

MERS-CoV causes death of about 35% of patients. Published studies showed that some coronaviruses are capable of suppressing interferon (IFN) expression in the early phase of infection and MERS-CoV proteins can modulate host immune response. In this study, we demonstrated that MERS-CoV nucleocapsid (N) protein suppresses the production of both type I and type III IFNs via sequestering TRIM25, an E3 ubiquitin ligase that is essential for activating the RIG-I signaling pathway. Ectopic expression of TRIM25 rescues the suppressive effect of the N protein. In addition, the C-terminal domain of the viral N protein plays a pivotal role in the suppression of IFN-β promoter activity. Our findings reveal how MERS-CoV evades innate immunity and provide insights into the interplay between host immune response and viral pathogenicity.
KEYWORDS: MERS-CoV, nucleocapsid protein, interferon-β, interferon-λ1, RIG-I, TRIM25
ABSTRACT
Type I and type III interferons (IFNs) are the frontline of antiviral defense mechanisms that trigger hundreds of downstream antiviral genes. In this study, we observed that MERS-CoV nucleocapsid (N) protein suppresses type I and type III IFN gene expression. The N protein suppresses Sendai virus-induced IFN-β and IFN-λ1 by reducing their promoter activity and mRNA levels, as well as downstream IFN-stimulated genes (ISGs). Retinoic acid-inducible gene I (RIG-I) is known to recognize viral RNA and induce IFN expression through tripartite motif-containing protein 25 (TRIM25)-mediated ubiquitination of RIG-I caspase activation and recruitment domains (CARDs). We discovered that MERS-CoV N protein suppresses RIG-I-CARD-induced, but not MDA5-CARD-induced, IFN-β and IFN-λ1 promoter activity. By interacting with TRIM25, N protein impedes RIG-I ubiquitination and activation and inhibits the phosphorylation of transcription factors IFN-regulatory factor 3 (IRF3) and NF-κB that are known to be important for IFN gene activation. By employing a recombinant Sindbis virus-EGFP replication system, we showed that viral N protein downregulated the production of not only IFN mRNA but also bioactive IFN proteins. Taken together, MERS-CoV N protein functions as an IFN antagonist. It suppresses RIG-I-induced type I and type III IFN production by interfering with TRIM25-mediated RIG-I ubiquitination. Our study sheds light on the pathogenic mechanism of how MERS-CoV causes disease.
IMPORTANCE MERS-CoV causes death of about 35% of patients. Published studies showed that some coronaviruses are capable of suppressing interferon (IFN) expression in the early phase of infection and MERS-CoV proteins can modulate host immune response. In this study, we demonstrated that MERS-CoV nucleocapsid (N) protein suppresses the production of both type I and type III IFNs via sequestering TRIM25, an E3 ubiquitin ligase that is essential for activating the RIG-I signaling pathway. Ectopic expression of TRIM25 rescues the suppressive effect of the N protein. In addition, the C-terminal domain of the viral N protein plays a pivotal role in the suppression of IFN-β promoter activity. Our findings reveal how MERS-CoV evades innate immunity and provide insights into the interplay between host immune response and viral pathogenicity.
INTRODUCTION
Coronaviruses are enveloped, positive-sense single-stranded RNA viruses that cause a variety of diseases in humans and domestic animals. Middle East respiratory syndrome coronavirus (MERS-CoV) represents a major public health threat that causes acute respiratory syndrome with a high mortality rate in humans. The first case of MERS-CoV was identified in September 2012 in Saudi Arabia (1). From the first outbreak to the end of November 2019, MERS-CoV spread through 27 countries and caused about 2,494 cases and 858 deaths (http://www.who.int/emergencies/mers-cov/en/). Studies looking for rapid and efficient treatment to reduce the probability of death of MERS-CoV-infected patients are ongoing.
Innate immune response is the first-line host defense against pathogens. Vertebrates have developed patterns recognition receptors (PRRs), including Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), to recognize different conserved molecular structures called pathogen-associated molecular patterns (PAMPs) on pathogens (2). These PRR families are diverse in ligand specificity and cellular localization, with the ability to activate different upstream signaling molecules, but converge into overlapping pathways to regulate the expression of downstream genes, such as interferons (IFNs). IFNs can be divided into three subfamilies: type I, II, and III. In humans, type I IFNs represent a large group that contains IFN-α and variants, IFN-β, and other subtypes. IFN-γ is the only member in the type II IFN group. The type III group consists of IFN-λ1 (IL-29), IFN-λ2 (IL28A), IFN-λ3 (IL28B), and IFN-λ4 (3–5). The IFN response includes the PRR signaling for IFN production in the early phase and IFN receptor signaling in the late phase. Upon virus infection, recognition of viral nucleic acids by host PRRs leads to activation of IFN promoters and subsequent IFN production.
RIG-I and MDA5 belong to the RLR family. Both RIG-I and MDA5 consist of two cysteine-aspartic protease (caspase) activation and recruitment domains (CARDs), a helicase domain, and a C-terminal domain (CTD; also known as the repressor domain or regulatory domain, RD). The N-terminal CARDs function as the activation domain. Binding of RIG-I onto viral RNA induces conformational change that exposes the CARDs of RIG-I, allowing their ubiquitination by the E3 ligase, tripartite motif protein 25 (TRIM25; also named estrogen-responsive finger protein, EFP), and subsequent oligomerization (6–8). Different from RIG-I signaling, MDA5 is activated by stacking cooperatively along viral RNA, which in turn promotes assembly of MDA5-CARD oligomers (9, 10). Both RIG-I-CARD and MDA5-CARD oligomers interact with the CARD of a downstream adaptor protein called mitochondrial antiviral signaling protein (MAVS) on the mitochondria/mitochondria-associated membrane, leading to phosphorylation and nuclear translocation of transcription factors, such as IFN-regulatory factor 3 (IRF3) and NF-κB, to induce type I and III IFN production. Ectopic expression of RIG-I-CARDs can partially bypass RNA stimulation to induce IFN expression (11). In addition, MAVS localized on peroxisomes particularly favors type III IFN induction (12).
The expression of type I IFN (IFN-β as an example) is induced by binding of IRFs (IRF3 and IRF7), NF-κB, and AP-1 to their corresponding promoter elements within the proximal region of approximately 120 bp (13–15). Similarly, type III IFN (IFN-λ1 as an example) is mediated by IRFs (IRF1, IRF3, and IRF7) and NF-κB (4, 16). By producing IFNs in an autocrine or a paracrine manner, type I and III IFNs bind to their specific receptors, IFNAR and IFNLR, respectively, to induce phosphorylation of signal transducer and activator of transcription (STAT) proteins by the receptor-associated Janus kinase 1 (JAK1)/tyrosine kinase 2 (TYK2). JAK2 is also involved in STAT phosphorylation via IFNLR (4). Furthermore, activated STAT1/STAT2 heterodimers collaborate with IRF9 to form IFN-stimulated gene factor 3 (ISGF3), which enters the nucleus to bind IFN-stimulated response elements (ISREs) in gene promoters and activate the transcription of IFN-stimulated genes (ISGs) (16–18).
By antagonizing host antiviral defenses, pathogenic viruses can evade immune attack. An early study of two MERS patients with distinct clinical outcomes demonstrated that the clinical outcome correlates well with distinct immune responses (19). Compared with the patient with a good clinical outcome, the patient who had a poor outcome had much lower type I IFN and eventually died of multiple organ failure. In addition, it was reported that MERS-CoV infection failed to induce expression of IFN-β and IFN-λ1 in human ex vivo respiratory organ cultures and in human lung adenocarcinoma cell line Calu-3 (20, 21). Analysis of transcription signatures of MERS-CoV infection on marmoset lungs showed a suppression of IFN-β expression (22). Analysis of immune-related gene expression profiles of SARS-CoV-infected human monocytic cells against those infected by coronavirus 229E also showed a downregulation of IFN-α/β-inducible genes (23). Furthermore, type I IFN signaling was shown to be suppressed by coronavirus proteins through inhibition of RIG-I/MDA-5 activation (24, 25), interference of RIG-I ubiquitination (26, 27), disruption of TBK1 complex formation and IRF3 phosphorylation (26, 28–30), and impeding the nuclear translocation of NF-κB (31). Suppression of type III IFN signaling was also demonstrated in porcine epidemic diarrhea virus (PEDV) (32) and MERS-CoV infection (20, 21), but the mechanistic bases are less documented.
Here, we investigated the effects of MERS-CoV structural proteins on IFN expression. We discovered that viral N protein suppresses RIG-I-CARD-induced type I and type III IFN promoter activities and RIG-I ubiquitination. The suppressive effect of viral N protein can be compensated by ectopic expression of TRIM25. In addition, the C-terminal domain of MERS-CoV N protein, designated N(237-413), interacts with TRIM25 and suppresses IFN promoter activity. These results suggest that MERS-CoV N protein suppresses IFN production and its C-terminal domain is sufficient for its antagonistic function.
RESULTS
MERS-CoV N protein suppresses the expression of IFN-α2, IFN-β, and IFN-λ1.
To study whether any MERS-CoV structural proteins play a role in IFN production, we transduced A549 cells with viral E, M, and N components separately and infected viral protein-expressing A549 cells with Sendai virus (SeV). The levels of IFN-α2, -β, and -λ1 mRNA were analyzed by quantitative real-time PCR (qRT-PCR). Figure 1A shows that SeV infection induced high levels of type I and III IFN mRNA in A549 cells. However, the SeV-induced IFN mRNA levels were affected in cells expressing viral structural proteins (Fig. 1B and C). While MERS-CoV E protein did not affect the level of SeV-induced IFN-α2, -β, or -λ1 mRNA, viral N protein significantly suppressed the mRNA levels of all three IFNs and M protein suppressed those of IFN-β and IFN-λ1. The expression of N protein dose-dependently inhibited the induction of IFN-β and IFN-λ1 mRNA (Fig. 1D).
FIG 1.

MERS-CoV N protein suppresses the mRNA levels of type I and III IFNs. (A) SeV-induced type I and type III IFN expression. Total RNAs were isolated from A549 cells with (+) or without (−) SeV infection and subjected to qRT-PCR analysis to determine the mRNA levels of IFN-α2, IFN-β, and IFN-λ1. (B to D) The effects of MERS-CoV structural proteins on IFN mRNA expression. A549 cells were transduced with lentivirus carrying MERS-CoV cDNA encoding V5- and His-tagged viral E, M, or N protein as indicated. At 64 h postransduction, the cells were treated with SeV for 8 h and then harvested for Western blotting (B and D) and qRT-PCR analysis (C and D). Control samples with (ctrl) or without (mock) SeV infection were analyzed in parallel. The mRNA levels of IFN-α2, IFN-β, and IFN-λ1 were quantified following qRT-PCR with specific primers as described in the Materials and Methods section. The immunoblot signals of V5-His-tagged MERS-CoV E, M, and N proteins were detected by anti-V5 antibody. The results are expressed as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We employed a luciferase assay to examine whether N protein also inhibited SeV-induced ISG expression. HEK293T cells were transfected with reporter plasmid pISRE-Luc or pBV-CXCL10(1200)-Luc, internal control pRL-TK, and titrated doses of pcDNA-MERS-N (expressing V5- and His-tagged N protein) before infection with SeV. The expression of C-X-C motif chemokine 10 (CXCL10; also known as interferon γ-induced protein 10, IP-10), an ISRE-dependent ISG was determined. As shown in Fig. 2A, expression of viral N protein dose-dependently reduced the luciferase activity of both ISRE-Luc and CXCL10-Luc. Transfected HEK293T cells were also treated with exogenous IFN-β to bypass the effect of viral N protein on IFN production and instead focus on the effect of N protein on downstream IFN receptor signaling. Results in Fig. 2B show that viral N protein failed to suppress IFN-β-induced ISG promoter activity. In addition, while N protein suppressed the expression of SeV-induced IFN-induced protein with tetratricopeptide repeats 3 (IFIT3), it did not affect IFN-β-induced IFIT3 expression (Fig. 2C and D). These data indicate that MERS-CoV N protein suppresses antiviral gene expression through inhibition of IFN production and its suppressive effect can be rescued by exogenous IFN-β.
FIG 2.
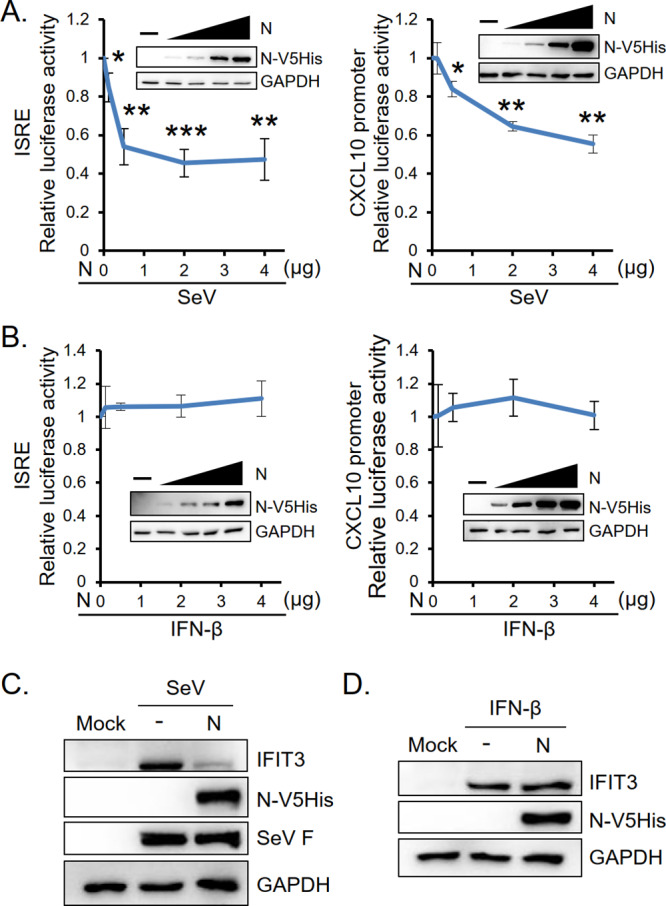
MERS-CoV N protein suppresses SeV-induced but not IFN-β-induced ISG promoter activity and IFIT3 protein expression. HEK293T cells in 6-well plates were transfected with plasmid encoding ISRE-Luc or CXCL10-Luc along with Renilla luciferase and titrated doses of V5-His-tagged MERS-CoV N-encoding plasmids as indicated and treated with SeV (A) or IFN-β (B) at 30 h posttransfection. Luciferase assay and Western blot analysis were performed. In addition, HEK293T cells were transfected with 2 μg of N protein-encoding plasmid and treated with SeV (C) or IFN-β (D). Cell lysates were harvested for Western blot analysis to detect the protein level of IFIT3. Control sample (mock) was analyzed in parallel. The immunoblot signals of V5-His-tagged MERS-CoV N was detected by anti-V5 antibody and IFIT3 protein level was detected by anti-IFIT3 antibody. SeV F protein and GAPDH served as the markers of SeV infection and loading control, respectively. The results are expressed as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
MERS-CoV N protein suppresses IFN-β/IFN-λ1 promoter and NF-ĸB/IRF3 activities.
We further cotransfected HEK293T cells with luciferase reporters driven by the IFN-β/IFN-λ1 promoter and titrated doses of viral N protein plasmids to investigate the involvement of MERS-CoV N protein in transcriptional regulation of IFNs. Results show that viral N protein significantly reduced IFN-β promoter (nucleotides [nt] −113 to +20; IFNB-113) and IFN-λ1 promoter (nt −1200 to +1; IFNL-1200) activities (Fig. 3A and B). Consistent with previous reports that IFN-λ1 transcription is highly dependent on the distal NF-κB-binding sites within the promoter upstream region between −1.2 and −1.0 kb (4, 16, 33, 34), we found that IFN-λ1 promoters of lengths 303 nt (IFNL-303) and 123 nt (IFNL-123) were only weakly activated by SeV. Since both IFNB-113 and IFNL-1200 contain potential binding sites for NF-κB, we further examined whether NF-κB contributes to N protein-mediated suppression of IFN-β/IFN-λ1 promoter activities. Cells were cotransfected with titrated doses of N protein and pNF-ĸB-Luc plasmids. Results in Fig. 3C show that N protein dose-dependently suppressed NF-ĸB-Luc luciferase activity. In addition, N protein suppressed the level of SeV-induced phosphorylated p65 (Fig. 3D). Since IRF3 has been reported to be essential for the activation of IFN-β and IFN-λ1 genes (16, 35, 36), we also investigated the effect of N protein on IRF3 activity. As shown in Fig. 3E and F, both IRF3-Luc luciferase activity and the level of SeV-induced phosphorylated IRF3 were reduced in the presence of N protein. These data indicate that both NF-κB and IRF3 are involved in MERS-CoV N protein-mediated suppression of IFN.
FIG 3.

MERS-CoV N protein suppresses SeV-induced IFN-β and IFN-λ1 promoter activities. HEK293T cells were cotransfected with a promoter reporter plasmid, the internal control Renilla luciferase plasmid, and titrated doses of MERS-CoV N protein-encoding plasmid. The promoter reporter plasmids used in this study included that carrying the IFN-β promoter (IFNB-113) (A), those carrying different lengths of the IFN-λ1 promoter (IFNL-123, IFNL-303, and IFNL-1200) (B), and those containing the binding sites for transcriptional factors NF-κB (pNF-κB-Luc) (C) and IRF3 (pIRF3-Luc) (E). Following transfection and SeV infection, cells were harvested for luciferase assay and Western blot analysis. Control samples without SeV infection (mock) were analyzed in parallel. Transcription factor binding sites present in the promoters are shown in panels A (top) and B (top). The results are expressed as the means ± SD from four (A and B) or three (C and E) independent experiments; *, P < 0.05; **, P < 0.01; ***, P < 0.001. The protein levels of the NF-κB p65 subunit, IRF3, and the phosphorylated forms were detected by Western blotting following transfection of HEK293T cells with MERS-CoV N protein-encoding plasmid and SeV infection at 40 h posttransfection (D and F). RI, relative intensity.
MERS-CoV N protein suppresses RIG-I-CARD-induced IFN promoter activity.
To understand the molecular mechanisms by which MERS-CoV N protein suppresses IFN expression, we first tested whether viral N protein interferes with the RLR pathway in which RIG-I and MDA5 bind to viral RNAs and activate MAVS to induce IFN expression. We found that the expression of N protein had little effect on the MAVS-induced IFN-β and IFN-λ1 promoter activities (Fig. 4A). Interestingly, however, expression of N protein suppressed RIG-I-CARDs-induced IFN-β and IFN-λ1 promoter activities (Fig. 4B). In contrast, N protein expression did not affect MDA5-CARD-induced IFN-β and IFN-λ1 promoter activities (Fig. 4C). Together, these data suggest that MERS-CoV N protein suppresses RIG-I pathway at a step prior to MAVS activation.
FIG 4.

MERS-CoV N protein suppresses RIG-I-CARD-induced IFN promoter activity. HEK293T cells were transfected with reporter plasmid carrying the IFN-β promoter (IFNB-113) or IFN-λ1 promoter (IFNL-1200), along with plasmids encoding Renilla luciferase, MAVS (FLAG-MAVS), RIG-I-CARDs (FLAG-CARD-V5His), or MDA5-CARD (FLAG-CARD), and titrated doses of N protein plasmid as indicated. Control samples prepared from cells transfected with the promoter reporter and Renilla luciferase reporter plasmids with (+) or without (−) coexpression of MAVS, RIG-I CARDs, or MDA5-CARD as indicated were analyzed in parallel; relative IFN promoter activities (fold) are shown. The immunoblot signals of MAVS (FLAG-MAVS) and MDA5-CARD (FLAG-CARD) were detected by anti-FLAG antibody, and MERS-CoV N protein (N-V5His) and RIG-I-CARDs (FLAG-CARD-V5His) were detected by anti-V5 antibody. The results are expressed as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01.
MERS-CoV N protein interferes with the ubiquitination of RIG-I.
TRIM25 E3 ligase is known to mediate K63-linked ubiquitination of RIG-I CARDs and is essential for the induction of IFN expression through the RIG-I signaling pathway (6, 37–39). We hypothesized that MERS-CoV N protein suppression of IFN promoter activity is the result of reduced RIG-I ubiquitination. HEK293T cells were cotransfected with RIG-I CARDs (FLAG-CARD-V5His)/full-length RIG-I (FLAG-RIG-I), HA-tagged ubiquitin, and titrated doses of N protein plasmids. As the expression of N protein increased, there was an N protein concentration-dependent decrease in ubiquitination on RIGI CARDs/full-length RIG-I (Fig. 5A and B). In addition, while ectopic overexpression of TRIM25 enhanced SeV-induced IFN-β/IFN-λ1 promoter activity, expression of N protein dose-dependently suppressed the promoter activity (Fig. 5C and D). We further cotransfected HEK293T cells with a luciferase reporter plasmid carrying the IFN-β/IFN-λ1 promoter, N-V5His plasmid, and titrated doses of FLAG-TRIM25 plasmids before infection with SeV. A luciferase assay demonstrated that ectopic overexpression of TRIM25 dose-dependently reversed N protein-mediated suppression of IFN promoter activity (Fig. 5E and F), indicating that N protein interferes with RIG-I signaling by sequestering functional TRIM25.
FIG 5.

MERS-CoV N protein blocks RIG-I ubiquitination and suppresses TRIM25-induced IFN promoter activity. (A and B) N protein suppresses RIG-I ubiquitination. HEK293T cells were cotransfected with plasmids encoding ubiquitin (HA-ubiquitin), titrated doses of N protein (N-V5His), and the RIG-I-CARDs (FLAG-CARD-V5His) (A) or the full-length RIG-I (FLAG-RIG-I) (B) and treated with SeV at 30 h posttransfection (B) as indicated. Cells were harvested at 48 h posttransfection for immunoprecipitation assay and Western blot analysis. Immunoblot signals of ubiquitinated FLAG-CARD-V5His (HA-Ub-FLAG-CARD-V5His) and FLAG-RIG-I (HA-Ub-FLAG-RIG-I) were detected by antibody against the HA-tag of ubiquitin. N-V5His and RIG-I-CARDs (FLAG-CARD-V5His) were detected by anti-V5 antibody. (C and D) N protein suppresses TRIM25-induced IFN promoter activity. HEK293T cells were cotransfected with reporter plasmid carrying the IFN-β/IFN-λ1 promoter (IFNB-113/IFNL-1200), along with plasmids encoding Renilla luciferase, FLAG-TRIM25, and titrated doses of N-V5His as indicated and treated with SeV at 30 h posttransfection. (E and F) Ectopic overexpression of TRIM25 rescues the N protein-suppressed IFN-β/IFN-λ1 promoter activity. HEK293T cells were cotransfected with reporter plasmid carrying the IFN-β/IFN-λ1 promoter (IFNB-113/IFNL-1200), plasmids encoding Renilla luciferase, N-V5His, and titrated doses of FLAG-TRIM25 as indicated and treated with SeV at 30 h posttransfection. Luciferase assay and Western blot analysis were performed. Immunoblot signal of N-V5His and FLAG-TRIM25 were detected by anti-V5 antibody and anti-FLAG antibody, respectively. The results of the luciferase assay are expressed as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01. Relative IFN promoter activities are shown. Control samples prepared from cells transfected with the promoter reporter and Renilla luciferase reporter plasmids with (+) or without (−) coexpression of exogenous TRIM25 (C and D) and the N protein (E and F) were analyzed in parallel.
MERS-CoV N protein competes with RIG-I for interaction with TRIM25.
Since interaction of RIG-I with TRIM25 is a prerequisite for RIG-I ubiquitination, we investigated whether MERS-CoV N protein interferes with RIG-I signaling by interacting with TRIM25. A reciprocal immunoprecipitation assay showed that viral N protein coimmunoprecipitated with TRIM25 (Fig. 6A). Expression of viral N protein dose-dependently diminished RIG-I-TRIM25 interaction (Fig. 6B). MERS-CoV N protein contains three conserved regions, the N-terminal domain (NTD; 1 to 164 aa), the intrinsically disordered linker region (LKR; 165 to 236 aa), and the C-terminal domain (CTD; 237 to 413 aa). Domain mapping and a reporter assay further demonstrated the association of TRIM25 with the CTD fragment, N(237-413), which alone was sufficient to suppress IFN-β promoter activity (Fig. 6C and D). Taken together, our results indicate that MERS-CoV N protein competes with RIG-I to interact with TRIM25, thereby inhibiting RIG-I ubiquitination and IFN production.
FIG 6.

MERS-CoV N protein interacts with TRIM25 and interferes with binding of TRIM25 to RIG-I. (A and C) Interactions between TRIM25 and N protein. HEK293T cells were cotransfected with plasmids encoding FLAG-TRIM25 and V5His-tagged N protein (A) or the N fragments N(1–164), N(165–236), and N(237–413) as indicated (C). At 48 h posttransfection, the cells were harvested for immunoprecipitation assay with anti-FLAG antibody for FLAG-TRIM25, anti-His antibody for N-V5His, or IgG antibody as a control followed by Western blotting. (B) N protein interferes with the interaction between TRIM25 and RIG-I. HEK293T cells were cotransfected with FLAG-RIG-I, HA-TRIM25, and titrated doses of N-V5His plasmids and treated with SeV at 30 h posttransfection. The cells were harvested at 48 h posttransfection for immunoprecipitation assay with anti-FLAG antibody for FLAG-RIG-I followed by Western blotting. (D) The C-terminal fragment of the N protein is sufficient to suppress IFN-β promoter activity. HEK293T cells were cotransfected with IFN-β promoter plasmid (IFNB-113), Renilla luciferase plasmid, and titrated doses of N(237–413)-V5His plasmid and treated with SeV at 30 h posttransfection. Luciferase assay and Western blot analysis were performed. The results are expressed as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01.
MERS-CoV N protein functions as an IFN antagonist to rescue IFN-suppressed SINV-EGFP replication.
The effect of MERS-CoV N protein on IFN production was functionally analyzed using IFN-sensitive recombinant Sindbis virus expressing enhanced green fluorescence protein (SINV-EGFP). The expression of EGFP in SINV-EGFP-infected cells is closely correlated with viral RNA replication (40). HEK293T cells with/without MERS-CoV N protein coexpression were infected with SeV (Fig. 7A). Culture supernatants were collected and used as conditioned media to culture naive HEK293T cells. Conditioned medium was replaced with fresh culture medium before infection with SINV-EGFP the next day. EGFP expression in SINV-EGFP-infected cells was an indicator of IFN levels in the conditioned media. A lower level of IFNs in the conditioned medium allows better replication of SINV-EGFP, thus a higher level of EGFP expression. Immunofluorescence imaging (Fig. 7B) and Western blotting (Fig. 7C) results show that while SINV-EGFP-infected cells cultured in mock medium expressed high levels of EGFP, EGFP expression was attenuated in cells cultured in conditioned medium collected from SeV-infected HEK293T cells that did not express viral N protein. EGFP expression was partially rescued when cells were cultured in conditioned medium harvested from SeV-infected HEK293T cells that expressed viral N protein. These results demonstrate that MERS-CoV N protein functions as an antagonist to suppress the production of not only IFN mRNA but also bioactive IFN protein.
FIG 7.

MERS-CoV N protein rescues SeV-suppressed SINV-EGFP replication. (A) Ectopic expression of N protein in SeV-infected HEK293T cells. HEK293T cells were transfected with control vector (−) or MERS-CoV N-V5His plasmid (+) and treated (+) with SeV (96 HA unit) at 24 h posttransfection as indicated. Cells were harvested at 48 h posttransfection for Western blot analysis and the culture supernatants were collected for rescue experiments. (B and C) Rescue of SINV-EGFP replication. Culture supernatants inactivated by UV radiation were used as conditioned media to culture newly seeded HEK293T cells. Conditioned medium was replaced with fresh culture medium before infection with SINV-EGFP the next day. EGFP expression in SINV-EGFP-infected cells was examined by immunofluorescence assay (B) and Western blot analysis (C) at 48 h postinfection.
DISCUSSION
We demonstrated in this study that MERS-CoV N protein antagonizes IFN induction by interfering with the RIG-I signaling pathway (Fig. 8). MERS-CoV N protein suppresses RIG-I-CARD-induced type I and III IFN promoter activities and RIG-I ubiquitination. It competes with RIG-I to interact with TRIM25 ubiquitin E3 ligase and the process can be reversed by overexpression of TRIM25. Further analyses revealed that viral N protein interacts with TRIM25 through its C-terminal domain to suppress IFN-β promoter activity. Since RIG-I signaling triggers IFN production, viral N protein, by blocking the RIG-I signaling pathway and inhibiting transcription activation, functions as an antagonist to suppress IFN production.
FIG 8.
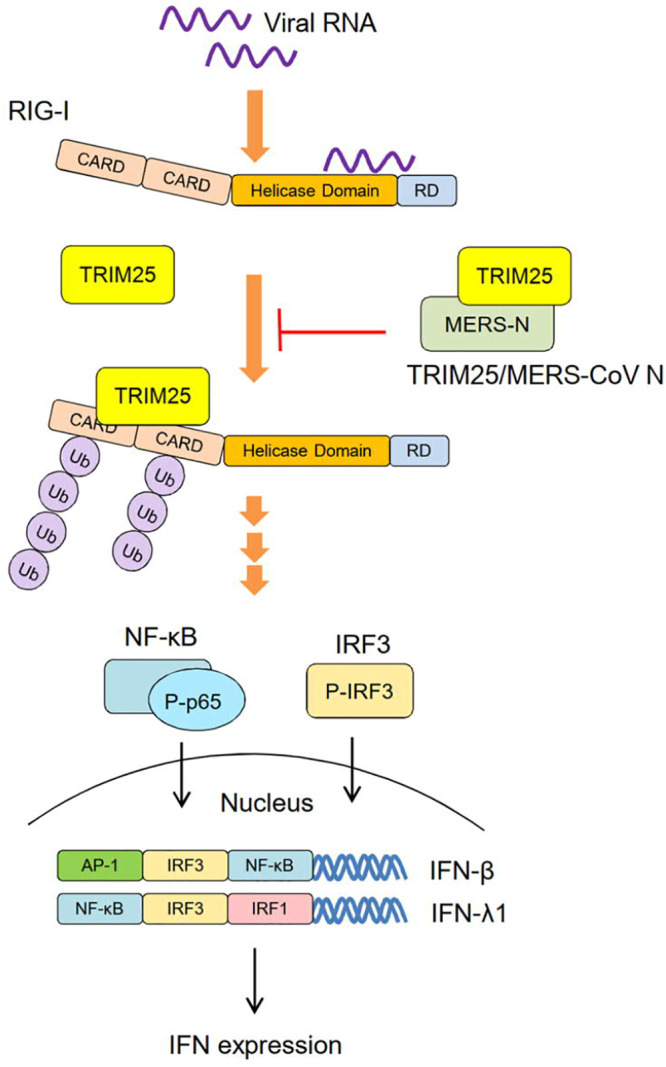
A current model for the suppressive effect of MERS-CoV N protein on the RIG-I-induced type I/III IFN expression. TRIM25 is an E3 ubiquitin ligase that ubiquitinates RIG-I and activates a downstream pathway to induce IFN production. The interaction of MERS-CoV N protein with TRIM25 blocks the ubiquitination of RIG-I and the activation of the RIG-I pathway. RIG-I signaling triggers the phosphorylation and translocation of transcription factors, including NF-κB and IRF3. In the presence of MERS-CoV N protein, phosphorylation of IRF3 and NF-κB p65 subunit is diminished, resulting in the suppression of IFN production.
Previous studies have demonstrated that IFN signaling can be induced by RIG-I and MDA5 during infection by a diverse range of RNA viruses (7, 41). RNAs of smaller than 300 bp with blunt ends containing a 5′ppp moiety are ideal ligands for RIG-I (7). In SeV- and influenza-infected cells, RIG-I preferentially interacts with short 5′ppp-RNA molecules and the genome of defective interfering particles but not the full-length virus genome (42). MDA5 recognizes long and higher-order structured single- or double-stranded viral RNA (43). The cellular RNA sensors for coronaviruses are not well defined. In mouse hepatitis virus (MHV)-infected oligodendrocytes, both RIG-I- and MDA5-dependent induction of IFN expression were observed (44). However, other reports showed that MHV failed to induce IFN production in fibroblast cells (45, 46) and MDA5 was demonstrated to be the RNA sensor in MHV-infected microglia/macrophage (47). We showed in the current study that MERS-CoV N protein functions as an antagonist against IFN production by interfering with the RIG-I pathway. We speculate that the capped 5′ terminal structure may prevent viral genomic RNA from being detected by RIG-I. The replication intermediates or negative-strand subgenomic RNAs of MERS-CoV are the likely ligands for RIG-I.
A previous study demonstrated that the MERS-CoV M protein interacts with TRAF3 to disrupt TRAF3-TBK1 association, which leads to reduced IRF3 phosphorylation and IFN-β expression (29). Activation of IFN-β and IFN-λ1 genes relies on transcription factor IRF3 (16, 35, 36), whereas activation of IFN-α2 gene mainly relies on TRAF6-mediated IRF7, but not IRF3, transcriptional regulation (48, 49). Consistent with these reports, we observed in Fig. 1C that both MERS-CoV M and N proteins suppress IFN-β and IFN-λ1 mRNA expression but that only N protein, not M protein, suppresses IFN-α2. Results in Fig. 3 also show that both NF-κB and IRF3, known to be activated by RIG-I through the MAVS pathway (50), are important for MERS-CoV N protein-mediated IFN antagonism.
It has been reported that coronavirus N protein antagonizes IFN production. MHV and SARS-CoV N proteins attenuate RIG-I/MDA5 activation by sequestering the protein activator of protein kinase R (PACT) (25). Porcine deltacoronavirus (PDCoV) N protein interaction with porcine RIG-I and TRAF3 proteins results in inhibition of porcine Riplet-induced RIG-I K63-linked ubiquitination and formation of TBK complex (51). PEDV N protein suppresses IFN-β expression by targeting TBK1 and interfering with TBK1-mediated IRF3 phosphorylation (30). TRIM25, an E3 ligase of RIG-I, is targeted by viral proteins, including SARS-CoV N protein (27) for immune evasion. We have shown in this study that MERS-CoV N protein, acting like SARS-CoV N protein, interacts with TRIM25 to inhibit RIG-I ubiquitination/activation (Fig. 5 and 6) and subsequent IFN production (Fig. 7).
TRIM25 consists of a RING-finger domain, two B-box domains, a coiled-coil dimerization domain, and a SPRY domain. The SPRY domain interacts with RIG-I CARDs and the RING-finger domain with E3 ligase activity ubiquitinates CARDs to activate the RIG-I pathway (6). In the case of influenza A virus, viral NS1 protein interacts with the coiled-coil domain of TRIM25 to disrupt TRIM25 multimerization and RIG-I ubiquitination (52). The V proteins of paramyxoviruses such as that of Nipha virus, measles virus, Sendai virus, and parainfluenza virus interact with both the SPRY domain of TRIM25 and the TRIM25-mediated ubiquitination sites of RIG-I CARDs (53). The interaction prevents RIG-I from becoming ubiquitinated. V proteins have affinity for the RIG-I/TRIM25 complex (53). In addition, V proteins of paramyxoviruses can function as MDA5 antagonists. Interaction of V proteins with the helicase domain of MDA5 results in inhibition of binding to double-stranded RNA (dsRNA) and subsequent MDA5 self-association (54). The TRIM25 SPRY domain is also a target of the SARS-CoV N protein. Interaction of TRIM25 with SARS-CoV N protein precludes TRIM25 binding to RIG-I and inhibits RIG-I ubiquitination/activation (27). Our results in this study indicate that MERS-CoV N protein interacts with TRIM25, however, it awaits to be determined which of the TRIM25 domains interacts with MERS-CoV N protein.
We showed in this study a competitive relationship between MERS-CoV N protein and RIG-I in their interaction with TRIM25, where binding of N protein to TRIM25 results in inhibition of RIG-I ubiquitination. Although it has been reported that the SPRY domain mediates the interaction of TRIM25 with RIG-I CARDs (6), structural analysis will be of value to determine the domain(s) that mediates TRIM25 interaction with MERS-CoV N protein. The web-based server UbiSite has revealed several potential ubiquitination sites in the MERS-CoV N protein. It will be of interest to examine whether the N protein can be ubiquitinated by TRIM25.
Evasion of innate immunity is a strategy exploited by viruses in order to survive and replicate in host cells. To antagonize IFN production is an efficient way to disarm the front-line alarm of host defenses. Without being able to act promptly, the immune system loses the opportunity to control viral replication in the early phase of infection. Recent studies of coronaviruses have revealed that SARS-CoV M, N, open reading frame (ORF) 3b, ORF6, PLpro, and nsp1, and MERS-CoV M, ORF4a, ORF4b, and PLpro, all target the IFN pathway (26, 28, 29, 55–59). It appears that MERS-CoV N protein is multifunctional; it not only serves as a structural protein critical for virus assembly but also participates in the regulation of viral replication.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney cell line HEK293T and adenocarcinomic human alveolar basal epithelial cell line A549 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640 medium (GIBCO), respectively, supplemented with 8% heat-inactivated fetal bovine serum plus 100 units of penicillin and 100 μg of streptomycin per ml in a humidified 5% CO2 atmosphere at 37°C.
Plasmids.
MERS-CoV structural protein expression plasmids pcDNA-MERS-N, pcDNA-MERS-M, and pcDNA-MERS-E encode V5- and His-tagged N, M, and E proteins, respectively, as previously described (60). Plasmids pcDNA-MERS-N(1-164), pcDNA-MERS-N(165-236), and pcDNA-MERS-N(237-413) encode V5- and His-tagged subdomains of N protein and were constructed by inserting their corresponding cDNA fragments into the EcoRI/XhoI sites of plasmid pcDNA-ctrl-V5HisTopo (60). Plasmids pLenti-MERS-X (where X represents N, M, or E) were constructed by replacing the mGFP gene sequence of pLenti-C-mGFP (OriGene) with the DNA fragments of the V5- and His-tagged MERS-CoV structural proteins (N, M, and E) following BamHI/PmeI restriction endonuclease digestion. These pLenti-MERS-X plasmids were used to produce pseudotyped lentivirus particles expressing individual MERS-CoV structural proteins as previously described (61).
Expression plasmids pEF-BOS-Flag-RIG-I (a gift from Curt Horvath at Northwestern University, Addgene plasmid number 52877; http://n2t.net/addgene:52877; RRID: Addgene_52877) (62) and pcDNA-Flag-RIG-I-N-V5HisTopo (kindly provided by Yi-Ling Lin at Academia Sinica, Taipei, Taiwan) encode the full-length RIG-I protein and the N-terminal CARDs, respectively. Plasmids pFlag-CMV2-TRIM25 (pFLAG-CMV2-EFP, a gift from Dong-Er Zhang at the University of California, San Diego, Addgene plasmid number 12449; http://n2t.net/addgene:12449; RRID: Addgene_12449) (63) and pcDNA-HA-TRIM25 (kindly provided by Rei-Lin Kuo at Chang Gung University, Taoyuan, Taiwan) encode Flag-tagged and HA-tagged TRIM25, respectively. Plasmid pCMV-HA-Ub (kindly provided by Show-Li Chen at National Taiwan University, Taipei, Taiwan) (64) encodes HA-tagged ubiquitin.
Luciferase reporter plasmids were constructed as follows. Plasmid pIFNB(113)-Luc encodes firefly luciferase driven by the IFN-β promoter from nucleotide (nt) −113 to +20 (13). Plasmid pBV-IFNL(1200)-Luc encodes firefly luciferase driven by the IFN-λ1 promoter from nt −1200 to +1 (33) and was generated by inserting the corresponding DNA fragment into the KpnI/EcoRI sites of plasmid pBV-Luc-wt-MBS1-4 (a gift from Bert Vogelstein at Johns Hopkins University, Addgene plasmid number 16564; http://n2t.net/addgene:16564; RRID:Addgene_16564) (65). Plasmids pGL3-IFNL(303)-Luc and pGL3-IFNL(123)-Luc that encode firefly luciferase driven by IFN-λ1 promoters from nt −303 to +1 and from −123 to +1, respectively, were generated by inserting the corresponding DNA fragments into the KpnI/HindIII sites of plasmid pGL3-Basic (Promega). Plasmid pBV-CXCL10(1200)-Luc that expresses firefly luciferase driven by the CXCL10 promoter from nt −1200 to +1 was generated by inserting the corresponding DNA fragment into the KpnI/EcoRI sites of pBV-Luc-wt-MBS1-4. Cis-reporter plasmids pISRE-Luc (Agilent Technologies), pNF-ĸB-Luc (Agilent Technologies), and pIRF3-Luc (Nova Lifetech) contain five copies of ISRE, five copies of NF-ĸB binding sites, and four copies of IRF3 binding sites, respectively, to regulate the transcription of the luciferase gene. Plasmid pRL-TK that encodes Renilla luciferase driven by the HSV TK promoter (Promega) was used to serve as a transfection internal control in the luciferase assay system.
Antibodies.
Monoclonal anti-V5 antibody (R960-25) was purchased from Invitrogen, anti-His antibody (70796-3) was from Novagen, anti-FLAG antibody (F1804) was from Merck, and antibody against the HA tag (H3663) was from Sigma. Monoclonal antibodies against NF-κB p65 (number 8242), phospho-NF-κB p65 (number 3033), IRF3 (number 4302), and phospho-IRF3 (number 4947) were from Cell Signaling Technology. Polyclonal anti-GAPDH antibody (sc-25778) was from Santa Cruz Biotech, and anti-Sendai virus (ab33988) and anti-IFIT3 (ab76818) antibodies were from Abcam. Horseradish peroxidase-conjugated antibodies against mouse IgG (115-035-003), rabbit IgG (sc-2004), and chicken IgY (GTX224126-01) were from Jackson, Santa Cruz Biotech, and GeneTex, respectively.
Transient transfection and pseudotyped lentivirus transduction.
Transient transfection was performed in HEK293T cells with T-Pro NTR II transfection reagent (T-Pro Biotechnology) according to the manufacturer’s protocol. Briefly, expression plasmid was diluted into Opti-MEM (GIBCO) and mixed with transfection reagent at the ratio of 1 μg DNA to 2 μl transfection reagent. The cells were harvested 2 days posttransfection. For lentivirus transduction of A549 cells, pseudotyped lentivirus particles expressing individual MERS-CoV structural proteins were produced as previously described (61). The cells were harvested 3 days postransduction.
Sendai virus infection and IFN-β stimulation.
SeV activates RIG-I signaling and is often used as a model virus to stimulate IFN responses. SeV (8,000 to 16,000 HA unit/ml) was obtained following amplification in fertilized specific pathogen-free (SPF) eggs for 3 days at 37°C with 70% humidity. SeV infection (at 96 HA unit/ml) was conducted at 30 h posttransfection or 64 h after transduction to induce the IFN response in cultured cells. Alternatively, direct IFN-β stimulation was applied at 30 h posttransfection with exogenous human IFN-β at 1,000 units/ml.
Harvest of cell lysates and Western blot analysis.
For harvest of protein lysates, cells were lysed using a radioimmunoprecipitation assay (RIPA) buffer consisting of 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS, and 1% complete EDTA-free protease inhibitor (Roche). The lysates were resolved by polyacrylamide gel electrophoresis and electrotransferred onto an Immobilon-P membrane (Millipore) in transfer buffer (25 mM Tris-HCl, 192 mM glycine, and 20% methanol) at 300 mA for 90 min. The membrane was then blocked in blocking buffer (5% nonfat milk in phosphate-buffered saline [PBS] consisting of 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4) for 1 h and incubated with primary antibody at 4°C overnight. Following the incubation, the membrane was rinsed with PBST buffer (0.5% Tween 20 in PBS buffer) and incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. Specific interactions between antigens and antibodies were detected by the enhanced chemiluminescence system (Advansta).
RNA isolation and quantitative real-time PCR.
Total RNA was isolated from culture cells with TRIzol reagent (Invitrogen). Reverse transcription was performed with the RNA templates, ProtoScript II First Strand cDNA Synthesis kit (New England Bioloabs), and oligo(dT) primer. The products were subjected to quantitative real-time PCR (qRT-PCR) with primer sets of specific genes and SensiFAST SYBR No-ROX kit (Bioline). The primer sets used were 5′-AAATACAGCCCTTGTGCCTGG-3′ and 5′-GGTGAGCTGGCATACGAATCA-3′ for IFN-α2, 5′-GTCTCCTCCAAATTGCTCTC-3′ and 5′-ACAGGAGCTTCTGACACTGA-3′ for IFN-β, and 5′-CGCCTTGGAAGAGTCACTCA-3′ and 5′-GAAGCCTCAGGTCCCAATTC-3′ for IFN-λ1. The primer set 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′ for GAPDH was used in parallel as an internal control. The results were analyzed with the CFX Connect Real-Time PCR Detection System (Bio-Rad).
Luciferase reporter assay.
For analysis of promoter activity, HEK293T cells were cotransfected with the firefly luciferase reporter plasmid driven by the IFN-β/IFN-λ1 promoter and the Renilla luciferase-expressing control plasmid pRL-TK in the presence or absence of specific effector plasmid such as pcDNA-MERS-N. At 48 h posttransfection, the luciferase reporter assay was conducted using the Dual-Glo luciferase assay system (Promega) and measured by luminometer. Where applied, SeV infection and IFN-β stimulation were conducted at 30 h posttransfection. The relative luciferase activity was determined by normalization of firefly luciferase activity against Renilla luciferase activity.
Immunoprecipitation and ubiquitination assay.
For immunoprecipitation, cells were harvested and homogenized in PBS with 0.5% NP-40. Protein A beads (GE Healthcare) and specific antibodies were then mixed with the cell lysates and incubated at 4°C overnight with gentle rotation. Following four washes in PBS with 0.5% NP-40, protein complexes conjugated on the beads were eluted by boiling in sample buffer and subjected to SDS-polyacrylamide gel electrophoresis and Western blotting. For the ubiquitination assay, immunoprecipitation was applied prior to Western blot analysis to enrich the ubiquitinated target proteins and exclude potential interference of ubiquitinated nonspecific proteins.
Rescue of suppressed replication of Sindbis virus-EGFP.
Recombinant Sindbis virus expressing enhanced green fluorescence protein (SINV-EGFP; kindly provided by Lih-Hwa Hwang at National Yang-Ming University, Taipei, Taiwan) was used to functionally examine the effect of MERS-CoV N protein on IFN production. The SINV-EGFP virus is an IFN-sensitive virus (66) for which it was demonstrated that SINV RNA species are more abundant in representative cell lines that have lower levels of IFNs. In addition, the expression of EGFP in SINV-EGFP-infected cells is closely correlated with the viral RNA replication (40). To perform the study, cultured supernatants were collected from HEK293T cells that had been transfected with plasmid encoding MERS-CoV N protein or control vector and infected with SeV. The cultured supernatants were exposed to UV radiation for 2 h and used as conditioned media to culture naive HEK293T cells seeded on coverslips. Following replacement of the conditioned medium with fresh medium the next day, the HEK293T cells were infected with SINV-EGFP at 3 MOI. At 48 h after the infection, cells fixed on the coverslips were immersed in Hoechst 33342 staining dye (Abnova) for 1 h and subjected to microscopy. Meanwhile, Western blot analysis was performed to examine the expression levels of MERS-CoV N protein and EGFP.
Statistical analysis.
Each experiment was repeated at least three times. Analysis was performed using Microsoft Excel 2016. Statistic comparisons were analyzed by two-tailed Student’s t tests and presented as means ± standard deviations (SD) with a P value of <0.05 considered statistically significant.
ACKNOWLEDGMENTS
We thank our colleagues Yi-Ling Lin (Academia Sinica, Taipei, Taiwan), Rei-Lin Kuo (Chang Gung University, Taoyuan, Taiwan), Lih-Hwa Hwang (National Yang-Ming University, Taipei, Taiwan), and Show-Li Chen (National Taiwan University, Taipei, Taiwan) for providing reagents. We are grateful to Betty A. Wu-Hsieh, Chien-Kuo Lee, and Shu-Chun Teng at the National Taiwan University College of Medicine for their helpful comments on the manuscript.
This study was supported in part by Research Grant 108-2321-B-002-014 from the Ministry of Science and Technology, Republic of China. The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
S.C.C. and M.-F.C. conceived the study. C.-Y.C. conducted the experiments. C.-Y.C., H.M.L., M.-F.C., and S.C.C. analyzed and interpreted the data. C.-Y.C. wrote the first draft of the manuscript, H.M.L., M.-F.C., and S.C.C. reviewed and edited the manuscript. All authors approved the final version. The study was supervised by S.C.C.
REFERENCES
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 2.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann HH, Schneider WM, Rice CM. 2015. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol 36:124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odendall C, Kagan JC. 2015. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol 12:47–52. doi: 10.1016/j.coviro.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-Lambda (IFN-λ) is expressed in a tissue dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 7.Kell AM, Gale M. Jr. 2015. RIG-I in RNA virus recognition. Virology 479–480:110–121. doi: 10.1016/j.virol.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. 2011. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 9.Lin J-P, Fan Y-K, Liu HM. 2019. The 14-3-3η chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog 15:e1007582. doi: 10.1371/journal.ppat.1007582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. 2013. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 12.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, Boulant S, Gehrke L, Cossart P, Kagan JC. 2014. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol 15:717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnefoy E, Bandu MT, Doly J. 1999. Specific binding of high-mobility-group I (HMGI) protein and histone H1 to the upstream AT-rich region of the murine beta interferon promoter: HMGI protein acts as a potential antirepressor of the promoter. Mol Cell Biol 19:2803–2816. doi: 10.1128/mcb.19.4.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renner I, Funk N, Geissler R, Friedrich S, Penzel A, Behrens SE. 2014. Antiviral interferon-beta signaling induced by designed transcription activator-like effectors (TALE). PLoS One 9:e114288. doi: 10.1371/journal.pone.0114288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thanos D, Maniatis T. 1995. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 16.Witte K, Witte E, Sabat R, Wolk K. 2010. IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev 21:237–251. doi: 10.1016/j.cytogfr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Lazear HM, Nice TJ, Diamond MS. 2015. Interferon-λ: immune functions at barrier surfaces and beyond. Immunity 43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 19.Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, Titecat M, Bortolotti P, Martinez L, Dubucquoi S, Dessein R, Gosset P, Mathieu D, Guery B. 2014. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS One 9:e88716. doi: 10.1371/journal.pone.0088716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan RW, Chan MC, Agnihothram S, Chan LL, Kuok DI, Fong JH, Guan Y, Poon LL, Baric RS, Nicholls JM, Peiris JM. 2013. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J Virol 87:6604–6614. doi: 10.1128/JVI.00009-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu DK, Hui KP, Perera RA, Miguel E, Niemeyer D, Zhao J, Channappanavar R, Dudas G, Oladipo JO, Traoré A, Fassi-Fihri O, Ali A, Demissié GF, Muth D, Chan MC, Nicholls JM, Meyerholz DK, Kuranga SA, Mamo G, Zhou Z, So RT, Hemida MG, Webby RJ, Roger F, Rambaut A, Poon LL, Perlman S, Drosten C, Chevalier V, Peiris M. 2018. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc Natl Acad Sci U S A 115:3144–3149. doi: 10.1073/pnas.1718769115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falzarano D, de Wit E, Feldmann F, Rasmussen AL, Okumura A, Peng X, Thomas MJ, van Doremalen N, Haddock E, Nagy L, LaCasse R, Liu T, Zhu J, McLellan JS, Scott DP, Katze MG, Feldmann H, Munster VJ. 2014. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog 10:e1004250. doi: 10.1371/journal.ppat.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu W, Yen YT, Singh S, Kao CL, Wu-Hsieh BA. 2012. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol 25:277–288. doi: 10.1089/vim.2011.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, Lui PY, Chan CP, Tse H, Woo PC, Yuen KY, Jin DY. 2014. Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol 88:4866–4876. doi: 10.1128/JVI.03649-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Ding Z, Fang L, Yuan S, Zhao L, Wang X, Long S, Wang M, Wang D, Foda MF, Xiao S. 2017. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-β antagonizing mechanism: attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 8:49655–49670. doi: 10.18632/oncotarget.17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. 2014. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5:369–381. doi: 10.1007/s13238-014-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P, Ma Q, Liu X, Cao C. 2017. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol 91:e02143–16. doi: 10.1128/JVI.02143-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siu KL, Kok KH, Ng MH, Poon VK, Yuen KY, Zheng BJ, Jin DY. 2009. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem 284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lui PY, Wong LY, Fung CL, Siu KL, Yeung ML, Yuen KS, Chan CP, Woo PC, Yuen KY, Jin DY. 2016. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg Microbes Infect 5:e39. doi: 10.1038/emi.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, Zhang H, Luo R, Chen H, Xiao S. 2014. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol 88:8936–8945. doi: 10.1128/JVI.00700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canton J, Fehr AR, Fernandez-Delgado R, Gutierrez-Alvarez FJ, Sanchez-Aparicio MT, García-Sastre A, Perlman S, Enjuanes L, Sola I. 2018. MERS-CoV 4b protein interferes with the NF-κB-dependent innate immune response during infection. PLoS Pathog 14:e1006838. doi: 10.1371/journal.ppat.1006838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng X, van Geelen A, Buckley AC, O’Brien A, Pillatzki A, Lager KM, Faaberg KS, Baker SC. 2019. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type I and type III interferon responses. J Virol 93:e02000–18. doi: 10.1128/JVI.02000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegel R, Eskdale J, Gallagher G. 2011. Regulation of IFN-λ1 promoter activity (IFN-λ1/IL-29) in human airway epithelial cells. J Immunol 187:5636–5644. doi: 10.4049/jimmunol.1003988. [DOI] [PubMed] [Google Scholar]
- 34.Thomson SJP, Goh FG, Banks H, Krausgruber T, Kotenko SV, Foxwell BMJ, Udalova IA. 2009. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci U S A 106:11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osterlund PI, Pietilä TE, Veckman V, Kotenko SV, Julkunen I. 2007. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol 179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 36.Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. 1998. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem 273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- 37.Ferrage F, Dutta K, Nistal-Villán E, Patel JR, Sánchez-Aparicio MT, De Ioannes P, Buku A, Aseguinolaza GG, García-Sastre A, Aggarwal AK. 2012. Structure and dynamics of the second CARD of human RIG-I provide mechanistic insights into regulation of RIG-I activation. Structure 20:2048–2061. doi: 10.1016/j.str.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martín-Vicente M, Medrano LM, Resino S, García-Sastre A, Martínez I. 2017. TRIM25 in the regulation of the antiviral innate immunity. Front Immunol 8:1187. doi: 10.3389/fimmu.2017.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez JG, Chiang JJ, Sparrer KMJ, Alam SL, Chi M, Roganowicz MD, Sankaran B, Gack MU, Pornillos O. 2016. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep 16:1315–1325. doi: 10.1016/j.celrep.2016.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang PY, Guo JH, Hwang LH. 2012. Oncolytic Sindbis virus targets tumors defective in the interferon response and induces significant bystander antitumor immunity in vivo. Mol Ther 20:298–305. doi: 10.1038/mt.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 42.Baum A, Sachidanandam R, García-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. 2009. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Liu Y, Zhang X. 2010. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol 84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou H, Perlman S. 2007. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J Virol 81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth-Cross JK, Bender SJ, Weiss SR. 2008. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 49.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett 441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 50.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 51.Likai J, Shasha L, Wenxian Z, Jingjiao M, Jianhe S, Hengan W, Yaxian Y. 2019. Porcine deltacoronavirus nucleocapsid protein suppressed IFN-β production by interfering porcine RIG-I dsRNA-binding and K63-linked polyubiquitination. Front Immunol 10:1024. doi: 10.3389/fimmu.2019.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sánchez-Aparicio MT, Feinman LJ, García-Sastre A, Shaw ML. 2018. Paramyxovirus V proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J Virol 92:e01960–17. doi: 10.1128/JVI.01960-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Childs KS, Andrejeva J, Randall RE, Goodbourn S. 2009. Mechanism of mda-5 inhibition by paramyxovirus V proteins. J Virol 83:1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng CT, Makino S. 2008. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol 82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niemeyer D, Zillinger T, Muth D, Zielecki F, Horvath G, Suliman T, Barchet W, Weber F, Drosten C, Müller MA. 2013. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J Virol 87:12489–12495. doi: 10.1128/JVI.01845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y, Ye F, Zhu N, Wang W, Deng Y, Zhao Z, Tan W. 2015. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci Rep 5:17554. doi: 10.1038/srep17554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang X, Chen X, Bian G, Tu J, Xing Y, Wang Y, Chen Z. 2014. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J Gen Virol 95:614–626. doi: 10.1099/vir.0.059014-0. [DOI] [PubMed] [Google Scholar]
- 60.Hsin WC, Chang CH, Chang CY, Peng WH, Chien CL, Chang MF, Chang SC. 2018. Nucleocapsid protein-dependent assembly of the RNA packaging signal of Middle East respiratory syndrome coronavirus. J Biomed Sci 25:47. doi: 10.1186/s12929-018-0449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen TI, Hsu YK, Chou CY, Chen YH, Hsu ST, Liou YS, Dai YC, Chang MF, Chang SC. 2019. Hepatitis C virus NS3 protein plays a dual role in WRN-mediated repair of nonhomologous end joining. J Virol 93:e01273–19. doi: 10.1128/JVI.01273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bamming D, Horvath CM. 2009. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J Biol Chem 284:9700–9712. doi: 10.1074/jbc.M807365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zou W, Zhang DE. 2006. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem 281:3989–3994. doi: 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]
- 64.Chang SW, Tsao YP, Lin CY, Chen SL. 2011. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J Virol 85:6750–6763. doi: 10.1128/JVI.02453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, Vogelstein B, Kinzler KW. 2000. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A 97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE, MacDonald MR, Klimstra WB. 2005. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J Virol 79:1487–1499. doi: 10.1128/JVI.79.3.1487-1499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]