

Natural killer (NK) cells are critically involved in the early immune response against various intracellular pathogens, including Coxiella burnetii and Chlamydia psittaci. Chlamydia-infected NK cells functionally mature, induce cellular immunity, and protect themselves by killing the bacteria in secreted granules. Here, we report that infected NK cells do not allow intracellular multiday growth of Coxiella, as is usually observed in other host cell types.
KEYWORDS: cell-autonomous immunity, Chlamydia psittaci, Coxiella burnetii, NK cells
ABSTRACT
Natural killer (NK) cells are critically involved in the early immune response against various intracellular pathogens, including Coxiella burnetii and Chlamydia psittaci. Chlamydia-infected NK cells functionally mature, induce cellular immunity, and protect themselves by killing the bacteria in secreted granules. Here, we report that infected NK cells do not allow intracellular multiday growth of Coxiella, as is usually observed in other host cell types. C. burnetii-infected NK cells display maturation and gamma interferon (IFN-γ) secretion, as well as the release of Coxiella-containing lytic granules. Thus, NK cells possess a potent program to restrain and expel different types of invading bacteria via degranulation. Strikingly, though, in contrast to Chlamydia, expulsed Coxiella organisms largely retain their infectivity and, hence, escape the cell-autonomous self-defense mechanism in NK cells.
INTRODUCTION
Natural killer cells (NK cells) belong to the subgroup of lymphocytes of the white blood cells along with B and T cells. They are part of the innate immune system (1) and play an important role in the early detection and lysis of pathogen-infected cells (2–5). NK cells make up about 5 to 10% of the recirculating lymphocyte population, and their induced gamma interferon (IFN-γ) secretion (6) is crucial in controlling various immunological events during infections with different microbes. The recognition of target cells is achieved via antibody-dependent cell-mediated cytotoxicity (ADCC), which is based on the recognition of antigen-specific antibodies on the target cells by Fc receptors (1). In addition, NK cells recognize infected target cells that have specifically downregulated major histocompatibility complex class I (MHC-I) (1). This is particularly important for infections that escape efficient recognition by T cells through reduced surface MHC-I expression (7). The lysis of target cells recognized by NK cells occurs via degranulation of cytoplasmic granules, which represent secretory lysosomes (8), leading to the release of perforins and granzymes into the extracellular space by targeted exocytosis (1, 9). The highly cytotoxic (10) and bactericidal (11) nature of these vacuolar compartments requires their release to be strictly regulated. Thus, degranulation occurs only by activating NK cells through contact with a target cell (1) or via direct infection (5, 12). Characteristic for the degranulation processes is the extensive remodeling of the cytoskeleton, which is accompanied by exocytosis of the granules (13). Different signaling pathways regulate the activation and function of NK cells. These signaling pathways are controlled by stimulatory/inhibitory receptors and/or through interactions with other immune cells (14–16). NK cell activation also depends on different Toll-like receptors (TLRs) (17–21) as well as on NOD (nucleotide-binding and oligomerization domain) receptors and NOD-like receptors (NLRs) (21). NK cell-activating receptors require protein kinase Cϴ (PKCϴ) for sustained signaling, transcriptional activation (22), secretion of IFN-γ (4, 23), and the release of lytic granules (1).
NK cells are essential for the immune defense against Chlamydia (3, 24–26). The absence of NK cells during chlamydial infection results in exacerbated histopathology, enhanced clinical disease, and a drastic increase in bacterial load (2, 3). Recently, we demonstrated that Chlamydia-infected NK cells functionally mature and prevent the intracellular establishment and growth of bacteria. Chlamydia organisms taken up by NK cells are subsequently released via degranulation in a noninfectious, highly immunogenic form, which then triggers a strong adaptive antichlamydial immune response (5).
Based on these previous observations, we asked whether this NK cell-mediated defense mechanism also targets other intracellular bacteria that localize in acidic cellular compartments during their intracellular life cycle. Such an invasion strategy is known for Coxiella burnetii, a zoonotic obligate intracellular Gram-negative bacterium which can infect a wide range of hosts, including ruminants, swine, cats, dogs, rabbits, rodents, and humans, as well as birds and ticks (27). It is typically transmitted by inhalation of aerosols containing it (28). The bacterium possesses the ability to infect different cell types, including monocytes, macrophages, dendritic cells (DCs), neutrophils, epithelial cells, and fibroblasts (29, 30). C. burnetii is a globally distributed pathogen that causes abortions in ruminant livestock. Zoonotic infections of humans cause Q fever, a flu-like illness whose symptoms typically include headache, myalgia, and fever (31). Most patients resolve the infection and gain long-lasting immunity to C. burnetii, but some are unable to clear the bacterium and develop a chronic infection (32).
C. burnetii targets lysosomal compartments of their host cells as a niche for replication (33, 34). After its uptake (29, 35), the organism exploits the autophagolysosomal pathway to establish acidic parasitophorous C. burnetii-containing vacuoles (CCVs) in the host cell (36), in which it replicates. Intracellular bacteria pose a unique set of challenges for the adaptive immune system. Infection with C. burnetii results in both humoral and cellular immune responses (37, 38). Cellular immunity is critical for this protection (39–41). Key Th1 cytokines like tumor necrosis factor alpha (TNF-α) and IFN-γ directly activate monocytes/macrophages and fibroblasts in order to control the intracellular growth of C. burnetii (29, 42).
The intracellular compartments established and occupied by Chlamydia and Coxiella differ fundamentally in their vesicular biogenesis and physiological properties. For instance, Chlamydia avoids phagosome-lysosome fusion and physical interaction with the endo-/lysosomal system. Chlamydial inclusions seem to have a neutral pH of 7 to 7.5 (43), and a low pH is apparently detrimental to the bacteria (44, 45). Indeed, in vitro studies have demonstrated a significant reduction of chlamydial replication when the organisms are exposed to acidic conditions (46, 47), and the growth of Chlamydia within epithelial cells is inhibited by low pH (48). While exocytic Rab GTPases are recruited to chlamydial inclusions, endocytic markers, like those of early (Rab5 and EEA1) and late (CD107a/LAMP1 and Rab7) endosomes, are excluded in epithelial host cells (49–51). In contrast, once Coxiella organisms are engulfed by host cells, the phagosome matures through the endocytic pathway, and the bacteria become resident in lysosomes (52–54). This compartment is then structurally and functionally remodeled by the pathogen to support intracellular replication (55). Features of the resulting parasitophorous vacuole, such as the presence of proteolytic and degradative enzymes as well as low pH of 4 to 5 (43), are closely related to those of lysosomes (56) and are essential for the metabolic activity of Coxiella (57, 58). Moreover, Coxiella’s protein translocation by the type IV secretion system seems to require endocytic maturation of the CCV (59). Thus, intracellular replication of C. burnetii and its effector translocation are efficiently blocked when the pH of the vacuoles is neutralized (49, 59).
The innate immune system seems to have sophisticated mechanisms to fight Coxiella infections. Accordingly, mast cells build extracellular actin filaments with microbicidal activity (so-called cytonemes), which mediate the capture and elimination of trapped bacteria before they can even enter host cells (60). Although C. burnetii infects a wide range of host cell types, including immune and nonimmune cells (61–64), it is not known whether it can infect NK cells, which play both supportive and harmful roles during bacterial infections (4, 5, 65). Based on our previous findings on Chlamydia (5) as well as the described differential interactions of Coxiella and Chlamydia with their parasitophorous vacuoles (PVs) in the endo- and exocytic pathway (49), we asked whether C. burnetii infects NK cells and, if so, whether the bacteria are able to escape from NK cell-mediated antibacterial immunity.
Here, we show that Coxiella not only is able to infect NK cells but also can actually escape the cellular self-defense mechanism in infected NK cells. Analogous to the situation in chlamydial infections (5), NK cells infected with Coxiella undergo functional activation, prevent the intracellular growth of bacteria, and incorporate the organisms into lytic granules. In contrast to our observation with Chlamydia, however, intracellular C. burnetii largely survives the hostile environment in NK cells granules and is released by degranulation as an infectious agent.
RESULTS
NK cells prevent the intracellular establishment and growth of C. burnetii.
NK cells function as essential sentinels and mediators of the immune system and are a primary innate source of IFN-γ produced before the development of an adaptive response (66). Investigation of the interaction between NK cells and C. burnetii could provide new insight into how the intracellular bacteria escape early immune responses. Using KY-2 cells, a murine NK cell line with homogeneous/consistent culture properties, and the C. burnetii strain NMII RSA 439 as a suitable model system for Coxiella infection, we first aimed to characterize whether and how Coxiella invades NK cells. To this end, KY-2 cells were incubated with C. burnetii for 24 h in the presence or absence of monodansylcadaverine (MDC), which blocks clathrin-mediated endocytosis. MDC sharply reduced Coxiella uptake as measured by a previously established flow cytometry-based assay (Fig. 1a) (5, 67), indicating that clathrin-mediated endocytosis is critically involved in Coxiella engulfment. This result is similar to previous observations in epithelial cells (68). Next, we analyzed the time course of Coxiella infection in KY-2 cells. KY-2 cells were infected with Coxiella for 3 h. Afterwards, they were extensively washed to remove all extracellular and cell surface-attached bacteria and then incubated for 0 to 72 h. To differentiate between intracellular and surface-bound bacteria, the ingestion of C. burnetii was analyzed by comparing anti-Coxiella stainings of permeabilized and nonpermeabilized NK cells (Fig. 1b, left). No cell surface-bound bacteria were found at 8 h postinfection (hpi), suggesting that the bacteria were efficiently engulfed by NK cells. The further course of infection was monitored via Western blotting using a Coxiella HSP60 (coxHSP60)-specific antibody (Fig. 1b, middle and right). The immunodetection of the bacterial molecular chaperone (69) (also termed GroEL [70]), which is essentially required for in vivo folding and assembly of proteins, was used as proxy to demonstrate the intracellular presence of bacteria in NK cells as well as their cellular expulsion into the culture supernatant. Interestingly, we observed a marked increase of intracellular coxHSP60 in cell extracts at 24 hpi, suggesting that KY-2 cells were indeed infected (Fig. 1b). However, at 72 hpi, the intracellular coxHSP60 signal had faded and was barely detectable (Fig. 1b). This is unlikely to reflect the degradative elimination of the pathogen in the infected cells, because the intracellular disappearance of coxHSP60 correlated tightly with a time-dependent increase of the bacterial chaperone in the extracellular medium (Fig. 1b). Thus, the total coxHSP60 levels (intracellular plus supernatant) remained nearly constant over the time course (Fig. 1b, right). Next, we analyzed the infection of KY-2 cells using immunofluorescence (Fig. 1c). In agreement with Fig. 1b, infected KY-2 cells did not establish a typical perinuclear Coxiella-containing vacuole (CCV) (67) during infection (Fig. 1c). While multiple peripheral small bacterium-positive vacuoles with diameters of 1 to 3 μm were observed at 24 hpi, at 72 hpi, only very few or no Coxiella structures were found (Fig. 1c). The time-dependent disappearance of bacteria in infected NK cells was further confirmed by flow cytometry (Fig. 1d). In line with the Western blot data (Fig. 1b), we observed a maximum of Coxiella-positive cells at 24 hpi, followed by a continuous reduction of intracellular bacteria between 48 and 72 hpi. During the time course, the number of living KY-2 cells remained largely constant (Fig. 1e), showing that these results cannot be explained by the death of infected host cells. Expulsion of tdTomato-expressing C. burnetii from infected KY-2 cells was observed by live-cell imaging (LCI) at ≥24 hpi (Fig. 1f and Fig. 2; see also Movies S1, S2, and S3 in the supplemental material). Time-lapse image acquisition with short intervals (∼1,700 ms, resulting in about 35 frames per minute) (Fig. 2; Movie S3) revealed that the entire bacterial release process takes about 15 min and is accompanied by a visible local and convex membrane protrusion on the cell surface. Released bacteria seem to remain on the outside of the cell membrane for some time before they are set free.
FIG 1.

Uptake and functional activation of KY-2 cells during C. burnetii infection. (a) Flow cytometric analysis of infected KY-2 cells (72 hpi; MOI, 10) in the absence or presence of MDC (200 μM). To quantify C. burnetii-positive NK cells (green), the negative cell population (black) was identified and gated via corresponding noninfected controls and then subtracted from the total cell population (left). The corresponding plot (right) shows the amount of positive KY-2 cells (***, P < 0.001 versus control [infected without treatment]; n = 3; mean ± SD). (b) To identify intra- and extracellular bacteria at 8 hpi, infected NK cells were permeabilized or not and then exposed to Coxiella-specific antiserum. Afterwards, cells were analyzed by immunofluorescence microscopy (left). The Western blot (middle) of infected KY-2 cells (0 to 72 hpi; MOI, 10) shows coxHSP60 in cell lysates (intracellular) and culture supernatants (extracellular). GAPDH served as a loading control. coxHSP60 signals were determined by densitometric analysis (right). The signal of total coxHSP60 at 24 hpi was set to 100. (c) KY-2 cells were infected or not with Coxiella (0 to 72 hpi; MOI, 10) and stained for Coxiella (green) and DNA (4′,6-diamidino-2-phenylindole [DAPI]; blue). (d) Flow cytometric analysis of C. burnetii infection in KY-2 cells (0 to 72 hpi; MOI, 10). The plot displays the relative amount of infected cells. The maximum mean value at 24 hpi was set to 1 (**, P < 0.01, and ***, P < 0.001, versus 24 hpi; n = 3; mean ± SD). (e) Analysis of necrotic/apoptotic KY-2 cells during Coxiella infection (0 to 72 hpi; MOI, 10) was performed via trypan blue staining (not significant [n.s.] versus 0 hpi; n = 3; mean ± SD). (f) Long-term time-lapse recording of Coxiella release from infected NK cells. A total of 1 × 105 NK cells were infected with C. burnetii (tdTomato; MOI, 30; washed 3 hpi), and LCI was started at 24 hpi. Bacterial movement was tracked at 37°C. Trajectories were determined from the image series. The timestamp is relative to the start of the respective image series and thus starts at 0 min. The upper and lower rows correspond to starting time points at 24 hpi and 26 hpi, respectively. The differently colored lines (green and magenta) show the individual Coxiella trajectories over periods of 510 and 800 min. The corresponding time-lapse movies are included in the supplemental material (Movies S1 and S2).
FIG 2.

Short-interval time-lapse image series of C. burnetii-release from an infected NK cell. A total of 1 × 105 NK cells were infected with C. burnetii (tdTomato; MOI, 30; washed 3 hpi) and overlaid with 0.6% low-melting-point-agarose-containing KY-2 cell culture medium at 20 hpi. The time-lapse image series shows a period of about 21.5 min, starting at about 31.5 hpi. Bacterial movement during release was tracked at 37°C, and the maximum z-projection of the fluorescence channel (C. burnetii, red) was merged with a single representative plane of the bright-field channel. Trajectories are shown as green lines. The first row displays selected bright-field images of the LCI analysis. The second row shows the overlay of the selected bright-field images with maximum z-projection of red fluorescent bacteria. The third and fourth rows depict corresponding detail views of the bacterial exit process as bright-field images and as overlays with red fluorescent bacteria. The time-lapse movie is included in the supplemental material (Movie S3).
In conclusion, our findings show that NK cells are able to impair the growth of internalized Coxiella, which is reminiscent of the previously described situation with Chlamydia (5). As with Chlamydia, NK cells expel Coxiella within 24 to 48 hpi by releasing them into the extracellular environment.
C. burnetii-infected NK cells display functional maturation and release infectious bacterial structures by degranulation.
Next, we examined whether the “transient” Coxiella infection, which is characterized by uptake and subsequent expulsion of the bacteria, also activates NK cells, as previously observed for Chlamydia-infected NK cells (5). To this end, KY-2 cells were infected with Coxiella for 48 h, and surface expression of CD107a was measured by flow cytometry (Fig. 3a). In parallel, we analyzed the release of granzyme B (Grzm B) and IFN-γ by enzyme-linked immunosorbent assay (ELISA) (Fig. 3b). All assays demonstrated that C. burnetii-infected KY-2 cells were activated and degranulated. Accordingly, PKCϴ underwent phosphoactivation (Fig. 3c), a step triggering downstream signaling, activation, and degranulation in NK cells (22), and the PKCϴ inhibitor sotrastaurin (71) strongly suppressed Coxiella release (Fig. 3d). To assess whether C. burnetii is able to grow at all inside NK cells when bacterial release is blocked, kinetics of Coxiella-infected cells were analyzed in the presence of sotrastaurin. In contrast to the case with Coxiella-infected L929 reporter cells, which showed a continuous growth of the bacteria over 72 h (Fig. S2), no increase in the bacterial load was observed for infected NK cells with blocked degranulation (Fig. S2). Thus, NK cells do apparently not provide the optimal vacuolar environment for the optimal intracellular growth of Coxiella.
FIG 3.

Activation of KY-2 cells during C. burnetii infection. (a) Flow cytometric analysis of NK cell degranulation via staining of surface CD107a/LAMP1 (left) on noninfected and C. burnetii-infected KY-2 cells (24 hpi; MOI, 10). KY-2 cell endocytosis was inhibited 12 hpi by MDC to stabilize surface-expressed CD107a/LAMP1. Cells were first stained for surface CD107a/LAMP1 and then fixed. After mild permeabilization, pretreated cells were stained for intracellular bacteria. The corresponding histogram plot (right) shows the relative increase of the surface CD107a/LAMP1 (noninfected controls were set to 1 (*, P < 0.05 versus control [noninfected]; n = 3; mean ± SD). (b) Secretion of Grzm B (left) and IFN-γ (right) of infected KY-2 cells (0 to 72 hpi; MOI, 10) measured by ELISA (**, P < 0.01, and ****, P < 0.0001, versus noninfected control; n = 3; mean ± SD). (c) Analysis of PKCϴ phosphoactivation during KY-2 cell infection. KY-2 cells were infected or not with C. burnetii (MOI, 10) for 72 h and analyzed by Western blots probed for P-PKCϴ, PKCϴ, and coxHSP60 (top). GAPDH served as a loading control. After densitometric analysis (bottom), the P-PKCϴ/PKCϴ ratio was plotted (gray histogram) and compared to the control of noninfected KY-2 cells (black histogram; controls were set to 1; *, P < 0.05, versus noninfected control; n = 3; ± SD). (d) Western blot analysis of coxHSP60 in infected KY-2 cells (MOI 10) (top) and culture supernatants (bottom) in the absence or presence of PKCϴ inhibitor sotrastaurin (250 nM) 24 hpi and 72 hpi. GAPDH and Ponceau-S staining served as loading controls for cell lysates and culture supernatants, respectively.
Degranulation of NK cells is a process of regulated secretion (72) of granules with sizes between 0.2 and 1 μm (73). This allows the incorporation of Coxiella small-cell variants (SCVs) (diameter, 0.2 to 0.3 μm) and large-cell variants (LCVs) (diameter, 0.5 to 1 μm) into these vacuoles. Thus, given a nearly simultaneous release of C. burnetii (Fig. 1) and Grzm B (Fig. 3b), we asked whether Coxiella structures are localized within secretory granules of infected NK cells, as is the case with Chlamydia (5). KY-2 cells were infected with Coxiella and analyzed by electron microscopy. In noninfected KY-2 cells (control), secretory granules were detectable as type II vacuolar structures (74) containing lamellar and vesicular material (Fig. 4a). In contrast, in infected cells (24 hpi), most of the secretory granules were filled with electron-dense material (Fig. 4a and b) and large round structures (diameter, 0.2 to 0.3 μm) corresponding in size to Coxiella SCVs or LCVs. A smaller bacterial fraction was associated with outer cell surface membrane structures (24 hpi), suggesting that they were on their way out or had already been released into the environment (Fig. 4a, middle, arrowheads). At 72 hpi, only a small fraction of bacteria remained inside the cells (Fig. 4a, right). This suggests that bacterial material was in physical contact with secretory granules and that the release of Coxiella was linked to degranulation in infected KY-2 cells. Consistent with this, C. burnetii structures colocalized with perforin-positive granules, whereas in noninfected cells, perforin resided in circular structures near the nucleus (Fig. 4c). Analysis of the expulsed Coxiella by immunofluorescence, Western blotting, and electron microscopy revealed that the extracellular bacteria were associated with perforin (Fig. 5a, left, and Fig. S1) and that some bacterial structures were characterized by aberrant large electron-dense material (Fig. 5a, middle). Nevertheless, a direct comparison with C. burnetii from infectious stock cultures (Fig. 5a, right) revealed that a well-detectable amount of intact Coxiella was present in the released pathogen fraction of NK cells. Chlamydia psittaci released by NK cells was also characterized by perforin association (Fig. 5b, left, inset). However, in contrast to the situation with Coxiella, the NK cell-released Chlamydia seemed to be structurally disordered (Fig. 5b, left) and, in contrast to C. psittaci from infectious chlamydial stocks (predominantly elementary bodies [EBs]), had a low electron density and no detectable intact outer membrane structures (Fig. 5b, right).
FIG 4.

Characterization of intracellular C. burnetii in infected KY-2 cells. (a) TEM of noninfected (control) and C. burnetii-infected (24 hpi and 72 hpi; MOI, 10) KY-2 cells. Arrowheads indicate cell surface membrane-associated Coxiella structures. (b) TEM of C. burnetii-infected (24 hpi; MOI, 10) KY-2 cells. Arrowheads indicate electron-dense intracellular Coxiella structures associated with secretory granules of KY-2 cells. (c) Immunofluorescence analysis of perforin (green) and C. burnetii (red) in infected (24 hpi; MOI, 10) and noninfected KY-2 cells. DNA was stained via DAPI (blue).
FIG 5.

Characterization of extracellular C. burnetii and C. psittaci released by infected KY-2 cells. (a) The left image depicts an immunofluorescence analysis showing colocalization of perforin (green) and KY-2 cell-released C. burnetii (red) 24 hpi (MOI, 10). A TEM analysis of NK cell-released bacterial structures (72 hpi; MOI, 10) is shown in the middle image. Black stars indicate intact C. burnetii, whereas black diamonds mark largely deformed, aberrant bacterial structures. The TEM analysis of purified C. burnetii stocks from infected L929 cells containing LCVs and SCVs of C. burnetii is shown in the right image. (b) The first image depicts a TEM analysis of NK cell-released chlamydial structures. The inset highlights an immunofluorescence analysis showing colocalization of perforin (green) and KY-2-released C. psittaci (red) at 24 hpi (MOI, 10). A TEM analysis of an infectious C. psittaci stock is shown in the second image from the left. The two images on the right show enlarged TEM pictures of Chlamydia, either released from NK cells by degranulation or harvested from an infectious stock culture.
Taken together, these findings suggest that a large proportion of structurally intact Coxiella survives the uptake into lytic granules and the subsequent export via degranulation. Accordingly, we found that NK cell-released C. burnetii, which is highly resistant to low pH (pH 4 to 5) (75) (Fig. 6a, top) and the bactericidal protease Grzm B (11) (Fig. 6b), maintained its infectivity when measured in standard reporter cells (Fig. 6c). These findings are in clear contrast to the pH and Grzm B sensitivities of Chlamydia (46–48) (Fig. 6a, bottom) and the situation with Chlamydia-infected NK cells, in which the released bacteria completely lose their infectious properties (Fig. 6c) (5).
FIG 6.

C. burnetii survives the cellular defense (granule uptake and release) of infected NK cells. (a) C. burnetii and C. psittaci were preincubated in citrate buffer at pH 4 and 5 or in Tris buffer at pH 7 (control) for 18 h at 4°C. After infection (MOI, 10) of suitable reporter cells (L929 and BGM; 72 hpi), infectivity was measured by flow cytometry (top). Bacterium-positive cells (green) were identified and gated (left) as described in the legend to Fig. 1. The corresponding histogram plots (right) show the number of positive cells (controls were set to 1 [infected without preincubation]; *, P < 0.05, **, P < 0.01, and ***, P < 0.001, versus control; n = 3; mean ± SD). (b) Western blot analysis of cell infection after treatment of purified Chlamydia or Coxiella with Grzm B. Both pathogens were incubated with Grzm B or left untreated. The bacteria were then washed and used for the infection of reporter cells (L929 for Coxiella and BGM for Chlamydia; 72 hpi; MOI, 30). Bacterial HSP60 (bactHSP60) signal intensities were measured by densitometric analysis (left). GAPDH served as a loading control. The obtained results of three independent experiments are depicted as a histogram plot (right) (signals of untreated samples were set to 1; n.s., ***, P < 0.001, versus untreated samples; n = 3; mean ± SD). (c) Flow cytometry of the infectivity of culture supernatants (sup.) from KY-2 cells. KY-2 cells were infected with bacteria (Chlamydia or Coxiella; MOI, 10 [10 IFU/cell]) or not for 72 h. GEs were determined by qPCR. To compare the infectivity of original stocks and NK cell-released bacteria, equal amounts of GEs (Coxiella and Chlamydia infections, 70 GEs/cell) were used in parallel experiments for reporter cell infection (L929 and BGM). Bacterium-positive cells (green) were identified and gated (upper panel) as described in the legend to Fig. 1. The corresponding histogram plot (bottom) shows the relative number of infected reporter cells (controls were set to 1 [original stocks]; *, P < 0.05, and ***, P < 0.001, versus controls; n = 3; mean ± SD).
To see whether NK cell-released C. burnetii is still able to grow and replicate in reinfected reporter cells, we analyzed the reinfection of L929 cells at different time points (24, 48, and 72 h) (Fig. 7a, top). Indeed, flow cytometry analysis to detect intracellular bacteria showed a time-dependent and continuous increase of cellular infection and bacterial load (relative mean fluorescence intensity [MFI] of bacterium-positive cells increased 5-fold between 24 and 72 hpi) (Fig. 7a, bottom). In addition, immunofluorescence studies revealed that CCV formation in reinfected reporter cells was comparable to that in cells infected with original Coxiella stocks (Fig. 7b). Consistent with this, we also observed that C. burnetii released by NK cells was able to initiate successive rounds of L929 reinfection (Fig. 7c).
FIG 7.

Growth/replication of Coxiella released from KY-2 cells. (a) Flow cytometry of the Coxiella growth/replication in L929 cells infected with bacteria expulsed by KY-2 (top). NK cells were infected with C. burnetii (MOI, 10 [10 IFU/cell]) or not for 72 h. Bacteria from corresponding culture supernatants were centrifuged and washed. GEs of the pellet fractions were determined by qPCR, and 70 GEs/cell were used for L929 cell infection and analyzed for different time points (control, 24 to 72 h). Bacterium-positive cells (green) were identified and gated (top) as described in the legend to Fig. 1. The corresponding histogram plots (bottom) show the relative number of infected reporter cells (control, 24 to 72 hpi) (values obtained for 72 hpi were set to 1; **, P < 0.01, and ***, P < 0.001, versus controls; n = 3; mean ± SD). (b) Immunofluorescence analysis of C. burnetii (green) in infected and reinfected (70 GEs/cell) L929 cells (72 hpi). DNA was stained via DAPI (blue). (c) Successive reinfection of L929 cells. Centrifuged/washed bacteria expulsed by infected KY-2 cells were used for an L929 cell reinfection assay (1. reinfection, 70 GEs/cell). After 72 h, one-half of the infected cells was examined for infection by flow cytometry. The other half was homogenized to harvest Coxiella for a second reinfection round in L929 cells (2. reinfection). As with the 1. reinfections, flow cytometry was performed after 72 hpi.
Our standard reporter cell line used for Coxiella infections and reinfections was L929 (Fig. 1 and 5). To rule out that the observed loss of infectivity of NK-cell released Chlamydia was a BGM cell line-specific phenomenon, additional experiments were carried out in which the L929 cells were also infected or reinfected with C. psittaci (Fig. S3). These studies confirmed further that regardless of the reporter cell line used, C. psittaci was sensitive to low pH (Fig. S3a) and Grzm B (Fig. S3b) treatment and when released by NK cells lost its infectivity (Fig. S3c).
Next, we examined whether primary NK and KY-2 cells show the same properties when infected with C. burnetii. To this end, primary NK cells were isolated from the spleens of C57BL/6 mice (with ≥96% purity) and then infected. coxHSP60-specific immunoblotting of cell extracts and culture supernatants demonstrated efficient Coxiella release into the environment (Fig. 8a). The time-dependent expulsion of bacteria in infected primary NK cells was furthermore confirmed by flow cytometry (Fig. 8b). As for KY-2 cells, infected primary NK cells displayed low numbers of dead cells at all analyzed time points, suggesting that their time-dependent reduction was not related to cell death (Fig. 8c). Moreover, in further agreement with the results obtained with infected KY-2 cells, the release was sotrastaurin sensitive (Fig. 8d), and no perinuclear CCV appeared during the infection (Fig. 8e). Peripheral small bacterium-containing structures colocalizing with perforin were observed inside and outside infected NK cells at 12 to 24 hpi (Fig. 8e). Following our previous observations on Chlamydia (5), this suggests that immortalized and primary NK cells use the same cellular defense strategy (uptake and subsequent degranulation) during infection. However, again as with KY-2 cells, Coxiella released from lytic granules of primary NK cells retained much of its infectivity (Fig. 8f), which was still not observed for NK cell-released chlamydia (regardless of the reporter cell system used) (Fig. 8f and Fig. S4). We also observed in this study that Coxiella released from primary NK cells can grow and multiply in reinfected reporter cells (Fig. 9a), form comparable CCVs there (Fig. 9b), and remain infectious in successive rounds of reporter cell infection (Fig. 9c).
FIG 8.

C. burnetii infection of primary NK cells. (a) Western blot of infected primary NK cells (0 to 72 hpi; MOI, 10) showing coxHSP60 in cell lysates (intracellular) and culture supernatants (extracellular) (left). GAPDH served as a loading control. coxHSP60 signals were determined by densitometric analysis (right). The signal of total coxHSP60 at 24 hpi was set to 100. (b) Flow cytometric analysis of C. burnetii infection in KY-2 cells (0 to 72 hpi; MOI, 10). The depicted plot displays the relative amount of infected cells. The maximum value at 24 hpi was set to 1 (**, P < 0.01, and ***, P < 0.001, versus 24 hpi; n = 3; ± SD). (c) Necrotic/apoptotic KY-2 cells during Coxiella infection (0 to 72 hpi; MOI, 10) were identified by trypan blue staining (n.s. versus 0 hpi; n = 3; mean ± SD). (d) Western blot and protein staining of coxHSP60 in infected primary NK cells (MOI, 10) as well as culture supernatants in the presence of sotrastaurin (72 hpi). Ponceau-S staining served as a loading control. (e) Immunofluorescence microscopy shows colocalization of perforin (green) and C. burnetii (red) in infected primary NK cells (24 hpi; MOI 20) (left) and for NK cell-released C. burnetii (12 hpi, MOI 20) (right). DNA was stained with DAPI (blue). (f) Flow cytometry of the infectivity of culture supernatants from primary NK cells. Primary NK cells were infected with bacteria (Chlamydia or Coxiella; MOI, 10) or not for 72 h. GEs were determined by qPCR. Equal amounts of GEs (Coxiella and Chlamydia infections, 70 GEs/cell) were used in parallel experiments for reporter cell infection. The corresponding histogram plot shows the relative number of infected reporter cells (controls were set to 1 [original stocks]; **, P < 0.01, and ***, P < 0.001, versus controls; n = 3; mean ± SD).
FIG 9.

Growth/replication of Coxiella released from primary NK cells. (a) Flow cytometry of the Coxiella growth/replication in L929 cells infected with bacteria expulsed by primary NK cells (top). NK cells were infected with C. burnetii (MOI, 10 [10 IFU/cell]) or not for 72 h. GEs were determined by qPCR, and 70 GEs/cell were used for L929 cell infection and analyzed for different periods (control, 24 to 72 h). Bacterium-positive cells (green) were identified and gated (top) as described in the legend to Fig. 1. The corresponding histogram plots (bottom) show the relative number of infected reporter cells (control, 24 to 72 hpi) (values obtained for 72 hpi were set to 1; **, P < 0.01, and ***, P < 0.001, versus controls; n = 3; mean ± SD). (b) Immunofluorescence analysis of C. burnetii (green) in infected and reinfected (70 GEs/cell) L929 cells (72 hpi). DNA was stained via DAPI (blue). c) Successive reinfection of L929 cells. Centrifuged/washed bacteria expulsed by infected primary NK cells were used for an L929 reinfection assay (1. Reinfection, 70 GEs/cell). After 72 h, one-half of the infected cells was examined for infection by flow cytometry. The other half was homogenized to harvest Coxiella for a second reinfection round in L929 cells (2. reinfection). As with the 1. reinfections, flow cytometry was performed after 72 hpi.
DISCUSSION
Recently, we demonstrated that NK cells are functionally activated by chlamydial infection. During this cellular self-defense process, intracellular bacteria are translocated into lytic granules and then released by infected NK cells via degranulation in an inactivated noninfectious form (5). Chlamydia normally replicates within nonacidified vacuoles. These vacuoles receive lipids from exocytic vesicles derived from the trans-Golgi network but are disconnected from the regular endo-/lysosomal pathway (49). In Chlamydia-infected NK cells, however, internalized bacteria are targeted to lytic granules (5), an acidic, protease-rich environment hostile to the organisms (5). Here, we show that C. burnetii invading NK cells is similarly targeted to lytic granules and leads to comparable NK cell activation (Fig. 1 and 5). However, Coxiella released via degranulation (Fig. 1 and 2) largely retains its infectivity (Fig. 6 to 9).
In contrast to Chlamydia, C. burnetii replicates in acidified compartments with (auto)phagolysosomal characteristics (76). Lysosomal membrane markers and enzymes, as well as molecules internalized by fluid-phase endocytosis, are found in CCVs (76). Moreover, bacterium-containing vacuoles fuse with organelles of the phagocytic/endocytic system (77–79). Acidic lysosomes contain a myriad of hydrolases and normally form a natural defense against phagocytosed bacteria. However, Coxiella is adapted to this environment and therein undergoes a biphasic development (80). Our experiments show that Coxiella survives an 18-h incubation at pH 4 and 5 (up to 75% survival), while Chlamydia seems to have only short-term pH stability (Fig. 6a). In fact, the acid milieu is essential for C. burnetii replication, as raising the lysosomal pH with lysosomotropic amines or proton pump V-ATPase inhibitors reduces the growth of the bacterium (49, 57). Thus, regarding their nature and properties, the distinct compartments occupied by Coxiella and Chlamydia represent polar opposites within the spectrum of the intracellular vacuolar system. C. burnetii enters NK cells via clathrin-mediated endocytosis (Fig. 1), which is reminiscent of Coxiella uptake into epithelial cells and/or mononuclear phagocytes (68, 81). Hence, it is tempting to speculate that targeting of the bacteria into lytic granules is related to the conventional endo-/lysosomal transport of C. burnetii into acidic compartments.
Lytic granules represent secretory lysosomes, have an acidic pH, and combine the degradative function of conventional lysosomes with the capacity to undergo regulated exocytosis (1). After entering NK cells, C. burnetii interacts with the host endo-/lysosomal transport machinery and its vacuole fuses with secretory instead of conventional lysosomes. Nevertheless, the intracellular LCV of Coxiella has a unique resistance to low pH as well as the highly degradative machinery of host cells (Fig. 6) (30, 82). C. burnetii does not actively inhibit lysosome function as an intracellular survival mechanism. Instead, Coxiella resists degradation and successfully replicates in digestive vacuoles that fully mature through the endo-/lysosomal pathway (56). Moreover, acidic vacuoles harboring Coxiella are proteolytically active and contain various lysosomal proteases, also present in lytic granules of NK cells (56, 83, 84). How C. burnetii resists low pH and the proteolytic environment within endo-/lysosomal compartments is still unclear. The resistance of the pathogen does not require pathogen metabolism, though, because chloramphenicol-treated organisms remain viable for several days in lysosome-like vacuoles of Vero cells (64). Furthermore, full-length lipopolysaccharide (LPS) of phase I Coxiella is also not required for protection (56). However, it might be possible that Coxiella combats the low pH and proteolytic environment via the synthesis of peptidoglycan-associated proteins, which are apparently protease resistant (85).
Recent findings of Samata and colleagues show that C. burnetii is able to inhibit endosomal/lysosomal maturation to reduce the number of proteolytically active lysosomes available for fusion with CCVs, probably as a mechanism to regulate the pH of bacterial compartments (86). No such vacuolar subversion strategy is known for Chlamydia, which diverges from the endocytic pathway early on, modifying its vacuole so that it resembles an exocytic vacuole (87). It is therefore tempting to speculate that this strategy of Coxiella could also be used to modulate the pH and environment of lytic granules in infected NK cells to allow survival/infectivity of C. burnetii during granule release (Fig. 6) but apparently not growth within these compartments (Fig. S2). It will, therefore, be interesting to see in future studies whether and to what extent there are differences in the compositions or activities of the lytic granules released by Coxiella- and Chlamydia-infected NK cells.
Our findings demonstrate that NK cells are directly activated during C. burnetii infection (Fig. 3), possibly a consequence of TLR2-dependent recognition of the bacteria (18). In line with this, our present studies show that Coxiella-infected NK cells display phosphoactivation of PKCϴ (Fig. 3). This kinase functions as a signaling intermediate downstream of surface receptors in the PKC pathway (88) and is critically involved in activation/degranulation processes (89, 90). Accordingly, the PKCϴ-inhibitor sotrastaurin (71) impairs the release of Coxiella via degranulation (Fig. 3d). It has been suggested that full-length LPS of phase I Coxiella acts as a virulence factor by shielding the outer membrane, thereby conferring resistance to complement-mediated killing (91), and by masking surface TLR ligands from innate immune recognition by human DCs (33, 92). In contrast, exposure of phase II TLR surface ligands is thought to stimulate potent activation and maturation of infected DCs as well as the release of proinflammatory cytokines (33, 92). Thus, it would be interesting to learn whether the infection of NK cells with phase I Coxiella also leads to PKCϴ-dependent NK cell activation and the release of infective C. burnetii via degranulation. Our laboratory is currently working on this question by comparing NK cell infection by phase I and phase II Coxiella.
In contrast to the case with B and T cells, which efficiently fight C. burnetii infections (39, 40, 93, 94), little is known about the functional role of NK cells during the immune response against Coxiella. Although NK cell deficiencies lead to enhanced histopathology during C. burnetii infection, it seems that NK cells have an only moderate overall impact on pathogen clearance (39). Thus, in line with our findings, it appears that Coxiella-infected NK cells can contribute to some extent to the immune response against the bacteria as well as to the control of infection by releasing IFN-γ (Fig. 3). However, they do not prevent the spread of cellular infection by killing the pathogen via granule-mediated self-defense, as NK cell-released C. burnetii largely remains infectious (Fig. 6 to 9).
Based on this, we postulate the following working model (Fig. 10). Upon uptake into NK cells, the infection activates the host cells via PKCϴ, driving increased IFN-γ secretion. The pattern recognition receptors (PRRs) NOD2 and TLR2, which are crucial for the induction of the immune response against C. burnetii (95, 96), are also involved in pathogen-mediated activation of NK cells (97), suggesting that they might trigger the respective pathways in Coxiella-infected NK cells. During this activation process, Coxiella structures fuse with lytic granules/secretory lysosomes that are then released via degranulation within 24 h. The release process is much shorter than the regular intracellular development cycle of C. burnetii (5 to 7 days). Therefore, it is rather unlikely that the bacteria have already transformed from SCVs to replicative LCVs. Together with intact Coxiella, deformed bacterial structures are released (Fig. 5). This may result from the failure of some of the organisms to complete the normal intracellular developmental cycle in infected NK cells before degranulation. Nevertheless, we assume that the majority of invading Coxiella organisms survive their transient inhabitance of these acidic and degrading compartments and largely retain infectivity upon release via degranulation (Fig. 6 to 9).
FIG 10.
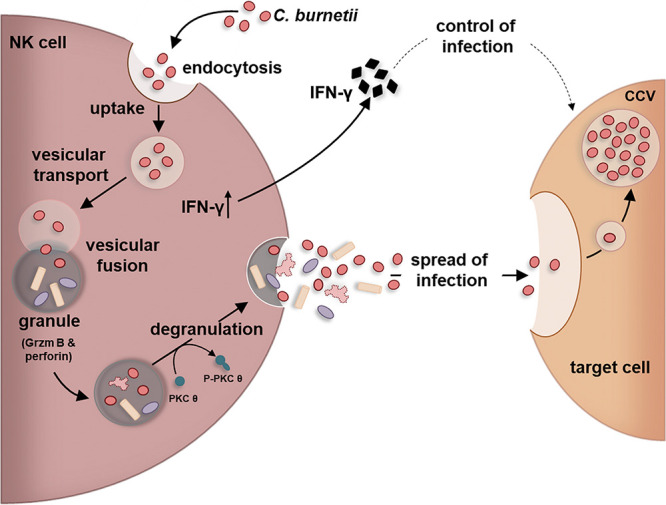
Working model of bacterial escape from cellular self-defense of C. burnetii-infected NK cells. The depicted model shows the transient C. burnetii-infection of NK cells triggering activation, cytokine secretion, bacterial granule fusion, and release of infectious Coxiella.
In Grzm B sensitivity, there is a clear contrast between Coxiella and Chlamydia (Fig. 6b and Fig. S3b), suggesting a difference in the protease sensitivities of the two bacterial pathogens. Indeed, chlamydial surface proteins are known to be protease sensitive (98). Moreover, the bacteria avoid any contact with the proteolytic machinery of lysosomes, which is used to eliminate the pathogen from host cells. For C. psittaci and C. trachomatis, it has been demonstrated that the protease sensitivities of their surface proteins have negative consequences for the attachment to host cells (99–101). In contrast, C. burnetii seems to have the property to resist proteolytic degradation by intracellular host cell proteases. This does not require bacterial metabolism (64) or full-length LPS (56). The results suggest that Grzm B deficiency in mice does not significantly alter their susceptibility to C. burnetii infection, indicating that the granule protease does not contribute to the control of Coxiella infections (102). One possible strategy of Coxiella could be the use of protease-resistant surface polypeptides (85), which may also affect the proteolytic attack by Grzm B and protect or shield the bacteria. Clearly, further studies are essentially required to understand the proteolytic resistance strategy of C. burnetii.
In noninfected NK cells, the perforin-containing granules were frequently present in perinucleus-centered radial arrangements (Fig. 4). In contrast, in Coxiella-infected NK cells, these characteristic intracellular structures were rearranged during degranulation, and the perforin-containing granules colocalized with the bacteria below the cell surface (Fig. 4). This behavior of infected NK cells strongly resembles the well-described granule export events in conventionally (receptor-) activated NK cells (1). Lytic granules are initially recruited along microtubules (MTs) that converge toward the microtubule-organizing center (MTOC) in the perinuclear region. On recognition of a target cell, NK cells are activated, and the cytoskeleton (including actin filaments and MTs) is reorganized. In consequence, granules move and polarize into close apposition with the cell surface, fuse with the plasma membrane, and release their lytic content of perforins and granzymes to the space between NK and target cells. Therefore, it is tempting to assume that the two degranulation scenarios differ only in that the release process takes place in infected NK cells without alignment to an extracellular target.
Interestingly, it seems that perforin (in contrast to IFN-γ and Grzm B) is physically attached to the released extracellular bacteria (Fig. 5 and 8; Fig. S1). This may have functional importance, because perforin is known to activate clathrin-dependent endocytosis (103), which is required for the uptake of Coxiella by the respective target cells (68). Hence, it is conceivable that this might even support the spread of bacteria from infected NK cells to neighboring target cells. Thus, our data suggest that NK cells could play a critical role in the early steps of C. burnetii infection. Studies based on this should examine the mechanisms by which Coxiella spreads from infected NK cells to other host cells. In this context, it is interesting that C. burnetii is also able to infect neutrophils (61). Moreover, the pathogen is viable within neutrophils, although it does not replicate. Coxiella inside neutrophils infects and replicates within macrophages, which engulf infected cells via phagocytosis, suggesting that neutrophils are unable to kill the bacteria and that Coxiella may use the infection of neutrophils as an evasive strategy to infect macrophages (61). In fact, this may be a survival mechanism of C. burnetii to transfer from a harsh, hostile environment (phagosome of a neutrophil) to a more preferred environment in macrophages (61). In addition to macrophages, neutrophils, and NK cells, other cells of the innate immune system might also be involved in the spread of the infection. These include phase I Coxiella-infected DCs, which are known to be impaired in their proper maturation and function (33, 104) and thus may contribute to tolerance (and/or ignorance) of bacterial antigens (29).
In summary, C. burnetii appears to subvert cell-autonomous resistance in NK cells. This may reflect a crucial Achilles heel in the innate immune response against the pathogen. Future experiments should aim to clarify the consequences of this during natural infections.
MATERIALS AND METHODS
Cell culture.
The murine NK cell clone KY-2 (a kind gift from W. Yokoyama, Washington University School of Medicine) (105) was cultivated at 37°C and 7.5% CO2 in RPMI 1640 medium supplemented with 2 mM l-glutamine, 10% fetal calf serum (FCS), β-mercaptoethanol (10 μM), and 200 U/ml of interleukin 2 (IL-2). Immortalized murine fibroblasts (L929) were used as reporter cells and were obtained from the ATCC (CCL-1). The epithelial African green monkey kidney cell line (BGM) was obtained from the National Reference Laboratory for Chlamydiosis of the Friedrich-Loeffler-Institut, Jena, Germany (CCLV-RIE 136). Cells were grown at 37°C and 7.5% CO2 in Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% FCS. Cellular uptake was analyzed by the use of a specific inhibitor (monodansylcadaverine [MDC] for inhibition of clathrin-dependent endocytosis) (Sigma-Aldrich). Sotrastaurin [3-(1H-indol-3-yl)-4-(2-(4-methylpiperazin-1-yl)quinazolin-4-yl)-1H-pyrrole-2,5-dione; Cayman Biochemical] as a pan-protein kinase C (PKC) inhibitor was used to block PKCϴ-dependent cellular degranulation.
C. burnetii NMII RSA 439.
Infection studies were performed with C. burnetii NMII RSA 439 (phase II, low virulence) and tdTomato-expressing C. burnetii NMII RSA 439 (phase II, low virulence). The tdTomato gene was amplified from pKM244mod-tdTomato (106) by standard PCR using the primers a533 (5′-GATTTAAGAAGGAGATCTGCAGATGGTGTCAAAAGGAG-3′) and a534 (5′-AAGCTTGCATGCCTCAGTCGACTTATTTATAAAGTTCATCCATGC-3′). The vector pJB-CAT-ProA-mCherry (kindly provided by Robert Heinzen and Paul Beare) was digested with PstI and SalI. Digested vector and the purified PCR product were ligated using the Gibson assembly method (107). The tdTomato-encoding plasmid pJB-CAT-ProA-TdTomatoCC was used to electroporate and transform the bacteria cultured in host-cell free medium ACCM-D (108). Transformation of C. burnetii was performed as described before (106, 109) and was approved by the government of Lower Franconia, Germany (8791.27.36.25). Selection was carried out on ACCM-D agar plates containing 3 μg/ml of chloramphenicol (CA). Screening for positive clones was performed after 10 to 14 days of growth by PCR using the primers a535 (5′-CACAGCTAACACCACGTCGTCC-3′) and a537 (5′-CTGCATCACTGGCCCATCGG-3′). Positive clones were expanded in liquid ACCM-D medium containing 3 μg/ml of CA and analyzed by fluorescence microscopy. For the preparation of bacterial stocks, murine fibroblasts (L929) were infected with C. burnetii at a multiplicity of infection (MOI) of 30 to 50, corresponding to 30 to 50 infection-forming units (IFU)/cell, and cultured at 37°C and 7.5% CO2 for 2 weeks. Subsequently, cell culture supernatants were collected and centrifuged at 5,000 × g for 30 min at 4°C and stored on ice. The pellet was washed with H2O and centrifuged again (5,000 × g for 30 min at 4°C). The remaining cell fraction was incubated with H2O for 45 min for hypoosmotic cell lysis, collected, and centrifuged at 250 × g for 10 min at 4°C. The supernatant was collected and stored on ice. The corresponding pellet was mixed with H2O, resuspended with different cannulas (G18 and G22), and centrifuged at 250 × g for 10 min at 4°C. All supernatants were collected and centrifuged at 5,000 × g for 30 min at 4°C. The resulting pellet containing purified C. burnetii was mixed with sterile phosphate-buffered saline (PBS) and stored at –80°C. The vital titer (IFU per milliliter) and MOI (IFU per cell) were determined as previously described (67).
Coxiella organisms released by infected NK cells were centrifuged to remove soluble proteins and nonassociated factors (10,000 × g for 30 min at 4°C). Coxiella-containing pellet fractions were then washed three times with large volumes of cold PBS. After resuspension in culture medium, the pellet fraction was used for GE determination and reporter cell infection. Immunoblots (Fig. S1) were used to analyze the pelleted bacteria from NK cell supernatants.
C. psittaci DC15.
The nonavian C. psittaci strain DC15 (110) was grown in BGM cells. Briefly, the cells were cultivated in antibiotic-free medium, and confluent cultures were infected with 5 × 107 IFU. After 48 h of cultivation at 37°C and 7.5% CO2, Chlamydia-containing cells were harvested, and the bacterial suspension was sonicated three times for 10 s at 100 W in an ultrasonic bath. The supernatant was carefully transferred to ultracentrifuge tubes after centrifugation (4,000 × g for 3 min at 4°C). Then, the suspension was underlaid with Visipaque (Nycomed) solutions of different concentrations (2 ml of 8% solution, 3 ml of 15% solution, and 5 ml of 30% solution) (111). Afterward, the tubes were centrifuged at 40,000 × g for 50 min and 4°C. The pellet fraction was resuspended in PBS and used for a second ultracentrifugation whereby the obtained fraction was again carefully underlaid with different Visipaque solutions (1 ml of 8%, 1 ml of 15%, 1 ml of 30%, 12 ml of 36%, 8 ml of 40%, and 5 ml of 47%). After the second ultracentrifugation (50,000 × g for 50 min at 4°C), infectious elementary bodies (EBs) were found between the 40% and 47% layers. This gradient fraction was diluted in PBS and centrifuged again (30,000 × g for 50 min at 4°C). Finally, the pellet of enriched EBs was resuspended in sucrose-phosphate-glutamic acid buffer (SPGA) and stored at –70°C.
Determination of genome equivalents (GEs).
For quantitative real-time PCR (qPCR) analysis, C. burnetii DNA was extracted from stocks, infected cells, and culture supernatants using the DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s instructions for Gram-negative bacteria. The dotA PCR assay amplifies a 65-bp fragment of the C. burnetii singular chromosomal dotA gene (GenBank accession no. ABS78182.2). dotA primers (forward, dotA_for [5′-GCGCAATACGCTCAATCACA-3′; positions 1336 to 1355], and reverse, dotA_rev [5′-CCATGGCCCCAATTCTCTT-3′; positions 1382 to 1401]; synthesized by Metabion) were adapted from the literature (33, 53). As a positive control, a pcDNA3.1_dotA vector was constructed by ligating a 109-bp fragment derived from C. burnetii dotA, which contains the primer binding sites, into pcDNA3.1 (Thermo Fisher Scientific). We used 10-fold serial dilutions of the dotA vector to generate standard curves for the determination of gene copy numbers. qPCR was performed using the iScript one-step real-time PCR kit in combination with SYBR green (Bio-Rad). All PCRs were carried out in duplicate in a Bio-Rad CFX96 real-time system (Bio-Rad) (10 s at 95°C, 30 s at 50°C, 44 cycles, followed by a melting-curve analysis: 52°C to 95°C with a temperature increase of 0.5°C). For C. psittaci, we amplified a 151-bp PCR fragment of the singular chromosomal gyrA gene (GenBank accession numbers CP002806.1 and AEG87507.1) with the following gyrA primers: forward, gyrA_for (5′‐GCGAAGCATCGTAAATGCGC‐3′ [positions 181 to 200]), and reverse, gyrA_rev (5′‐AGCCAAAGTTTCCTTGACCAT‐3′ [positions 311 to 331]).
Antibodies.
To verify bacterial infection in Western blots, immunofluorescence, and flow cytometry, anti-Coxiella HSP60 (anti-coxHSP60; Enzo Life Science) and an anti-C. burnetii antiserum (67) were used. Further, antibodies against P-PKCϴ/PKCϴ, NK1.1, perforin, CD107a/LAMP1, IFN-γ, Grzm B, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Abcam, Cell Signaling, and Millipore. Secondary antibodies were purchased from Dianova, Thermo Fisher, and BioLegend.
NK cell isolation.
Spleens were isolated from uninfected female C57BL/6 mice (8 weeks old). These were transferred to 15-ml tubes and stored on ice. To release cells, spleens were cut into sections and passed through a cell sieve (70 μm) with slight pressure. The resultant cell suspension was centrifuged (10 min at 300 × g and 4°C) and washed with sterile PBS. Afterward, NK cells were isolated by using the mouse NK cell isolation kit from Miltenyi Biotec. This isolation was followed by a negative-selection process in which all “non-NK cells” were bound by a mix of biotin-coupled antibodies and depleted with anti-biotin antibodies from the cell suspension. The separation was carried out by applying a magnetic field through a magnetic activated cell sorter (MACS) from Miltenyi Biotec. Eluted NK cells were collected in culture medium containing IL-2 (200 U/ml). To determine the purity of the isolated NK cells, the cells were stained with a phycoerythrin (PE)-coupled anti-NK1.1 antibody and measured in a MACSQuant flow cytometer (Miltenyi Biotec). The final control revealed that 96% of the purified cells were positive for NK1.1.
Western blotting.
Cells were lysed on ice in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, cOmplete protease inhibitor [Roche], and 50 mM NaF) with 4 M urea. After centrifugation (14,000 rpm for 30 min at 4°C), postnuclear supernatants were analyzed in Western blots as described before (112). SDS-PAGE protein markers were obtained from Serva and Thermo Fisher Scientific. Fluorographs were scanned and quantified with GelEval 1.32 (FrogDance Software).
Flow cytometry.
For intracellular bacterial staining and titer determination, cells were collected, once washed, and fixed with 2% paraformaldehyde (PFA; 2% in PBS) for 10 min at room temperature (RT). Fixed cells were permeabilized in flow cytometric washing buffer (0.5% bovine serum albumin [BSA; Carl Roth], 0.5% saponin [Sigma-Aldrich] in PBS) at RT for 30 min. Subsequently, cells were incubated with the primary (1 h) and the respective secondary (30 min) antibodies (diluted in flow cytometric washing buffer) at RT. Staining of surface molecules occurred without PFA fixation and permeabilization. Antibody staining of cells was performed at 4°C and analyzed via a MACSQuant analyzer and MACSQuantify software (version 2.11; Miltenyi Biotec).
Immunofluorescence microscopy.
For fluorescence microscopy, cells were grown on coverslips and fixed for 20 min in 2% paraformaldehyde, blocked with 3% BSA, permeabilized with 0.1% saponin (Sigma-Aldrich), and incubated serially with the desired primary and corresponding secondary antibodies. Images were taken with an Axiovert 200M/ApoTome microscope. Colocalization was measured using AxioVision colocalization and Zen 2009 software (Zeiss).
IFN-γ and Grzm B ELISA.
To detect the amounts of IFN-γ and granzyme B (Grzm B) in the cell culture supernatant of C. burnetii-infected or uninfected NK cells, the mouse IFN-γ Platinum ELISA and the mouse Grzm B Platinum ELISA from eBioscience were used. For the analysis, 1 × 105 immortalized NK cells were cultured in 6-well plates and infected with C. burnetii (MOI, 10) for 0 to 72 h. Cell culture supernatants were collected at the respective time points, centrifuged for 30 min at 14,000 rpm (4°C), and analyzed via ELISA according to the manufacturer’s instructions for culture supernatants (50 μl of undiluted supernatants as well as supernatants diluted 1:50 and 1:100 in PBS).
Transmission electron microscopy.
For transmission electron microscopy (TEM) analysis, 5 × 105 NK cells were infected with C. burnetii (MOI, 10; washed 3 hpi). At different time points (0, 24, and 72 hpi), either cells or cell culture supernatants were processed further. Cells were carefully removed from the culture flasks and centrifuged at 300 × g for 5 min at 4°C. The resulting pellet was treated with fixing solution (2.5% glutaraldehyde buffered in 0.1 M sodium cacodylate [pH 7.2], 300 mosmol; Merck) and embedded in 1.8% low-melting-point agarose (Biozym). Small pieces were postfixed in 1.0% aqueous OsO4 and stained for contrast with uranyl acetate. After stepwise dehydration in ethanol, cells were cleared in propylene oxide, embedded in Glycid Ether 100 (Serva), and polymerized at 60°C for 3 days. Ultrathin sections counterstained with uranyl acetate and lead salts were analyzed with a Tecnai-Spirit TEM (FEI). For the collection and analysis of released bacteria, cell culture supernatants were centrifuged at 5,000 × g for 60 min at 4°C. Resulting bacterial pellets were directly embedded in 1.8% low-melting-point agarose (Biozym) and afterwards treated with a respective PFA fixing solution. The remainder of the procedure was carried out as described above.
Grzm B assay.
For activation, recombinant mouse Grzm B (100 μg/ml; R&D Systems) was first incubated with 10 μg/ml of recombinant mouse cathepsin C (R&D Systems) in activation buffer (50 mM morpholineethanesulfonic acid [MES], 50 mM NaCl [pH 5.5]) at 37°C for 4 h. The activated Grzm B was then diluted to 1 μg/ml in assay buffer (50 mM Tris [pH 7.5]) containing 106 IFU of Coxiella or Chlamydia. A corresponding experimental approach without Grzm B was chosen for the controls. After incubation for 4 h at 37°C, preincubated bacteria were directly used for reporter cell infection and then analyzed by flow cytometry.
Live-cell imaging.
A total of 1 × 105 NK cells were seeded in μ-dishes (ibidi; 35 mm, low) and infected with C. burnetii (tdTomato; MOI, 30; washed 3 hpi) to perform live-cell imaging (LCI). During the LCI experiments, the culture medium was supplemented with 20 mM HEPES. Time-lapse image series were acquired with a Leica DMi8 microscope (objective, HC PL APO 63×/1.30 GLYC UV; camera, DFC7000GT) and a Leica DMI6000 TCS SP5 confocal laser-scanning microscope (objective, HCX PL APO lambda blue 63×/1.40 oil UV; detection via hybrid detector [HyD]; scan speed, 1,400 Hz; pinhole, 280 μm; z-step size, 2 μm) at 37°C using a heating chamber (HT200; ibidi) and a heated incubation chamber, respectively. Time-lapse imaging was started at 24 hpi and continued up to 48 hpi. The frame rates are indicated in the respective figure legends. In the case of confocal microscopy, intra- and extracellular localization of the bacteria was checked by z-stack analysis at each recorded time point. A full z-stack (8 planes; z-step size, 2 μm) was acquired every 1,690 ms, resulting in a frame rate of about 35 per minute (frame = maximum z-projection of the entire stack per time point). To reduce the mobility of the examined cells during high-speed image acquisition at the confocal laser-scanning microscope, they were overlaid at 20 hpi with 0.6% low-melting-point agarose in regular KY-2 cell culture medium, supplemented with 20 mM HEPES as indicated above. Postprocessing, analysis, and annotations were performed with Fiji (113).
Statistical analysis.
Analysis of the obtained data is shown as the means ± standard deviations (SD) of three individual experiments and was estimated using GraphPad Prism 6 (GraphPad Software). Data were analyzed by t test.
Data availability.
All data generated or analyzed during this study are included in this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ralf M. Leonhardt for helpful comments and critically reading the manuscript. Robert Heinzen and Paul Beare are acknowledged for providing the pJB-CAT-ProA_mCherry plasmid. Petra Meyer, Mandy Jörn, and Stefanie Knöfel are acknowledged for their technical assistance.
The BMBF under the project numbers 01KI1726A/01KI1726C of “Q-GAPS” as part of the Research Network Zoonotic Infectious Diseases is gratefully acknowledged for financial support (A.L. and M.R.K.). The work was also financially supported by the Federal Excellence Initiative of Mecklenburg Western Pomerania and the European Social Fund (ESF) Grant KoInfekt (ESF/14-BM-A55-0002/16) (L.Z. and S.F.).
S.M. and M.R.K. directed and planned the studies. S.M. and M.R.K. designed and performed the experiments with the support of L.Z., K.F., R.J., C.F., and S.F. M.M. and A.L. created the tdTomato-plasmid, provided tdTomato-expressing C. burnetii, and contributed to bacterial stock preparation. Live-cell imaging experiments were performed by S.M., L.Z., R.J., and S.F. S.M. and K.F. carried out the electron microscopic studies. S.M., L.Z., K.F., S.F., A.L., and M.R.K. contributed to the interpretation of the results. S.M. and M.R.K. wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Topham NJ, Hewitt EW. 2009. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128:7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiao L, Gao X, Joyee AG, Zhao L, Qiu H, Yang M, Fan Y, Wang S, Yang X. 2011. NK cells promote type 1 T cell immunity through modulating the function of dendritic cells during intracellular bacterial infection. J Immunol 187:401–411. doi: 10.4049/jimmunol.1002519. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Dong X, Zhao L, Wang X, Wang Y, Yang X, Wang H, Zhao W. 2016. Natural killer cells regulate Th1/Treg and Th17/Treg balance in chlamydial lung infection. J Cell Mol Med 20:1339–1351. doi: 10.1111/jcmm.12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tseng CT, Rank RG. 1998. Role of NK cells in early host response to chlamydial genital infection. Infect Immun 66:5867–5875. doi: 10.1128/IAI.66.12.5867-5875.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radomski N, Franzke K, Matthiesen S, Karger A, Knittler MR. 2019. NK cell-mediated processing of Chlamydia psittaci drives potent anti-bacterial Th1 immunity. Sci Rep 9:4799. doi: 10.1038/s41598-019-41264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paolini R, Bernardini G, Molfetta R, Santoni A. 2015. NK cells and interferons. Cytokine Growth Factor Rev 26:113–120. doi: 10.1016/j.cytogfr.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Hewitt EW. 2003. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110:163–169. doi: 10.1046/j.1365-2567.2003.01738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shafer WM, Pohl J, Onunka VC, Bangalore N, Travis J. 1991. Human lysosomal cathepsin G and granzyme B share a functionally conserved broad spectrum antibacterial peptide. J Biol Chem 266:112–116. [PubMed] [Google Scholar]
- 9.Trapani JA, Smyth MJ. 2002. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 10.Smyth MJ, Kelly JM, Sutton VR, Davis JE, Browne KA, Sayers TJ, Trapani JA. 2001. Unlocking the secrets of cytotoxic granule proteins. J Leukoc Biol 70:18–29. [PubMed] [Google Scholar]
- 11.Dotiwala F, Sen Santara S, Binker-Cosen AA, Li B, Chandrasekaran S, Lieberman J. 2017. Granzyme B disrupts central metabolism and protein synthesis in bacteria to promote an immune cell death program. Cell 171:1125–1137.e11. doi: 10.1016/j.cell.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertzbach LD, van Haarlem DA, Hartle S, Kaufer BB, Jansen CA. 2019. Marek’s disease virus infection of natural killer cells. Microorganisms 7:588. doi: 10.3390/microorganisms7120588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul S, Lal G. 2017. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol 8:1124. doi: 10.3389/fimmu.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. 2011. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 89:216–224. doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 15.Cooper MA, Colonna M, Yokoyama WM. 2009. Hidden talents of natural killers: NK cells in innate and adaptive immunity. EMBO Rep 10:1103–1110. doi: 10.1038/embor.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zwirner NW, Domaica CI. 2010. Cytokine regulation of natural killer cell effector functions. Biofactors 36:274–288. doi: 10.1002/biof.107. [DOI] [PubMed] [Google Scholar]
- 17.Chalifour A, Jeannin P, Gauchat JF, Blaecke A, Malissard M, N'Guyen T, Thieblemont N, Delneste Y. 2004. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood 104:1778–1783. doi: 10.1182/blood-2003-08-2820. [DOI] [PubMed] [Google Scholar]
- 18.Marcenaro E, Ferranti B, Falco M, Moretta L, Moretta A. 2008. Human NK cells directly recognize Mycobacterium bovis via TLR2 and acquire the ability to kill monocyte-derived DC. Int Immunol 20:1155–1167. doi: 10.1093/intimm/dxn073. [DOI] [PubMed] [Google Scholar]
- 19.Lindemann RA. 1988. Bacterial activation of human natural killer cells: role of cell surface lipopolysaccharide. Infect Immun 56:1301–1308. doi: 10.1128/IAI.56.5.1301-1308.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hart OM, Athie-Morales V, O'Connor GM, Gardiner CM. 2005. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol 175:1636–1642. doi: 10.4049/jimmunol.175.3.1636. [DOI] [PubMed] [Google Scholar]
- 21.Qiu F, Maniar A, Diaz MQ, Chapoval AI, Medvedev AE. 2011. Activation of cytokine-producing and antitumor activities of natural killer cells and macrophages by engagement of Toll-like and NOD-like receptors. Innate Immun 17:375–387. doi: 10.1177/1753425910372000. [DOI] [PubMed] [Google Scholar]
- 22.Anel A, Aguilo JI, Catalan E, Garaude J, Rathore MG, Pardo J, Villalba M. 2012. Protein kinase c-theta (pkc-theta) in natural killer cell function and anti-tumor immunity. Front Immunol 3:187. doi: 10.3389/fimmu.2012.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. 2008. Functions of natural killer cells. Nat Immunol 9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 24.Vasilevsky S, Greub G, Nardelli-Haefliger D, Baud D. 2014. Genital Chlamydia trachomatis: understanding the roles of innate and adaptive immunity in vaccine research. Clin Microbiol Rev 27:346–370. doi: 10.1128/CMR.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong GM, de la Maza LM. 1988. Activation of mouse peritoneal macrophages in vitro or in vivo by recombinant murine gamma interferon inhibits the growth of Chlamydia trachomatis serovar L1. Infect Immun 56:3322–3325. doi: 10.1128/IAI.56.12.3322-3325.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roan NR, Starnbach MN. 2008. Immune-mediated control of Chlamydia infection. Cell Microbiol 10:9–19. doi: 10.1111/j.1462-5822.2007.01069.x. [DOI] [PubMed] [Google Scholar]
- 27.Eldin C, Melenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, Mege JL, Maurin M, Raoult D. 2017. From Q fever to Coxiella burnetii infection: a paradigm change. Clin Microbiol Rev 30:115–190. doi: 10.1128/CMR.00045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McQuiston JH, Childs JE. 2002. Q fever in humans and animals in the United States. Vector Borne Zoonotic Dis 2:179–191. doi: 10.1089/15303660260613747. [DOI] [PubMed] [Google Scholar]
- 29.Shannon JG, Heinzen RA. 2009. Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol Res 43:138–148. doi: 10.1007/s12026-008-8059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 31.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. doi: 10.1128/CMR.12.4.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frankel D, Richet H, Renvoise A, Raoult D. 2011. Q fever in France, 1985–2009. Emerg Infect Dis 17:350–356. doi: 10.3201/eid1703.100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shannon JG, Howe D, Heinzen RA. 2005. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A 102:8722–8727. doi: 10.1073/pnas.0501863102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benoit M, Barbarat B, Bernard A, Olive D, Mege JL. 2008. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol 38:1065–1070. doi: 10.1002/eji.200738067. [DOI] [PubMed] [Google Scholar]
- 35.Capo C, Lindberg FP, Meconi S, Zaffran Y, Tardei G, Brown EJ, Raoult D, Mege JL. 1999. Subversion of monocyte functions by Coxiella burnetii: impairment of the cross-talk between alphavbeta3 integrin and CR3. J Immunol 163:6078–6085. [PubMed] [Google Scholar]
- 36.Colombo MI, Gutierrez MG, Romano PS. 2006. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy 2:162–164. doi: 10.4161/auto.2827. [DOI] [PubMed] [Google Scholar]
- 37.Zhang G, Samuel JE. 2004. Vaccines against Coxiella infection. Expert Rev Vaccines 3:577–584. doi: 10.1586/14760584.3.5.577. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Russell-Lodrigue KE, Andoh M, Zhang Y, Hendrix LR, Samuel JE. 2007. Mechanisms of vaccine-induced protective immunity against Coxiella burnetii infection in BALB/c mice. J Immunol 179:8372–8380. doi: 10.4049/jimmunol.179.12.8372. [DOI] [PubMed] [Google Scholar]
- 39.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Read AJ, Erickson S, Harmsen AG. 2010. Role of CD4+ and CD8+ T cells in clearance of primary pulmonary infection with Coxiella burnetii. Infect Immun 78:3019–3026. doi: 10.1128/IAI.00101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Dow C, Wang P, Sidney J, Read A, Harmsen A, Samuel JE, Peters B. 2011. Identification of CD4+ T cell epitopes in C. burnetii antigens targeted by antibody responses. PLoS One 6:e17712. doi: 10.1371/journal.pone.0017712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turco J, Thompson HA, Winkler HH. 1984. Interferon-gamma inhibits growth of Coxiella burnetii in mouse fibroblasts. Infect Immun 45:781–783. doi: 10.1128/IAI.45.3.781-783.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grieshaber S, Swanson JA, Hackstadt T. 2002. Determination of the physical environment within the Chlamydia trachomatis inclusion using ion-selective ratiometric probes. Cell Microbiol 4:273–283. doi: 10.1046/j.1462-5822.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- 44.Lampe MF, Rohan LC, Skinner MC, Stamm WE. 2004. Susceptibility of Chlamydia trachomatis to excipients commonly used in topical microbicide formulations. Antimicrob Agents Chemother 48:3200–3202. doi: 10.1128/AAC.48.8.3200-3202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong Z, Luna Y, Yu P, Fan H. 2014. Lactobacilli inactivate Chlamydia trachomatis through lactic acid but not H2O2. PLoS One 9:e107758. doi: 10.1371/journal.pone.0107758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahmoud EA, Svensson LO, Olsson SE, Mardh PA. 1995. Antichlamydial activity of vaginal secretion. Am J Obstet Gynecol 172:1268–1272. doi: 10.1016/0002-9378(95)91491-9. [DOI] [PubMed] [Google Scholar]
- 47.Yasin B, Pang M, Wagar EA, Lehrer RI. 2002. Examination of Chlamydia trachomatis infection in environments mimicking normal and abnormal vaginal pH. Sex Transm Dis 29:514–519. doi: 10.1097/00007435-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 48.Schramm N, Bagnell CR, Wyrick PB. 1996. Vesicles containing Chlamydia trachomatis serovar L2 remain above pH 6 within HEC-1B cells. Infect Immun 64:1208–1214. doi: 10.1128/IAI.64.4.1208-1214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. 1996. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun 64:796–809. doi: 10.1128/IAI.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA. 2003. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect Immun 71:5855–5870. doi: 10.1128/iai.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun HS, Eng EW, Jeganathan S, Sin AT, Patel PC, Gracey E, Inman RD, Terebiznik MR, Harrison RE. 2012. Chlamydia trachomatis vacuole maturation in infected macrophages. J Leukoc Biol 92:815–827. doi: 10.1189/jlb.0711336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. 2002. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun 70:5816–5821. doi: 10.1128/iai.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulze-Luehrmann J, Eckart RA, Olke M, Saftig P, Liebler-Tenorio E, Lührmann A. 2016. LAMP proteins account for the maturation delay during the establishment of the Coxiella burnetii-containing vacuole. Cell Microbiol 18:181–194. doi: 10.1111/cmi.12494. [DOI] [PubMed] [Google Scholar]
- 54.Newton HJ, Roy CR. 2011. The Coxiella burnetii Dot/Icm system creates a comfortable home through lysosomal renovation. mBio 2:e00226-11. doi: 10.1128/mBio.00226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lührmann A, Newton HJ, Bonazzi M. 2017. Beginning to understand the role of the type IV secretion system effector proteins in Coxiella burnetii pathogenesis. Curr Top Microbiol Immunol 413:243–268. doi: 10.1007/978-3-319-75241-9_10. [DOI] [PubMed] [Google Scholar]
- 56.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hackstadt T, Williams JC. 1981. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A 78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maurin M, Benoliel AM, Bongrand P, Raoult D. 1992. Phagolysosomes of Coxiella burnetii-infected cell lines maintain an acidic pH during persistent infection. Infect Immun 60:5013–5016. doi: 10.1128/IAI.60.12.5013-5016.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Newton HJ, McDonough JA, Roy CR. 2013. Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS One 8:e54566. doi: 10.1371/journal.pone.0054566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mezouar S, Vitte J, Gorvel L, Ben Amara A, Desnues B, Mege JL. 2019. Mast cell cytonemes as a defense mechanism against Coxiella burnetii. mBio 10:e02669-18. doi: 10.1128/mBio.02669-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elliott A, Peng Y, Zhang G. 2013. Coxiella burnetii interaction with neutrophils and macrophages in vitro and in SCID mice following aerosol infection. Infect Immun 81:4604–4614. doi: 10.1128/IAI.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baca OG, Akporiaye ET, Aragon AS, Martinez IL, Robles MV, Warner NL. 1981. Fate of phase I and phase II Coxiella burnetii in several macrophage-like tumor cell lines. Infect Immun 33:258–266. doi: 10.1128/IAI.33.1.258-266.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schoenlaub L, Elliott A, Freches D, Mitchell WJ, Zhang G. 2015. Role of B cells in host defense against primary Coxiella burnetii infection. Infect Immun 83:4826–4836. doi: 10.1128/IAI.01073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howe D, Melnicakova J, Barak I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell Microbiol 5:469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- 65.Clark SE, Filak HC, Guthrie BS, Schmidt RL, Jamieson A, Merkel P, Knight V, Cole CM, Raulet DH, Lenz LL. 2016. Bacterial manipulation of NK cell regulatory activity increases susceptibility to Listeria monocytogenes infection. PLoS Pathog 12:e1005708. doi: 10.1371/journal.ppat.1005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tato CM, Martins GA, High FA, DiCioccio CB, Reiner SL, Hunter CA. 2004. Cutting Edge: innate production of IFN-gamma by NK cells is independent of epigenetic modification of the IFN-gamma promoter. J Immunol 173:1514–1517. doi: 10.4049/jimmunol.173.3.1514. [DOI] [PubMed] [Google Scholar]
- 67.Rebbig A, Matthiesen S, Lührmann A, Knittler MR. 2018. Flow cytometry as a new complementary tool to study Coxiella burnetii in cell cultures. J Microbiol Methods 151:39–43. doi: 10.1016/j.mimet.2018.05.022. [DOI] [PubMed] [Google Scholar]
- 68.Latomanski EA, Newton HJ. 2018. Interaction between autophagic vesicles and the Coxiella-containing vacuole requires CLTC (clathrin heavy chain). Autophagy 14:1710–1725. doi: 10.1080/15548627.2018.1483806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vodkin MH, Williams JC. 1988. A heat shock operon in Coxiella burnetii produces a major antigen homologous to a protein in both Mycobacteria and Escherichia coli. J Bacteriol 170:1227–1234. doi: 10.1128/jb.170.3.1227-1234.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elsa J, Duron O, Severine B, Gonzalez-Acuna D, Sidi-Boumedine K. 2015. Molecular methods routinely used to detect Coxiella burnetii in ticks cross-react with Coxiella-like bacteria. Infect Ecol Epidemiol 5:29230. doi: 10.3402/iee.v5.29230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Evenou JP, Wagner J, Zenke G, Brinkmann V, Wagner K, Kovarik J, Welzenbach KA, Weitz-Schmidt G, Guntermann C, Towbin H, Cottens S, Kaminski S, Letschka T, Lutz-Nicoladoni C, Gruber T, Hermann-Kleiter N, Thuille N, Baier G. 2009. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J Pharmacol Exp Ther 330:792–801. doi: 10.1124/jpet.109.153205. [DOI] [PubMed] [Google Scholar]
- 72.Krzewski K, Coligan JE. 2012. Human NK cell lytic granules and regulation of their exocytosis. Front Immunol 3:335. doi: 10.3389/fimmu.2012.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS. 2011. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol 9:e1001151. doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burkhardt JK, Hester S, Lapham CK, Argon Y. 1990. The lytic granules of natural killer cells are dual-function organelles combining secretory and pre-lysosomal compartments. J Cell Biol 111:2327–2340. doi: 10.1083/jcb.111.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Samanta D, Gilk SD. 2017. Measuring pH of the Coxiella burnetii parasitophorous vacuole. Curr Protoc Microbiol 47:6C.3.1–6C.3.11. doi: 10.1002/cpmc.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heinzen RA, Hackstadt T, Samuel JE. 1999. Developmental biology of Coxiella burnetii. Trends Microbiol 7:149–154. doi: 10.1016/s0966-842x(99)01475-4. [DOI] [PubMed] [Google Scholar]
- 77.Gomes MS, Paul S, Moreira AL, Appelberg R, Rabinovitch M, Kaplan G. 1999. Survival of Mycobacterium avium and Mycobacterium tuberculosis in acidified vacuoles of murine macrophages. Infect Immun 67:3199–3206. doi: 10.1128/IAI.67.7.3199-3206.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veras PS, de Chastellier C, Moreau MF, Villiers V, Thibon M, Mattei D, Rabinovitch M. 1994. Fusion between large phagocytic vesicles: targeting of yeast and other particulates to phagolysosomes that shelter the bacterium Coxiella burnetii or the protozoan Leishmania amazonensis in Chinese hamster ovary cells. J Cell Sci 107:3065–3076. [DOI] [PubMed] [Google Scholar]
- 79.Veras PS, Moulia C, Dauguet C, Tunis CT, Thibon M, Rabinovitch M. 1995. Entry and survival of Leishmania amazonensis amastigotes within phagolysosome-like vacuoles that shelter Coxiella burnetii in Chinese hamster ovary cells. Infect Immun 63:3502–3506. doi: 10.1128/IAI.63.9.3502-3506.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol 186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Larson CL, Beare PA, Howe D, Heinzen RA. 2013. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci U S A 110:E4770–E4779. doi: 10.1073/pnas.1309195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCaul TF, Williams JC. 1981. Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J Bacteriol 147:1063–1076. doi: 10.1128/JB.147.3.1063-1076.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ginsburg I, Neeman N, Duchan Z, Sela MN, James J, Lahav M. 1975. The effect of leukocyte hydrolases on bacteria: IV. The role played by artificial enzyme “cocktails” and tissue enzymes in bacteriolysis. Inflammation 1:41–56. doi: 10.1007/BF00918058. [DOI] [PubMed] [Google Scholar]
- 84.Bangalore N, Travis J, Onunka VC, Pohl J, Shafer WM. 1990. Identification of the primary antimicrobial domains in human neutrophil cathepsin G. J Biol Chem 265:13584–13588. [PubMed] [Google Scholar]
- 85.Amano K, Williams JC, McCaul TF, Peacock MG. 1984. Biochemical and immunological properties of Coxiella burnetii cell wall and peptidoglycan-protein complex fractions. J Bacteriol 160:982–988. doi: 10.1128/JB.160.3.982-988.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Samanta D, Clemente TM, Schuler BE, Gilk SD. 2019. Coxiella burnetii type 4B secretion system-dependent manipulation of endolysosomal maturation is required for bacterial growth. PLoS Pathog 15:e1007855. doi: 10.1371/journal.ppat.1007855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Di Russo Case E, Samuel JE. 2016. Contrasting lifestyles within the host cell. Microbiol Spectr 4:VMBF-0014-2015. doi: 10.1128/microbiolspec.VMBF-0014-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tassi I, Cella M, Presti R, Colucci A, Gilfillan S, Littman DR, Colonna M. 2008. NK cell-activating receptors require PKC-theta for sustained signaling, transcriptional activation, and IFN-gamma secretion. Blood 112:4109–4116. doi: 10.1182/blood-2008-02-139527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aguilo JI, Garaude J, Pardo J, Villalba M, Anel A. 2009. Protein kinase C-theta is required for NK cell activation and in vivo control of tumor progression. J Immunol 182:1972–1981. doi: 10.4049/jimmunol.0801820. [DOI] [PubMed] [Google Scholar]
- 90.Bonnema JD, Rivlin KA, Ting AT, Schoon RA, Abraham RT, Leibson PJ. 1994. Cytokine-enhanced NK cell-mediated cytotoxicity. Positive modulatory effects of IL-2 and IL-12 on stimulus-dependent granule exocytosis. J Immunol 152:2098–2104. [PubMed] [Google Scholar]
- 91.Vishwanath S, Hackstadt T. 1988. Lipopolysaccharide phase variation determines the complement-mediated serum susceptibility of Coxiella burnetii. Infect Immun 56:40–44. doi: 10.1128/IAI.56.1.40-44.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shannon JG, Howe D, Heinzen RA. 2005. Lack of dendritic cell maturation following infection by Coxiella burnetii synthesizing different lipopolysaccharide chemotypes. Ann N Y Acad Sci 1063:154–160. doi: 10.1196/annals.1355.024. [DOI] [PubMed] [Google Scholar]
- 93.Islam A, Lockhart M, Stenos J, Graves S. 2013. The attenuated Nine Mile phase II clone 4/RSA439 strain of Coxiella burnetii is highly virulent for severe combined immunodeficient (SCID) mice. Am J Trop Med Hyg 89:800–803. doi: 10.4269/ajtmh.12-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kishimoto RA, Rozmiarek H, Larson EW. 1978. Experimental Q fever infection in congenitally athymic nude mice. Infect Immun 22:69–71. doi: 10.1128/IAI.22.1.69-71.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ammerdorffer A, Schoffelen T, Gresnigt MS, Oosting M, den Brok MH, Abdollahi-Roodsaz S, Kanneganti TD, de Jong DJ, van Deuren M, Roest HJ, Rebel JM, Netea MG, Joosten LA, Sprong T. 2015. Recognition of Coxiella burnetii by Toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J Infect Dis 211:978–987. doi: 10.1093/infdis/jiu526. [DOI] [PubMed] [Google Scholar]
- 96.Martinez J, Huang X, Yang Y. 2010. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog 6:e1000811. doi: 10.1371/journal.ppat.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Athie-Morales V, O’Connor GM, Gardiner CM. 2008. Activation of human NK cells by the bacterial pathogen-associated molecular pattern muramyl dipeptide. J Immunol 180:4082–4089. doi: 10.4049/jimmunol.180.6.4082. [DOI] [PubMed] [Google Scholar]
- 98.Hackstadt T, Caldwell HD. 1985. Effect of proteolytic cleavage of surface-exposed proteins on infectivity of Chlamydia trachomatis. Infect Immun 48:546–551. doi: 10.1128/IAI.48.2.546-551.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gutiérrez-Martín CB, Ojcius DM, Hsia R, Hellio R, Bavoil PM, Dautry-Varsat A. 1997. Heparin-mediated inhibition of Chlamydia psittaci adherence to HeLa cells. Microb Pathog 22:47–57. doi: 10.1006/mpat.1996.0090. [DOI] [PubMed] [Google Scholar]
- 100.Ting LM, Hsia RC, Haidaris CG, Bavoil PM. 1995. Interaction of outer envelope proteins of Chlamydia psittaci GPIC with the HeLa cell surface. Infect Immun 63:3600–3608. doi: 10.1128/IAI.63.9.3600-3608.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Su H, Watkins NG, Zhang YZ, Caldwell HD. 1990. Chlamydia trachomatis-host cell interactions: role of the chlamydial major outer membrane protein as an adhesin. Infect Immun 58:1017–1025. doi: 10.1128/IAI.58.4.1017-1025.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buttrum L, Ledbetter L, Cherla R, Zhang Y, Mitchell WJ, Zhang G. 2018. Both major histocompatibility complex class I (MHC-I) and MHC-II molecules are required, while MHC-I appears to play a critical role in host defense against primary Coxiella burnetii infection. Infect Immun 86:e00602-17. doi: 10.1128/IAI.00602-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, Boucrot E, Kirchhausen T, Lieberman J. 2010. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115:1582–1593. doi: 10.1182/blood-2009-10-246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gorvel L, Textoris J, Banchereau R, Ben Amara A, Tantibhedhyangkul W, von Bargen K, Ka MB, Capo C, Ghigo E, Gorvel JP, Mege JL. 2014. Intracellular bacteria interfere with dendritic cell functions: role of the type I interferon pathway. PLoS One 9:e99420. doi: 10.1371/journal.pone.0099420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karlhofer FM, Orihuela MM, Yokoyama WM. 1995. Ly-49-independent natural killer (NK) cell specificity revealed by NK cell clones derived from p53-deficient mice. J Exp Med 181:1785–1795. doi: 10.1084/jem.181.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schafer W, Eckart RA, Schmid B, Cagkoylu H, Hof K, Muller YA, Amin B, Lührmann A. 2017. Nuclear trafficking of the anti-apoptotic Coxiella burnetii effector protein AnkG requires binding to p32 and Importin-alpha1. Cell Microbiol 19:e12634. doi: 10.1111/cmi.12634. [DOI] [PubMed] [Google Scholar]
- 107.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A 106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goellner S, Schubert E, Liebler-Tenorio E, Hotzel H, Saluz HP, Sachse K. 2006. Transcriptional response patterns of Chlamydophila psittaci in different in vitro models of persistent infection. Infect Immun 74:4801–4808. doi: 10.1128/IAI.01487-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schachter J, Wyrick PB. 1994. Culture and isolation of Chlamydia trachomatis. Methods Enzymol 236:377–390. doi: 10.1016/0076-6879(94)36028-6. [DOI] [PubMed] [Google Scholar]
- 112.Fiegl D, Kägebein D, Liebler-Tenorio EM, Weisser T, Sens M, Gutjahr M, Knittler MR. 2013. Amphisomal route of MHC class I cross-presentation in bacteria-infected dendritic cells. J Immunol 190:2791–2806. doi: 10.4049/jimmunol.1202741. [DOI] [PubMed] [Google Scholar]
- 113.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article.