

Abstract
Optimal CD8 T cell immunity is orchestrated by signaling events initiated by T cell receptor (TCR) recognition of peptide antigen in concert with signals from molecules such as CD28 and 4–1BB. The molecular mechanisms underlying the temporal and spatial signaling dynamics in CD8 T cells remain incompletely understood. Here we show that stimulation of naïve CD8 T cells with agonistic CD3 and CD28 antibodies, mimicking TCR and costimulatory signals, coordinately induces 4–1BB and cRel to enable elevated cytosolic cRel:IκBα complex formation and subsequent 4–1BB-induced IκBα degradation, sustained cRel activation, heightened IL-2 production and T cell expansion. NfkbiaNES/NES CD8 T cells harboring a mutated IκBα nuclear export sequence abnormally accumulate inactive cRel:IκBα complexes in the nucleus following CD3+CD28 stimulation, rendering them resistant to 4–1BB induced signaling and a disrupted chain of events necessary for efficient T cell expansion. Consequently, CD8 T cells in NfkbiaNES/NES mice poorly expand during viral infection, and this can be overcome by exogenous IL-2 administration. Consistent with cell-based data, adoptive transfer experiments demonstrated that the antiviral CD8 T cell defect in NfkbiaNES/NES mice was cell intrinsic. Thus, these results reveal that IκBα, via its unique nuclear export function, enables, rather than inhibits 4–1BB-induced cRel activation and IL-2 production to facilitate optimal CD8 T cell immunity.
Introduction
CD8 T cells are important effectors in immune responses to viral infections and cancer (1–3). Following antigen recognition, along with signaling emanating from the T cell receptor (TCR) complex and CD28, engagement of additional molecules construct a progressive wave of signaling events to ensure a contextually appropriate response (4). Within this complex signaling environment, dysregulated signals can result in suboptimal responses (4, 5). However, the temporal and spatial regulatory mechanisms that coordinate numerous events necessary for robust CD8 T cell immunity in vivo are incompletely understood.
Members of the tumor necrosis factor receptor (TNFR) superfamily, such as 4–1BB, OX40 and CD27, provide signals to T cells beyond CD3 and CD28 signaling for optimal cytokine production, survival, and memory formation (4). Unlike CD28, these TNFR superfamily members are expressed at low levels in naïve cells and are significantly induced following initial TCR stimulation in vivo (2). Similarly, their ligands are transiently upregulated following inflammatory signaling in professional antigen presenting cells as well as on B and T cells (6). Thus, the coordinated expression of TNFR superfamily members and their ligands tune the dynamics of CD8 T cell signaling and responses. However, whether intracellular signaling pathways are also dynamically modulated to coordinate with their respective receptor systems is unknown.
The NF-κB pathway is a key cell signaling pathway that is rapidly engaged following TCR/CD3, CD28, and TNFR superfamily activation. In mammals, the NF-κB family of transcription factors is composed of 5 family members, RelA (p65), cRel, RelB, p100/p52, and p105/p50, which form cell- and context-specific homo- and heterodimers (7–9). Canonical NF-κB complexes frequently consist of RelA:p50 or cRel:p50 dimers, while RelB:p52 dimers represent non-canonical complexes. Normally, canonical dimers are held in stable inactive cytoplasmic complexes by the IκB family of inhibitor proteins, of which the main family member, IκBα, preferentially binds RelA and cRel dimers, but not RelB:p52 dimers (10). Upon stimulation, IκB proteins are phosphorylated and degraded allowing free NF-κB dimers to enter the nucleus. Free, nuclear NF-κB then regulates expression of its target genes to modulate biological processes, including immune and inflammatory responses. RelA and cRel play particularly critical roles in T cell signaling and function (11–15). However, how NF-κB activities are dynamically and spatially regulated specifically during CD8 T cell immune responses remains incompletely understood.
Among the many NF-κB target genes is the Nfkbia gene which encodes the inhibitor IκBα (16, 17). A negative feedback loop of inhibition occurs when newly synthesized IκBα enters the nucleus, removes NF-κB from DNA, and exports inactive NF-κB complexes to the cytoplasm. This latter process is driven by the nuclear export sequence (NES) of IκBα (18–21). IκBβ, another commonly expressed IκB family member, does not have an NES (18, 21). In addition, both RelA:IκBα and cRel:IκBα complexes shuttle between the nucleus and the cytoplasm in unstimulated cells, while those complexed with IκBβ do not (19, 22). A classical NES is also present in RelA, but not cRel (23, 24); however, the physiological significance of the nuclear shuttling or nuclear export regulation of NF-κB:IκB complexes in specific biological contexts remains mostly unclear. To begin to address this, we previously created the NfkbiaNES/NES mouse model harboring a germline IκBα NES mutation and reported defects in B cell development and secondary lymphoid tissue formation (25). Contrary to B cells, however, T cell development and mature T cell populations in the periphery remained relatively unaffected in NfkbiaNES/NES mice. However, it remains unknown whether CD8 T cell activation and functions are disrupted in these mutant mice.
In this study, we investigated NfkbiaNES/NES CD8 T cell responses and found that following stimulation of the TCR and costimulatory receptors by means of agonistic antibodies against CD3 and CD28, respectively, the expression of inducible TNFR superfamily members occurred in tandem with a temporally timed cRel induction to assemble increased levels of cytosolic cRel:IκBα complexes in newly activated T cells relative to naive cells. These concurrent events set the stage for magnified and sustained cRel activation in CD8 T cells through the engagement of 4–1BB, resulting in heightened IL-2 production and CD8 T cell expansion. However, such a chain of events was disrupted in NfkbiaNES/NES CD8 cells due to abnormal accumulation of inactive cRel:IκBα complexes in the nucleus following CD3+CD28 stimulation. Consequently, NfkbiaNES/NES mice displayed defects in CD8 T cell expansion following infection with lymphocytic choriomengitis virus (LCMV) which induces a well-defined virus-specific T cell response (26, 27). Overall, our findings highlight how IκBα, a major NF-κB inhibitory protein, governs signaling dynamics and CD8 T cell immunity by temporally and spatially regulating critical accessory signaling events necessary for proper T cell immunity.
Materials and Methods
Mice
NfkbiaNES/NES mice were generated as previously described (25). Six- to twelve-week-old littermate NfkbiaWT/WT (WT) and NfkbiaNES/NES mice were age and sex matched for all experiments. Male host mice (B6.SJL-Ptprca Pepcb/BoyJ, Ly5.1) utilized for mixed bone marrow were obtained from The Jackson Laboratory (Bar Harbor, ME). Rel−/− mice were obtained from Dr. Jonathan Levenson (28) with permission from Dr. Hsiou-Chi Liou. The P14 LCMV GP33-specific TCR transgenic mice on the Ly5.1 background (provided by Dr. Rafi Ahmed, Emory University) were bred in University of Wisconsin–Madison. P14 mice were crossed with NfkbiaNES/NES mice to generate P14/NfkbiaWT/WT and P14/NfkbiaNES/NES mice. All experiments were approved and conducted in accordance by the Institutional Animal Care and Use Committee of the University of Wisconsin–Madison. All mice were housed and bred in specific pathogen-free conditions at the University of Wisconsin–Madison.
In vitro T cell stimulation and ELISA
Naive T cells were isolated from spleens using negative selection with the Pan T cell Isolation Kit II (Miltenyi Biotec) in addition to anti-CD44 microbeads (Miltenyi Biotec). Naïve CD8 T cells were isolated from spleens using negative selection with the Naive CD8a+ T cell Isolation kit (Miltenyi Biotec) and purity was confirmed by flow cytometry to be >95% CD8 CD44lo cells. Cells were cultured in RPMI medium supplemented with 10% FBS, 50 uM β-mercaptoethanol, 1X GlutaMAX (Life Technologies), 100 U/mL Penicillin and 100 ug/mL Streptomycin. Cell culture plates were coated with anti-CD3 (Bio X Cell, Clone 17A2) and anti-CD28 (Bio X Cell, Clone 37.51) antibodies overnight at 4°C or for 2 hours at 37°C. For 4–1BB and OX40 stimulation, soluble anti-4–1BB (Bio X Cell, Clone 3H3) or anti-OX40 (Bio X Cell, Clone OX86) antibodies were added at the start of stimulation with anti-CD3 and anti-CD28. For CD27 stimulation, soluble anti-CD27 antibody (eBioscience, Clone LG.3A10) was crosslinked with anti-Rat and anti-Hamster Ig κ Light Chain (BD, #550336) and added at the start of stimulation. For the IL-2 ELISA (eBioscience), cells were cultured in 96-well plates for 48 hours and supernatants were collected and frozen at −80°C until ready to assay. ELISA data are represented as the mean +/− SEM of at least three technical replicates and significance was determined using an unpaired Student’s t test. Generated p values are two-tailed and p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) was considered statistically significant. For proliferation assays, splenocytes were stained with 10 uM CFSE (Invitrogen) in RPMI with 10% FBS for 10 minutes at room temperature. Naive CD8 T cells were then isolated as described above. For cell survival assays, 1×106 naive CD8 T cells were stimulated with plate bound 5 μg/mL each of anti-CD3 and anti-CD28 antibodies, and soluble anti-4–1BB antibody for 48 hours in 24-well plates. Following this initial 48 hours, viable cells were quantified by the MACSquant flow cytometer and 4×105 cells were re-plated in 24 well plates in RPMI media supplemented with 10% FBS, 50 uM β-mercaptoethanol, 1X GlutaMAX (Life Technologies), 100 U/mL Penicillin and 100 ug/mL Streptomycin. Viable cells were quantified by the MACSquant flow cytometer 48 hours following re-plating. For cell survival studies, data are represented as the mean +/− SEM of at least three technical replicates and significance was determined using an unpaired Student’s t test. Generated p values are two-tailed and p < 0.001 (***) was considered statistically significant.
Electrophoretic mobility shift assay (EMSA) and supershift assay
T cells were lysed in Totex buffer (20 mM HEPES [pH 7.9], 350 mM NaCl, 20% glycerol, 1% NP-40, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT) with 1X HALT protease inhibitor cocktail (Thermo Fisher Scientific) and 1 uM DTT. Cell extracts (6–10 μg) were incubated with 1 μg of poly(dI-dC) and EMSA binding buffer (75 mM NaCl, 15 mM Tris HCl [pH 7.5], 1.5 mM EDTA, 1.5 mM DTT, 7.5% glycerol, 0.3% NP-40, 20 μg/mL BSA) in a total reaction volume of 9 uL for 20 minutes at 4°C. For supershift assays, 1 μL of each antibody (0.2 μg) for RelA (Santa Cruz, clone C-20), cRel (Santa Cruz, clone C) or RelB (Santa Cruz, clone C-19) was added to cell extracts, with the reaction volume not exceeding 10 μL. One μL of 32P labeled double stranded oligonucleotides containing the κB site from the Igκ gene (5’-TCAACAGAGGGGACTTTCCGAGAGGCC-3’), the IL-2 gene (5’-GACCAAGAGGGATTTCACCTAAATCCA-3’), and the Oct-1 consensus site (Promega) was added to the reaction and incubated for 20 minutes at room temperature and spun down at 14,000 RPM prior to sample loading and separation on a 4% native polyacrylamide gel, which was then dried and exposed to film or a storage phosphor screen. A Typhoon Biomolecular Imager (GE Healthcare) and accompanying ImageQuant TL software were used to scan and quantify resulting EMSAs. NF-κB binding was first normalized to Oct-1 binding and fold change was determined. Data is represented as the mean +/− SEM of at least three biological replicates and significance was determined using a paired Student’s t test. Generated p values are two-tailed and p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) was considered statistically significant.
Immunoblotting and immunoprecipitation analyses
Samples were lysed in Totex buffer and normalized by total protein amount before being diluted 1:1 with 2X Laemmli sample buffer and boiled for 10 minutes. Protein extracts were separated on a 10% SDS-PAGE gel and then transferred to a PVDF membrane (Millipore). Membranes were blocked in PBS containing 0.2% Tween 20 (PBST) and 5% milk for 1 hour. The membrane was then probed with the antibodies against RelA (Santa Cruz, clone C-20), RelB (Santa Cruz, clone C-19), cRel (Santa Cruz, clone C), p105/p50 (Santa Cruz, clone NLS), p100/p52 (Santa Cruz, clone C-5), IκBα (Santa Cruz, clone C-21), IκBβ (Santa Cruz, clone C-20) or tubulin (Calbiochem, clone DM1A) diluted in PBST overnight. The following day, membranes were washed 3 times in PBST and incubated with the secondary anti-rabbit IgG (GE, NA934V) or anti-goat IgG antibody (GE, NA931V) conjugated to horseradish peroxidase. Membranes were washed 3 times in PBST and developed using Amersham ECL Western Blotting Detection Reagents (GE Healthcare). To detect degradation of IκBα and IκBβ, cells were incubated with the protein synthesis inhibitor, cycloheximide, at 20 μg/mL for 30 minutes at 37°C prior to stimulation. For IκBα:NF-κB co-immunoprecipitation (IP) analyses, samples were lysed in IP buffer (20 mM Tris, 250 mM NaCl, 3 mM EDTA, 3mM EGTA, 0.5% NP-40, 1mM DTT, and 1X HALT) for 20 minutes at 4°C. Cells were then completely lysed by 3 cycles of freezing by incubating tubes containing cells in 70% ethanol in dry ice for 2 minutes and thawing in a 37°C water bath for 2 minutes. This was followed by an incubation at 4°C for 10 minutes and centrifugation at 14,000 RPM for 10 minutes at 4°C. Total protein concentration in lysates was determined and approximately 150–200 ug of protein was brought to a final volume of 500 μL in IP buffer. Samples were rotated at 4°C with 2 μg of anti-IκBα antibody (Santa Cruz, clone C-21) for 1 hour. In the meantime, protein A sepharose beads (GE Healthcare) were equilibrated with IP buffer. Approximately 50 μL of 1:1 equilibrated beads were added to each IP sample. Samples were rotated overnight at 4°C. Beads were washed with IP buffer 3 times and then resuspended in 100 μL Laemmli buffer, boiled for 10 minutes, and loaded onto a 10% SDS-PAGE gel for analysis by immunoblotting.
Immunofluorescence staining and ImageStream acquisition and analysis
To induce maximal RelA, cRel, and IκBα nuclear localization, naïve and activated CD8 T cells and B cells were incubated with 20 ng/mL Leptomycin B (LMB) (Cayman Chemical) for 40 minutes at 37°C. Activated CD8 T cells were stained with Fixable Viability Dye (FVD) eFluor 780 (eBioscience) prior to fixation. CD8 T cells and B cells were fixed with 4% paraformaldehyde for 10 minutes and permeabilized with 0.3% Triton X-100 for 30 minutes. Following permeabilization, cells were stained with anti-RelA (Cell Signaling #D14E12, 1:200), anti-cRel (Santa Cruz #SC-71, 1:150), or anti-IκBα (Santa Cruz #SC-371,1:200) antibodies with 1.5% normal goat serum overnight at 4°C. The following day, primary antibody was removed and cells were incubated with a goat anti-rabbit secondary antibody directly conjugated to Alexa Fluor 488 (Cell Signaling) in 1.5% normal goat serum for 1 hour at 4°C. Nuclear staining was achieved with 5 uM DRAQ5 (BD Biosciences) for 10 minutes at 37°C immediately prior to acquisition. Cells were collected at 60X magnification and the low speed/high sensitivity setting using an ImageStream flow cytometer (Amnis ImageStreamX Mk II, 2 lasers [488 nm and 642 nm] and 6 channels [457/45, 533/55, 577/35, 610/30, 702/85, 762/35]). Single, focused, and viable cells were collected based on Area and Aspect Ratio in the brightfield channel, Gradient RMS in the nuclear channel, and exclusion of FVD eFluor 780. Utilizing the IDEAS software package, the “similarity score” was calculated utilizing at least 1500–2000 cells based on positive staining for RelA, cRel, or IκBα and DRAQ5.
Gene expression analysis and RNA sequencing
Naïve WT and NfkbiaNES/NES CD8 T cells stimulated with plate bound 5 μg/mL anti-CD3 and 5 μg/mL anti-CD28 antibodies with or without soluble anti-4–1BB antibody for 12 hours. Cells were lysed in TRIzol reagent (Invitrogen) and frozen at −80° C until total RNA was extracted with the Direct-zol RNA MiniPrep Plus (Zymo Research) kit. For RNA-seq, libraries were generated with the TruSeq Stranded mRNA Library Prep Kit for NeoPrep (Illumina) and sequencing was performed on an Illumina HiSeq 2000.
RNA-seq processing and analysis
Adapter-trimmed reads were aligned to the mouse (mm10) genome using STAR with default parameters (29). Only primary mapped reads with alignment score >30 were selected by using “samtools view” with “–F 2820” and “–q 30” and then input into featureCounts (30) to get counts per gene using GENCODE M6 (GRCm38.p4) annotation. The number of raw reads from sequencing is approximately 22 million per sample and the final number of reads after filtering was approximately 16 million reads per sample. The average Pearson’s correlation between biological replicates was approximately 0.995 using log2 CPM (counts per million reads).
To obtain the final “NES dependent” gene list, we performed two rounds of differential gene expression analysis by using R package EdgeR (glmFit and glmLRT functions) (31). First, we selected 4–1BB induced genes by testing between 12 hr 4–1BB stimulated samples (12hr+) against 12 hr unstimulated samples (12hr-) (N = 3 for each time point) in WT (Log2 fold change > 1 and FDR < 0.01). A second test between WT vs. NES at 12 hours of 4–1BB stimulation was performed to determine NES dependency (LFC < −0.5 and FDR < 0.05). Lastly, by intersecting the genes from this two-round analysis, we divided these genes into three categories: 1) induced in WT but induction is not NES dependent (“NES independent”); 2) induced in WT and induction is NES dependent (“NES dependent”); 3) not induced in WT and expression is reduced in NES mutant (“NES reduced”). Of these, NES-dependent genes’ Ensembl IDs were entered into DAVID 6.8 to perform functional enrichment analysis on KEGG_PATHWAY. The RPKM for these genes were normalized to [0,1] range for each gene and then used to produce an expression heatmap. The top 50 induced genes in WT were ranked based on induction fold change in WT and selected after filtering genes with average RPKM < 3 for WT 12hr-.
Motif enrichment analysis
To study the binding motif enrichment of RelA:p50, cRel:p50, and cRel:cRel (32), position weight matrixes (PWMs) (33) were inputted into HOMER motif discovery software (34) and their occurrence was scanned for in the promoter regions of “NES dependent” and “NES independent” genes (promoters defined as −1000bp to 300bp of TSS). Fisher’s exact test was performed to calculate the fold enrichment and p-value for each motif in “NES dependent” against “NES independent” genes.
Viral infections
Six- to eight- week old WT and NfkbiaNES/NES mice were infected i.p. with 2×105 PFU of the Armstrong strain of lymphocytic choriomeningitis virus (LCMV) to induce an acute infection. For re-infection of mixed bone marrow chimeras, LCMV immunized mice were infected i.v. with 2×106 PFU of the clone 13 strain of LCMV. Plaque assays on Vero cells were used for quantification of infectious LCMV, as previously described (27).
Flow cytometry and cell staining
Peripheral blood was collected in 4% sodium citrate and mononuclear cells were isolated following density centrifugation with Ficoll Histopaque (Sigma). Single cell suspensions of mononuclear cells from spleens were obtained using a standard procedure. Approximately 1×106 cells were stained per sample. Cells were first stained with Fixable Viability Dye eFluor 780 or eFluor 450 (eBioscience), followed by staining with MHC Class I tetramers specific for the LCMV epitope H2-Db NP396–404 and/or H2-Db GP33–41 (NIH Tetramer Facility) and antibodies against cell surface markers, CD8 (BD Biosciencies, Clone 53–6.7), CD44 (BD Biosciencies, Clone IM7), IFN-γ (BD Biosciences, Clone XMG1.2), CD62L (BD Biosciences, Clone MEL-14), KLRG (BD Biosciences, Clone 2F1), Ly5.1 (BD Biosciences, Clone A20), Ly5.2 (BD Biosciences, Clone 45.2), Thy1.1 (BD Biosciences, Clone OX-7) and Thy1.2 (BD Biosciences, Clone 53–2.1). Cells were then fixed with 4% paraformaldehyde. For intracellular cytokine staining, splenocytes were stimulated ex vivo with LCMV epitope NP396–404 (200 ng/mL), Golgi Plug (BD Biosciences), and IL-2 (2.5 ng/mL) for 5 hours at 37°C. Following incubation, cells were stained with cell surface markers first, fixed and permeabilized utilizing the Intracellular Fixation & Permeabilization Buffer Set (eBioscience) and then stained with anti-IFN-γ antibody. For LCMV studies, data are represented as the mean +/− SEM of biological replicates and significance was determined using an unpaired Student’s t test. Generated p values are two-tailed, and p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) was considered statistically significant.
Generation of mixed bone marrow chimeras
Bone marrow cells were obtained from the femurs and tibias of host strain male (B6.SJL-Ptprca Pepcb/BoyJ, Ly5.1) mice, WT (Ly5.2) and NfkbiaNES/NES mice (Ly5.2). An equal number of bone marrow cells from host strain mice (Ly5.1) was mixed with an equal number of cells from either WT (Ly5.2) or NfkbiaNES/NES (Ly5.2) mice. A total of 8×106 cells were adoptively transferred into lethally irradiated (450 rads x 2) male host mice (Ly5.1). Following reconstitution, host mice were given neomycin (25 μg/mL) and Polymyxin B (13 μg/mL) in drinking water every other day for 6 weeks. Reconstitution of the hematopoietic compartment was assessed 6 weeks following reconstitution by analysis of peripheral blood. Mice were infected with LCMV (Armstrong strain) one week later.
P14/NfkbiaWT/WT and P14/NfkbiaNES/NES adoptive cell transfer assay
For P14 adoptive transfer studies, naïve CD8 T cells were purified from P14/NfkbiaWT/WT and P14/NfkbiaNES/NES mice spleens and 104 cells of the appropriate genotype were transferred into recipient mice (Jackson Laboratory, B6.PL-Thy1a/CyJ). Mice were infected with LCMV (Armstrong strain) 24 hours later as above.
Results
IκBα NES is not required for acute NF-κB activation and proliferation in CD3+CD28-stimulated CD8 T cells
To begin to analyze the activation and functional status of T cells in NfkbiaNES/NES mice (25), we first investigated the responses of naïve splenic T cells to TCR and costimulatory CD28 signaling by stimulating them on culture plates coated with agonistic anti-CD3 and anti-CD28 antibodies (referred from here on as “CD3+CD28”). The amounts of antibodies used were based on preliminary titration studies (not shown). Electrophoretic mobility shift assay (EMSA) demonstrated that basal and CD3+CD28-induced NF-κB activities were partially reduced in NfkbiaNES/NES T cells relative to NfkbiaWT/WT (WT) littermate control cells (Supplemental Figure 1A). To specifically investigate the impact of mutant IκBα on CD8 T cells, naïve splenic CD8 T cells were analyzed as above. We evaluated the 2 hour time point to examine acute CD3+CD28 induced activation potential and further performed supershift assays to decipher which of the NF-κB subunits was involved. Basal NF-κB activity, primarily composed of RelA, was significantly reduced in NfkbiaNES/NES CD8 cells relative to WT counterparts (Figure 1A). Reduced basal NF-κB activity suggested the possibility that expression of NF-κB-regulated NF-κB family members might be reduced in NfkbiaNES/NES CD8 T cells as in the case with splenic mutant B cells (25), which was confirmed by western blot analysis showing reduced expression of cRel, RelB, p105/p50 and p100, but not RelA (Figure 1B). IκBα expression was slightly increased while IκBβ expression was slightly reduced in NfkbiaNES/NES cells, as was the case in mutant splenic B cells (25). Despite these expression differences of NF-κB and IκB proteins, activation of both RelA and cRel complexes following CD3+CD28 stimulation was not significantly inhibited in the NfkbiaNES/NES CD8 T cells (Figure 1A). Stimulation of NfkbiaNES/NES and WT CD8 T cells with CD3+CD28 induced comparable levels of: (i) IL-2 receptor α chain (CD25) and TNFR family member (4–1BB, CD27, OX40) expression, as analyzed by flow cytometry (Figures 1C), (ii) IL-2 secretion, as assessed by ELISA with Rel−/− CD8 T cells serving as a control (Figures 1D), and (iii) cell proliferation, as measured with a CFSE dilution assay (Figures 1E). Thus, contrary to B cells whose maturation, activation and proliferation were significantly defective in NfkbiaNES/NES mice (25), the IκBα NES was not necessary for optimal acute CD3+CD28-induced NF-κB activation, activation marker expression, IL-2 secretion and proliferation in CD8 T cells.
Figure 1. IκBα NES is not necessary for efficient CD3+CD28 induced NF-κB activation and proliferation in CD8 T cells.
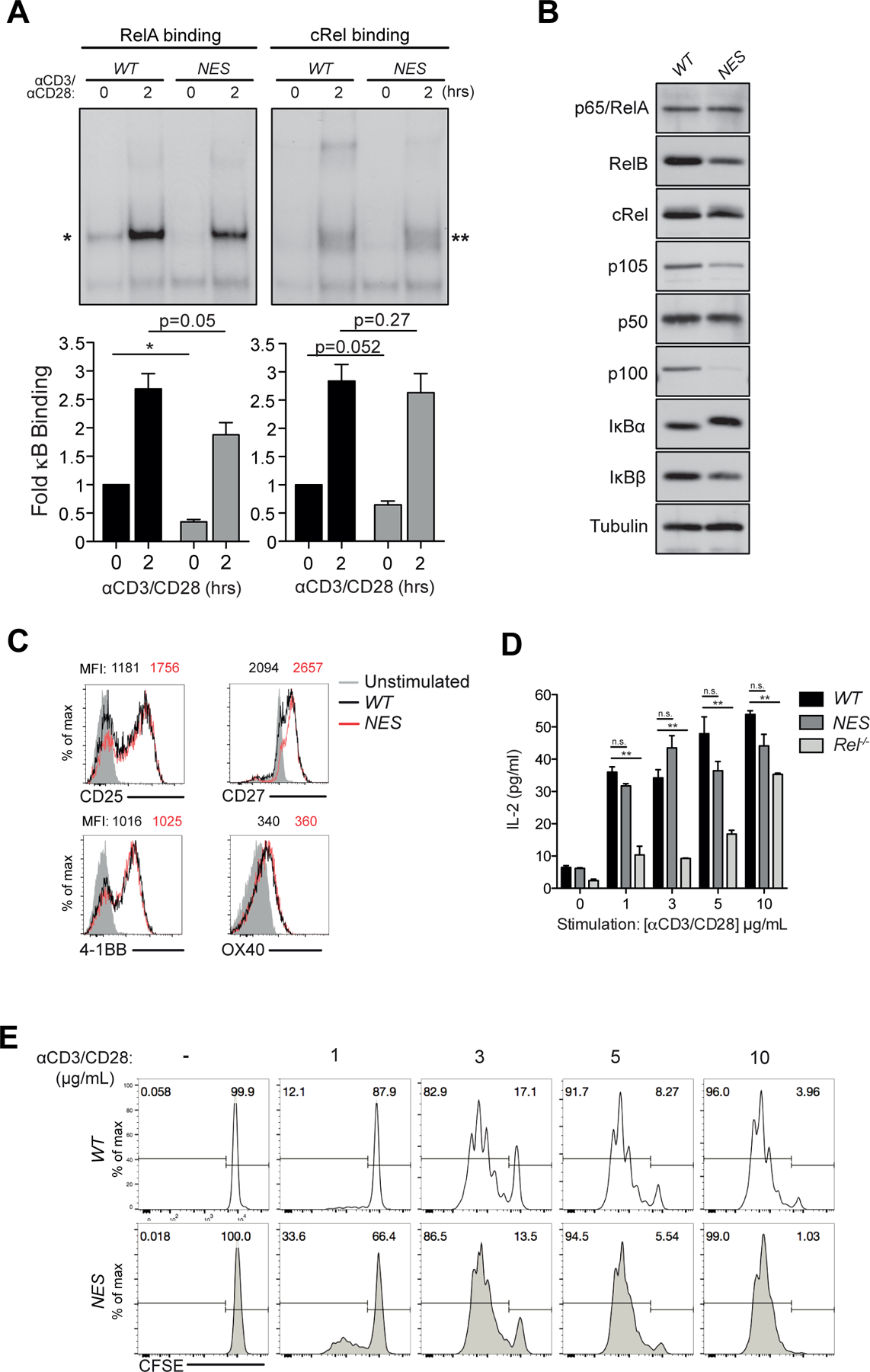
(A) Naive NfkbiaWT/WT (WT) and NfkbiaNES/NES (NES) T cells were stimulated with 5 μg/mL each anti-CD3 and anti-CD28 antibodies and cell lysates were prepared. Supershift EMSA analysis with anti-cRel and anti-ReA antibodies was used to detect RelA and cRel complexes, respectively. Below, quantification of RelA (left, *) and cRel (right, **) binding, relative to unstimulated WT T cells and normalized to Oct-1 binding, is shown as fold change in comparison to unstimulated samples from three independent experiments with the mean +/− SEM with statistical significance at *p < 0.05 or as indicated.
(B) Naive CD8 T cells were purified from WT and NfkbiaNES/NES spleens and total cell extracts were made. Immunoblot analysis of indicated NF-κB and IκB family members is shown with anti-tubulin as a loading control.
(C) Naive WT and NfkbiaNES/NES CD8 T cells were stimulated with 5 µg/mL each of anti-CD3 and anti-CD28 antibodies for 24 hours. Cell were analyzed by flow cytometry with unstimulated CD8 T cells shown in shaded grey in each histogram. Median fluorescence intensity (MFI) of WT (black) and NfkbiaNES/NES (red) CD8 T cells is shown on top of each histogram. Data is representative of experiments performed independently at least three times.
(D) WT, NfkbiaNES/NES, Rel−/− CD8 T cells stimulated for 48 hours with 5 µg/mL anti-CD3 and anti-CD28 antibodies. Supernatants were analyzed by ELISA for IL-2. Data is the mean +/− SEM of three technical replicates with statistical significance at *p < 0.05 and ** p < 0.01 and is representative of experiments performed independently at least three times.
(E) Naive WT and NfkbiaNES/NES CD8 T cells were labeled with CFSE and stimulated with the indicated concentrations of anti-CD3 and anti-CD28 antibodies for 72 hours. Percentages of divided cells are shown. Data is representative of experiments performed independently at least five times.
IκBα NES mutation causes 4–1BB signaling defects and functional deficits in CD8 T cells
Proper T cell responses require signaling through not only TCR activation but also additional co-stimulatory signals (4). Because 4–1BB was expressed at low to undetectable levels in naïve cells, but was robustly induced relative to other TNFR family members in both WT and NfkbiaNES/NES CD8 T cells (Figures 1C), we next focused our analyses on 4–1BB signaling. Stimulation of naïve CD8 T cells with an agonistic 4–1BB antibody elicited no measurable NF-κB signaling (not shown). Next, in order to measure the impact of the NES mutation on 4–1BB signaling, we stimulated naïve CD8 T cells with CD3+CD28 antibodies in the presence or absence of 4–1BB antibody. We reasoned that 4–1BB would be induced in CD3+CD28 activated CD8 T cells, which would then enable stimulation via the 4–1BB antibody, simulating the dynamics of naïve CD8 T cell stimulation by antigen presenting cells (35). Stimulation with CD3+CD28+4–1BB activated both cRel and RelA complexes in WT T cells (Figure 2A), but the dominant NF-κB complexes shifted from RelA in naïve cells activated by CD3+CD28 to cRel in CD3+CD28+4–1BB-stimulated CD8 T cells (Figure 2B). In striking contrast, activation of RelA and cRel in NfkbiaNES/NES cells stimulated with 4–1BB was significantly reduced relative to WT controls and the impact of inhibition was greater for cRel than RelA (an average of 2.3-fold vs 1.5-fold, respectively) (Figure 2B). Similar differences were also evident at 12 hours of CD3+CD28+4–1BB stimulated T cells (Supplemental Figure 2A). Although certain members of the TNFR superfamily, such as CD40, can strongly activate non-canonical NF-κB (RelB:p52 complex) in B cells (Supplemental Figure 2B) (36), supershift analyses of 4–1BB-stimulated WT and NfkbiaNES/NES CD8 T cells revealed the lack of detectable RelB complexes (Supplemental Figure 2C). In addition, while the processing of the precursor protein p100 to its active p52 form was nearly completely induced by CD40 engagement in WT and NfkbiaNES/NES B cells (Supplemental Figure 2D), the majority of p100 remained unprocessed in 4–1BB-stimulated CD8 T cells of both genotypes (Supplemental Figure 2E). Thus, activation of only canonical NF-κB complexes was significantly inhibited in 4–1BB-stimulated NfkbiaNES/NES CD8 T cells.
Figure 2. IκBα nuclear export culminates in 4–1BB signaling defects in NfkbiaNES/NES CD8 T cells.
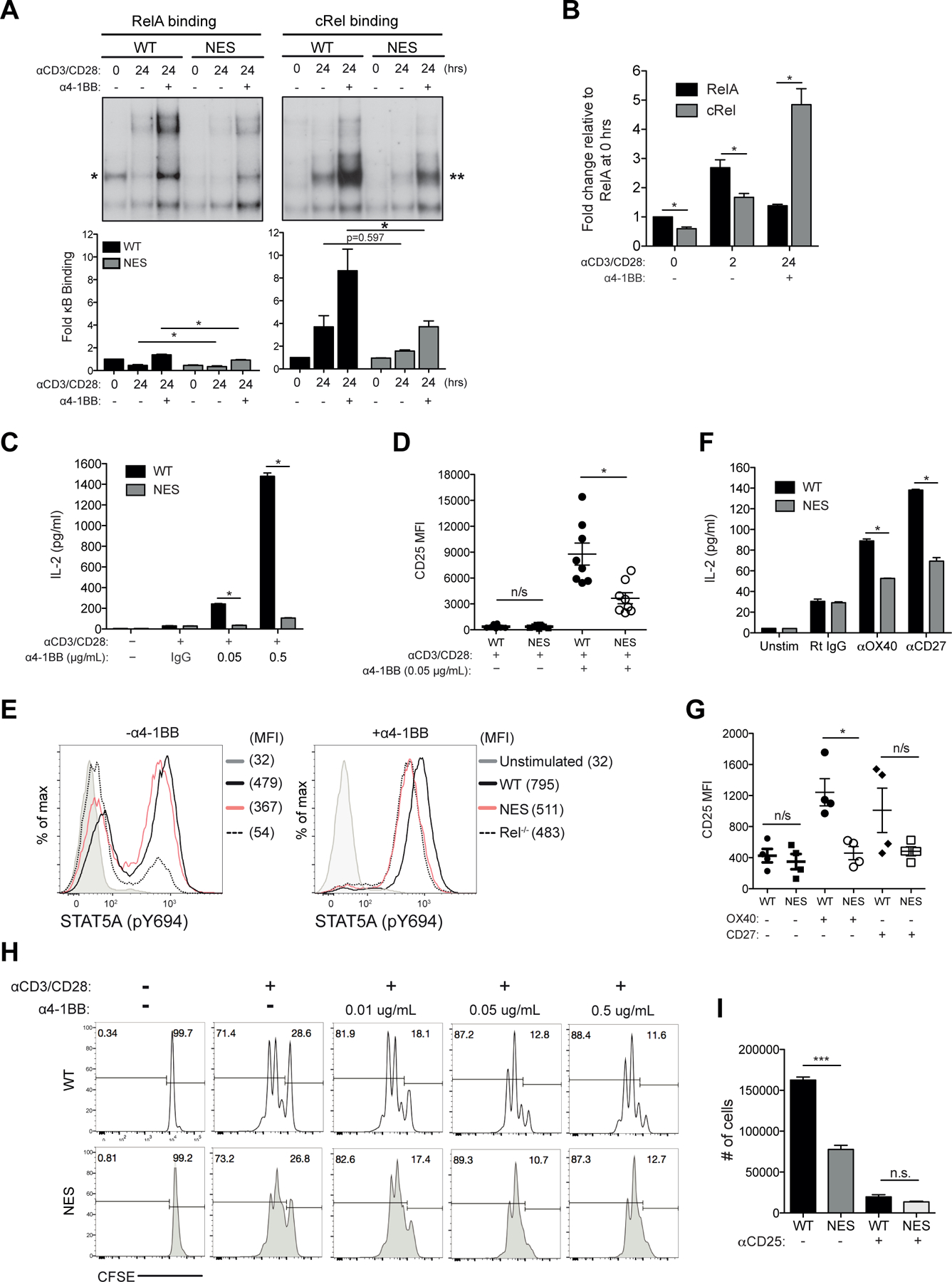
(A) Naïve WT and NfkbiaNES/NES CD8 T cells were stimulated for 24 hours with 5 μg/mL anti-CD3 and anti-CD28 antibodies with or without 0.05 μg/mL anti-4–1BB antibody. Supershift analysis using anti-cRel and anti-RelA antibodies was performed to detect RelA (left, *) and cRel (right, **) complexes, respectively. Below, quantification of indicated complexes is shown as fold change in comparison to unstimulated samples and normalized to Oct-1 binding. Data plotted is the mean +/− SEM and includes 3 independent experiments with statistical significance at *p < 0.05.
(B) Absolute RelA and cRel binding levels in Figure 1A & 2A relative to the absolute RelA binding level at 0 hrs (Figure 2A) and normalized to Oct-1 binding are shown with SEM and statistical significance at *p < 0.05.
(C) Naïve WT and NfkbiaNES/NES CD8 T cells were stimulated for 48 hours with 5 μg/mL anti-CD3 and anti-CD28 antibodies with or without increasing concentrations of anti-4–1BB antibody or normal rat IgG (IgG) control. Supernatants were analyzed by ELISA for IL-2. Data is the mean +/− SEM of three technical replicates and *p < 0.05 and representative of three independent experiments.
(D) Samples stimulated as in (C) were analyzed by flow cytometry for CD25 expression. Each data point represents one independent biological replicate with the SEM and *p < 0.05.
(E) Naïve WT, NfkbiaNES/NES, and Rel−/− CD8 T cells were stimulated as in (C) for 48 hours and analyzed by flow cytometry for phosphorylated STAT5.
(F) Naïve WT and NfkbiaNES/NES CD8 T cells were unstimulated or stimulated with 5 μg/mL each of anti-CD3 and anti-CD28 antibodies with control Rat IgG, anti-OX40 antibody (5 μg/mL), or anti-CD27 antibody (5 ug/mL) crosslinked by anti-rat and anti-hamster Ig for 48 hours. Supernatants were analyzed by ELISA for IL-2. Data is the mean +/− SEM of three technical replicates and *p < 0.05 and representative of three independent experiments.
(G) Samples stimulated as in (F) analyzed by flow cytometry for CD25 expression. Each data point represents one independent biological replicate and SEM is indicated with *p<0.05.
(H) Naive WT and NfkbiaNES/NES CD8 T cells isolated from the spleen were labeled with CFSE and stimulated with 5 µg/mL of anti-CD3 and anti-CD28 antibodies along with the indicated concentrations of anti-4–1BB for 72 hours. Percentages of undivided cells (right) and divided cells (left) are shown. Data is representative of experiments performed independently at least 3 times.
(I) Naïve WT and NfkbiaNES/NES CD8 T cells were stimulated as in Supplemental Figure 2F. Absolute live cells numbers were quantified by flow cytometry. Data is representative of experiments performed independently at least 3 times.
The change in cRel dominance of activation following 4–1BB stimulation in WT CD8 T cells (Figure 2B) correlated with significantly larger amounts of IL-2 production in response to CD3+CD28+4–1BB stimulation in comparison to CD3+CD28 alone (Figure 2C). 4–1BB-induced IL-2 production was significantly defective in NfkbiaNES/NES CD8 T cells, approaching the level of the CD3+CD28 alone condition (Figure 2C). Furthermore, upregulation of CD25 (IL-2Rα) by CD3+CD28+4–1BB stimulation, which was again much higher than that induced by CD3+CD28, was also significantly reduced in NfkbiaNES/NES CD8 T cells (Figure 2D). The level of phosphorylated STAT5A (pY694) downstream of CD25 signaling under CD3+CD28+4–1BB stimulation in WT T cells was reduced in NfkbiaNES/NES cells, which was similar to that detected in similarly stimulated Rel−/− cells (Figure 2E, right). Thus, the CD25 induction defect in the mutant cells likely stemmed from reduced feed forward signaling mediated by IL-2 (37). Moreover, exposure of naïve NfkbiaNES/NES CD8 T cells to CD3+CD28 and agonistic antibodies to OX40 or CD27 also demonstrated defective IL-2 production (Figure 2F) and CD25 expression (for OX40 stimulation) (Figure 2G). These results suggested that NfkbiaNES/NES CD8 T cell defects were not limited only to 4–1BB but were shared by other TNFR superfamily members.
Based on the above results, we expected that CD8 T cell proliferation in response to CD3+CD28+4–1BB would be significantly reduced in NfkbiaNES/NES CD8 T cells relative to the WT cells. However, while both WT and mutant cells stimulated with CD3+CD28+4–1BB proliferated more than CD3+CD28 stimulation alone, NfkbiaNES/NES CD8T cells proliferated similarly to the WT cells under the same conditions (Figure 2H). We therefore next considered the possibility of higher IL-2 secretion induced by 4–1BB stimulation in WT CD8 T cells relative to mutant cells affecting cell survival. To test this, CD8 T cells were first stimulated by CD3+CD28+4–1BB for 48 hours and then cultured for additional 48 hours without any stimuli (Supplemental Figure 2F for schematic). The recovery of NfkbiaNES/NES CD8 T cells was less than 50% of WT cells (Figure 2I). When a CD25 blocking antibody was included during the first 48 hour activation phase to block IL-2 signaling, cell recovery was nearly completely eliminated for both WT and NfkbiaNES/NES T cells (Figure 2I). Thus, IL-2 signaling was required for the accumulation of activated CD8 T cells upon withdrawal of activation signals. Based on these results, we propose that a major function of secreted IL-2 and CD25 signaling in response to 4–1BB stimulation was to promote survival and continued proliferation of activated CD8 T cells. Perhaps, this phenomenon reflects programmed proliferation and accumulation of activated CD8 T cells, following withdrawal of TCR signaling (38). This aspect of CD8 T cell biology was significantly defective in NfkbiaNES/NES CD8 T cells.
IκBα NES is necessary for CD3+CD28 induced cytoplasmic localization of IκBα:cRel complexes to enable efficient 4–1BB signaling
Given that the NES of IκBα promotes its own cytoplasmic localization with its associated NF-κB proteins, in particular cRel (25), we next examined subcellular localization of IκBα, RelA and cRel in naïve versus CD3+CD28-stimulated NfkbiaNES/NES and WT CD8 T cells. After fixation, permeabilization and staining with appropriate antibodies against these proteins (see Materials and Methods), we utilized ImageStream flow cytometry and the IDEAS software to calculate a log transformed Pearson correlation coefficient between the pixel values of two images to derive a “similarity score” (39). We used DRAQ5 nuclear stain along with NF-κB/IκBα staining to obtain similarity scores such that a higher similarity score denoted higher nuclear localization of the protein of interest. We further employed the nuclear export inhibitor, leptomycin B (LMB) (40), as a control to demonstrate maximal nuclear localization of NF-κB and IκBα proteins. We first tested the utility of this assay using splenic B cells isolated from WT and NfkbiaNES/NES hosts. The similarity scores for IκBα and cRel were higher in NfkbiaNES/NES B cells compared to WT cells, which were similar to those induced by LMB in WT cells (Supplemental Figure 3). However, RelA in both WT and mutant cells showed lower similarity scores than LMB conditions. These results are consistent with the NES-mutant IκBα causing nuclear localization of associated cRel, but not RelA, complexes, as reported previously (25).
Having validated the assay, we next evaluated subcellular localization of IκBα, RelA and cRel in naïve WT and NfkbiaNES/NES CD8 T cells. Analysis of fluorescence imaging indicated that IκBα is largely cytoplasmic in T cells of both genotypes (Figure 3A, IκBα, upper). Correspondingly, the similarity scores for IκBα in both genotypes were similar and lower than that induced by LMB treatment in WT cells (Figure 3A, IκBα, lower). The similarity score for RelA in NfkbiaNES/NES CD8 T cells was slightly higher than for WT cells but was still much lower than that induced by LMB (Figure 3A, RelA, lower). For cRel, the similarity score was slightly higher in the mutant cells than WT cells and similar to that increased with LMB treatment in WT cells (Figure 3A, cRel, lower). Following CD3+CD28 stimulation, expression of not only TNFR family members (Figure 1C) but also cRel and p50 increased in WT CD8 T cells (Figure 3B), which was accompanied by increased formation of IκBα/cRel complexes (Figure 3C). In addition, IκBα and RelA levels were slightly reduced in activated WT cells (Figure 3B). Now, LMB treatment of CD3+CD28 activated WT CD8 T cells demonstrated a greater increase in the similarity scores in both IκBα and cRel, similar to RelA, relative to those in naïve cells (Figure 3A and 3D, compare WT +/− LMB). In NfkbiaNES/NES CD8 T cells, cRel and p50 levels also increased with a further reduction in RelA, but not IκBα (Figure 3B); IκBα and cRel, but not RelA, displayed high similarity scores that approached those scores induced by LMB in WT cells (Figure 3D, NES). These results suggested that newly formed IκBα/cRel complexes in activated T cells were mis-localized to the nucleus in NfkbiaNES/NES CD8 T cells while RelA remained cytoplasmic. Accordingly, 4–1BB stimulation of CD3+CD28 activated CD8 T cells caused rapid IκBα degradation in WT cells but much less so in the mutant cells (Figure 3E). Degradation of IκBβ was barely detectable in both WT and mutant cells. This 4–1BB-induced resistance of IκBα to degradation (due to nuclear accumulation) could explain the significant cRel activation defects observed in NfkbiaNES/NES CD8 T cells (Figure 2A–2B).
Figure 3. IκBα NES is necessary for CD3+CD28 induced cytoplasmic localization of IκBα:cRel complexes to enable efficient 4–1BB signaling.
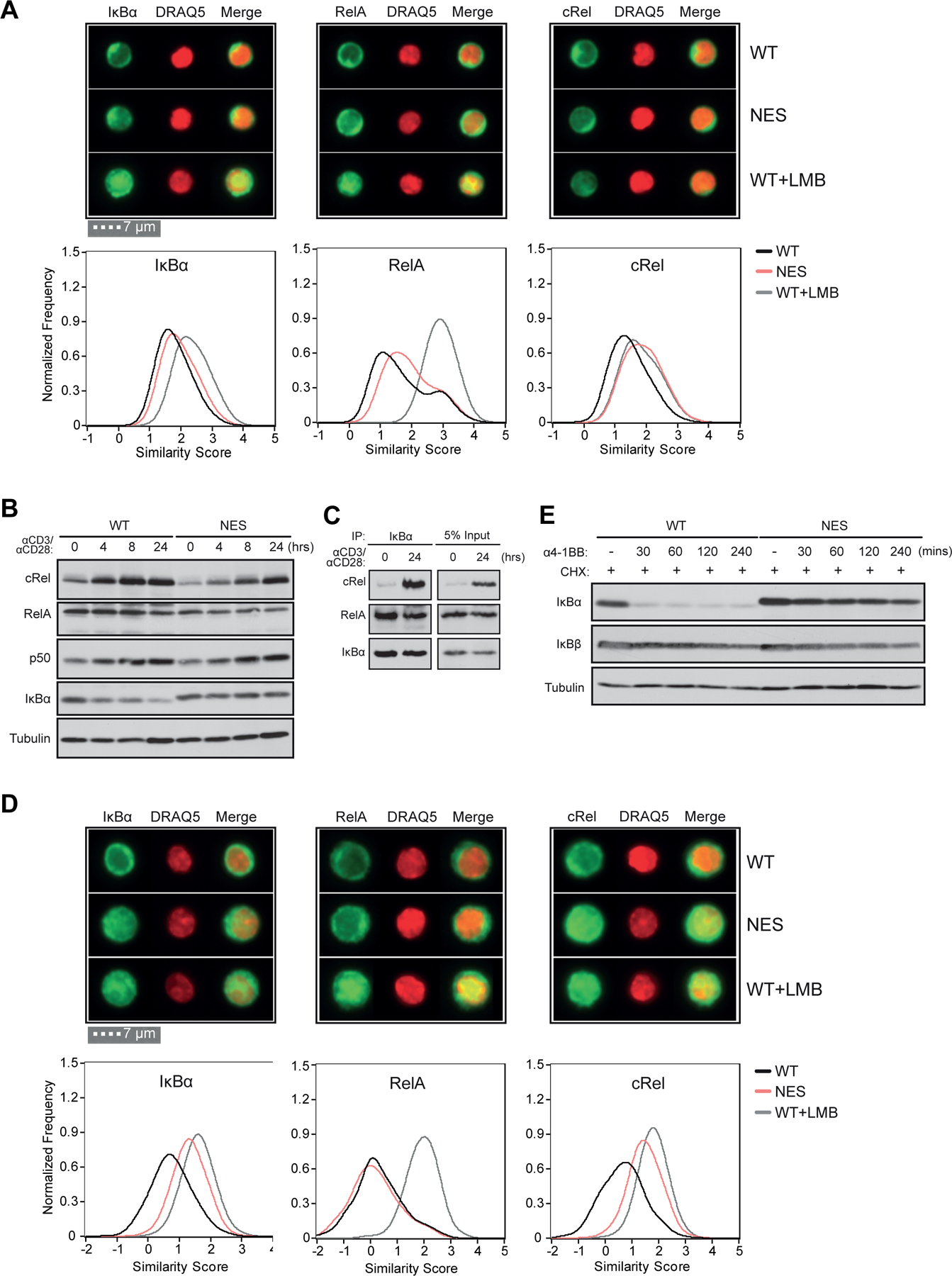
(A) Naive WT and NfkbiaNES/NES CD8 T cells were fixed, permeabilized, and stained with anti-IκBα, RelA or cRel antibodies and the DRAQ5 nuclear dye. WT cells were treated with 20 ng/ml of Leptomycin B for 45 minutes as a control. Single cell image analysis by ImageStream flow cytometry. Representative images from each group are shown at an equivalent magnification (above) and the similarity score for each antibody staining is plotted. Data plotted includes 3 independent experiments (see Methods).
(B) Naive WT and NfkbiaNES/NES CD8 T cells were stimulated for the indicated times with 5 μg/mL each anti-CD3 and anti-CD28. Immunoblot analysis of cRel, RelA, p50, IκBα, and tubulin is shown. Data is representative of 3 independent experiments.
(C) Cell extracts from naive WT CD8 T cells and those unstimulated or stimulated as in (B) for 24 hours were immunoprecipitated with an antibody specific for IκBα. Immunoblot analysis of cRel, RelA, and IκBα is shown.
(D) Naive WT and NfkbiaNES/NES CD8 T cells were stimulated for 24 hours with 5 μg/mL each anti-CD3 and anti-CD28 and stained and analyzed as in (A). Representative images for each antibody staining are shown at the equivalent magnification as in (A). Note the larger cell size of the stimulated cells relative to naïve cells in (A).
(E) Naive WT and NfkbiaNES/NES CD8 T cells were stimulated for 24 hours with anti-CD3 and anti-CD28 antibodies as in (A). Following stimulation, cells were pre-treated with 20 ug/mL cycloheximide (CHX) for 30 minutes and then stimulated with 0.05 μg/mL anti-4–1BB antibody for the indicated times. Immunoblot analysis of IκBα, IκBβ, and tubulin is shown.
IκBα nuclear export is required for inducing a 4–1BB-mediated NF-κB transcriptional program in CD8 T cells
The biochemical and functional data thus far suggested that a 4–1BB-induced transcriptional program would likely be altered in NfkbiaNES/NES CD8 T cells. To define a 4–1BB induced transcriptional program that requires proper IκBα nuclear export in CD8 T cells, we next performed RNA-seq analyses on WT and NfkbiaNES/NES CD8 T cells stimulated with CD3+CD28 with or without 4–1BB stimulation (Figure 4A). We chose a 12-hour time point for analysis to capture early changes induced by 4–1BB signaling as 4–1BB mediated disruption of NF-κB signaling was already evident at this time point (Supplemental Figure 2A). Stimulation of WT CD8 T cells with CD3+CD28+4–1BB altered expression of a set of up- and down- regulated genes relative to CD3+CD28 alone (Figure 4B, left). Moreover, analysis of similarly stimulated NfkbiaNES/NES CD8 T cells revealed 3 distinct groups of genes (Figure 4C); 1) “NES independent”: genes induced by 4–1BB in WT cells and similarly in NfkbiaNES/NES (110 genes), 2) “NES dependent”: genes induced by 4–1BB in WT cells but less induced in NfkbiaNES/NES (88 genes), and 3) “NES reduced”: genes not induced by 4–1BB in WT cells but reduced in NfkbiaNES/NES (114 genes) (Figure 4C). Of these, the “NES dependent” category was of particular interest because these genes were candidates whose transcriptional programs were controlled by IκBα nuclear export in response to 4–1BB stimulation over CD3+CD28 alone. KEGG pathway analysis of the “NES dependent” category revealed an enrichment of pathways such as the TNF and Jak-STAT signaling pathways and processes such as cytokine signaling (Figure 4D). A ranking of the most upregulated genes by 4–1BB based on their fold induction in WT CD8 T cells revealed those relevant to 4–1BB biology, such as Icam1, Fas, and Jun (Figure 4E). This ranking also demonstrated that Il2 was the 2nd most highly induced gene. This corroborates our previous finding that IL-2 secretion was significantly reduced in 4–1BB-stimulated NfkbiaNES/NES CD8 T cells (Figure 2C).
Figure 4. IκBα nuclear export is required for inducing a 4–1BB-mediated NF-κB transcriptional program in CD8 T cells.
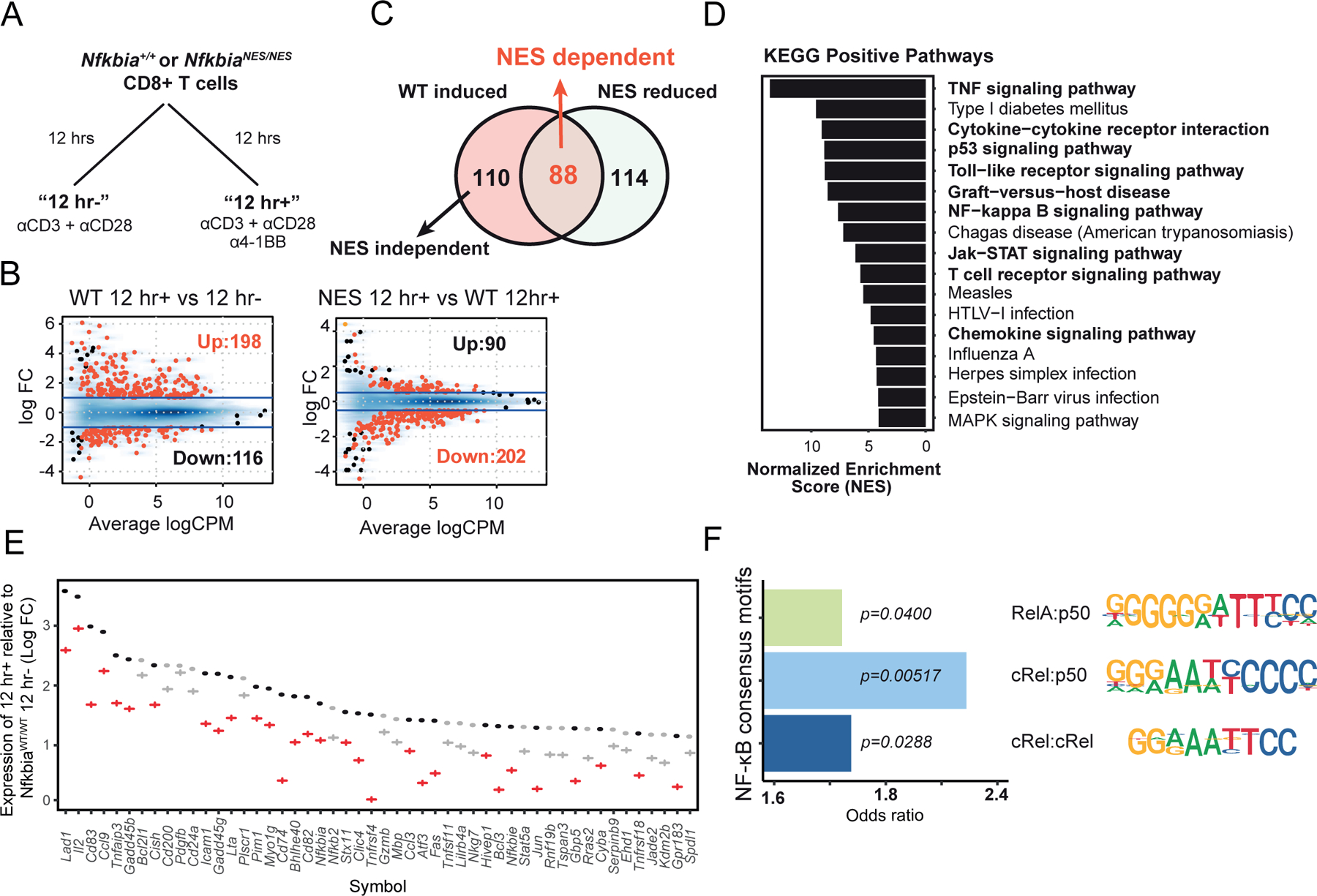
(A) The RNA sequencing study design is shown with naïve Nfkbia+/+ and NfkbiaNES/NES CD8 T cells stimulated with 5 μg/mL each of anti-CD3 and anti-CD28 antibodies for 12 hours without (12hr-) or with (12 hr+) 0.05 μg/mL anti-4–1BB antibody.
(B) MA plots showing differentially expressed genes (up and down) between “12 hr+” vs. “12 hr-” WT CD8 T cells (left, absolute Log2 fold change > 1 and FDR < 0.01) and between WT “12 hr+” versus NfkbiaNES/NES “12 hr+” (right, absolute Log2 fold change > 0.5 and FDR < 0.05).
(C) Venn diagram illustrating the number of genes regulated by 4–1BB and by NfkbiaNES/NES in CD8 T cells.
(D) Bar graph showing ranked fold enrichment of enriched KEGG pathways (p-value < 0.05) in “NES dependent” genes.
(E) Comparison of expression of the top 20 WT 12 hr+ vs. 12 hr- genes. Dots and crosses represent Log2 fold change comparing WT “12 hr+” vs. WT “12 hr-” and NfkbiaNES/NES “12 hr+” vs. WT “12 hr-”, respectively with red and grey colors indicating “NES dependent” and “NES independent,” respectively.
(F) Bar graph showing the odds ratios for the occurrence of NF-κB dimer binding motifs (p65:p50, cRel:p50, cRel:cRel) in the promoter regions (−1kb to 300bp) of “NES dependent” genes. P-values from the same Fisher’s exact test are shown.
To assess whether “NES dependent” genes were under the control of the NF-κB pathway, in particular, cRel complexes, as our previous biochemical and imaging data suggest (Figures 2 and 3), promoter regions of these genes were tested for the enrichment of NF-κB consensus motifs for RelA:p50, cRel:p50, and cRel:cRel complexes (32, 33). We found the greatest enrichment of cRel:p50 motifs in “NES dependent” genes when compared to “NES independent” genes, along with less significant enrichment for RelA:p50 and cRel:cRel motifs (Figure 4F). Despite a cRel:p50 binding site was not detected in the IL-2 gene by the above bioinformatic analysis, we confirmed cRel binding (16.5-fold over basal) to an NF-κB consensus sequence from the IL-2 gene promoter in WT T cells stimulated with CD3+CD28+4–1BB for 12 hours, which was reduced (to 5.2-fold) in NfkbiaNES/NES T cells (Supplemental Figure 2A). RelA binding also increased in CD3+CD28+4–1BB stimulated WT cells but was much less than cRel binding. These results suggested that 4–1BB stimulation induced a prominent cRel-dependent transcriptional program in CD3+CD28 activated CD8 T cells, which was enabled by IκBα NES-mediated proper subcellular distribution of cRel complexes.
The IκBα NES is necessary for cell-intrinsic anti-viral CD8 T cell responses in vivo
These results thus far demonstrated that CD8 T cells in NfkbiaNES/NES mice have 4–1BB-mediated NF-κB signaling, transcriptomic and functional defects ex vivo. To test whether IκBα nuclear export was also necessary for activation and expansion of CD8 T cells in vivo, we utilized the Armstrong strain of lymphocytic choriomengitis virus (LCMV) that induces a well-defined virus-specific T cell response (26). At the peak of the anti-viral CD8 T cell response (i.e. day 8 after infection), the accumulation of activated (CD8 CD44hi) and LCMV NP396-specific CD8 T cells in the spleen of NfkbiaNES/NES mice was significantly lower than in WT mice (Figure 5A). This data suggested that loss of IκBα nuclear export reduced expansion of CD8 T cells during an acute viral infection. Our ex vivo results demonstrated that NfkbiaNES/NES CD8 T cells produced significantly lower levels of Il2 transcripts (Figure 4E) and IL-2 protein (Figure 2C), and displayed defects in IL-2-dependent survival, following activation (Figure 2I). In order to test the hypothesis that a defective production of IL-2 was at least in part responsible for a defect in CD8 T cell accumulation in LCMV-infected NfkbiaNES/NES mice in vivo, IL-2 was administered for 6 days following LCMV infection. Although IL-2 administration of LCMV-infected WT mice increased the percentage of CD8 T cells, it did not increase the percentages or the total number of LCMV NP396- or GP33-specific CD8 T cells in the spleen (Figures 5B & 5C). Strikingly, IL-2 administration completely restored the accumulation of LCMV-specific CD8 T cells in the spleens of NfkbiaNES/NES mice (Figures 5B & 5C). These data suggested that exogenous IL-2 administration could overcome defective accumulation of CD8 T cells in LCMV-infected NfkbiaNES/NES mice.
Figure 5. NfkbiaNES/NES mice have a primary CD8 T cell expansion defect following LCMV infection which can be restored by exogenous IL-2 administration.
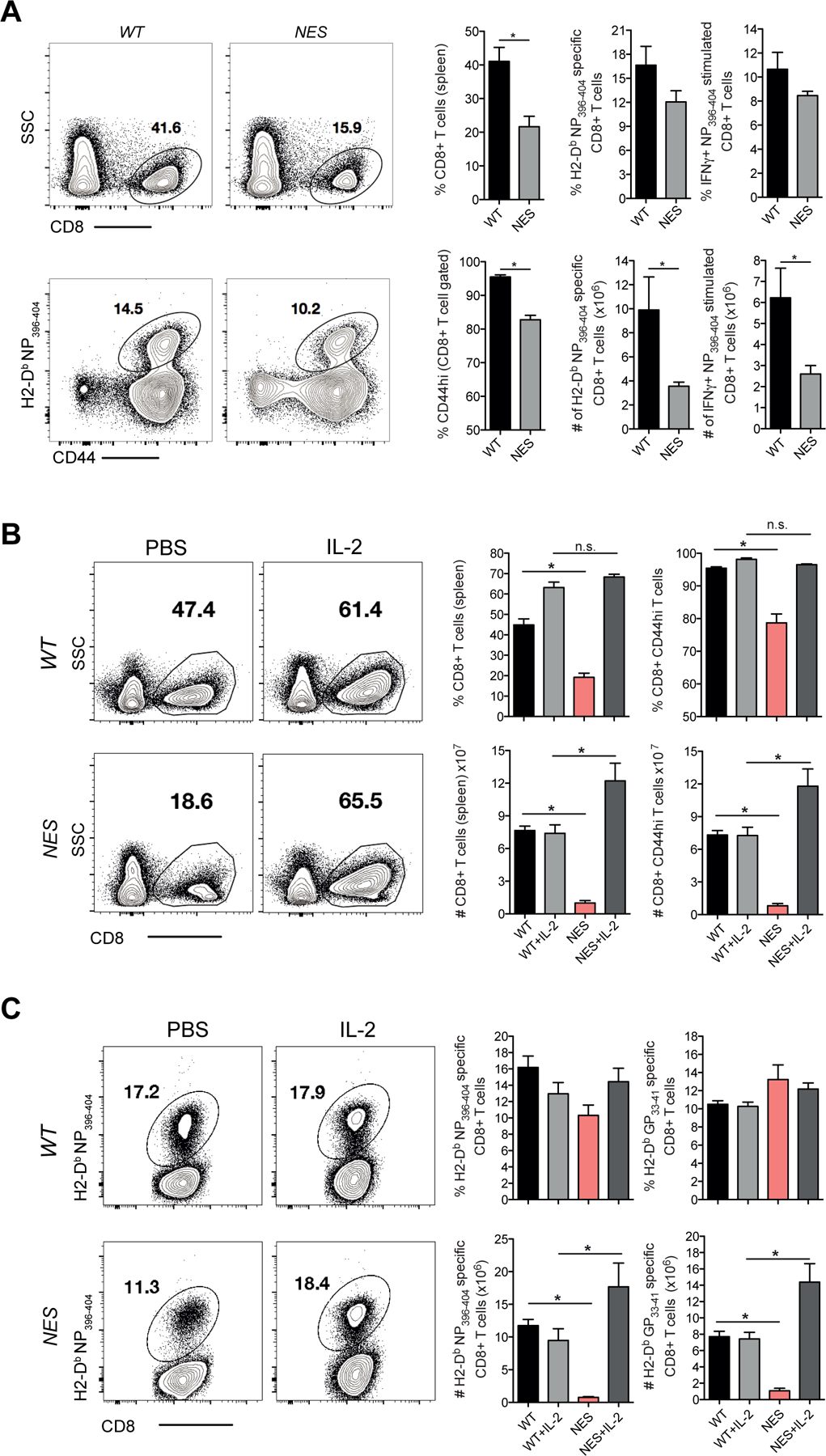
(A) WT and NfkbiaNES/NES mice were infected with LCMV Armstrong. Eight days post-infection, splenocytes from each group were assessed by flow cytometry for CD8 T cells staining positive for CD44 and H2-Db specific NP396–404 tetramer and staining positive for IFN-γ following restimulation with NP396–404 peptide.
(B) WT and NfkbiaNES/NES mice were infected with LCMV Armstrong. Mice were injected with recombinant human IL-2 (10,000 IU) or PBS twice a day for 6 days following infection. Eight days post-infection, splenocytes were analyzed by flow cytometry and percentages and absolute numbers of cells staining positive for CD8 and CD44 are shown.
(C) WT and NfkbiaNES/NES mice from (B) were also analyzed by flow cytometry with percentages and absolute numbers staining positive for GP33–41 and NP396–404 tetramers are shown. Data shown in (A)–(C) are the mean +/− SEM of five mice per group and representative of two independent experiments with *p<0.05.
Although our data above demonstrated that virus-specific CD8 T cell responses were suboptimal in NfkbiaNES/NES mice and could be restored by exogenous IL-2 administration, this defect could be cell extrinsic due to the presence of the IκBα NES mutation in the germline. Moreover, NfkbiaNES/NES mice also harbor B cell and secondary tissue disruptions (25), which might have impacted anti-viral CD8 T cell responses. Thus, we further performed three different in vivo studies to address this question. First, to address whether defects in anti-viral CD8 T cell responses in NfkbiaNES/NES mice originated from hematological cell populations, we generated mixed bone marrow chimeras. Lethally irradiated Ly5.1 host mice were reconstituted with a mix of bone marrow cells from congenically marked host strain (Ly5.1) and Ly5.2 NfkbiaWT/WT or NfkbiaNES/NES mice, generating control and experimental chimeras (CC and EC, respectively) (Supplemental Figure 3A). As expected, the NfkbiaNES/NES compartment of CC and EC had normal T cell populations (Supplemental Figure 3B). At day 8 after infection, we quantified LCMV-specific CD8 T cell responses in control and experimental chimeras. The activation of WT (Ly5.1+) host CD8 T cell responses were comparable in control and experimental chimeras; percentages of WT NP396-specific CD8 T cells among Ly5.1+ CD8 T cells were comparable between control and experimental chimeras (Supplemental Figure 3C). CD8 T cells derived from NfkbiaWT/WT (Ly5.2+) bone marrow cells showed strong activation in control chimeras; >10% of the CD8 T cells were specific to the immunodominant epitope NP396–404 (Figure 6A). By contrast, the percentages of NfkbiaNES/NES (Ly5.2) NP396–404-specific CD8 T cells in experimental chimeras were lower, as compared to their Ly5.2 WT counterparts in control chimeras (Figure 6A). We also calculated the ratios of the percentages of Ly5.1+ to Ly5.2+ NP396-specific CD8 T cells in CCs and ECs. The Ly5.1:Ly5.2 ratio of NP396-specific CD8 T cells in CCs and ECs were 0.83 and 1.72, respectively and this was statistically significant (p=0.029). The significant reduction of NP396–404 specific cells in the NfkbiaNES/NES compartment persisted through the memory phase at 64 days post-infection to a re-challenge with LCMV clone 13 (Supplemental Figure 3D). These results support the conclusion that the anti-viral CD8 T cell defect in NfkbiaNES/NES mice was of hematological origin and likely cell intrinsic.
Figure 6. The antiviral defect in NfkbiaNES/NES mice is CD8 T-cell intrinsic.
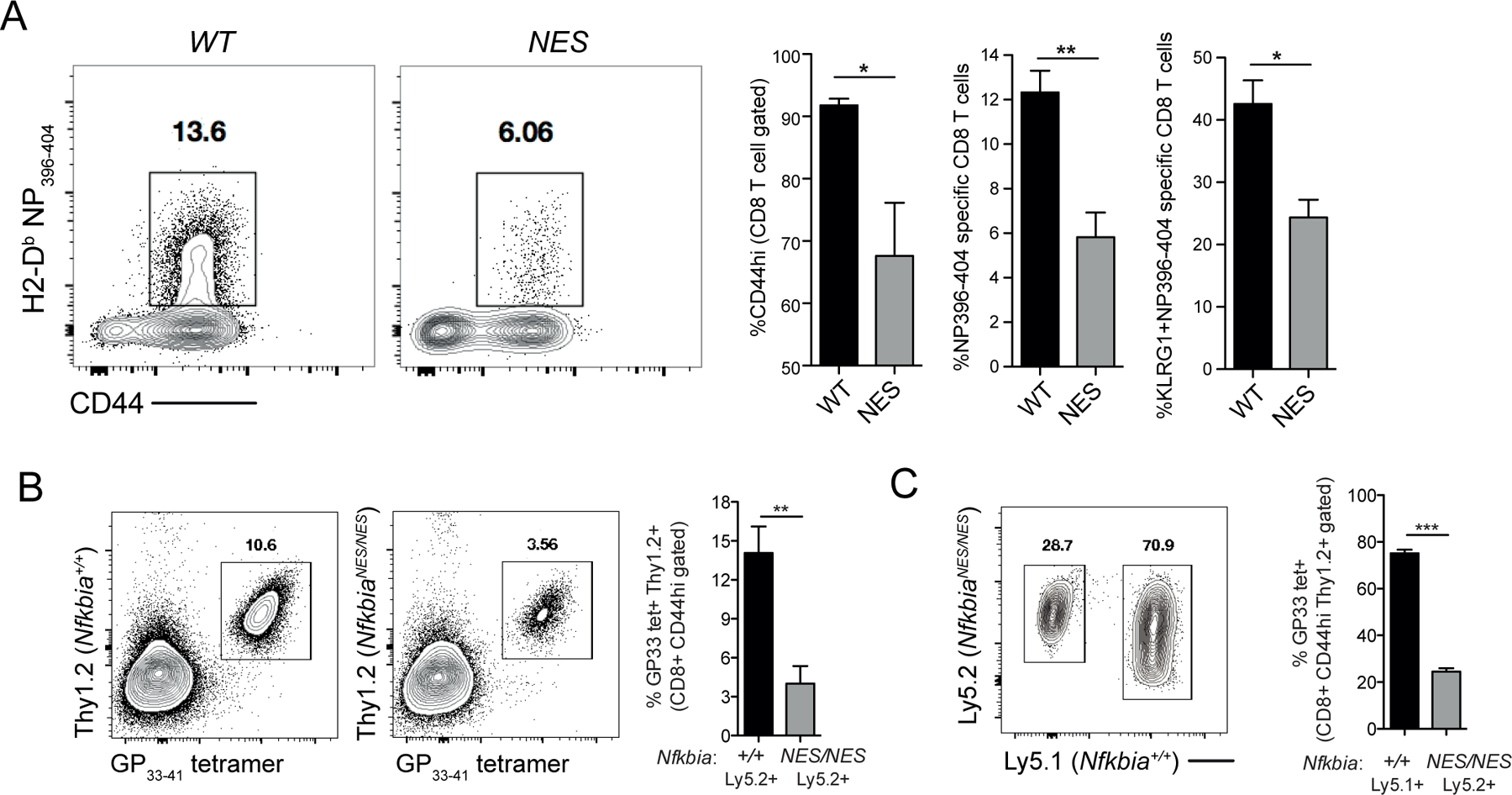
(A) Lethally irradiated Ly5.1 host mice were reconstituted with a mix of bone marrow to generate control chimeras, CC (Ly5.1+ host strain cells and Ly5.2+ NfkbiaWT/WT) or experimental chimeras, EC (Ly5.1+ host strain cells and Ly5.2+ NfkbiaNES/NES) (see Supplemental Figure 3A for schematic) and infected with LCMV Armstrong. Eight days post-infection, splenocytes from control and experimental chimeras were assessed by flow cytometry for Ly5.2+ CD8 T cells staining positive for CD44, CD62L, NP396–404 tetramer, and KLRG1. Data are the mean +/− SEM and representative of 3–4 mice per group with statistical significance at *p<0.05 and **p < 0.01.
(B) Thy1.1 host mice were adoptively transferred with either P14 transgenic Thy1.2+ NfkbiaWT/WT (Ly5.2+) or P14 transgenic Thy1.2+ NfkbiaNES/NES (Ly5.2+) CD8 T cells. Mice were infected with LCMV Armstrong and analyzed 8 days post-infection for P14 transgenic T cell expansion. Data are the mean +/− SEM and representative of 6 mice per group with statistical significance at **p < 0.01.
(C) Thy1.1 host mice were adoptively transferred with a 1:1 mix of P14 transgenic Thy1.2+ Nfkbia+/+ (Ly5.1+) and Thy1.2+ NfkbiaNES/NES (Ly5.2+) cells. Twenty-four hours later, mice were infected with LCMV Armstrong and analyzed 8 days post-infection for P14 transgenic T cell expansion. Data are the mean +/− SEM and representative of 7 mice per group with statistical significance at ***p < 0.001.
Next, to specifically address whether the above NfkbiaNES/NES mouse defects were CD8 T cell-intrinsic, we crossed NfkbiaNES/NES mice with LCMV glycoprotein (GP) 33-specific TCR transgenic P14 mice to generate P14/NfkbiaNES/NES mice. Thy1.1+ hosts were adoptively transferred with either P14/NfkbiaWT/WT (Ly5.2+/Thy1.2+) or P14/NfkbiaNES/NES (Ly5.2+/Thy1.2+) naïve CD8 T cells and then infected with LCMV to test the responses of donor mutant CD8 T cells (Supplemental Figure 3E, non-competitive environment). Analysis of mice 8 days post-infection revealed a significantly reduced expansion of P14/NfkbiaNES/NES cells in comparison to P14/NfkbiaWT/WT transgenic cells (Figure 6B). Thus, these results indicated that the anti-viral CD8 T cell expansion defect of NfkbiaNES/NES mice was cell intrinsic. Finally, to further validate this, we performed the same experiment in a competitive environment in which an equal number of P14/NfkbiaWT/WT (Ly5.1+/Thy1.2+) and P14/NfkbiaNES/NES (Ly5.2+/Thy1.2+) naïve CD8 T cells were adoptively transferred into Thy1.1 mice and then infected with LCMV (Supplemental Figure 3F, competitive environment). Examination of mice after establishment of transferred cells prior to infection showed a slightly higher level of the NfkbiaNES/NES CD8 T cells relative to NfkbiaWT/WT cells in the hosts (Supplemental Figure 3F, right). However, at 8 days post-infection, P14/NfkbiaNES/NES T cell expansion was significantly reduced in comparison to P14/NfkbiaWT/WT cells (Figure 6C), demonstrating that the NfkbiaNES/NES CD8 T cells were unable to effectively compete with NfkbiaWT/WT cells in the identical host environment. Our data collectively supports the critical role of IκBα NES to promote proper cell-intrinsic anti-viral CD8 T cell responses in vivo.
Discussion
We have now uncovered an unexpected new physiological role for IκBα, as a signal-specific amplifier, not just an inhibitor, of the NF-κB pathway in CD8 T cells, mediated by its unique nuclear export function. Our studies revealed that initial TCR (CD3) and CD28 engagement increased the expression of 4–1BB (and other TNFR superfamily members), increased expression of cRel, and resulted in an upregulation of IκBα:cRel cytosolic complexes. This coordinated receptor and NF-κB pathway reconfiguration sets the stage for 4–1BB-mediated cRel activation due to selective targeting of IκBα degradation by 4–1BB signaling. Sustained cRel activation was dependent on continual IκBα-mediated nuclear export of inactive cRel and was necessary for a dominant cRel transcriptional program, maximal production of IL-2, and primary accumulation of virus-specific CD8 T cells. There was no measurable impact on 4–1BB induced non-canonical NF-κB signaling by the IκBα NES mutation. Thus, our data suggest a spatial and temporal regulation of canonical NF-κB signaling that is dynamically sustained by IκBα nuclear export to enable optimal CD8 T cell expansion in vivo.
Our findings uncovered aspects of CD8 T cell biology that have not been revealed by previous NF-κB and IκB family member genetic knockout mouse studies (14, 15, 41–48). This study, together with our prior work (25), demonstrated that although there was reduced basal RelA activity and reduced steady-state expression of several NF-κB family members, NES-mutated IκBα has minimal impact on the number of CD4 and CD8 T cell populations in the periphery. CD3+CD28-induced acute RelA and cRel activation, IL-2 secretion, expression of several key cell surface activation markers, and proliferation were all normal in NfkbiaNES/NES CD8 T cells. The lack of an acute RelA activation defect in naïve NfkbiaNES/NES CD8 T cells is likely a consequence of the presence of a compensating NES on RelA. In contrast, the lack of an acute CD3+CD28-induced cRel activation defect in naïve NfkbiaNES/NES CD8 T cells is likely due to the primary association of cRel with IκBβ, not mutant IκBα, in naïve T cells (24, 49). A minor level of cRel associated with IκBα in naïve NfkbiaNES/NES cells appears to be subjected to alteration in subcellular localization detected by ImageStream flow cytometry. However, this fraction of cRel and IκBα is too small to have a major impact on acute CD3+CD28-induced signaling in naïve cells. In contrast, cRel becomes highly susceptible to IκBα nuclear export-mediated regulation after increased cRel synthesis and subsequent increased cRel:IκBα complex formation following CD3+CD28 stimulation. Since cRel does not have its own NES sequence and function, its subcellular localization is dominantly controlled by IκBα. Thus, we now show that the major physiological role of IκBα NES is realized primarily when the fraction of cRel:IκBα complexes increase significantly following TCR+CD28 stimulation in CD8 T cells.
How TNFR family members coordinate their expression and cell signaling processes to functional transcriptional programs has remained undefined (50). 4–1BB expression is itself known to be regulated by NF-κB signaling following T cell signaling (51), however, NfkbiaNES/NES CD8 T cells set the same stage with coordinated expression of 4–1BB and cRel as in WT cells following CD3+CD28 stimulation. Consequently, in WT cells, cRel complexes are potently activated by subsequent 4–1BB signaling due to the selective degradation of IκBα over IκBβ, followed by a 4–1BB-dependent transcriptional program, heightened IL-2 production and cell proliferation. In contrast, in NfkbiaNES/NES CD8 T cells, due to defective localization of cRel:IκBα complexes to the nucleus which shields them from 4–1BB-induced signaling, the chain of events necessary for IL-2 production and activation is disrupted. Our data further suggested that such 4–1BB regulation is not unique to this TNFR family member but is also likely shared by OX40 and CD27. Thus, IκBα-mediated nuclear export of cRel ensures a spatial regulation of NF-κB dimers, particularly cRel dimers, to sustain the TNFR co-stimulatory signaling necessary for proper T cell immunity.
Of the many genes whose expression is altered by the IκBα NES mutation in CD8 T cells, IL-2 appears to be one of the most critical cytokines to control their accumulation in response to viral infection. On first glance, this appears unsurprising given 1) the significant inhibition of cRel dimer activity in NfkbiaNES/NES CD8 T cells and 2) the fact that Rel−/− T cells have an IL-2 synthesis defect and fail to proliferate in vitro (14, 15). However, despite the significant reduction of 4–1BB-induced IL-2 secretion, NfkbiaNES/NES CD8 T cells proliferated surprisingly normally in vitro, making it distinct from the total cRel-deficient T cell phenotype. In contrast, withdrawal of TCR and co-stimulatory signals revealed that NfkbiaNES/NES CD8 T cells depended on autocrine IL-2 secretion to sustain survival in vitro. This suggests the possibility that TCR and co-stimulatory signals trigger a program of CD8 T cell expansion that is dependent on autocrine IL-2 even after exogenous stimuli are removed. Previous studies have suggested that depending on the route or model of infection, autocrine IL-2 is necessary for expansion of viral specific CD8 T cells (52–54); however, how autocrine IL-2 is regulated in activated CD8 T cells in vivo remained unclear. Our biochemical and transcriptomic analyses demonstrated that the nuclear export function of IκBα assumes an unexpectedly significant cell autonomous role in the regulation of cRel dimer activity to likely maintain autocrine IL-2 production. Similar to a previously published study investigating the role of IL-2 in effector differentiation (55), a significant reduction in KLRG1hi NfkbiaNES/NES CD8 T cells provided evidence that IL-2 production was defective following LCMV infection. Thus, while other aspects of CD8 T cell function may require additional factors, the ability of exogenous IL-2 to restore CD8 T cell accumulation defects in LCMV infected NfkbiaNES/NES mice, coupled with chimera and adoptive transfer studies showing CD8 T cell-intrinsic defects, supports the biological significance of autocrine IL-2 production by CD8 T cells in vivo. Also, it shows that defects in autocrine IL-2 production by CD8 T cells can be overcome by supra-physiological levels of exogenous IL-2.
The newly uncovered role of nuclear export-dependent regulation of CD8 T cell immunity may have implications beyond anti-viral responses. Exportin-1/chromosome region maintenance 1 (XPO1/CRM1) mediates the nuclear export of classical NES containing proteins, including IκBα and RelA. Many tumors overexpress XPO1 and pharmacologic inhibition of XPO1 mediated nuclear export can result in tumor cell death or sensitivity to chemotherapies (56). Recently, utilization of selective inhibitors of nuclear export (SINE) compounds have been shown to be tolerated in clinical trials for treatment of hematologic and solid tumors (57). However, thorough mechanistic studies investigating the physiological consequences of nuclear export inhibition in the immune system are lacking. To our knowledge, the NfkbiaNES/NES mouse represents one of only two genetically modified mouse models to reveal insights into the physiological role of nuclear export in vivo (25, 58). Given the increased focus on the significance of anti-tumor CD8 T cell immune responses and utilization of chimeric antigen receptor (CAR) T cells containing 4–1BB signaling domains (59), our study provides significant aspects of CD8 T cell biology that may be considered to improve anti-tumor immunity and CAR T cell strategies, including timing of the administration of SINE compounds with immunotherapy approaches (60).
Collectively, our study has provided new insights into how CD8 T cell immunity is dependent on the coordinated reconfiguration of an intracellular signaling system and cognate cell surface receptors to enable signaling through co-stimulatory receptors. While it is well appreciated that fine adjustments of extracellular physical interactions, such as antigen-receptor and ligand-receptor interactions, govern one part of T cell function (61), our study uncovered that regulation of intracellular cell signaling dynamics is another critical point of regulation. We now provide a plausible model that links dynamic CD8 T cell signaling through nuclear export of specific NF-κB family members to three key events in CD8 T cell immunity in vivo: sustained signaling through co-stimulatory TNFR members, autocrine IL-2 production, and maximal accumulation of effector cells.
Supplementary Material
Key Points:
4–1BB signaling activates NF-κB/cRel and is dependent on IκBα nuclear export
IκBα nuclear export is necessary for efficient IL-2 production and T cell immunity
Acknowledgements
We thank the UWCCC Flow cytometry core facility, the NIH tetramer core facility (Atlanta, GA) for H-2D(b) / KAVYNFATM and H-2D(b) / FQPQNGQFI tetramers, the University of Wisconsin-Madison Biomedical Research Model Services Mouse Breeding Core for mouse colony maintenance, Dr. Hsiou-Chi Liou for Rel−/− mice, Dr. Rafi Ahmed for P14 TCR transgenic mice, and members of the Miyamoto lab for thoughtful discussions.
This work was supported in part by NIH grants T32 CA157322 and F32 AI112206 to DNL, U01 AI124299 and R21 AI118326 to MS, R01 CA081065 and R56 CA081065 to SM, Wisconsin Partnership Program award to JWW and SM, NIH small instrument grants #1S10RR025483-01 and #1S100OD018202-01, and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Duttagupta PA, Boesteanu AC, and Katsikis PD. 2009. Costimulation signals for memory CD8+ T cells during viral infections. Crit Rev Immunol 29: 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croft M 2009. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 9: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jameson SC, and Masopust D. 2009. Diversity in T cell memory: an embarrassment of riches. Immunity 31: 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, and Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13: 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, and Katsikis PD. 2007. Memory CD8+ T cells require CD28 costimulation. J Immunol 179: 6494–6503. [DOI] [PubMed] [Google Scholar]
- 6.Wortzman ME, Clouthier DL, McPherson AJ, Lin GH, and Watts TH. 2013. The contextual role of TNFR family members in CD8(+) T-cell control of viral infections. Immunol Rev 255: 125–148. [DOI] [PubMed] [Google Scholar]
- 7.Oeckinghaus A, Hayden MS, and Ghosh S. 2011. Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12: 695–708. [DOI] [PubMed] [Google Scholar]
- 8.Hayden MS, and Ghosh S. 2011. NF-kappaB in immunobiology. Cell Res 21: 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Napetschnig J, and Wu H. 2013. Molecular basis of NF-kappaB signaling. Annu Rev Biophys 42: 443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobrzanski P, Ryseck RP, and Bravo R. 1994. Differential interactions of Rel-NF-kappa B complexes with I kappa B alpha determine pools of constitutive and inducible NF-kappa B activity. EMBO J 13: 4608–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerondakis S, and Siebenlist U. 2010. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2: a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beg AA, Sha WC, Bronson RT, Ghosh S, and Baltimore D. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 376: 167–170. [DOI] [PubMed] [Google Scholar]
- 13.Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, Hayden MS, Klein U, Rabadan R, and Ghosh S. 2017. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 47: 450–465 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doi TS, Takahashi T, Taguchi O, Azuma T, and Obata Y. 1997. NF-kappa B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med 185: 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, and Gerondakis S. 1995. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 9: 1965–1977. [DOI] [PubMed] [Google Scholar]
- 16.Sun SC, Ganchi PA, Ballard DW, and Greene WC. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915. [DOI] [PubMed] [Google Scholar]
- 17.Chiao PJ, Miyamoto S, and Verma IM. 1994. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci U S A 91: 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang TT, Kudo N, Yoshida M, and Miyamoto S. 2000. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci U S A 97: 1014–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang TT, and Miyamoto S. 2001. Postrepression activation of NF-kappaB requires the amino-terminal nuclear export signal specific to IkappaBalpha. Mol Cell Biol 21: 4737–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson C, Van Antwerp D, and Hope TJ. 1999. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J 18: 6682–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tam WF, Lee LH, Davis L, and Sen R. 2000. Cytoplasmic sequestration of rel proteins by IkappaBalpha requires CRM1-dependent nuclear export. Mol Cell Biol 20: 2269–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tam WF, and Sen R. 2001. IkappaB family members function by different mechanisms. J Biol Chem 276: 7701–7704. [DOI] [PubMed] [Google Scholar]
- 23.Harhaj EW, and Sun SC. 1999. Regulation of RelA subcellular localization by a putative nuclear export signal and p50. Mol Cell Biol 19: 7088–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tam WF, Wang W, and Sen R. 2001. Cell-specific association and shuttling of IkappaBalpha provides a mechanism for nuclear NF-kappaB in B lymphocytes. Mol Cell Biol 21: 4837–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wuerzberger-Davis SM, Chen Y, Yang DT, Kearns JD, Bates PW, Lynch C, Ladell NC, Yu M, Podd A, Zeng H, Huang TT, Wen R, Hoffmann A, Wang D, and Miyamoto S. 2011. Nuclear export of the NF-kappaB inhibitor IkappaBalpha is required for proper B cell and secondary lymphoid tissue formation. Immunity 34: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed R, Salmi A, Butler LD, Chiller JM, and Oldstone MB. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160: 521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, and Ahmed R. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8: 177–187. [DOI] [PubMed] [Google Scholar]
- 28.O’Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, Desai P, Fitch O, Malone L, Ahn HJ, Liou HC, Sweatt JD, and Levenson JM. 2006. Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci 26: 4870–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao Y, Smyth GK, and Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- 31.Robinson MD, McCarthy DJ, and Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alves BN, Tsui R, Almaden J, Shokhirev MN, Davis-Turak J, Fujimoto J, Birnbaum H, Ponomarenko J, and Hoffmann A. 2014. IkappaBepsilon is a key regulator of B cell expansion by providing negative feedback on cRel and RelA in a stimulus-specific manner. J Immunol 192: 3121–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, and Bulyk ML. 2011. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol 13: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan JT, Whitmire JK, Ahmed R, Pearson TC, and Larsen CP. 1999. 4–1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J Immunol 163: 4859–4868. [PubMed] [Google Scholar]
- 36.Sun SC 2017. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol 17: 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakajima H, Liu XW, Wynshaw-Boris A, Rosenthal LA, Imada K, Finbloom DS, Hennighausen L, and Leonard WJ. 1997. An indirect effect of Stat5a in IL-2-induced proliferation: a critical role for Stat5a in IL-2-mediated IL-2 receptor alpha chain induction. Immunity 7: 691–701. [DOI] [PubMed] [Google Scholar]
- 38.Kaech SM, and Ahmed R. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol 2: 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maguire O, Collins C, O’Loughlin K, Miecznikowski J, and Minderman H. 2011. Quantifying nuclear p65 as a parameter for NF-kappaB activation: Correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A 79: 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolff B, Sanglier JJ, and Wang Y. 1997. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol 4: 139–147. [DOI] [PubMed] [Google Scholar]
- 41.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, and Siebenlist U. 1998. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med 187: 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, and Baltimore D. 2002. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med 195: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, and Bravo R. 1995. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell 80: 331–340. [DOI] [PubMed] [Google Scholar]
- 44.Guerin S, Baron ML, Valero R, Herrant M, Auberger P, and Naquet P. 2002. RelB reduces thymocyte apoptosis and regulates terminal thymocyte maturation. Eur J Immunol 32: 1–9. [DOI] [PubMed] [Google Scholar]
- 45.Das J, Chen CH, Yang L, Cohn L, Ray P, and Ray A. 2001. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol 2: 45–50. [DOI] [PubMed] [Google Scholar]
- 46.Beg AA, Sha WC, Bronson RT, and Baltimore D. 1995. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev 9: 2736–2746. [DOI] [PubMed] [Google Scholar]
- 47.Boothby MR, Mora AL, Scherer DC, Brockman JA, and Ballard DW. 1997. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med 185: 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao P, Hayden MS, Long M, Scott ML, West AP, Zhang D, Oeckinghaus A, Lynch C, Hoffmann A, Baltimore D, and Ghosh S. 2010. IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature 466: 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banerjee D, Liou HC, and Sen R. 2005. c-Rel-dependent priming of naive T cells by inflammatory cytokines. Immunity 23: 445–458. [DOI] [PubMed] [Google Scholar]
- 50.Mbanwi AN, and Watts TH. 2014. Costimulatory TNFR family members in control of viral infection: outstanding questions. Semin Immunol 26: 210–219. [DOI] [PubMed] [Google Scholar]
- 51.Kim JO, Kim HW, Baek KM, and Kang CY. 2003. NF-kappaB and AP-1 regulate activation-dependent CD137 (4–1BB) expression in T cells. FEBS Lett 541: 163–170. [DOI] [PubMed] [Google Scholar]
- 52.Boyman O, Cho JH, and Sprent J. 2010. The role of interleukin-2 in memory CD8 cell differentiation. Adv Exp Med Biol 684: 28–41. [DOI] [PubMed] [Google Scholar]
- 53.Feau S, Arens R, Togher S, and Schoenberger SP. 2011. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol 12: 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams MA, Tyznik AJ, and Bevan MJ. 2006. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441: 890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, and Ahmed R. 2010. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32: 91–103. [DOI] [PubMed] [Google Scholar]
- 56.Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, Villalobos P, Neggers JE, Mendiratta S, Wei S, Landesman Y, Senapedis W, Baloglu E, Chow CB, Frink RE, Gao B, Roth M, Minna JD, Daelemans D, Wistuba II, Posner BA, Scaglioni PP, and White MA. 2016. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 538: 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Senapedis WT, Baloglu E, and Landesman Y. 2014. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol 27: 74–86. [DOI] [PubMed] [Google Scholar]
- 58.Biondi CA, Das D, Howell M, Islam A, Bikoff EK, Hill CS, and Robertson EJ. 2007. Mice develop normally in the absence of Smad4 nucleocytoplasmic shuttling. Biochem J 404: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, and Mackall CL. 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tyler PM, Servos MM, de Vries RC, Klebanov B, Kashyap T, Sacham S, Landesman Y, Dougan M, and Dougan SK. 2017. Clinical Dosing Regimen of Selinexor Maintains Normal Immune Homeostasis and T-cell Effector Function in Mice: Implications for Combination with Immunotherapy. Mol Cancer Ther 16: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malissen B, and Bongrand P. 2015. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol 33: 539–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.