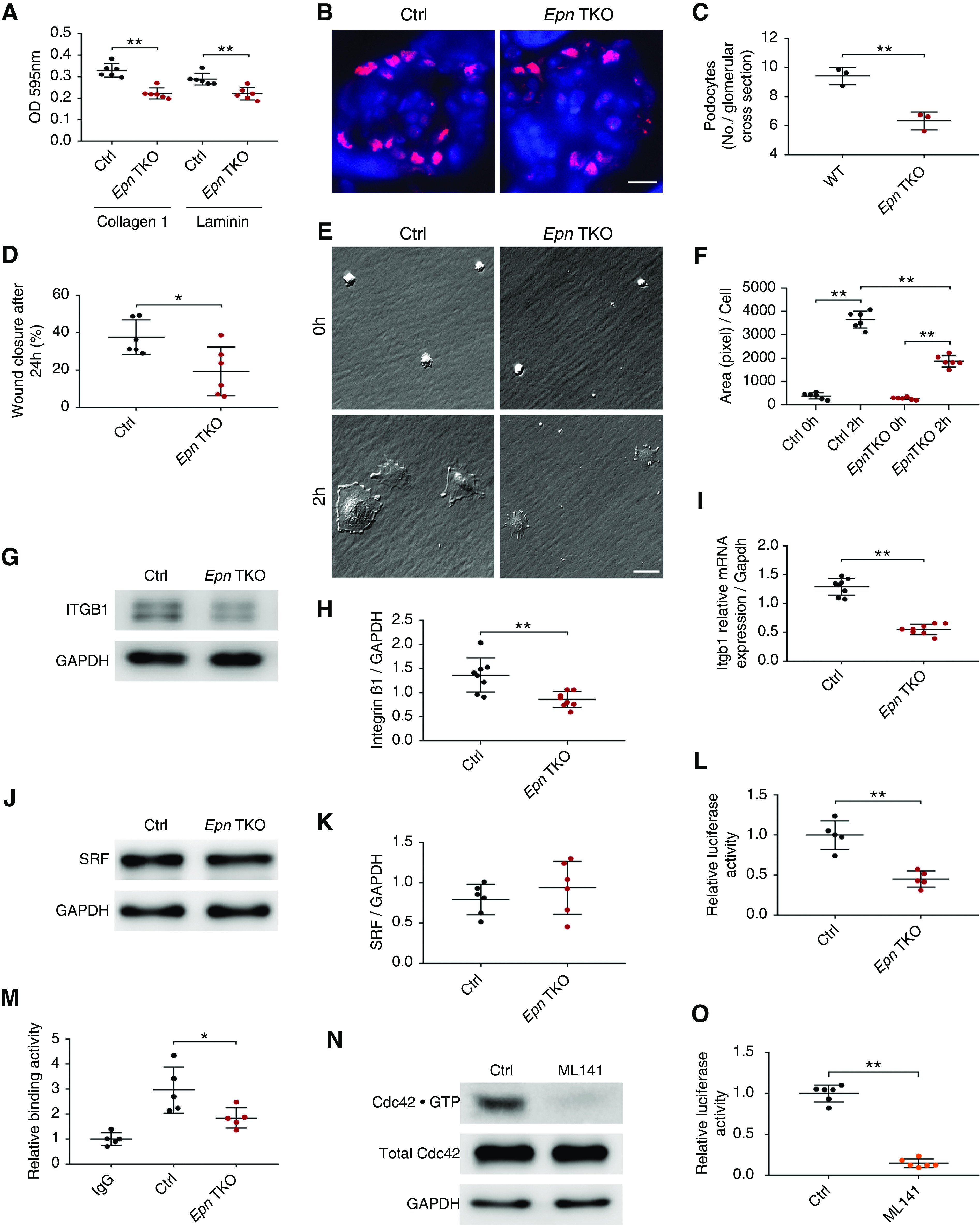
Pod-Epn TKO podocytes demonstrate reduced cell adhesion, migration, spreading, and integrin β1 expression. (A) Adhesion to type-1 collagen and laminin in control (Ctrl) (black) and Pod-Epn TKO (red) primary podocytes. n=6. (B) Representative immunofluorescence images of WT1 (red) and 4′,6-diamidino-2-phenylindole (blue) in Ctrl and Pod-Epn TKO mice glomeruli. Scale bar, 10 μm. (C) Quantification of the number of WT1 stained nucleus in (B). n=3 different mice. (D) Quantification of wound-healing assay in Ctrl (black) and Pod-Epn TKO (red) mice primary podocytes. n=6. (E) Representative images of cell spreading in Ctrl and Pod-Epn TKO mice primary podocytes. Scale bar, 20 μm. (F) Quantification of podocyte cell surface area in (E). n=6. (G) Representative immunoblot images for integrin β1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in Ctrl and Pod-Epn TKO primary podocytes. (H) Quantification of immunoblots in (G). n=8. (I) Igtb1 mRNA expression levels in Ctrl (black) and Pod-Epn TKO (red) primary podocytes. n=8. (J) Representative immunoblot images for SRF and GAPDH in Ctrl and Pod-Epn TKO primary podocytes. (K) Quantification of immunoblots in (J). n=6. (L) Quantification of relative SRF activity using luciferase reporter assay in control (black) and Pod-Epn TKO (red) primary podocytes. n=5. (M) Quantification of ChIP-qPCR using SRF Ab and primer sets of Itgb1 promoter region in Ctrl (black) and Pod-Epn TKO (red) primary podocytes with negative control as IgG. n=5. (N) Cdc42 activity in primary podocytes examined with or without ML 141 pretreatment. (O) Quantification of relative SRF activity using luciferase reporter assay in primary podocytes treated with or without ML 141. n=6. *P<0.05, **P<0.01.
Pod-Srf KO Mice Display Albuminuria, Interstitial Fibrosis, and Kidney Failure
The Pod-Epn TKO mice exhibit profound defects in their ability to maintain a working glomerular filtration barrier. The reduced β1 integrin expression in these mice, as mediated by Cdc42 and SRF, prompted us to examine the effects of SRF loss on podocyte function and overall filtration-barrier maintenance. Srf-floxed mice were mated with podocin-Cre and Rosa-Dtrfl/fl transgenic mice to generate podocyte-specific Srf KO (Pod-Srf KO) mice (Supplemental Figure 2A). Confirmation of the mutant mice was verified via PCR-based tail genotyping of the Srf, Podocin-Cre, and the Rosa-Dtrfl/fl transgene (Supplemental Figure 2B). Deletion of podocyte Srf was confirmed by immunoblot in primary podocytes and immunofluorescence in both kidney sections and primary podocytes (Supplemental Figure 2, C–E). These mice developed albuminuria by 6 weeks of age, which progressed to severe albuminuria by 12 weeks of age. Quantitative analysis of urine by ELISA validated that albuminuria started as early as 3 weeks of age, and increased progressively to 12 weeks of age. Furthermore, these mutant mice displayed kidney failure with elevated plasma creatinine and BUN at 12 weeks of age (Figure 6, B and C). Although the glomeruli appeared normal at birth (data not shown), histologic examination of Pod-Srf KO mice kidneys by PAS revealed an onset of severe glomerulosclerosis by 12 weeks of age (Figure 6D and quantified in Figure 6G). Low-power microscopic examination of the renal interstitium by TRI showed evidence of interstitial fibrosis, tubular dilation, and proteinaceous casts starting at 6 weeks of age and progressing till 12 weeks of age. (Figure 6E and quantified in Figure 6H). Ultrastructural examination of Pod-Srf KO mice by TEM revealed severe foot process effacement at 6 weeks of age, which further progressed by 12 weeks of age (Figure 6F and quantified in Figure 6I).
Figure 6.
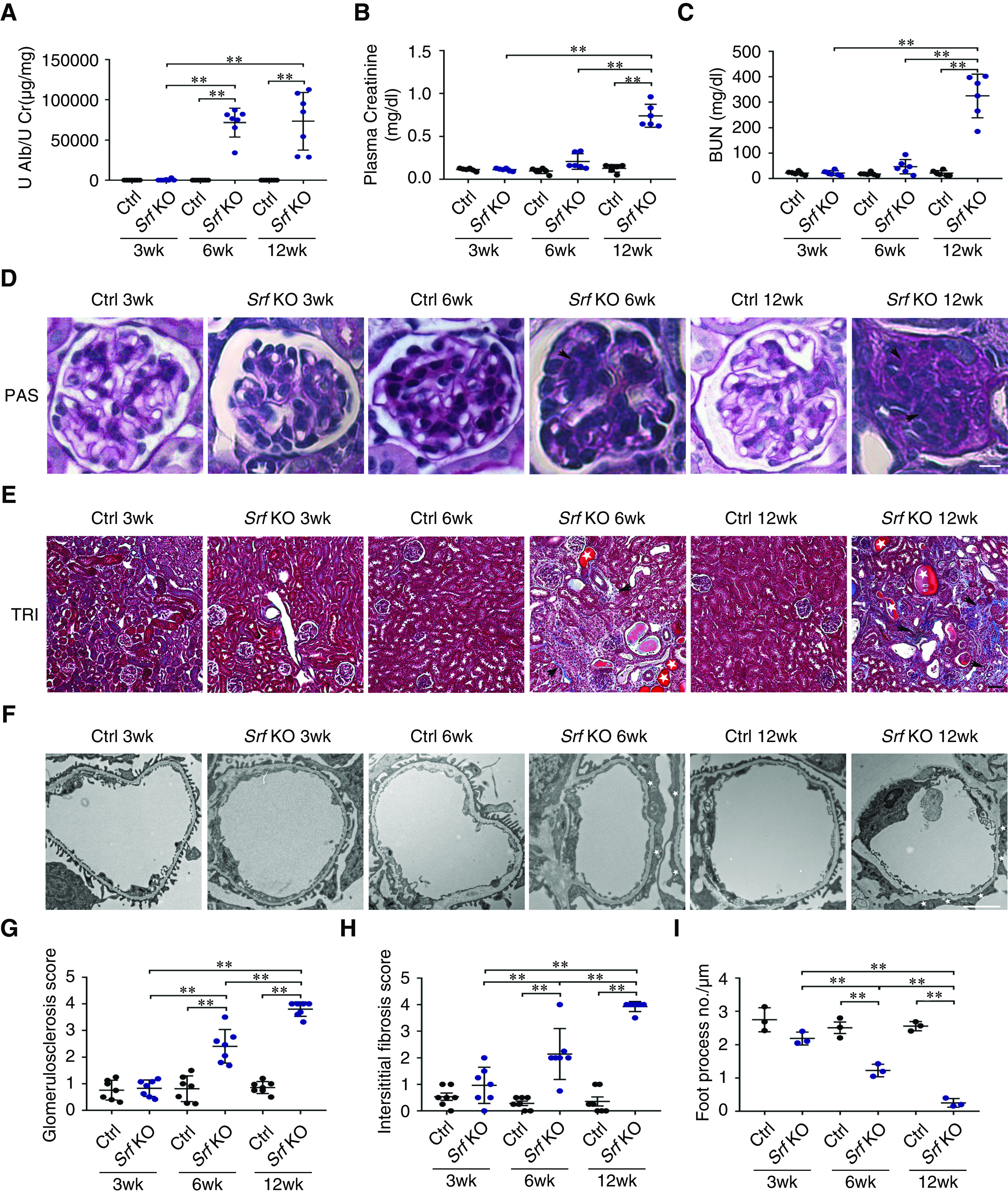
Podocyte-specific deletion of Srf results in proteinuria, glomerulosclerosis, and kidney failure. (A) Quantification of urine albumin normalized to urine creatinine in control (Ctrl) (black) and Pod-Srf KO (blue) mice at 3, 6, and 12 weeks of age. n=7. (B) Plasma creatinine in Ctrl (black) and Pod-Srf KO (blue) mice at 3, 6, and 12 weeks of age. n=6. (C) Plasma BUN in Ctrl (black) and Pod-Srf KO (blue) mice at 3, 6, and 12 weeks of age. n=6. (D) Representative light microscopic images of PAS staining in Ctrl and Pod-Srf KO mice glomeruli at 3, 6, and 12 weeks of age. Arrowheads (black) show mesangial matrix accumulation. Scale bar, 25 μm. (E) Representative images of TRI staining in Ctrl and Pod-Srf KO mice kidney interstitium at 3, 6, and 12 weeks of age. Arrowheads (black) display interstitial fibrosis, and asterisk (white) represents dilated tubules and proteinaceous casts. Scale bar, 50 μm. (F) Transmission electron micrographs in Ctrl and Pod-Srf KO mice foot process at 3, 6, and 12 weeks of age. Asterisk (white) depict foot process effacement. Scale bar, 2 μm. (G) Quantification of glomerulosclerosis in (D). n=7 different mice. (H) Quantification of interstitial fibrosis in (E). n=7 different mice. (I) Quantification of the number of foot processes per micrometer of GBM in Ctrl (black) and Pod-Srf KO (blue) mice at 3, 6, and 12 weeks of age. n=3 different mice. **P<0.01. Alb, albumin; Cr, creatinine; U, urinary.
Pod-Srf KO Podocytes Exhibit Reduced Cell Adhesion, Spreading, and Migration, Accompanied with Diminished β1 Integrin Expression
Because the phenotype of the Pod-Srf KO mice was much more severe than the Pod-Epn TKO mice, we speculated the Pod-Srf KO podocytes would present with similar morphologic defects in cell adhesion, spreading, and migration, along with reduced β1 integrin expression. To investigate this hypothesis, primary podocytes isolated from control, Pod-Epn TKO, and Pod-Srf KO mice were seeded on laminin and type-1 collagen–coated cell-culture plates. We observed significant impairment in podocyte adhesion to both laminin and collagen type 1 substrates in Pod-Srf KO podocytes when compared with control and with Pod-Epn TKO purified primary podocytes (Figure 7A). Meanwhile, immunofluorescence staining with WT1 (a podocyte-specific transcription factor) revealed fewer podocytes in Pod-Srf KO mice kidney sections compared with the control conditions (Figure 7B and quantified in Figure 7C). Furthermore, we noted reduced cell spreading in Pod-Srf KO podocytes when compared with control and with Pod-Epn TKO primary podocytes (Figure 7D and quantified in Figure 7E). Cell migration, as measured by a wound-healing assay, was considerably reduced in Pod-Srf KO podocytes compared with both control and Pod-Epn TKO podocytes (Figure 7F). Additionally, Pod-Srf KO podocytes showed a decrease in β1 integrin protein expression compared with both control and Pod-Epn TKO primary podocytes (Figure 7H and quantified in Figure 7I). Immunostaining for β1 integrin recapitulated these findings: Pod-Srf KO podocytes and Pod-Srf KO glomeruli showed a more pronounced reduction in β1 integrin expression when compared with Pod-Epn TKO and control podocytes and glomeruli (Figure 7G). To further clarify the link between epsins and SRF, we examined the predicted SRF target genes with the published mouse podocyte mRNA expression database.55 We selected 15 of the most highly expressed genes in the podocyte mRNA expression database that were also identified as SRF target genes. By performing qPCR, we observed reductions in mRNA expression of Myl6, Tpm4, Ier2, Fos, Hs3st6, Cd55, Ube2s, Tspan15, Metrnl, and Pdk2 in both Pod-Epn TKO podocytes and Pod-Srf KO podocytes (Supplemental Figure 3; primers for those genes were presented in Supplemental Table 1).
Figure 7.
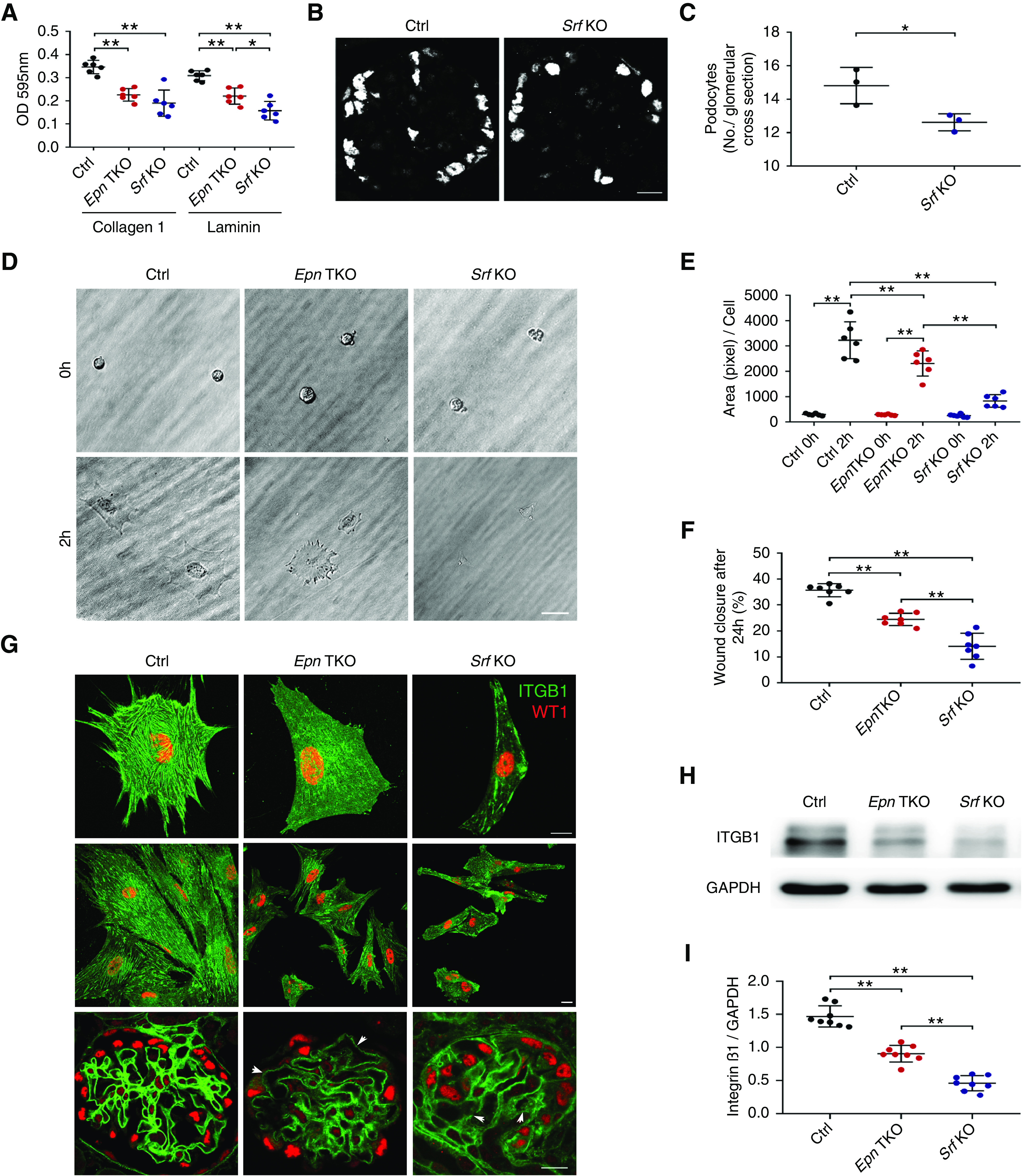
Pod Srf KO podocytes demonstrate reduced cell adhesion, spreading, and migration accompanied with diminished β1 integrin expression. (A) Adhesion to type-1 collagen and laminin in control (Ctrl) (black), Pod-Epn TKO (red), and Pod-Srf KO (blue) primary podocytes. *P<0.05, **P<0.01, compared with Ctrl and Pod-Epn TKO. n=6. (B) Representative immunofluorescence images of WT1 in Ctrl and Pod-Srf KO mice glomeruli at 6 weeks of age. Scale bar, 10 μm. (C) Quantification of the number of WT1 stained nucleus (B). n=3 different mice. (D) Representative images of cell spreading in Ctrl, Pod-Epn TKO, and Pod-Srf KO mice primary podocytes. Scale bar, 20 μm. (E) Quantification of podocyte cell surface area in (D). n=6. (F) Quantification of wound-healing assay in Ctrl (black), Pod-Epn TKO (red), and Pod-Srf KO (blue) mice primary podocytes. n=7. (G) Representative immunofluorescence images of integrin β1 (green) and WT1 (red) in Ctrl, Pod-Epn TKO, and Pod-Srf KO primary podocytes and glomeruli. (H) Representative immunoblot images of integrin β1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in Ctrl, Pod-Epn TKO, and Pod-Srf KO primary podocytes. (I) Quantification of immunoblots in (H). n=8. *P<0.05, **P<0.01.
Discussion
Three Epn genes (Epn1, -2, and -3) with overlapping functions have been recognized in mammalian cells.31,32,56 They appear to be critical to development and survival, because simultaneous inactivation of both Epn1 and Epn2 results in embryonic lethality.16 In this investigation, we generated mice lacking constitutive expression of all three epsins in podocytes, resulting in severe proteinuria and foot process effacement.
The most highly conserved domain of epsins is the ENTH domain. Besides its well-studied role in triggering and promoting membrane curvature to initiate endocytosis,22,23,25,57,58 the ENTH domain also exerts a remarkable function on actin and signaling regulation.12,24,59 Epsin’s role in signaling activation has been linked to the Notch developmental pathway16,60,61 and Rho-GTPase signaling.24,26,62
Unexpectedly, we did not observe significant differences in clathrin accumulation or α-adaptin staining patterns in Pod-Epn TKO podocytes, as found in our Pod-Dnm1/2, synaptojanin1, and endophilin TKO podocytes.7 Although epsins contain clathrin- and AP2-binding aspartate, proline, and tryptophan motifs, in podocytes, epsins may function differently by playing a more central role, mechanistically, by cell signaling. This is corroborated by the absence of epsin 1, 2, and 3 in clathrin-coated vesicles of podocytes on mass spectrometry.63 Thus, we focused on examining the small Rho-GTPase signaling pathway, because it has been shown previously that mammalian epsin binds RalBP1/RLIP76, a GAP that acts on Cdc42 and Rac1 through epsin’s ENTH domain.24,26,62 In addition, the podocyte-associated Cdc42 has been shown to induce severe albuminuria in mice.41,42 We hypothesize that loss of epsins would release RalBP1 from binding to the ENTH domain, thereby enhancing their suppressive function when bound to the Rho GTPases, inducing dephosphorylation from the GTP to the GDP state, abrogating Rho-GTPase activity. Our results showed that, among the three Rho GTPases, only Cdc42 activity was dramatically reduced in the Pod-Epn TKO primary podocytes compared with the control condition, which corresponded to our prediction that Cdc42 activation would decrease once the interaction between epsin’s ENTH domain and Cdc42 GAPs was interrupted. Consistent with our findings, an epsin-2 deletion has been shown to markedly decrease the activity of Cdc42 in oocytes.62 Yet it is unclear in our study whether epsin’s ENTH domain interacts with the GAPs for Cdc42 primarily or exclusively in the podocytes. The interaction of GAPs with Rac1 or RhoA is still a possibility, but our data showed that loss of epsins did not alter the activation of Rac1 and RhoA. Our Pod-Epn TKO mice exhibited a milder phenotype than the podocyte-specific loss of Cdc42, most likely because our mutant mice only showed reduced Cdc42 activity but normal expression levels when compared with littermate controls. We assessed filopodia formation, cell adhesion, and cell migration in both Pod-Epn TKO and control podocytes. Our results indicated Pod-Epn TKO podocytes displayed reduced cellular adhesion and migration, phenotypes that were consistent with decreased Cdc42 activity. However, there was no significant difference in filopodia formation between Pod-Epn TKO and control primary podocytes. We speculate the negative results may be due to two reasons: one possibility is that the podocyte fixation procedures destroyed the thin structure of the filopodia, and the second is that the highly dynamic characteristics of filopodia make it difficult to analyze at a single time point.
Podocyte-ECM interactions play an important role in cellular adhesion and spreading, events that are mainly mediated by a large family of transmembrane receptors called integrins.52,64 The most abundant subunit among this family is β1 integrin, which causes severe proteinuria when deleted in podocytes.65 Our data implies that, in Pod-Epn TKO podocytes, β1 integrin expression is significantly reduced at both transcriptional and protein levels. Lower β1 integrin expression levels likely contribute to the reduction in cell spreading and adhesion observed in our Pod-Epn TKO primary podocytes, which likely explains the decreased number of podocytes observed in our Pod-Epn TKO mice glomeruli.
Additionally, the activity of the transcription factor SRF was also reduced in the Pod-Epn TKO primary podocytes. We also observed less SRF binding to the Itgb1 promoter compared with control podocytes. Cdc42 can transcriptionally regulate β1 integrin expression via SRF activation in podocytes, which is consistent with recently published data showing that Cdc42 can transcriptionally regulate β1 integrin expression through SRF activation in different cancer cells.51,53,54 However, the exact mechanism of Cdc42-mediated SRF activation is not perspicuous. Although it has been shown that Cdc42 can affect SRF activity by regulating actin polymerization,66 we observed no differences in the F-/G-actin ratios between Pod-Epn TKO podocytes and control podocytes (Supplemental Figure 4A; quantified in Supplemental Figure 4B). Additionally, although RhoA has also been reported to stimulate SRF activation,66,67 our results show that RhoA activation is not affected in the Pod-Epn TKO podocytes.
To further investigate the role of SRF in podocytes, we generated podocyte-specific Srf KO (Pod-Srf KO) mice, which exhibited severe albuminuria and kidney failure at 12 weeks of age, along with severe glomerulosclerosis and interstitial fibrosis, validating a phenotype of Pod-Srf KO mice that has been previously reported.68 As we expected, the podocytes isolated from Pod-Srf KO mice displayed considerably diminished β1 integrin expression, with an accompanying reduction in cell adhesion, spreading, and migration when compared with both Pod-Epn TKO and control primary podocytes. The severely nephrotic phenotype of Pod-Srf KO mice, and the abnormalities visualized in Pod-Srf KO primary podocytes, suggest that Cdc42-mediated regulation of β1 integrin expression via SRF activity is partly responsible for the underlying pathogenic/pathologic phenotypes observed in our Pod-Epn TKO mice (Figure 8). Because there was a modest reduction in the SRF promoter activity, compared with the degree of reduction of β1 integrin expression, other factors may also be regulating transcription of β1 integrin. It has been shown in human tumors that suppressor of cancer cell invasion serves as a cofactor that can bind the SRF cofactor myocardin-related transcription factor to inhibit Itgb1 expression.53 The ability of SRF to interact with >60 cofactors regulating gene transcription may partially explain the conundrum of why SRF activity is reduced, but its expression level is not, in the Pod-Epn TKO podocytes. Furthermore, B1-adjacent long non-coding RNA (BLNCR) and N-Myc epxression may also play a potential role in pod-Epn TKO podocytes to regulate β1 integrin expression, independent of SRF activity.69,70 These findings motivate future studies to understand Itgb1 transcriptional regulation. Although differences exist in the precise timing of the podocyte dysfunction between these mutant mice, the pathology appears similar. Compared with the Pod-Epn TKO mice, Pod-Srf KO mice have a worse phenotype because the total ablation of Srf may differ from just partially losing its activity, which was observed in the Pod-Epn TKO mice. Moreover, one can speculate that the total loss of Srf may result in the loss of other critical podocyte genes under the regulation of this transcription factor, and the loss of interactions with its known binding partners. To further verify the upstream role of Epn in relation to Srf, we examined the mRNA expression levels of 15 podocyte-enriched SRF targets genes in both Pod-Epn TKO and Pod-Srf KO primary podocytes. Within those genes, Myl6 and Tpm4 have been implicated in regulating the actin cytoskeletal organization, whereas Ier2 and Tpm4 have been shown to be critical for cell adhesion. Interestingly, Ier2 has been shown to regulate integrin β1–mediated signaling pathways.71 Conversely, FosB was not reduced in the Pod-Srf KO podocytes, suggesting regulation may potentially be independent of SRF, because cAMP response element–binding protein can also regulate FosB expression.72 Hence, further exploration would be of interest in understanding the relevance of these differentially expressed genes in the future. Lastly, the importance of EPN and SRF in human proteinuric kidney diseases was examined. In Nephroseq databases, microarrays from glomerular samples of patients with FSGS and those with nephrotic syndrome suggest EPN1 and SRF expression are reduced with worsening proteinuria, whereas EPN2 expression is reduced with declining eGFR in diabetic kidney disease glomerular samples (data not shown). More studies will be needed to validate these associations.
Figure 8.

Cartoon schematic showing that loss of epsins in podocytes results in impaired Cdc42 activation and reduced integrin β1 expression through reduced SRF binding to the Itgb1 promoter. (A) Cartoon schematic of normal podocyte with Epn regulating CDC42 activation to induce Srf regulated Itgb1 expression. (B) Loss of Epn results in impaired CDC42 activation resulting in reduced Itgb1 expression resulting in podocyte loss.
In conclusion, our study supports the functional importance of podocyte-resident epsins in maintaining the integrity of the glomerular filtration barrier, which, when compromised, results in severe proteinuria and kidney failure that is partially due to the reduction of SRF activity.
Disclosures
C. Pedigo is currently working at Angion Biomedica, a late-stage pharmaceutical company. All work was done during his tenure at Yale and has no relevance to his current position. All remaining authors have nothing to disclose.
Funding
This work was supported in part by George O’Brien Kidney Center at Yale School of Medicine grant P30DK078310; National Institutes of Health grants DK083294 (to S. Ishibe), DK093629 (to S. Ishibe), and DK118863 (to C. Pedigo); China Scholarship Council grant 201606555023 (to Y. Wang); and U.S. Department of Defense grants W81XWH-17-1-0662 (to S. Ishibe).
Supplementary Material
Acknowledgments
We would like to thank Dr. Mirko Messa for providing advice for the epsin 1 and epsin 2 Abs used in this manuscript and Dr. Pietro De Camilli for the mice used in the study.
Dr. Ying Wang, Dr. Christopher E. Pedigo, Dr. Kazunori Inoue, Dr. Xuefei Tian, Dr. Elizabeth Cross, Dr. Karen Ebenezer, Dr. Wei Li, Dr. Eike Schwartze, Dr. Zhen Wang, and Dr. Marwin Groener performed the research; Dr. Ying Wang, Dr. Kazunori Inoue, Dr. Christopher E. Pedigo, and Dr. Shuta Ishibe designed the research and analyzed the data; Dr. Ying Wang, Dr. Karen Ebenezer, Dr. Wei Li, and Dr. Shuta Ishibe wrote the manuscript; and all authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020050691/-/DCSupplemental.
Supplemental Figure 1. Pod-Epn TKO podocytes does not have clathrin coated pit accumulation distribution.
Supplemental Figure 2. Generation of the Podocyte specific Srf KO mice.
Supplemental Figure 3. Transcriptional levels of genes regulated by Epn and Srf in podocytes.
Supplemental Figure 4. Pod-Epn KO podocytes does not affect F and G actin ratio.
Supplemental Table 1. List of primers used in Supplemental Figure 3.
References
- 1.Nagata M: Podocyte injury and its consequences. Kidney Int 89: 1221–1230, 2016. [DOI] [PubMed] [Google Scholar]
- 2.Schiffer M, Teng B, Gu C, Shchedrina VA, Kasaikina M, Pham VA, et al. : Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat Med 21: 601–609, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen P, Zhang F, Fu Y, Zhu JY, Han Z: Exocyst genes are essential for recycling membrane proteins and maintaining slit diaphragm in Drosophila nephrocytes. J Am Soc Nephrol 31: 1024–1034, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin CE, New LA, Phippen NJ, Keyvani Chahi A, Mitro AE, Takano T, et al. : Multivalent nephrin-Nck interactions define a threshold for clustering and tyrosine-dependent nephrin endocytosis. J Cell Sci 133: jcs236877, 2020. [DOI] [PubMed] [Google Scholar]
- 5.Inoue K, Tian X, Velazquez H, Soda K, Wang Z, Pedigo CE, et al. : Inhibition of endocytosis of clathrin-mediated angiotensin II receptor type 1 in podocytes augments glomerular injury. J Am Soc Nephrol 30: 2307–2320, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kampf LL, Schneider R, Gerstner L, Thünauer R, Chen M, Helmstädter M, et al. : TBC1D8B mutations implicate RAB11-dependent vesicular trafficking in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol 30: 2338–2353, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al. : Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest 122: 4401–4411, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue K, Ishibe S: Podocyte endocytosis in the regulation of the glomerular filtration barrier. Am J Physiol Renal Physiol 309: F398–F405, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertelsen V, Sak MM, Breen K, Rødland MS, Johannessen LE, Traub LM, et al. : A chimeric pre-ubiquitinated EGF receptor is constitutively endocytosed in a clathrin-dependent, but kinase-independent manner. Traffic 12: 507–520, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Kazazic M, Bertelsen V, Pedersen KW, Vuong TT, Grandal MV, Rødland MS, et al. : Epsin 1 is involved in recruitment of ubiquitinated EGF receptors into clathrin-coated pits. Traffic 10: 235–245, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, et al. : Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci U S A 102: 2760–2765, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen A, Madhivanan K, Mukherjee D, Aguilar RC: The epsin protein family: Coordinators of endocytosis and signaling. Biomol Concepts 3: 117–126, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messa M, Fernández-Busnadiego R, Sun EW, Chen H, Czapla H, Wrasman K, et al. : Epsin deficiency impairs endocytosis by stalling the actin-dependent invagination of endocytic clathrin-coated pits. eLife 3: e03311, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sochacki KA, Dickey AM, Strub MP, Taraska JW: Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat Cell Biol 19: 352–361, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Overstreet E, Fitch E, Fischer JA: Fat facets and liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development 131: 5355–5366, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, et al. : Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci U S A 106: 13838–13843, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langridge PD, Struhl G: Epsin-dependent ligand endocytosis activates Notch by force. Cell 171: 1383–1396.e12, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wendland B: Epsins: Adaptors in endocytosis? Nat Rev Mol Cell Biol 3: 971–977, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Itoh T, Koshiba S, Kigawa T, Kikuchi A, Yokoyama S, Takenawa T: Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science 291: 1047–1051, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Capraro BR, Yoon Y, Cho W, Baumgart T: Curvature sensing by the epsin N-terminal homology domain measured on cylindrical lipid membrane tethers. J Am Chem Soc 132: 1200–1201, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holkar SS, Kamerkar SC, Pucadyil TJ: Spatial control of epsin-induced clathrin assembly by membrane curvature. J Biol Chem 290: 14267–14276, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, et al. : Curvature of clathrin-coated pits driven by epsin. Nature 419: 361–366, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Alai MM, Heidemann J, Skruzny M, Gieras A, Mertens HDT, Svergun DI, et al. : Epsin and Sla2 form assemblies through phospholipid interfaces. Nat Commun 9: 328, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguilar RC, Longhi SA, Shaw JD, Yeh LY, Kim S, Schön A, et al. : Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc Natl Acad Sci U S A 103: 4116–4121, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon BG, Burgner J, Camonis JH, Aguilar RC: The epsin family of endocytic adaptors promotes fibrosarcoma migration and invasion. J Biol Chem 285: 33073–33081, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossé C, L’Hoste S, Offner N, Picard A, Camonis J: RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate epsin during the switch off of endocytosis in mitosis. J Biol Chem 278: 30597–30604, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Barry DM, Xu K, Meadows SM, Zheng Y, Norden PR, Davis GE, et al. : Cdc42 is required for cytoskeletal support of endothelial cell adhesion during blood vessel formation in mice. Development 142: 3058–3070, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridley AJ: Life at the leading edge. Cell 145: 1012–1022, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Vega FM, Ridley AJ: SnapShot: Rho family GTPases. Cell 129: 1430, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, et al. : Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 394: 793–797, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Spradling KD, McDaniel AE, Lohi J, Pilcher BK: Epsin 3 is a novel extracellular matrix-induced transcript specific to wounded epithelia. J Biol Chem 276: 29257–29267, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Ko G, Paradise S, Chen H, Graham M, Vecchi M, Bianchi F, et al. : Selective high-level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proc Natl Acad Sci U S A 107: 21511–21516, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, et al. : Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest 122: 4424–4438, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo JK, Shi H, Koraishy F, Marlier A, Ding Z, Shan A, et al. : The Terminator mouse is a diphtheria toxin-receptor knock-in mouse strain for rapid and efficient enrichment of desired cell lineages. Kidney Int 84: 1041–1046, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halene S, Gao Y, Hahn K, Massaro S, Italiano JE Jr., Schulz V, et al. : Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood 116: 1942–1950, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma H, Togawa A, Soda K, Zhang J, Lee S, Ma M, et al. : Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J Am Soc Nephrol 21: 1145–1156, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian X, Kim JJ, Monkley SM, Gotoh N, Nandez R, Soda K, et al. : Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J Clin Invest 124: 1098–1113, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue K, Gan G, Ciarleglio M, Zhang Y, Tian X, Pedigo CE, et al. : Podocyte histone deacetylase activity regulates murine and human glomerular diseases. J Clin Invest 129: 1295–1313, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassan H, Tian X, Inoue K, Chai N, Liu C, Soda K, et al. : Essential role of X-box binding protein-1 during endoplasmic reticulum stress in podocytes. J Am Soc Nephrol 27: 1055–1065, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals : Guide for the Care and Use of Laboratory Animals 8th Ed., Washington (DC): National Academies Press (US), 2011 [PubMed] [Google Scholar]
- 41.Scott RP, Hawley SP, Ruston J, Du J, Brakebusch C, Jones N, et al. : Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J Am Soc Nephrol 23: 1149–1154, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, et al. : Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 84: 920–930, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Z, Zhang L, Chen Y, Zhang H, Zhang Q, Li R, et al. : Cdc42 deficiency induces podocyte apoptosis by inhibiting the Nwasp/stress fibers/YAP pathway. Cell Death Dis 7: e2142, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nobes CD, Hall A: Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62, 1995. [DOI] [PubMed] [Google Scholar]
- 45.Mattila PK, Lappalainen P: Filopodia: Molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9: 446–454, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Govind S, Kozma R, Monfries C, Lim L, Ahmed S: Cdc42Hs facilitates cytoskeletal reorganization and neurite outgrowth by localizing the 58-kD insulin receptor substrate to filamentous actin. J Cell Biol 152: 579–594, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kliewe F, Scharf C, Rogge H, Darm K, Lindenmeyer MT, Amann K, et al. : Studying the role of fascin-1 in mechanically stressed podocytes. Sci Rep 7: 9916, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Machesky LM, Li A: Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol 3: 263–270, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nobes CD, Hall A: Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144: 1235–1244, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaffe AB, Hall A: Rho GTPases: Biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Reymond N, Im JH, Garg R, Vega FM, Borda d’Agua B, Riou P, et al. : Cdc42 promotes transendothelial migration of cancer cells through β1 integrin. J Cell Biol 199: 653–668, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hynes RO: Integrins: Bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Brandt DT, Baarlink C, Kitzing TM, Kremmer E, Ivaska J, Nollau P, et al. : SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of beta1-integrin. Nat Cell Biol 11: 557–568, 2009. [DOI] [PubMed] [Google Scholar]
- 54.Xu Y, Zhang H, Lit LC, Grothey A, Athanasiadou M, Kiritsi M, et al. : The kinase LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin β1. Sci Signal 7: ra58, 2014. [DOI] [PubMed] [Google Scholar]
- 55.Kann M, Ettou S, Jung YL, Lenz MO, Taglienti ME, Park PJ, et al. : Genome-wide analysis of wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J Am Soc Nephrol 26: 2097–2104, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, et al. : The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. J Biol Chem 274: 33959–33965, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Stahelin RV, Long F, Peter BJ, Murray D, De Camilli P, McMahon HT, et al. : Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J Biol Chem 278: 28993–28999, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Kweon DH, Shin YK, Shin JY, Lee JH, Lee JB, Seo JH, et al. : Membrane topology of helix 0 of the Epsin N-terminal homology domain. Mol Cells 21: 428–435, 2006. [PubMed] [Google Scholar]
- 59.De Camilli P, Chen H, Hyman J, Panepucci E, Bateman A, Brunger AT: The ENTH domain. FEBS Lett 513: 11–18, 2002. [DOI] [PubMed] [Google Scholar]
- 60.Fischer JA, Leavell SK, Li Q: Mutagenesis screens for interacting genes reveal three roles for fat facets during Drosophila eye development. Dev Genet 21: 167–174, 1997. [DOI] [PubMed] [Google Scholar]
- 61.Tian X, Hansen D, Schedl T, Skeath JB: Epsin potentiates Notch pathway activity in Drosophila and C. elegans. Development 131: 5807–5815, 2004. [DOI] [PubMed] [Google Scholar]
- 62.Li L, Han L, Zhang J, Liu X, Ma R, Hou X, et al. : Epsin2 promotes polarity establishment and meiotic division through activating Cdc42 in mouse oocyte. Oncotarget 7: 50927–50936, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Groener M, Wang Y, Cross E, Tian X, Ebenezer K, Baik E, et al. : Identification of podocyte cargo proteins by proteomic analysis of clathrin-coated vesicles. Kidney360 1: 480–490, 2020. 10.34067/KID.0000212020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sachs N, Sonnenberg A: Cell-matrix adhesion of podocytes in physiology and disease. Nat Rev Nephrol 9: 200–210, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Pozzi A, Jarad G, Moeckel GW, Coffa S, Zhang X, Gewin L, et al. : Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev Biol 316: 288–301, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson EN, Nordheim A: Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11: 353–365, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill CS, Wynne J, Treisman R: Serum-regulated transcription by serum response factor (SRF): A novel role for the DNA binding domain. EMBO J 13: 5421–5432, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo B, Lyu Q, Slivano OJ, Dirkx R, Christie CK, Czyzyk J, et al. : Serum response factor is essential for maintenance of podocyte structure and function. J Am Soc Nephrol 29: 416–422, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanis SEJ, Köksal ES, van Buggenum JAGL, Mulder KW: BLNCR is a long non-coding RNA adjacent to integrin beta-1 that is rapidly lost during epidermal progenitor cell differentiation. Sci Rep 9: 31, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Judware R, Culp LA: Concomitant down-regulation of expression of integrin subunits by N-myc in human neuroblastoma cells: Differential regulation of alpha2, alpha3 and beta1. Oncogene 14: 1341–1350, 1997. [DOI] [PubMed] [Google Scholar]
- 71.Xu Z, Zhu L, Wu W, Liao Y, Zhang W, Deng Z, et al. : Immediate early response protein 2 regulates hepatocellular carcinoma cell adhesion and motility via integrin β1-mediated signaling pathway. Oncol Rep 37: 259–272, 2017. [DOI] [PubMed] [Google Scholar]
- 72.Vialou V, Feng J, Robison AJ, Ku SM, Ferguson D, Scobie KN, et al. : Serum response factor and cAMP response element binding protein are both required for cocaine induction of ΔFosB. J Neurosci 32: 7577–7584, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.