

Abstract
Background:
Mechanisms governing the induction of heart failure by the impairment of autophagy and the ubiquitin-proteasome system (UPS) and the molecular pathways to cardiomyocyte (CM) necrosis remain incompletely understood. COPS8 is an essential subunit of the COP9 signalosome (CSN), a key regulator of ubiquitination. Mice with CM-restricted knockout of Cops8 (Cops8-cko) show autophagic and UPS malfunction and massive CM necrosis followed by acute heart failure and premature death, providing an excellent animal model to address the mechanistic gaps specified above. This study was conducted to determine the nature and underlying mechanisms of the CM necrosis in Cops8-cko mice.
Methods and Results:
Compared with littermate control mice, myocardial protein levels of key factors in the necroptotic pathway (RIPK1, RIPK3, MLKL, the RIPK1-bound RIPK3), protein carbonyls, full-length caspase 8, and BCL2, as well as histochemical staining of superoxide anions were significantly higher but the cleaved caspase 8 and the caspase 8 activity were significantly lower in Cops8-cko mice. In vivo CM uptake of Evans blue dye (EBD) was used as an indicator of necrosis. Cops8-cko mice treated with a RIPK1 kinase inhibitor (necrotstatin-1) showed less EBD uptake (0.005% vs. 0.20%; p<0.0001) and longer median lifespan (32.5 vs. 27 days; p<0.01) than those treated with vehicle control. RIPK3 haploinsufficiency showed similar recuing effects on Cops8-cko but Cyclophilin D deficiency did the opposite.
Conclusions:
Cardiac Cops8/CSN malfunction causes RIPK1-RIPK3 dependent, but mitochondrial permeability transition pore independent, CM necroptosis in mice and the CSN plays an indispensable role in suppressing CM necroptosis.
Keywords: COPS8, necroptosis, RIPK1, RIPK3, caspase 8, Cyclophilin D
Introduction
Heart failure is a leading cause of disability and death in humans. Inadequate protein quality control (PQC) as a result of impaired functioning in the ubiquitin-proteasome system (UPS) and in the autophagic lysosomal pathway (ALP) has been implicated as a major pathogenic factor in human heart failure,1, 2 and so does the loss of cardiomyocytes (CMs) in the forms of apoptosis and regulated necrosis.3, 4 Hence a better understanding of the molecular and cellular mechanisms by which PQC impairment contributes to heart failure genesis and of the molecular pathways to CM necrosis are expected to identify potentially new therapeutic targets.
Morphologically, cell death can be generally classified into necrosis (AKA, lytic cell death) and apoptosis (AKA, non-lytic cell death).3, 4 Necrosis is featured by the loss of cell membrane integrity, which allows free entry of extracellular fluid into the cell. This process leads to cell swelling, rupturing, and subsequent releasing of cellular contents into the extracellular space; hence, necrosis will inevitably trigger inflammation. Conversely, apoptosis is a well-known and well-characterized form of programmed or regulated cell death that requires caspase activation via either the mitochondrial or the extrinsic pathway. When undergoing apoptosis in a tissue, the cell keeps its membrane sealed well and, even at the late stage, the apoptotic cell breaks into smaller pieces known as apoptotic bodies, each of which is capsuled by membrane. Hence, apoptosis generally does not trigger inflammation and is a much cleaner form of cell death than necrosis.3 Recent advances in cell death research have further unveiled that a significant portion of necrosis can also be regulated cell death, known as regulated necrosis, of which death receptor-triggered necrosis is known as necroptosis.3 Originally identified in caspase 8 deficient or inhibited cells, the induction of necroptosis by TNFα is now known to require the formation of necrosomes consisting of receptor interacting protein kinase 1 (RIPK1), RIPK3, and a pseudo-kinase termed mixed lineage kinase-like protein (MLKL). In the canonical pathway by which the activation of TNFα receptor 1 (TNFR1) induces necroptosis, the kinase activities of both RIPK1 and RIPK3 are required to phosphorylate MLKL. Phosphorylated MLKL forms amyloid-like oligomers, which will then translocate and incorporate into the plasma membrane; ultimately, this will produce pores on the membrane, leading to the cell swelling and plasma membrane rupture.3 Ubiquitination plays an essential role in the regulation of both the kinase activity of RIPK1 and the activation of caspase 8. For example, in TNFR1 signaling, both K63-linked and methionine 1 linear ubiquitination of RIPK1 are required for the incorporation of RIPK1 into the complex 1 and thereby promote NFκB activation and cell survival,5, 6 whereas K48-linked polyubiquitination of RIPK1 mediates its proteasomal degradation.7, 8 Cullin3 (Cul3)-based polyubiquitination of caspase 8 drives full activation and processing of caspase 8, which leads to activation of the extrinsic apoptotic pathway.9 However, it remains unclear how the malfunction of the COP9 signalosome (CSN), a major regulator of Cullin-RING ligases (CRLs), impacts these cell death pathways although ablation of various Cops genes and the chemical inhibition of the CSN are known to induce cell death.10–14
CM apoptosis and various forms of regulated necrosis contribute to heart failure.3, 15 Analyses of necroptotic biochemical markers in the myocardium of humans with end-stage heart failure resulting from myocardial infarction (MI) or dilated cardiomyopathy indicate an involvement of necroptosis in heart failure.16 A genetic variant in the RIPK3 promoter region associated with increased RIPK3 transcription may contribute to the poor prognosis of heart failure patients.17 Animal experiments demonstrated an important role for necroptosis in post-MI remodeling,18 myocardial ischemia/reperfusion (I/R) injury, cardiotoxicity of doxorubicin treatment,19, 20 and paraquat-induced cardiac contractile dysfunction.21 By definition, necroptosis and the necrosis-driven by the mitochondrial permeability transition pore (MPT) opening are two different types of regulated necrosis.3 MPT opening requires Cyclophilin D that is encoded by the Ppif gene;22 hence MPT-dependent necrosis should not occur in Ppif knockout cells. Interestingly, Zhang et al. reported that cardiac necroptosis induced by I/R injury or doxorubicin treatment requires RIPK3 but not RIPK1 and MLKL; the upregulated RIPK3 phosphorylates and activates the calcium/calmodulin-dependent protein kinase II (CaMKII) and thereby opens MPT to induce CM necroptosis.19 However, more recent evidence suggests that the RIPK3-MLKL axis may still be important for myocardial necroptosis during I/R injury.20 Myocardial I/R was shown to induce myocardial dysregulation of both strands (5p and 3p) of miR-223 in mice and this dysregulation induces cardiac necroptosis during I/R by acting on TNFR1 and other points upstream of RIPK3.23 Consistent with the crucial role of transforming growth factor beta-activated kinase 1 (TAK1) and TNFR-associated protein 2 (TRAF2) in TNFR1-triggered survival signaling, CM-restricted ablation of the gene encoding TAK1 or TRAF2 in mice causes CM apoptosis and necroptosis and thereby increases the propensity for heart failure.24, 25 Taken together, these studies strongly support the proposition that CM necroptosis plays an important role in the development of heart failure from common etiologies such as ischemic heart disease, dilated cardiomyopathy, and perhaps hypertensive heart disease. Therefore, a better understanding of the molecular mechanisms governing CM necroptosis may provide new therapeutic strategies to prevent or more effectively treat heart failure.
The CSN is a highly conserved protein complex formed by 8 unique protein subunits (COPS1 through COPS8). The known biochemical activity of the CSN is to serve as the deneddylase to remove NEDD8 from a neddylated cullin in the CRLs via a process known as deneddylation,26 which is further enhanced by association with the 9th subunit CSN9.27 The catalytic center of the CSN is harbored in COPS5 but COPS5 exerts proper deneddylating activity only when it is incorporated into the CSN holocomplex formed by all 8 subunits;26 hence, loss of any of the COPS subunits will impair Cullin deneddylation. Cullin functions as a scaffold in CRLs that are the largest family of ubiquitin E3s and, by estimate, responsible for the ubiquitin-dependent degradation of approximately 20% of cellular proteins.28 CRLs play an important role in the degradation of misfolded proteins in the heart.29 The Skp1-Cul1-F-box (SCF) E3s are the prototype of CRLs and classified as the CRL1 class. There are at least 7 other classes of CRLs.30 Cullin neddylation and deneddylation regulate the cyclic assembly and disassembly of CRLs, which is essential for remodeling CRLs to meet timely the need to ubiquitinate specific substrate proteins within the cell.30 Thus the CSN by virtue of Cullin deneddylation plays an indispensable role in regulating the ubiquitination of a significant proportion of cellular proteins. We have previously reported that cardiomyocyte (CM)-restricted knockout (KO) of the Cops8 gene (Cops8CKO) in mice initiated at the perinatal period leads to massive CM necrosis rather than apoptosis, dilated cardiomyopathy, and mouse premature death, which is preceded by perturbation of not only the UPS but also the ALP.13, 14 Similar findings were also observed in mice with adult-onset Cops8CKO.12
The present study determined the nature and underlying mechanisms of the CM necrosis in Cops8CKO mice. It revealed that CM necrosis induced by Cops8 deficiency or CSN impairment was associated with increased interaction of RIPK1 with RIPK3, decreases in caspase 8 activation, and increased oxidative stress. Moreover, inhibition of RIPK1 kinase activity and the haploinsufficiency of RIPK3, but not ablation of the Ppif gene were able to significantly attenuate Cops8CKO-induced CM necrosis and delay mouse premature death. Here we demonstrate that COPS8 deficiency or CSN impairment causes CM necroptosis in mice through activating the RIPK1-RIPK3 pathway and impairing caspase 8 activation, which establishes Cops8/the CSN as a crucial suppressor of CM necroptosis and unravels novel mechanisms for cardiac UPS and ALP malfunction in injuring the heart.
Materials and Methods
A full description of Materials and Methods can be found in online supplements. The authors declare that all supporting data are available within the article and its online supplementary files.
Animal models
Perinatal cardiomyocyte-restricted ablation of the Cops8 gene (Cops8CKO) was achieved in C57BL/6J inbred mice as we previously reported.13 The creation of RIPK3 null mice was previously described.31 Mice with germline knockout of the Ppif gene (encoding Cyclophilin D) were provided by Dr. Jeffrey Molkentin of University of Cincinnati.22
The animal care and use protocols for this study were approved by the Institutional Animal Care and Committee of the University of South Dakota and followed the NIH guide for the care and use of laboratory animals.
Evan’s blue dye (EBD) uptake assay
After absorbed into the circulatory system, EBD is bound by albumin in the blood. Therefore, in this assay, EBD does not enter the cell with intact plasma membrane; EBD only enters a cell whose membrane permeability is abnormally increased, a key feature that distinguishes necrosis from apoptosis.3 Hence, the in vivo EBD uptake assay was performed to detect CM necrosis in mice at 3 weeks of age as we reported.13, 14
Necrostatin-1 (Nec-1) treatment
At 2 weeks of age, 19 Cops8CKO mice were continuously administered Nec-1 (BML-AP309, Enzo Life Science; 1.56 mg/kg/day) or vehicle (10% DMSO in PBS) by intraperitoneal implantation of osmotic mini-pumps (Alzet Model 1002, designed for continuous drug delivery for 2 weeks). Two cohorts of mice were included. For CM necrosis analysis using the EBD uptake assay as described above, one cohort (3 mice per group) was sacrificed 7 days after implantation of the mini-pump. The other cohort was used for Kaplan-Meier survival analysis.
Dihydroethidium (DHE) staining for reactive oxygen species (ROS)
Three mouse hearts per group were perfused in situ and excised in PBS, embedded in OCT and rapidly frozen. Serial cryosections (10 μm thick) were mounted onto glass slides. The slides were air-dried and incubated with 2.5 μM DHE in PBS at 37°C for 30 min. When oxidized to ethidium bromide by the superoxide anion, DHE produces a red fluorescence.32 The red fluorescence derived from DHE was measured with confocal microscopy and is used as the indicator of ROS content.
Western blot analyses and co-immunoprecipitation assays (Co-IP)
Western blotting and Co-IP were performed as described.33, 34
Protein carbonyl assays
Protein carbonyls, biomarkers of oxidative stress, were assessed as described.35 Briefly, Ventricular protein samples were subjected to 2,4-dinitrophenylhydrazine (DNPH) derivatization. The carbonyl groups in the protein side chains are derivatized to 2,4-dinitrophenylhydrazone (DNP-hydrazone). The DNP-derivatized proteins were then immuno-detected with an anti-DNP antibody.
Caspase 8 activity assays
The activities of caspase 8 in myocardial crude protein extracts were measured using the Caspase-8 Colorimetric Assay Kit (BioVision, Inc., USA).
Statistical analyses
GraphPad Prism software (San Diego, CA) was used. All continuous variables are presented as mean±SEM unless indicated otherwise. All data were examined for normality with the Shaprio Wilk’s test prior to application of parametric statistical tests. Those that failed this test were analyzed with non-parametric methods. Tests are specified in figure legends. In all cases where 1-way ANOVA was used, post hoc Tukey’s test was performed for pairwise comparisons only when ANOVA showed an overall significant effect. A p value <0.05 is considered statistically significant.
Results
Key proteins of the necroptotic pathway are increased in Cops8CKO mouse hearts
To explore the mechanism governing the CM necrosis in Cops8CKO mice, we examined the potential involvement of the RIPK1-RIPK3 pathway. Western blot analyses revealed that myocardial protein levels of RIPK1, RIPK3, and MLKL in Cops8CKO mice were significantly higher than in littermate control mice (Figure 1A, 1B). Co-immunoprecipitation of RIPK1 detected greater association of RIPK3 with RIPK1 in Cops8CKO hearts compared with littermate controls (Figure 1C, 1D).
Figure 1. Increases in myocardial RIPK1, RIPK3 and MLKL and in RIPK1-bound RIPK3 proteins in Cops8CKO mice.
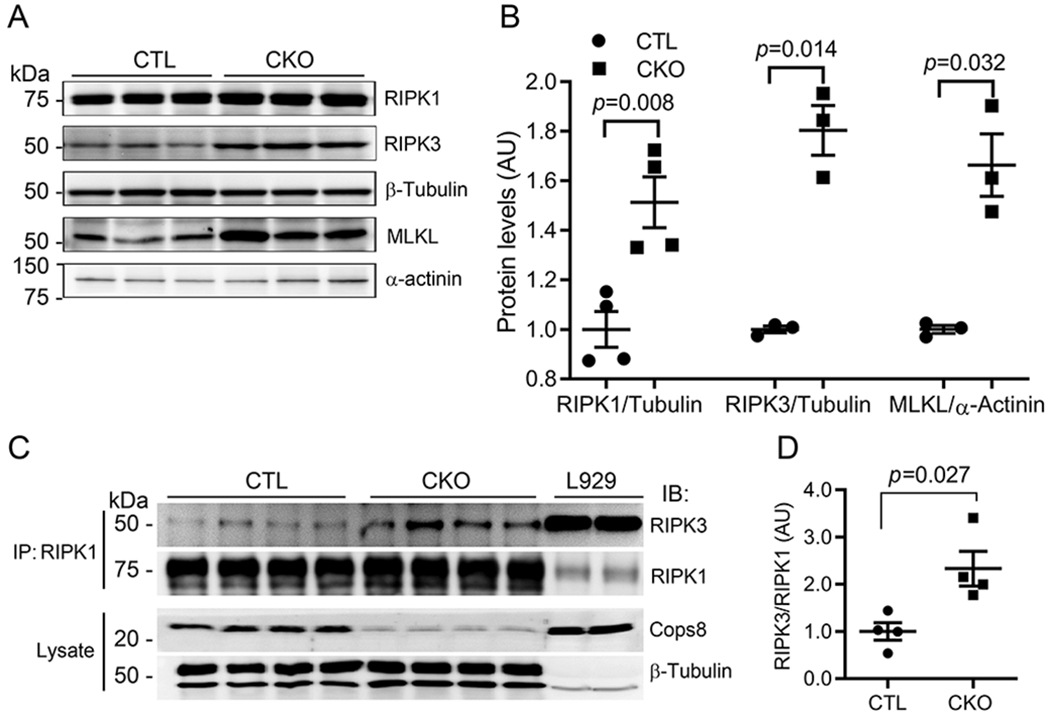
A and B, Representative images (A) and pooled densitometry data (B) of western blot analysis for the indicated proteins in the myocardial extracts of 3-week-old Cops8CKO (CKO) and littermate control (CTL) mice. β-Tubulin (for RIPK1, RIPK3) and α-actinin (for MLKL) were probed as loading controls. C, Western blot (IB) analyses for RIPK1 and RIPK3 in the RIPK1 immuno-precipitates (IP) from the protein lysate of ventricular myocardium from 3-week-old CTL and Cops8CKO mice. L929 cell lysates were used as positive controls. D, RIPK1/RIPK1 ratios in the RIPK1 IP. The density of RIPK3 and RIPK1 bands for individual samples shown in panel C was used for the calculation of RIPK3 to RIPK1 ratios, the mean of the ratios of the CTL group is defined as 1 arbitrary unit (AU). The p values shown in this figure are derived from two-side unpaired t-test with Welch’s correction. Each lane and each dot represents a unique mouse.
Cops8 deficiency increases myocardial oxidative stress
Next, we examined whether oxidative stress was increased in CMs of Cops8CKO mice. The level of superoxide anion (O2−) in myocardial sections was probed with the DHE staining assay. The red fluorescence intensity of the DHE-probed myocardial sections from homozygous Cops8CKO mice was greater than that from either heterozygous Cops8CKO or Cops8FL/FL control mice (Figure 2A, 2B), indicating that Cops8 deficiency increases myocardial superoxide levels. Myocardial ROS were also assessed via immunoblotting for DNPH-derivatized protein carbonyls. Immuno-probing of DNP in protein dot blots revealed that myocardial protein carbonyls were higher in the homozygous Cops8CKO mice compared with heterozygous Cops8CKO, Cops8FL/FL, or Myh6-CreTG mice (Figure 2C, 2D). Western blot analyses further showed that the increased carbonyls were mainly on proteins of a molecular weight ranging from 25 to 37 kDa (Figure 2E). These findings indicate that Cops8 deficiency in CMs increases myocardial oxidative stress.
Figure 2. Changes of myocardial reactive oxygen species (ROS) in Cops8CKO mice.

A and B, Detection of ROS in myocardial sections by dihydroethidium (DHE) staining (red). Four to six representative sections per mouse and 5 mice per genotype were analyzed. Panel A shows representative confocal fluorescence images of DHE stained myocardial sections from homozygous Cops8FL/FL, heterozygous Cops8CKO (Het-KO), and homozygous Cops8CKO (Hom-KO) mice. Scale bar=100µm. Panel B presents a scatter dot plot of individual average fluorescent intensity values for each sections, superimposed by median with 95% CI. P values are derived from the Kruskal-Walls test followed by Dunn’s pairwise comparison tests. C and D, Dot blot analyses for DNP-derivatized protein carbonyls. Equal amounts of proteins were subject to DNP-derivatization and equal proportions of the DNP-derivatized preparation were used for dot blot and subsequent immunoprobing for DNP. α-Actinin was probed as a loading control (see Supplementary Figure S1 for densitometry data). Shown are representative images (C) and pooled densitometry data (D). Mean±SEM are superimposed. Cre, Myh6-CreTG only; n = 3 mice for each group; one way ANOVA followed by Tukey’s test. E, Representative image of western blot analysis for DNP-derivatized protein carbonyls (upper image) and a-actinin (bottom). The opening curly brace demarcates the protein molecular weight range where carbonyls were increased most in the Hom-KO group. NC, negative control where equal amount of a Hom-KO myocardial protein sample that were not subject to DNP derivatization was loaded. Each lane represents a unique mouse.
Impaired caspase 8 activation and upregulated BCL2 expression in Cops8CKO hearts
Since necroptosis was originally observed in TNFα-treated cells whose caspase 8 is defective or suppressed, we sought to examine myocardial expression and activity of caspase 8 in Cops8CKO mice. Both the cleaved/activated form of caspase 8 and the activities of caspase 8 were significantly lower but the abundance of the full-length caspase 8 was discernibly greater in Cops8CKO mice than in littermate controls at 3 weeks of age (Figure 3A ~ 3C). Myocardial protein levels of BCL2, a key inhibitor of the mitochondrial apoptotic pathway, were significantly greater in 3-week-old homozygous Cops8CKO mice, compared with heterozygous Cops8CKO and Cops8FL/FL littermates (2.98±0.10 vs. 1.97±0.20, 1.00±0.16; p=0.010, <0.001, respectively; Figure 3D). Myocardial BCL2 mRNA levels were also greater in homozygous Cops8CKO mice than littermate controls at both 2 and 3 weeks of age (Figure 3E).
Figure 3. Changes in both protein expression and activities of caspases 8 as well as BCL2 protein and mRNA levels in Cops8CKO mouse hearts.

A and B, Representative images (A) and scatter dot plots of pooled densitometry data (B) of western blot analyses for caspase 8 (Casp8). L.C., Loading control which is a portion of the image from stain-free in-gel imaging of total proteins that was used to normalize caspase 8 western blot signals. F-, full length; C-, cleaved form. C, Changes in myocardial caspase 8 activities in Cops8CKO mice at 3 weeks. CTL, littermate control; KO, homozygous Cops8CKO. D, Representative images of western blot analyses for myocardial BCL2 in homozygous Cops8FL/FL (Floxed), heterozygous Cops8CKO (Het-CKO), and homozygous Cops8CKO (Hom-CKO) mice at 3 weeks of age. E, Changes in myocardial BCL2 mRNA levels in mice at 2 and 3 weeks of age. Each scatter dot plot is superimposed by mean±SEM; each dot represents a mouse; p values are derived from unpaired t-tests with Welch’s correction (B, C) or one way ANOVA followed by Tukey’s test (E).
Suppression of CM necrosis and delay of premature death by RIPK1 inhibition in Cops8CKO mice
To determine whether RIPK1 kinase activity is required for CM necrosis in Cops8CKO hearts, we tested the impact of necrostatin-1 (Nec-1), a RIPK1 kinase-specific inhibitor.36 Since CM necrosis is detectable in Cops8CKO mice at 3 weeks, but not at 2 weeks of age, the administration of Nec-1 or vehicle control via intraperitoneal implantation of osmotic mini-pumps was initiated at 2 weeks. CM necrosis was assessed using the in vivo EBD uptake assay in the heart harvested 7 days after mini-pump implantation. EBD positive CMs were not detectable in mice with control genotypes (Myh6-CreTG, Cops8FL/FL, and Cops8+/+; data not shown) but were readily detectable in homozygous Cops8CKO mice treated with vehicle control. Strikingly, the CM EBD positivity was strikingly less in the Nec-1 treated Cops8CKO mice than in the vehicle treated (0.005% vs. 0.20%, p<0.001; Figure 4A, 4B), indicating that RIPK1 kinase activity is required for Cops8 deficiency to induce CM necrosis in mice. Moreover, Kaplan-Meier survival analyses revealed that Nec-1 treatment significantly delayed the premature death observed in Cops8CKO mice (p=0.007; Figure 4C).
Figure 4. Necrostatin-1 (Nec-1) treatment markedly reduces cardiomyocyte necrosis and delays premature death of Cops8CKO mice.
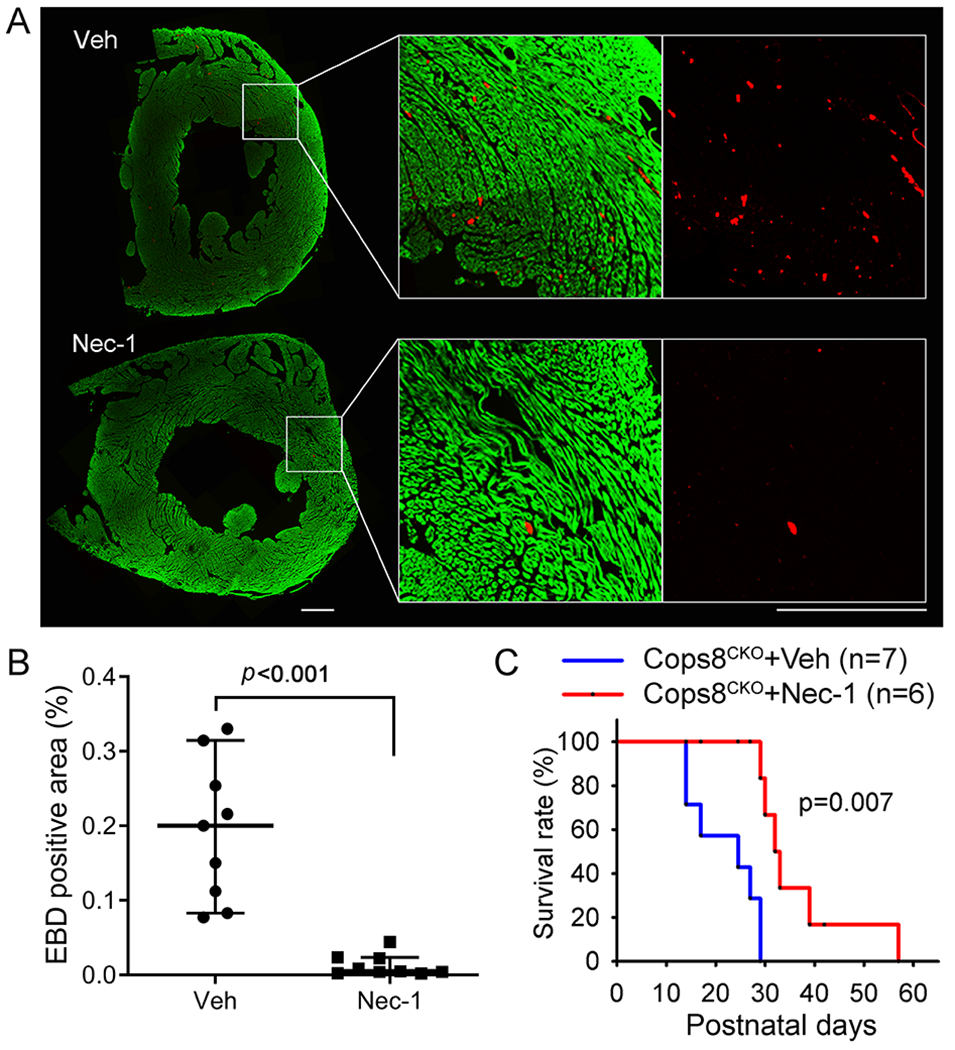
Cohorts of Cops8CKO mice at 2 weeks of age were treated with necrostatin-1 (Nec-1, 1.56 mg/kg/day) or vehicle (Veh) via intraperitoneal osmatic mini-pumps for 1 week (A, B) or continued for >2 weeks for the Kaplan-Meier survival analysis (C). A and B, At day 6, after min-pump implantation, mice were treated with one dose of Evan’s blue dye (EBD; 100 mg/kg, i.p.) 18 hours before they were anesthetized and perfusion-fixed in situ. Cryosections from the fixed heart were stained with Alexa488-conjugated phalloidin to identify cardiomyocytes (green) and subjected to fluorescence confocal microscopy. The images of each ventricular tissue ring were reconstructed and used for quantification of EBD-positive area (red) and total green area. Panel A shows representative reconstructed images from a pair of Cops8CKO hearts treated with Veh or Nec-1; scale bar=0.5 mm. Individual percent values of average EBD positive area in the 3 representative sections/mouse from 3 mice of each group are plotted in panel B, superimposed by median with 95% CI; Mann Whitney test. C, Kaplan-Meier survival curve of Cops8CKO mice treated with Veh or Nec-1. Nec-1 treatment significantly increased lifespan of Cops8CKO mice compared with the vehicle-treated group (median lifespan: 32.5 vs. 27 days); Log-rank Test.
Requirement of RIPK3 for CM necrosis in Cops8CKO mice
To test the role of RIPK3 in the CM necrosis of Cops8CKO mice, RIPK3 germline knockout (RIPK3−/−) mice were cross-bred with Cops8CKO mice and the resultant Cops8CKO::RIPK3+/+ and Cops8CKO::RIPK3+/− littermate mice were subjected to EBD CM necrosis assessment at 3 weeks of age as well as Kaplan-Meier survival analysis. The prevalence of EBD-positive CMs in Cops8CKO::RIPK3+/− mice was lower than that of littermate Cops8CKO::RIPK3+/+ mice (p<0.001; Figure 5A, 5B;); also, the lifespan of the former was longer than that of the latter (p<0.001; Figure 5C). These analyses show that RIPK3 haploinsufficiency is capable of suppressing CM necrosis and delaying premature death in Cops8CKO mice.
Figure 5. RIPK3 haploinsufficiency significantly reduces cardiomyocyte necrosis and delays premature death of Cops8CKO mice.
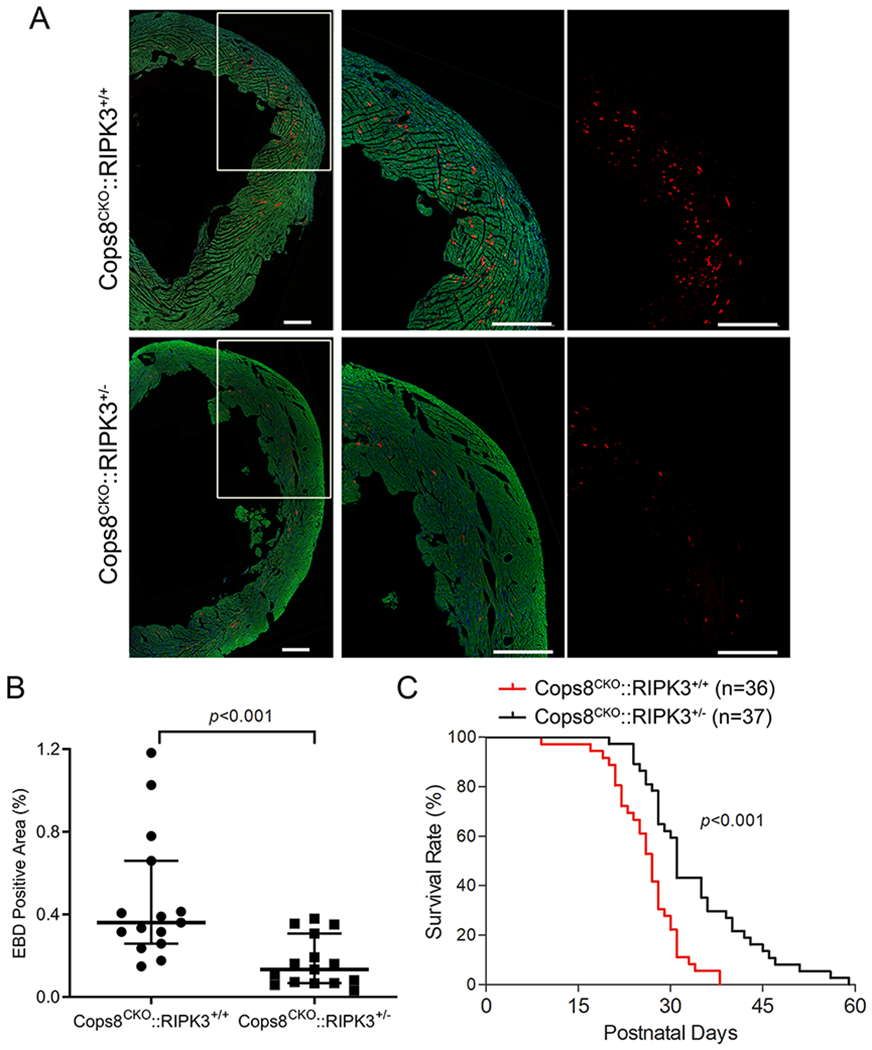
A, Representative confocal micrographs of EBD assays. Littermate mice of the indicated genotypes at 3 weeks of age were subjected to the EBD assays in the same way as described in Figure 4. EBD positive cells display autofluorescence (red) and F-actin was stained using Alexa-488-conjugated phalloidin (green). Shown are representative composed images for the entire cross-section of the left ventricle or a higher magnification view of the marked portion of the composed image (A). Scale bar=500µm. B, Scatter dot plots of the individual percent values of EBD positive area in the 5 representative sections/mouse of 3 mice of each group, , superimposed by median with 95% CI. Mann Whitney test. C. Kaplan-Meier survival curve. The median lifespan for Cops8CKO::RIPK3+/− was significantly longer than that of Cops8CKO::RIPK3+/+ mice (31 vs. 27 days; hazard ratio=3.581). Log-Rank Test.
CM necroptosis in Cops8CKO mice is independent of mitochondrial permeability transition (MPT)
To determine whether MPT-driven necrosis contributes to CM necrosis in Cops8CKO mice, we cross-bred Cops8CKO mice with Ppif gene knockout mice to test whether ablation of the Ppif gene would mitigate the CM necrosis and mouse premature death induced by Cops8CKO. Compared to mice harboring Cops8CKO alone, mice harboring Cops8CKO coupled with either heterozygous or homozygous knockout of the Ppif gene did not show a delay in premature death; on the contrary, homozygous Ppif knockout moderately increased CM necrosis (p=0.010) and accelerated mouse premature death caused by Cops8CKO (p=0.007; Figure 6). These results indicate that MPT is not a mediator for CM necrosis in Cops8CKO mice.
Figure 6. Cyclophilin D knockout exacerbates cardiomyocyte necrosis and premature death of Cosp8CKO mice.
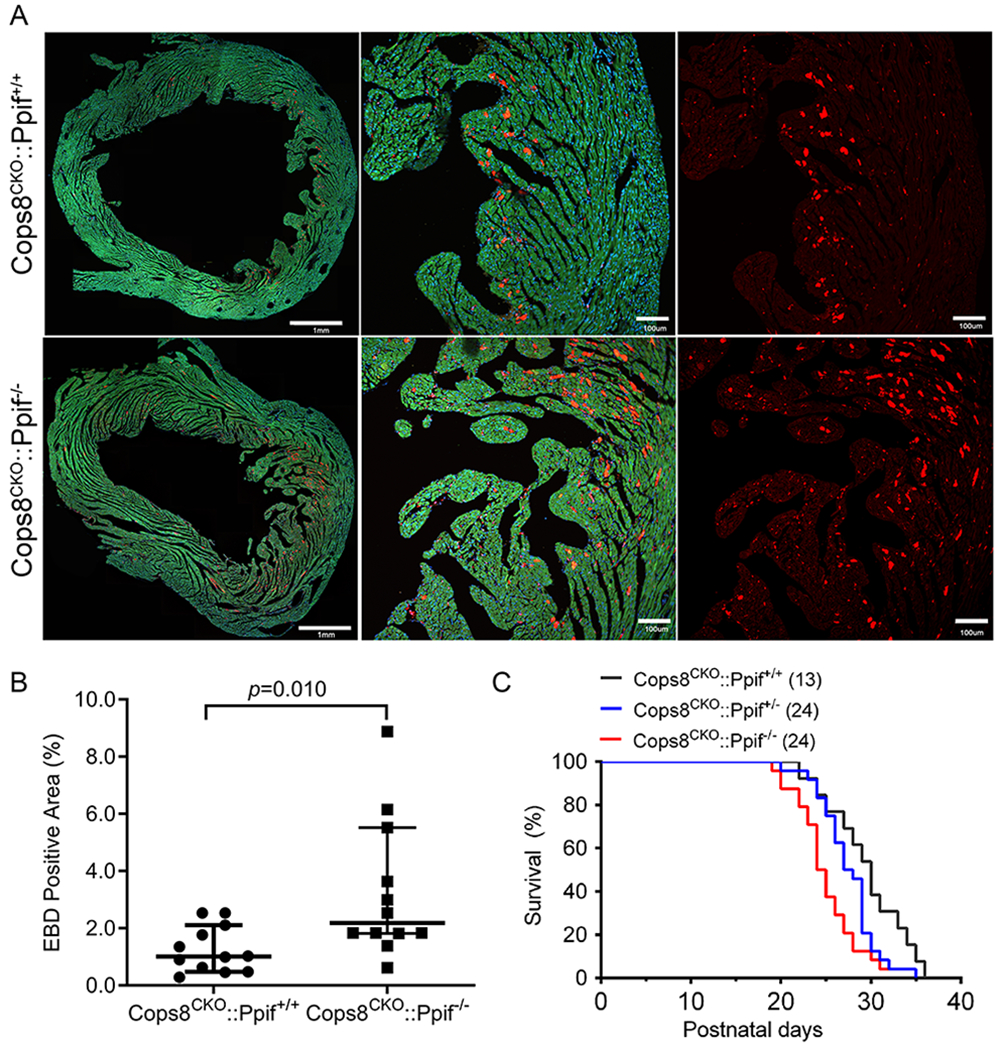
The Ppif gene encodes Cyclophilin D. Cops8CKO was induced in the Ppif wild type as well as hemizygous and homozygous null background through cross-breeding. A and B, in vivo EBD uptake assays were performed on mice at 3 weeks of age as described in Figure 4. Shown are representative confocal micrographs of myocardial sections from mice of the indicated genotypes (A) and scatter dot plot of the individual percent values of average EBD positive area in the 3 representative sections per mouse and 4 mice per group, superimposed by median with 95% CI (B). Mann Whitney test. C, Kaplan-Meier survival curves of littermate mice with the indicated genotypes. Median lifespans for the Cops8CKO::Ppif+/+ (n=13 mice), Cops8CKO::Ppif+/− (n=24), and Cops8CKO::Ppif−/− (n=24) groups were 30, 27.5 and 24.5 days, respectively. p=0.001 for all 3 groups, Log-rank test for trend; p=0.006 and hazard ratio=2.238 for Cops8CKO::Ppif−/− vs. Cops8CKO::Ppif+/+, Log-rank (Mantel-Cox) test.
Discussion
The present study unveils for the first time that CMs deficient of Cops8 die primarily in the form of necroptosis. Mechanistically, likely impairing CRL-mediated ubiquitination, Cops8 deficiency impairs caspase 8 activation and increases BCL2 expression, thereby suppressing both extrinsic and intrinsic apoptotic pathways, which steers the death receptor-mediated signaling towards activation of the RIPK1-RIPK3-mediated necroptotic pathway (Figure 7). Findings of this study also demonstrate that the MPT does not play an important role in the induction of CM necroptosis by Cops8CKO. These discoveries not only establish the CSN as a crucial factor to suppress CM necroptosis but also suggest that combined UPS and autophagy impairment may be a proximal causing factor for CM necroptosis, a major form of cell death linked to heart failure.
Figure 7. A working model for induction of cardiomyocyte necroptosis by Cops8 deficiency.
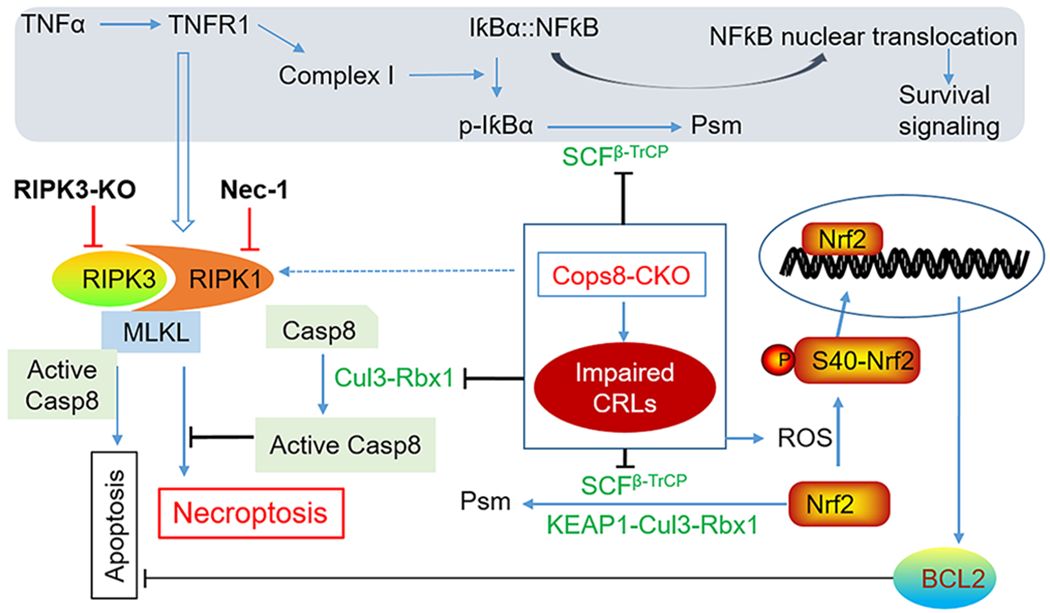
The main interrogations of this study marked with bold black font. Loss of function of Cops8/CSN is expected to suppress the CRLs shown in green font, which in turn disrupts the respective ubiquitination events and thereby blocks NFκB-mediated survival signaling and inhibits the apoptotic pathway via increasing BCL2 and hindering the activation of caspase 8 (Casp8), steering the death receptor triggered pathway to the RIPK1-RIPK3 mediated necroptosis. The pathways in the shaded zone are likely involved but not directly examined in Cops8 deficient mice. We hypothesize that TFNα comes from autocrinal or paracrinal routes from myocardium with cardiomyocyte-restricted Cops8 knockout. Dot line denotes a potential link not tested yet. Psm, proteasomal degradation.
Cops8 deficient or CSN inhibited CMs die primarily from necroptosis
Massive CM necrosis occurs in Cops8CKO mice, which is evidenced by increased EBD uptake by CMs in the absence of increased TUNEL positivity, and by the ultrastructural features like CM swelling and a broken plasma membrane.12–14 Activation of RIPK3 is the centerpiece of necroptotic pathway although RIPK1 is also required in the induction of necroptosis by TNFα at least.37 The detection of necroptosis currently requires a combination of rather sophisticate tests to reveal both the necrotic feature (e.g., loss of plasma membrane integrity) and the dependence on RIPK3 activation, according to recently published guidelines.38 Here we found that CM necrosis in Cops8CKO mice was associated with increases in RIPK1, RIPK3, MLKL, and RIPK1-bound RIPK3 protein levels (Figure 1) and depended on both RIPK1 kinase activity (Figure 4) and increased expression of RIPK3 (Figure 5). These new findings demonstrate unequivocally that the massive CM necrosis induced by Cops8CKO in mice belongs to necroptosis and the RIPK1-RIPK3 pathway mediates its occurrence. Notably, in contrast to a recently delineated RIPK3-CamKII-MPT pathway to cardiac necroptosis,19 MPT does not play a major role in the execution of CM necroptosis in Cops8CKO mice. This is because Cyclophilin D knockout, which is known to inhibit MPT, did not attenuate but rather exacerbated CM necrosis and premature death in Cops8CKO mice (Figure 6). Indeed, MPT- and even mitochondria-independent but RIPK3-depdent necroptosis was reported for several other cell types.39–41 It will be interesting to test whether CaMKII is involved in CM necrosis in Cops8CKO mice.
How does Cops8 deficiency cause CM necroptosis?
The requirement of both RIPK1 and RIPK3 by the CM necrosis observed here suggests that the induction of CM necroptosis by Cops8CKO shares the same pathway taken by TNFR1 activation (Figure 7). The ligation of TNFR1 by TNFα can lead to at least 3 possible downstream events: (1) formation of complex 1 where RIPK1 serves as a scaffold in a manner independent of its kinase activity, which provides survival signals via activation of nuclear factor κB (NFκB) and mitogen-activated protein kinases (MAPKs), (2) formation of complex 2a, which induces apoptosis via caspase 8 and downstream cascade, and (3) formation of complex 2b (i.e., the RIPK1-RIPK3-MLKL) and thereby induction of necroptosis when caspase 8 is defective or inhibited.3 The kinase activity of RIPK1 is required for RIPK1 to induce cell death in complex 2. UPS-dependent degradation of IκBα is a key step in the activation of NFκB by TNFα where the ubiquitination of IκBα is driven by Skp1-Cul1-β-TrCP (SCFβ-TrCP),42 a member of the CRL1 family E3 ligases whose assembly and disassembly are regulated by the CSN. Hence, the survival signaling from NFκB is likely suppressed by impairment of IκBα ubiquitination due to defective Cullin deneddylation resulting from Cops8 deficiency. Our prior study detected decreases in myocardial F-box protein β-TrCP protein levels in Cops8CKO mice,13 adding a reason to predict a reduction of SCFβ-TrCP ligase activities. Thus, Cops8 deficiency swings TNFR1 signaling towards the cell death direction.
Then, the next question is why necroptosis instead of apoptosis takes place. At least in the case of induction of necroptosis by death receptor activation, two prerequisites must be met. First, the formation of the so-called complex 2 containing RIPK1 and RIPK3 and second, the failure of caspase 8 to activate.3 Indeed, both prerequisites were met in the Cops8CKO hearts. Not only were RIPK1, RIPK3, and MLKL protein levels markedly increased but also RIPK1-RIPK3 interaction was significantly augmented as indicated by the RIPK1-RIPK3 Co-IP results (Figure 1); and importantly the cleaved form of caspase 8 as well as caspase 8 activity were substantially lower in the homozygous Cops8CKO hearts compared with CTL hearts (Figure 3). It is very likely that this impairment of caspase 8 activation directly result from the loss of Cullin deneddylation because a prior study has established that Cul3-RBX1 mediated polyubiquitination of caspase 8 is required for further processing and activation of caspase 8 and the signaling of the extrinsic apoptotic pathway.9 Both neddylation and deneddylation of Cullins are required for proper functioning of CRLs; hence, the ubiquitination of caspase 8 by Cul3-RBX1 is very likely suppressed by Cops8 deficiency. This argument is strongly supported by a recent report that inhibition of CRLs with the neddylation inhibitor MLN4924 sensitizes monocyte necroptosis in vitro.43 Besides caspase 8 which is essential to the extrinsic pathway of apoptosis, as discussed below, the mitochondrial pathway is likely suppressed by increased BCL2 (Figure 3).13
We have previously observed a suppressed autophagic flux in Cops8CKO mice. This could probably be due to impairment in autophagosome-lysosome fusion that occurs before impairment in the UPS degradation of a surrogate misfolded protein as well as CM necrosis become discernible.14 We propose dual impairment of both the UPS and the ALP plays an overall causative role in the CM necrosis that now proves to be necroptosis. This proposition now has support from two recent studies that collected evidence from cultured H9c2 cells suggesting a major contribution from impaired autophagy to the induction of necroptosis by TNFα.44, 45 According to these reports, the induction of RIPK1-RIPK3 interaction and necroptosis by the combined treatment with TNFα and a broad spectrum caspase inhibitor was associated with suppression of autophagic flux;44 improving autophagic flux via mTORC1 inhibition suppressed the necroptosis in an autophagy- and transcription factor EB (TFEB; a master regulator of the ALP)-dependent manner;44, 45 and MPT did not play a major role in the execution of necroptosis.44 This scenario starkly resembles what we have unveiled in the Cops8CKO mouse myocardium. Reduced myocardial autophagic flux was found to occur in the Cops8CKO mice at as young as 1 week of age,14 which is two weeks earlier than CM necroptosis becomes discernible.13 Hence, it will be interesting and important to test whether the autophagic impairment has exacerbated activation of the RIPK1-RIPK3-MLKL necroptotic pathway in Cop8CKO mice.
Previous reports have shown an important role of increased reactive oxygen species (ROS) in RIPK3-mediated necroptosis in cultured cells.46, 47 In TNFα induced necroptosis, the RIPK3-centered necrosome increases ROS production through stimulating aerobic metabolism and RIPK3 does so probably by activating key enzymes of metabolic pathways including glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), glutamate dehydrogenase 1 (GLUD1),46 and more recently pyruvate dehydrogenase (PDH) which is a rate-limiting enzyme linking glycolysis to aerobic respiration.48 The increased ROS further promotes necrosome formation and yields cytotoxicity during necroptosis.47 As reflected by increased DHE staining and the elevated levels of protein carbonyls in Cops8CKO hearts (Figure 2), increases in ROS or oxidative stress are indeed associated with CM necroptosis in Cops8CKO mice. Consistent with increased oxidative stress, our prior transcriptome analysis revealed that the Nrf2 pathway, the master regulator of antioxidant and defensive responses, was markedly upregulated in Cops8CKO hearts even before CM necrosis becomes discernible.49 The exact role of altered redox state in the induction of CM necroptosis by Cops8 deficiency remains to be determined.
Clinical implications
First of all, inadequate cardiac PQC due to UPS malfunction and ALP impairment has been implicated in the progression from a large subset of heart disease to heart failure;1, 50 however, the mechanistic link between impaired PQC and heart failure remains obscure. The discoveries of the present study implicate that CM necroptosis could be one of the missing links, because cardiac PQC impairment is obviously the apical defect in Cops8CKO mice. Accordingly, targeting the necroptotic pathway could potentially help alleviate the adverse outcome of cardiac PQC impairment. Second, a small molecule inhibitor of the CSN (CSN5i) has shown great promise in anti-tumor effects in experimental studies.11 Hence, there is a good possibility for this compound to move into clinical trials for cancer treatment. The findings of the present study caution that cardiac function should be closely monitored should CSN5i or alike be moved into clinical trials.
Supplementary Material
Clinical Perspective.
What Is New?
Myocardial caspase 8 activation is impaired in mice with cardiomyocyte-restricted ablation of the Cops8 gene encoding an essential subunit of the COP9 signalosome.
Cardiomyocyte-restricted Cops8 knockout (Cops8-cko) induces massive cardiomyocyte necroptosis.
Induction of cardiomyocyte necroptosis by Cops8-cko requires both RIPK1 and RIPK3 but is independent of mitochondrial permeability transition pore.
Cyclophilin D deficiency exacerbates cardiomyocyte necroptosis in Cops8-cko mice.
What Are the Clinical Implications?
Impairment of protein degradation and ultimately loss of cardiomyocytes in Cops8-cko mice are two major pathogenic factors also associated with most human heart diseases and heart failure. The present study suggests that the RIPK1-RIPK3 pathway constitutes a mechanistic link between the two pathogenic factors and both RIPK1 and RIPK3 represent potentially new drug targets for the intervention of these cardiac disorders.
A small molecule inhibitor of the COP9 signalosome has shown great promise to become an anticancer agent. Findings of the present study caution that cardiac function should be closely monitored should CSN5i or alike be moved into clinical trials.
Acknowledgments:
We thank Dr. Yibin Wang of University of California at Los Angeles (Los Angeles, CA) for his kind assistance in obtaining the RIPK3 knockout mouse for this study. Author contributions: Conception and experimental design (X.W., C.W., J. Liu), data collection and interpretation (P.X., C.W., J. Li, H.S., L.Y., P.W., M.T.L.), and manuscript preparation (P.X., X.W.).
Source of Funding: This study is in part supported by NIH grants HL072166, HL085629, and HL131667 (to X.W.), as well as HL124248 (to H.S.).
Nonstandard Abbreviations and Acronyms
- ALP
autophagic-lysosomal pathway
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CM
cardiomyocyte
- Cops8CKO
cardiomyocyte-restricted Cops8 knockout
- CRLs
Cullin-RING ligases
- CSN
COP9 signalosome
- EBD
Evan’s blue dye
- I/R
ischemia/reperfusion
- MPT
mitochondrial permeability transition pore
- PQC
protein quality control
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor α
- TNFR1
TNFα receptor 1
- UPS
ubiquitin-proteasome system
Footnotes
Disclosures: None.
References
- 1.Wang X, Wang H. Priming the proteasome to protect against proteotoxicity. Trends Mol Med. 2020. (in press). doi: 10.1016/j.molmed.2020.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Cui T. Autophagy modulation: A potential therapeutic approach in cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2017;313:H304–H319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight. 2019;4:e128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Zhang H, Xu C, Li X, Li M, Wu X, Pu W, Zhou B, Wang H, Li D, Ding Q, Ying H, Wang H, Zhang H. Ubiquitination of ripk1 suppresses programmed cell death by regulating ripk1 kinase activation during embryogenesis. Nat Commun. 2019;10:4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang Y, Tu H, Zhang J, Zhao X, Wang Y, Qin J, Lin X. K63-linked ubiquitination regulates ripk1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat Commun. 2019;10:4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. [DOI] [PubMed] [Google Scholar]
- 8.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of a20 downregulate nf-kappab signalling. Nature. 2004;430:694–699. [DOI] [PubMed] [Google Scholar]
- 9.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. [DOI] [PubMed] [Google Scholar]
- 10.Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY. Multiple functions of jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279:43013–43018. [DOI] [PubMed] [Google Scholar]
- 11.Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, Jones M, Hassiepen U, Schaefer M, Kiffe M, Weiss A, Wiesmann C, Sedrani R, Eder J, Martoglio B. Targeted inhibition of the cop9 signalosome for treatment of cancer. Nat Commun. 2016;7:13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su H, Li J, Osinska H, Li F, Robbins J, Liu J, Wei N, Wang X. The cop9 signalosome is required for autophagy, proteasome-mediated proteolysis, and cardiomyocyte survival in adult mice. Circ Heart Fail. 2013;6:1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, Wang X. Perturbation of cullin deneddylation via conditional csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su H, Li F, Ranek MJ, Wei N, Wang X. Cop9 signalosome regulates autophagosome maturation. Circulation. 2011;124:2117–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. [DOI] [PubMed] [Google Scholar]
- 16.Szobi A, Goncalvesova E, Varga ZV, Leszek P, Kusmierczyk M, Hulman M, Kyselovic J, Ferdinandy P, Adameova A. Analysis of necroptotic proteins in failing human hearts. J Transl Med. 2017;15:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu D, Huang J, Hu S, Zhang Y, Li S, Sun Y, Li C, Cui G, Wang DW. A common variant of rip3 promoter region is associated with poor prognosis in heart failure patients by influencing sox17 binding. J Cell Mol Med. 2019;23:5317–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe HJ, Linkermann A, Wolf MJ, Rose-John S, Lullmann-Rauch R, Adam D, Flogel U, Heikenwalder M, Luedde T, Frey N. Rip3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103:206–216. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. Camkii is a rip3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–182. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Li C, Wang Y, Yang J, Yin Y, Liu M, Shi Z, Mu N, Yu L, Ma H. Melatonin attenuates chronic pain related myocardial ischemic susceptibility through inhibiting rip3-mlkl/camkii dependent necroptosis. J Mol Cell Cardiol. 2018;125:185–194. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Feng Q, Wang T. Necrostatin-1 protects against paraquat-induced cardiac contractile dysfunction via rip1-rip3-mlkl-dependent necroptosis pathway. Cardiovasc Toxicol. 2018;18:346–355. [DOI] [PubMed] [Google Scholar]
- 22.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin d reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- 23.Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, Essandoh K, Mu X, Peng T, Han Q, Yu KJ, Fan GC. Microrna-223-5p and -3p cooperatively suppress necroptosis in ischemic/reperfused hearts. J Biol Chem. 2016;291:20247–20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo X, Yin H, Li L, Chen Y, Li J, Doan J, Steinmetz R, Liu Q. Cardioprotective role of tumor necrosis factor receptor-associated factor 2 by suppressing apoptosis and necroptosis. Circulation. 2017;136:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Chen Y, Doan J, Murray J, Molkentin JD, Liu Q. Transforming growth factor beta-activated kinase 1 signaling pathway critically regulates myocardial survival and remodeling. Circulation. 2014;130:2162–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thoma NH. Crystal structure of the human cop9 signalosome. Nature. 2014;512:161–165. [DOI] [PubMed] [Google Scholar]
- 27.Gutierrez C, Chemmama IE, Mao H, Yu C, Echeverria I, Block SA, Rychnovsky SD, Zheng N, Sali A, Huang L. Structural dynamics of the human cop9 signalosome revealed by cross-linking mass spectrometry and integrative modeling. Proc Natl Acad Sci U S A. 2020;117:4088–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of nedd8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. [DOI] [PubMed] [Google Scholar]
- 29.Su H, Li J, Zhang H, Ma W, Wei N, Liu J, Wang X. Cop9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ Res. 2015;117:956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by f-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newton K, Sun X, Dixit VM. Kinase rip3 is dispensable for normal nf-kappa bs, signaling by the b-cell and t-cell receptors, tumor necrosis factor receptor 1, and toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wojtala A, Bonora M, Malinska D, Pinton P, Duszynski J, Wieckowski MR. Methods to monitor ros production by fluorescence microscopy and fluorometry. Methods Enzymol. 2014;542:243–262. [DOI] [PubMed] [Google Scholar]
- 33.Hu C, Tian Y, Xu H, Pan B, Terpstra EM, Wu P, Wang H, Li F, Liu J, Wang X. Inadequate ubiquitination-proteasome coupling contributes to myocardial ischemia-reperfusion injury. J Clin Invest. 2018;128:5294–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan B, Zhang H, Cui T, Wang X. Tfeb activation protects against cardiac proteotoxicity via increasing autophagic flux. J Mol Cell Cardiol. 2017;113:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Powell SR, Wang X. Enhancement of proteasome function by pa28α overexpression protects against oxidative stress. FASEB J. 2011;25:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of rip1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to tnf-alpha. Cell. 2009;137:1100–1111. [DOI] [PubMed] [Google Scholar]
- 38.Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, Narula J, Takahashi M, Abbate A, Piristine HC, Kar S, Su S, Higa JK, Kawasaki NK, Matsui T. Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol. 2019;317:H891–H922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013;110:12024–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in t cells. J Exp Med. 2011;208:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanarek N, Ben-Neriah Y. Regulation of nf-kappab by ubiquitination and degradation of the ikappabs. Immunol Rev. 2012;246:77–94. [DOI] [PubMed] [Google Scholar]
- 43.El-Mesery M, Seher A, Stuhmer T, Siegmund D, Wajant H. Mln4924 sensitizes monocytes and maturing dendritic cells for tnf-dependent and -independent necroptosis. Br J Pharmacol. 2015;172:1222–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogasawara M, Yano T, Tanno M, Abe K, Ishikawa S, Miki T, Kuno A, Tobisawa T, Muratsubaki S, Ohno K, Tatekoshi Y, Nakata K, Ohwada W, Miura T. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J Mol Cell Cardiol. 2017;108:203–213. [DOI] [PubMed] [Google Scholar]
- 45.Abe K, Yano T, Tanno M, Miki T, Kuno A, Sato T, Kouzu H, Nakata K, Ohwada W, Kimura Y, Sugawara H, Shibata S, Igaki Y, Ino S, Miura T. Mtorc1 inhibition attenuates necroptosis through rip1 inhibition-mediated tfeb activation. Biochim Biophys Acta Mol Basis Dis. 2019;1865:165552. [DOI] [PubMed] [Google Scholar]
- 46.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. Rip3, an energy metabolism regulator that switches tnf-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. [DOI] [PubMed] [Google Scholar]
- 47.Schenk B, Fulda S. Reactive oxygen species regulate smac mimetic/tnfalpha-induced necroptotic signaling and cell death. Oncogene. 2015;34:5796–5806. [DOI] [PubMed] [Google Scholar]
- 48.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, Chen X, Liang Y, Wu J, Zhao S, Zhou D, Han J. Rip3 targets pyruvate dehydrogenase complex to increase aerobic respiration in tnf-induced necroptosis. Nat Cell Biol. 2018;20:186–197. [DOI] [PubMed] [Google Scholar]
- 49.Abdullah A, Eyster KM, Bjordahl T, Xiao P, Zeng E, Wang X. Murine myocardial transcriptome analysis reveals a critical role of cops8 in the gene expression of cullin-ring ligase substrate receptors and redox and vesicle trafficking pathways. Front Physiol. 2017;8:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pan B, Li J, Parajuli N, Tian Z, Wu P, Lewno MT, Zou J, Wang W, Bedford L, Mayer RJ, Fang J, Liu J, Cui T, Su H, Wang X. The calcineurin-tfeb-p62 pathway mediates the activation of cardiac macroautophagy by proteasomal malfunction. Circ Res. 2020. May 5. doi: 10.1161/CIRCRESAHA.119.316007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.