

Abstract
Ligand–receptor interactions are customarily described by equations that apply to solutes. Yet, most receptors are present in cell membranes so that sufficiently lipophilic ligands could reach the receptor by a two-dimensional approach within the membrane. As summarized in this review, this may affect the ligand–receptor interaction in many ways. Biophysicians calculated that, compared to a three-dimensional approach from the liquid phase, such approach could alter the time the ligands need to find a receptor. Biochemists found that ligand incorporation in lipid bilayers modifies their conformation. This, along with the depth at which the ligands reside in the bilayer, will affect the probability of successful receptor interaction. Novel mechanisms were also introduced, including “exosite” binding and ligand translocation between the receptor's α-helical transmembrane domains. Pharmacologists focused attention at ligand concentrations in membrane, their adsorption and release rates and the effects thereof on ligand potency and residence time at the receptor.
Keywords: Ligands, Receptors, Plasma membranes, Partitioning, Diffusion, Binding kinetics, Affinity, Conformation, Residence time, Exosites
Ligand–receptor interactions are customarily described by equations that apply to solutes, i.e. molecules that are homogeneously distributed in a solvent and free to move therein. Yet, neurotransmitter receptors, channel-associated receptors and most of the hormone receptors are integral components of the cell plasma membrane. This not only creates an in-homogeneity in the receptor distribution but, above all, it may profoundly affect the molecular and kinetic characteristics of their interaction with ligands provided that these are sufficiently lipophilic. Over the past forty years, experts belonging to disciplines like biophysics, biochemistry, pharmacology, biology and chemistry have contributed to a better knowledge of how membranes may affect ligand–receptor (as well as substrate–enzyme) interactions at the molecular level. The ambition of this review is to integrate the outcomes of these studies in a single comprehensive monograph and, by doing so, to stimulate interdisciplinary cross-talks on this far from obsolete topic.
1. Diffusion !
Diffusion plays an eminent role in biology. From the microscopic viewpoint, it reflects Brownian motion; i.e. random walks of molecules in solution resulting from their continuous collisions with molecules of the solvent such as water. After statistical treatment, the behavior of a large set of such random walks can be formulated by the classical equations of macroscopic diffusion first introduced by Adolf Fick in 1855. In vivo as well as in appropriate surrogate in vitro experimental systems, such soluble molecules (solutes) will occasionally collide with the obstructions like cell membranes and the extracellular matrix. In the early mathematical treatments, cell membranes were considered to constitute perfectly reflective barriers so that solutes should bounce-off immediately after such a collision. Such calculations revealed that, after this first collision, it is most likely for solutes to collide with that surface more than once again before drifting away to the bulk of the solution (Berg and Purcell, 1977).
Most receptors for natural messengers like hormones and neurotransmitters are embedded in the cell plasma membrane and their interaction with these messengers or related ligands is customarily described by equations that apply to the interaction between two solutes, i.e. molecules that are homogeneously distributed in and free to move in solution. Yet, because of the comparatively enormous size of cells and even of isolated membrane fragments thereof, it is only the diffusion of the ligand molecules that needs to be taken into account. Moreover, since most of the receptors only cover a minute fraction of total surface of a cell plasma membrane, a direct hit between a free ligand and such receptor is probably a rare event. In their pioneering calculations on the influence of surfaces on heterogeneous reactions, Berg and Purcell (1977) concluded that it is most likely for the ligand to make a few encounters with non-specific areas of the membrane (which was considered to represent a reflective surface) before it interacts with an embedded receptor target or drifts away (Fig. 1 , left panel). Because of this initial tendency of the ligand to remain in close proximity of the membrane surface, it has a higher probability to hit a receptor within a given time-span as compared to the situation in where both ligand and isolated receptor molecules are homogeneously distributed and freely diffusing in solution (Berg and Purcell, 1977). In other words, merely based on their physical characteristics, membranes are already able to boost the rate of ligand–receptor encounters. Of note is that, in those as well as in many ensuing calculations, receptors were represented as traps/“perfect sinks”: i.e. somewhat similar to the holes in a snooker table, each hit with a receptor should result in the immediate and irreversible disappearance of the ligand molecule in question.
Fig. 1.
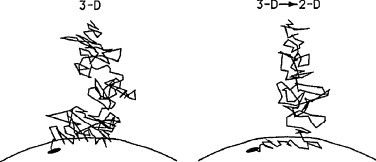
Representative example of the path taken by a ligand to reach its receptor target at the surface of a membrane (black dot) via an exclusive 3D approach (left panel) or via a mixed 3D–2D approach (right panel) (Wang et al., 1992).
In practice, cell membranes are able to undergo electrostatic and hydrophobic interactions with solutes. In many instances, small ligand molecules and even peptides become absorbed to the membrane–solution interface and in the case of highly hydrophobic ligands, they can even become completely embedded within the membrane (Sargent et al., 1988, Herbette et al., 1988). Hence, the behavior of membranes is quite distinct from that of perfectly reflective barriers. Based thereon, Adam and Delbrück (1968) first introduced the idea that the rate by which a membrane-associated traps reacts with a dissolved ligand can be enhanced if the ligand first absorbs to the surface of the membrane and then laterally diffuses to this “trap”. This “reduction of dimensionality” theory implies that, rather than approaching the receptor by pure three-dimensional (3D) diffusion, the ligand should first be directed by 3D diffusion to a “non-specific” region of the membrane surface followed by its adsorption and further two-dimensional (2D) diffusion at the surface of or even within the membrane to the receptor (Fig. 1, right panel). These considerations obviously imply that the ligands in question should be able to perform their final approach to the receptor by a 2D route, i.e. without the need for temporarily leaving the lipid bilayer to reach the receptor from within the aqueous phase.
Conclusive evidence for 2D surface diffusion to a receptor target has been provided by patch clamp experiments with dihydropyridine (DHP) derivatives on cultured rat myocardial cells and neonatal rat or adult guinea pig ventricular cells. It was indeed shown that, when added to medium outside the patch, some of these ligands are anyhow able to affect single calcium channels within the patch (Kokubun and Reuter, 1984, Brown et al., 1984). Since the receptors make part of calcium channels that are physically isolated from the bulk solvent (i.e. because of their presence within the patch), the DHPs have to reach these receptors through the lipid bilayer instead of an aqueous 3D approach. Moreover, and in agreement with a thermodynamically highly unfavorable “flip-flop” of amphiphilic molecules between both leaflets of the plasma membrane, ionized DHPs only interacted with the calcium channel when applied from the correct side of the membrane (Bangalore et al., 1994).
The major outcome of Adam and Delbrück's calculations was indeed that, on average, such mixed 3D–2D approach should allow the ligand to reach the receptor by a shorter route than in the case of an exclusive 3D approach. Based on the consideration that this increases the efficiency of multimolecular reaction processes at low concentration, Eigen (1974) went even one step further by suggesting that “reduction of dimensionality” is a nature's trick to overcome the barrier of diffusion control. This could explain the prevalence of membrane-bound enzymes in living systems.
Subsequent calculations were done with the aim to find out under which conditions “reduction of dimensionality” has the most favorable impact on the collision rate between a ligand/substrate and its membrane-associated receptor/enzyme. In this respect, the most important parameters to be taken into consideration were the ligand concentration in solution, the receptor concentration (i.e. amount of receptors per unit of membrane surface area), the distribution pattern of the receptors (such as homogenous dispersion vs. their accumulation in discrete areas such as lipid rafts), the 3D and 2D diffusion rate constants of the ligand (denoted by D 3 and D 2 respectively), the ligand's affinity for non-specific/non-target sites at the membrane and occasionally also the corresponding non-specific adsorption and release rate constants of the ligand. In general, the capture rate (i.e. steady-state flux of adsorbed ligands to the receptors/traps) via the combined 3D–2D pathway was found to increase upon increasing the ligand's affinity for non-specific sites at the membrane (given by the equilibrium dissociation constant K eq, in moles l−1 of aqueous solution) and the surface diffusion coefficient D 2. In this respect, it is noteworthy that the ligand does not need to possess overly high affinity for the membrane to favor the combined 3D–2D approach (Berg and Purcell, 1977, Wang et al., 1992). For example, the relatively low affinity of ACTH1–24 for non-specific sites at the membrane (K eq of 40 μM) was calculated to be already strong enough to increase the speed/likelihood of this ligand to find a receptor upon switching its route from a 3D random diffusion in solution to a 2D search within the plane of the membrane (Sargent et al., 1988). Moreover, a two-step searching was also found to yield a rate advantage in the case of low bulk ligand and receptor target concentrations (Adam and Delbrück, 1968, Berg and Purcell, 1977, Rhodes et al., 1985, McCloskey and Poo, 1986, Wang et al., 1992). For example, because of the very low DHP receptor density in cardiac sarcolemmal membranes (i.e. 1 site/μm2), Rhodes et al. (1985) calculated that the mean time for ligand–receptor collisions via 3D–2D combination should be about 1000 times smaller than the mean time for collisions via the 3D pathway only.
2. Diffusion?
Yet, because of differences in the models and in the restrictive assumptions, the calculations did not always yield the same outcome (Wang et al., 1992). This led some to proclaim that there is no strict guarantee for a ligand to find a membrane-associated receptor more quickly with the aid of surface diffusion, even when D 2 is nearly as high as D 3 and when the ligand possesses moderate affinity for the membranes (McCloskey and Poo, 1986). In this respect, 2D diffusion rates of small molecules may vary widely: from 1000 × less than the 3D rate for a fluorescently labelled β-adrenoceptor antagonist in membranes to as high as the 3D rate for molecules which diffuse in the central (most disorganized) part of artificial membranes (McCloskey and Poo, 1986). An additional caveat with most of these models is that they only paid attention to diffusion-limited ligand–receptor interactions and that, to facilitate calculations, the receptors were regarded to act as irreversibly absorbing perfect sinks. A distinctive feature of a perfect sink is that, even in steady state, it induces a local depletion of soluble ligand molecules around itself (Axelrod and Wang, 1994). Yet, this assumption does not fit with experimental findings indicating that receptors usually bind their ligands in a reversible fashion, releasing them unaltered back into solution. This implies that local depletion zones rarely exist under steady state conditions and, hence, that “perfect sink” models do not accurately describe real-world situations (Wang et al., 1992).
An even more important conceptual turning point was provided by Rhodes et al. (1985). These authors noticed that experimental DHP–receptor association rates were far lower than the ones calculated on the basis of diffusion only. It was therefore concluded that the binding of such ligands is reaction limited (reflecting the small probability of a diffusive ligand–receptor encounter to result in successful binding) rather than merely diffusion limited. Subsequent to these observations, Axelrod and Wang (1994) explored the outcomes of a model in where collision with a ligand only rarely leads to binding and in where receptors do not create significant local depletion zones of the ligand. Quite similar to the outcomes of the previous calculations based on “perfect sink” models, it was shown that “reduction in dimensionality” could exhibit significant rate enhancement when the drug concentration in solution and K eq are low, D 2 is large and the receptor targets are far apart.
The relative contribution of ligand diffusion and the probability of successful binding to the formation (and dissociation) of bimolecular ligand–receptor complexes is addressed in a comprehensive model first introduced by DeLisi and coworkers (DeLisi, 1981, DeLisi and Wiegel, 1981). This model stipulates that binding between a ligand (L) and its receptor (R) proceeds according to a two-step process. Diffusion of the ligand will first bring them to within a very short distance of one another (to the so-called “reaction distance”) and it is only when this “encounter complex” ([L…R]) is formed that the binding process (formation of L·R) can take place with a specific “reaction forward rate constant” (k 1) (Fig. 2 , left panel). This model neatly separates the ligand–receptor binding in two parts: one depends on the viscosity of the medium (manifested by the diffusion rate constants k + and k −) while the other depends on the reaction mechanism (manifested by k 1 and k −1). It can also be represented by a single-step process with the “effective” forward rate constant k f = k + k 1/(k − + k 1) and the “effective” reverse rate constant k r = k − k −1/(k − + k 1) (Fig. 2, right panel). Inherently, this model implies that the mixed 3D–2D approach could provide a rate advantage over the 3D approach if partitioning of the ligand in the membrane allows it to adopt a more adequate position, orientation and/or conformation (Rhodes et al., 1985, Chester et al., 1987). These issues are addressed in more detail below.
Fig. 2.
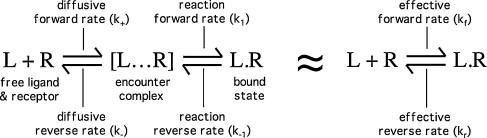
Schematic representation of drug (L)–receptor (R) binding with the formation of an ephemeric encounter complex from where the drug can either bind with the reaction forward rate constant k1 or diffuse away from the receptor.
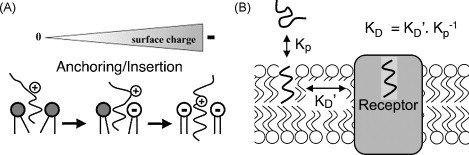
3. Ligand partitioning and penetration into the membrane
When obtained after equilibrating the ligand between the aqueous solvent (buffer, medium) and membrane fraction, the equilibrium partition coefficient, K p, can be calculated by dividing the ligand concentration in the membrane (in moles l−1 of membrane) by the ligand concentration in the solvent (in moles l−1 of solution). K p is dimensionless and should not be confused with the ligand's equilibrium dissociation constant K eq for binding to non-specific sites in the membrane. With respect to the experimental models that are used to estimate K p values, it is now widely accepted that partitioning of ligands in biological membranes and artificial phospholipid bilayer systems is more adequate than the previously widely used isotropic two-phase bulk solvent systems such as the octanol/buffer combination. Indeed, these latter experiments only provide information about the hydrophobicity of the ligand without taking account of the ability of the ligands to interact with the polar head groups of membrane lipids (Mason et al., 1991). In line with this theoretical consideration, partition coefficients of DHPs and D2-dopamine receptor antagonists for membranes and membrane lipids are very different from those reported for the octanol/water combination (Fig. 3 ) (Oliveira et al., 1989, Mason et al., 1991). However, when using synthetic membranes for K p determinations, one also needs to be aware of the important contribution of their physical state (and e.g. the influence of the temperature thereon), their phospholipid composition and even their cholesterol content (Mason et al., 1990). For example, the K p's for DHP incorporation into lipid bilayers were found to be inversely related to their cholesterol content (Herbette, 1994).
Fig. 3.
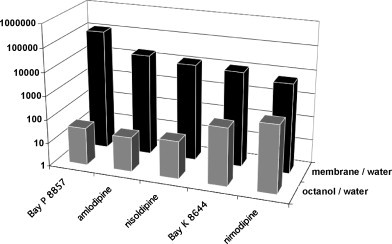
Dihydropyridine partition coefficients into biological (sarcoplasmic reticulum) membranes and octanol: adapted form Mason et al. (1991).
When K p is elevated, it is conceivable that the high concentration of ligand molecules in the membrane plays a more eminent role in boosting its rate of receptor association (i.e. the association rate constant multiplied by the local ligand concentration) than the faster accessibility of the receptors by 2D diffusion (Mason et al., 1991). In other words, even if 2D diffusion is essentially nonexistent, increasing the local ligand concentration by membrane partitioning could produce a rate advantage compared to a pure 3D approach. In the same line, high affinity binding could merely be the consequence of an elevated concentration of ligand in the membrane (Sargent and Schwyzer, 1986). Indeed, conventional equilibrium dissociation constants for ligand–receptor interaction (K D, expressed in moles l−1 of aqueous solution) could be defined as where is the local equilibrium dissociation constant (Mason et al., 1991, Castanho and Fernandes, 2006).
However, K p-based calculations would still underestimate the true concentration of a ligand in the vicinity of the receptor if the ligand molecules were confined to a discrete portion of the bilayer (Mason et al., 1991). Whatever the pertinence of this assertion, it points at a crucial aspect of membrane partitioning, namely that because of the highly ordered structure of the lipid bilayer, amphiphilic and lipophilic ligands align themselves within a membrane bilayer in a preferred orientation and location in accordance to their electronic and stereochemical properties. Hence, the physical characteristics of these ligands may contribute to the discrete depth by which they tend to reside within the membrane. This depth has been defined as the center of a Gaussian curve reflecting the distribution profile of the drug in the lipid bilayer (Mason et al., 1991). That this location may affect K p is suggested by a comparative study by Herbette et al. (1988) (Fig. 4 ) in where (a) amiodarone was found to be located deep within the bilayer close to the end of the phospholipid fatty acid tails at the bilayer center and to have the highest K p value and (b) propranolol and nimodipine (a DHP) were closer to the hydrocarbon core–water interface of the bilayer and exhibited smaller K p values. This location facilitates both a hydrophobic interaction with the phospholipid acyl chains and electrostatic interactions between the amino functions of the ligands and the phosphate head groups.
Fig. 4.
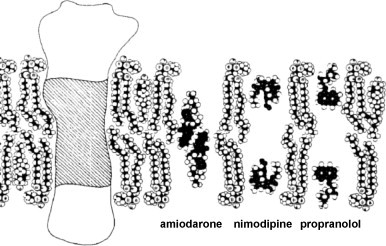
Locations of a membrane-spanning protein (hydrophobic TM domains are shaded) and the ligands amiodarone, nimodipine and propranolol at different depths within the lipid bilayer (from Herbette et al., 1988).
Based on observations that hydrophobic and amphiphilic ligands tend to reside at a discrete depth within the membrane, it has been proposed that the binding domains on their receptor should face the bilayer at the same “depth” as the active portion of those ligands (Rhodes et al., 1985, Chester et al., 1987, Castanho and Fernandes, 2006). In line with this view, structure–activity relationship and mutation studies suggest that the DHP binding site is located on the α-helical transmembrane (TM) domains of the L-type calcium channel approximately 11–14 Å from the external membrane surface (Bangalore et al., 1994). This site is located allosterically with respect to the calcium-binding site of the channel (Peterson and Catterall, 2006).
4. Lateral translocation between the receptor's transmembrane α-helices?
Mutation experiments with G protein-coupled receptors (GPCRs) have pointed out that their binding pocket for small natural messengers and small competitive ligands is deeply embedded within the central cleft formed by their seven membrane-spanning α-helical domains (TM domains). Whereas such binding pockets are traditionally considered to be accessible to hydrophilic ligands that reside in the aqueous phase, it has already been proposed at several occasions that membrane-associated amphiphilic and lipophilic ligands could still gain access to them via lateral diffusion between the receptor's α-helical TM domains of that receptor (Fig. 5A). Such pathway has been proposed for a number of ligands (Fig. 5B) like the endogenous CB1 cannabinoid receptor agonist anandamide, the synthetic β2-adrenergic receptor agonist salmeterol, the AT1-type angiotensin II receptor antagonist losartan and the D2-dopamine receptor antagonist spiperone (Anderson, 1993, Anderson et al., 1994, Theodoropoulou and Marsh, 1999, Zoumpoulakis et al., 2003, Mavromoustakos et al., 2004, Makriyannis et al., 2005, Tian et al., 2005, Packeu et al., 2008). Anadamantine (Fig. 5B) is not stored in a cellular compartment but is produced upon demand from cell membrane phospholipid components. It is then conveyed by specialized carrier proteins to presynaptic nerve terminals where it incorporates in the plasma membrane. In these membranes, anadamantine is likely to adopt an extended conformation with its head group near the lipid–water interface and the end of its fatty acid tail near the bilayer center (Barnett-Norris et al., 2005, Lynch and Reggio, 2005). This allows anandamide to engage the CB1 receptors through a fast lateral diffusion within the membrane (Makriyannis et al., 2005, Tian et al., 2005). In this respect, a bXXb motif (formed by beta branching amino acids, V6.43 and I6.46) on the lipid face of the CB1 receptor in its inactive state has been proposed to serve as an initial interaction site for anandamide (Tian et al., 2005, Lynch and Reggio, 2006). The fatty acid tail of this agonist is indeed located at the correct depth in the bilayer to interact with this motif. Other cannabinergic compounds are generally amphiphilic in nature as well and, although they are likely to bind to distinct pockets of the CB1 receptor, they are also thought to reach those pockets by lateral diffusion within the membrane (Xie et al., 1996, Murphy and Kendall, 2003, Makriyannis et al., 2005, Tian et al., 2005, Price et al., 2005, Lynch and Reggio, 2006, Kapur et al., 2007). Interestingly, a survey of sequence data by Lynch and Reggio (2006) indicates that a matching bXXb motif is also borne by several other GPCRs for lipid-derived ligands like the CB2 cannabinoid receptor, an oxoeicosanoid receptor and several lysosphingolipid and prostanoid receptors.
Fig. 5.
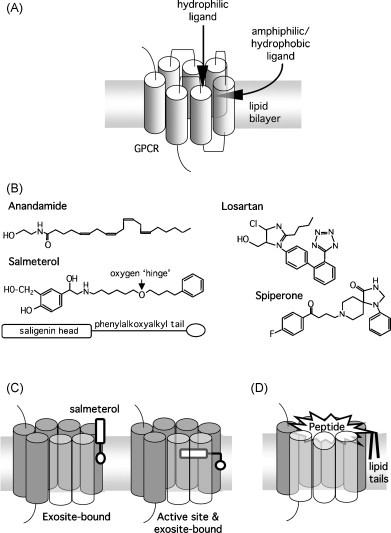
Panel A, proposed model in where hydrophilic ligands approach a GPCR from the aqueous phase while amphiphilic/hydrophobic ligands approach the receptor by lateral diffusion within the membrane and then translocate via the receptor's TM domains to their central binding pocket (TM domain in front of translocation is semi-transparent). Panel B, structure of amphiphilic ligands that allegedly reach their receptors via a combined 3D–2D approach. Panel C, binding of salmeterol via its hydrophobic phenylalkoxyalkyl tail to an alleged exosite in the vicinity of the β2-adrenergic receptor with occupancy (right side) or not (left side) of the receptor's central binding pocket by the saligenin head (TM domains in front of the saligenin head are semi-transparent). Panel D, binding of a lipid-conjugated gastrin molecule to its receptor according to a molecular modeling study by Lutz et al. (1997) (TM domains in front of the peptide section are semi-transparent).
Salmeterol (Fig. 5B) is an amphiphilic synthetic β2-adrenergic receptor agonist. It consists of a hydrophilic saligenin head which is responsible for receptor activation and an extended lipophilic phenylalkoxyalkyl side chain which is responsible for its very high partitioning in synthetic membranes (Rhodes et al., 1992, Bergendal et al., 1996). Similar to phospholipids, salmeterol is about 25 Å long, it assumes the same specific orientation in membranes and it is not prone to ‘flip-flop’ to the other face of the membrane (Rhodes et al., 1992, Johnson et al., 1993). This agonist is considered to accomplish a 2D approach as well as a lateral diffusion between the receptor's TM domains (Johnson et al., 1993, Anderson et al., 1994, Coleman et al., 1996, Teschemacher and Lemoine, 1999). As only the hydrophilic saligenin head is necessary for receptor activation, it has been proposed that the phenylalkoxyalkyl side chain interacts with a non-polar region in the cell membrane in the close vicinity of the receptor, the ‘exosite’ (Johnson et al., 1993, Clark et al., 1996). This should allow the saligenin head to freely reach (and leave) the central core of the β2-adrenergic receptor by a Charnière (hinge) principle (Fig. 5C). This model provides also an elegant explanation for the very fast termination of the salmeterol response in the presence of hydrophilic antagonists like sotalol and the fast reappearance/“’reassertion” of the response when the antagonist is washed away even though no the wash-out medium is salmeterol-free (Ball et al., 1991, Lindén et al., 1991, Voss, 1994). The oxygen atom in the alkyloxalkyl side chain has been proposed to act as the point of support for the pivoting saligenin head (Johnson, 2006) and, in this respect, structure–activity relationship studies have pointed at an important role of its position in the alkyloxalkyl side chain. It has therefore been proposed that the efficiency of the saligenin head pivoting/docking process is dictated by the average depth of the “hinge” in the membrane (Herbette, 1994, Chester et al., 1987, Mason et al., 1991, Castanho and Fernandes, 2006).
The combined 3D–2D approach has to do with ligands that are able to perform their final approach to the receptor without the need for temporarily leaving the lipid bilayer. Yet, this also applies to ligands whose reactive groups need to approach the receptor from the aqueous phase provided that another portion of these molecules is still tethered to the membrane (Axelrod, 1994). This has been elegantly demonstrated with experiments involving fatty acid attachment to peptide hormones and analogues like CCK-9, [Thr28,Nle31]-CCK-(25-33) and [Nle15]-human-gastrin-(2-17) (Romano et al., 1992, Moroder et al., 1993, Moroder, 1997). Starting with short lipid chains, initial elongation decreased the affinity of these lipo-gastrin peptide constructs for their respective receptor. As these lipid chains are still too short to permit a firm attachment of the construct to the membrane, they were still supposed to escape from the membrane first and then to reach the receptor's binding site via the aqueous phase. Hence, the effect of initial lipid chain elongation on the affinity of the constructs was related the fact that it became more and more difficult for them to escape from the membrane. However escape is no longer possible with lipid chain lengths beyond C10. This explains why further chain elongation no longer produced a substantial decrease in receptor affinity. These latter findings suggest that the extracellular loop domains of the involved GPCRs are sufficiently flexible and mobile to permit a lateral penetration of the peptide head groups at the water/lipid interface so that they can reach their binding site in the central cleft of the receptor (Fig. 5D) (Lutz et al., 1997). This also implies that if a segment of a peptide is sufficiently hydrophobic in character, it is likely to provoke the spontaneous partitioning of that peptide in the membrane (Sargent et al., 1988). Hence, the combined 3D–2D approach could also apply to certain peptide ligands.
5. Conformational modification of the ligand in the lipid bilayer
The so-called ‘non-specific’ nature of ligand–membrane interactions is likely to be a misconception. Indeed, rather than merely controlling ligand approach kinetics and local accumulation at the plasma membrane, these interactions could also affect the receptor binding process by exerting translational, conformational and orientational constraints on each ligand on a very specific structural basis (Sargent and Schwyzer, 1986, Rhodes et al., 1992, Schwyzer, 1995, Bader et al., 2001). In this respect, it is noteworthy that this not only applies to small amphiphilic ligand molecules but also to peptide messengers like gastrin, CCK and NPY (Bader et al., 2001, Castanho and Fernandes, 2006, Stone et al., 2007). While such regulatory peptides have no unique 3D structure in solution, they acquire well-defined conformations upon interacting with the membrane (Sargent et al., 1988, Contreras et al., 2001). Hydrophobic interactions certainly play an important role in this process and for peptides it is generally assumed that amino acids with hydrophobic residues form alpha helices that penetrate perpendicularly into the lipid bilayer (Gremlich et al., 1983, Sargent and Schwyzer, 1986, Bokvist et al., 2004). In the same line, ACTH1–24, substance P and dynorphin1–13 have been found to adopt a partially helical structure in 2,2,2-trifluoroethanol, a solvent that mimics the lipid environment (Greff et al., 1976, Erne et al., 1986, Sargent et al., 1988). Yet, peptide–membrane interactions could also comprise an important electrostatic component. This has clearly been demonstrated in the case of peptideSARSIFP (severe acute respiratory syndrome coronavirus sequence, amino acids 873–888), which interacts differently with the membrane depending on the charge of the phospholipids (Bokvist et al., 2004). In the same line, the 39–42 amino acid long amphiphilic amyloid-β peptide (Aβ), the main constituent found in extracellular amyloid plaques in the brain of Alzheimer Disease patients, is likely to be inserted in the plasma membrane by its hydrophobic domain prior to its release. In this respect, experiments with Aβ1–40 revealed that only a short part of this peptide is inserted in neutral lipid bilayers (Fig. 6A). This is because the hydrophilic part of these peptides is only stabilized in the bulk solution. However, the insertion becomes more pronounced upon increasing the membrane surface potential because of the emerging electrostatic anchoring of charged peptide residues close to the membrane surface (Bokvist et al., 2004).
Fig. 6.
Panel A, insertion of the hydrophobic Aβ1–40 peptide segment into membranes (Bokvist et al., 2004). Panel B, simplified scheme for the “membrane catalysis” model of Sargent and Schwyzer (1986) in where partitioning and conformational change of a peptide ligand is described as a single equilibrium step defined by Kp. is the local equilibrium dissociation constant and refers to the ligand concentration in the membrane at which half of the receptors are occupied. The overall equilibrium dissociation constant for the ligand–receptor interaction (KD, expressed in moles l−1 of aqueous solution) corresponds to .
Electrostatic and hydrophobic components of the ligand–membrane interaction may produce a favorable enthalpic contribution capable of compensating the entropic penalty resulting from the reduced number of conformations and orientations that the ligand can adopt within the membrane (Sargent et al., 1988, Moroder and Romano, 1994). Provided that some of the conformations and orientations of the membrane-associated ligand are favorable for receptor binding, this process will have less entropy requirements. In other words, there should be an entropic advantage for the 2D approach when compared to a situation in which the ligand approaches its binding site at the receptor via a 3D random walk in solution (Castanho and Fernandes, 2006). An interesting aspect of dividing the binding process in several steps is that, compared to a one-step model with equivalent total free energy (ΔG ≈ observed K D), the lower energetic requirements of the individual steps should significantly increase the overall reaction rate (Sargent and Schwyzer, 1986). For the same reason, there should also be a significant increase in the overall dissociation rate, implying that ligands with experimental equilibrium dissociation constants of 1 nM may have dissociation half-lives in the minute range. These considerations constitute the rationale for the “membrane catalysis” model (Schwyzer, 1995, Sargent and Schwyzer, 1986), which stipulates that a flexible peptide ligand binds to the membrane in the first step. After acquiring a membrane-induced conformation, the peptide then binds to the receptor in the second step (Fig. 6B). According to this view, the most important role attributed to membranes is their ability to optimize the conformation of the peptide ligands, so that they enter and fit the binding site of the receptor with greater ease. The membrane catalysis model is obviously also applicable to non-peptide ligands but, irrespective of the ligand, it is only applicable if the receptor possesses a docking site at the required depth in the lipid bilayer and if the ligand acquires a binding-prone conformation in the membrane (Aiello et al., 1998, Castanho and Fernandes, 2006).
The membrane catalysis model is supported by several experimental observations. For example, side chain deployments in the membrane-bound state of substance P analogues are found to be similar to those in the biologically active chemically constrained states (Schwyzer, 1995) and peptide hormones such as glucagon, ACTH, calcitonin, and β-endorphin also have amphiphilic structures that are essential for biological activity (Kaiser and Krzdy, 1984). On the other hand, [L-Ala2]Leu-enkephalin may be inactive because its orientation on membranes is different from that of the active compounds (Schwyzer, 1995).
6. Ligand partitioning and in vivo residence time
In drug screening studies the interaction between a receptor and its ligands is traditionally quantified in terms of affinity only. Yet, there is increasing awareness that the in vivo effectiveness and duration of ligand action is also dictated by the time period over which this ligand resides at its receptor (Copeland et al., 2006, Tummino and Copeland, 2008, Vauquelin and Van Liefde, 2006). This in vivo residence half-life should not be confused with the half-life by which ligand–receptor complexes dissociate. Indeed the half-life of the ligand–receptor complexes is measured under conditions in which no new complexes can be formed, e.g. by adding an excess of unlabelled ligand in the case of radioligand dissociation experiments (Vauquelin and Szczuka, 2007). Instead, the in vivo residence half-life refers to the time needed to halve the receptor occupancy when the concentration of free and membrane-associated ligand is more or less slowly declining (via desorption, clearance, degradation,…). This implies that, at any time, complexes can still be formed by binding of native ligand molecules as long as they are still around as well as by rebinding of ligand molecules that previously dissociated from the same or neighboring receptor molecules (Vauquelin and Szczuka, 2007). In this respect membrane partitioning could contribute to a long residence time by a number of different mechanisms.
From an equilibrium point of view, very lipophilic molecules have a high membrane/water partition coefficient K p because they prefer to reside in the lipid bilayer hydrocarbon core. Without considering other parameters, the accumulation of such ligands in the membrane may already increase their in vivo residence half-life because of the hyperbolic relationship between the free ligand concentration and receptor occupancy (such as shown by a typical radioligand saturation binding curve). This implies that, when starting with an already low free ligand concentration (i.e. when only part of the receptors are occupied), the in vivo residence half-life will be very close to the half-life by which the free ligand concentration declines (Fig. 7A). However, when the initial ligand concentration is elevated (i.e. when most of the receptors are occupied), the in vivo residence half-life will markedly exceed the half-life of the free ligand.
Fig. 7.
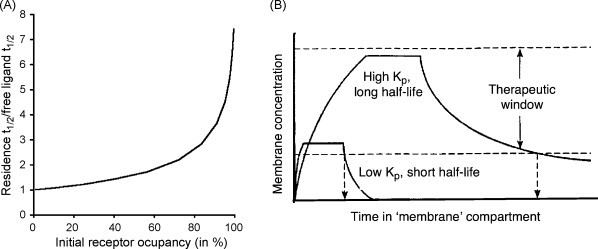
Panel A, influence of the initial receptor occupancy (abscissa, in percent of total receptor concentration) on the residence half-life (ordinate, the free ligand half-life being taken as unit) when the free ligand concentration decreases exponentially with time. Panel B, a high Kp and a slow ligand desorption rate act in concert to prolong the concentration of a membrane-associated ligand within the therapeutic window (adapted from Herbette, 1994).
While K p refers to the ratio of the adsorption and release/desorption rates of a ligand, it is obvious that these pharmacokinetic parameters may also affect the ligand's therapeutic action in their own right. In this respect, slow release from the plasma membrane is widely considered to play a preponderant role in the long duration of action of highly lipophilic ligands. This is typically illustrated by the hydrophobic β2-adrenergic receptor agonist salmeterol (Fig. 5B) whose aptness to produce pseudo-irreversible relaxation of guinea pig trachea and human bronchi at all concentrations (Johnson et al., 1993, Nials et al., 1994) has been linked by some to its slow release from airway smooth muscle membranes (Anderson, 1993, Anderson et al., 1994, Austin et al., 2003). Such link is embodied in the “diffusion microkinetic” model that stipulates that the plasma membrane can act as a depot/reservoir for the ligand rather than merely functioning as an inert substratum for the receptor (Anderson, 1991, Coleman et al., 1996). Obviously, slow desorption will act in concert with a high K p to prolong the effect duration of a ligand (Fig. 7B). This has been proposed to be the case for the highly lipophilic DHP lacidipine (Herbette, 1994). Lacidipine has a higher K p than many other DHPs (so that a higher local concentration can be attained), it is located deeper in the lipid bilayer and it partitions slower into and out of the membrane. While the high K p establishes a high concentration of this ligand within the membrane, it slow release ensures that its concentration in the membrane remains above its therapeutic threshold for a long period of time (Herbette, 1994). This combination of properties may explain the comparatively gentle onset and long duration of clinical action of this drug despite its short plasma half-life (Herbette, 1994).
However, membrane partitioning could also increase the effect duration of a ligand by alternative mechanisms. An intriguing possibility is that some ligands could undergo specific and long-lasting binding to ‘exosites’ in the membrane. These auxiliary sites could be located either at the receptors themselves (i.e. an anchoring site that is different from the active site but is not supposed to affect the conformation of the active site via an allosteric mechanism) or in their immediate vicinity in the membrane. The exosite theory was originally proposed by Rocha e Silva (1969) to explain the persistent antagonistic activity at histamine H1 receptors and it has subsequently been adopted to explain the long duration of action of the muscarinic acetylcholine receptor agonist xanomeline and the β2-adrenergic receptor agonist salmeterol (Fig. 5C) (Johnson et al., 1993, Johnson and Coleman, 1995, Coleman et al., 1996, Christopoulos et al., 1998). The presence of an exosite implies that, once dissociated from the active site of the receptor, the ligand is not free to diffuse away from that receptor. This constitutes an important difference with the microkinetic theory that permits ligands to freely interchange between the receptor's active site and the lipid bulk phase. Hence, while the concentration of free ligand should be prone to decline due to redistribution, degradation and/or other elimination pathways, the concentration of ligand available for active site binding should be maintained for a longer time period in the case of exosite binding (Green et al., 1996).
A major handicap of the exosite theory is that such auxiliary binding sites have hitherto never been positively identified (e.g. by radioligand binding studies) and that a number of experimental findings even cast doubt on the validity of this theory (Bergendal et al., 1996, Teschemacher and Lemoine, 1999, Jakubík et al., 2002). For example, it was argued that if the exosite for salmeterol displays pharmacological specificity and high affinity for its phenylalkoxyalkyl side chain, other molecules with the same side chain would attenuate its reassertion behavior of salmeterol. Yet such structural mimetics failed to do so (Bergendal et al., 1996). Moreover, in disagreement with the exosite theory, reassertion of the muscarinic acetylcholine receptor agonist xanomeline only took place at high concentrations in cell systems and, in the same vein, persistent reassertion of the salmeterol-mediated relaxation of strips from guinea pig trachea was only observed when its initial concentration was sufficiently elevated (Bergendal et al., 1996, Jakubík et al., 2002). This provides support to those who claim that the reassertion of an agonist's effect is simply related to its long-lasting association with the plasma membrane (i.e. the ‘microkinetic’ theory). The term “reassertion” has hitherto only been linked to the reappearance of an agonist's effect. Yet, this term could apply to all ligand types if, by definition, it refers to the reestablishment of receptor occupancy (rather than solely receptor activation) by a ligand after wash-out of the aqueous ligand-containing solution and temporary occupancy of the receptor's binding pocket by a competitive ligand molecule. Based on this more general definition, “reassertion” is likely to apply all kinds of amphiphilic/hydrophobic ligands. In this respect, it could well explain some of the behavioral aspects of D2-dopamine receptor antagonist [3H]-spiperone in radioligand binding studies on intact recombinant cells (Fig. 8A).
Fig. 8.
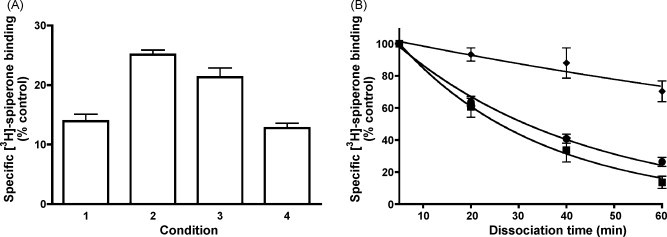
Panel A, translocation of [3H]-spiperone from “non-specific” sites in the plasma membrane from recombinant intact Chinese hamster ovary cells (CHO cells) to the therein-expressed D2L-receptors. Experiments were done as in Packeu et al. (2008). In short, intact cells were pre-incubated for 30 min at 37 °C with 1 nM [3H]-spiperone after which its specific (i.e. 1 μM (+)-butaclamol-displaceable) binding is measured (here denoted as control binding). Co-addition of 10−5 M raclopride reduces the [3H]-spiperone binding to 15% of the control (Lane 1). When [3H]-spiperone + raclopride-pretreated cells are washed twice (to remove the free ligands present in the aqueous solution) and finally incubated for 10 min at 37 °C with buffer alone, specific binding almost doubles (Lane 2). This increase reflects genuine receptor occupancy as it can be blocked by raclopride in a concentration-dependent fashion (10−7 M in Lane 3 and 10−5 M in Lane 4) and with the same potency as in competition binding experiments. N = 3–5. Panel B, different rates of [3H]-spiperone dissociation from D2L-receptors in recombinant CHO cells and membrane preparations thereof. Intact cells (♦) and membrane preparations thereof (●) were incubated for 30 min at 37 °C with 1 nM [3H]-spiperone. [3H]-Spiperone dissociation at 37 °C was initiated with 1 μM (+)-butaclamol and, at the times indicated, its remaining specific binding was measured. The same dissociation experiments were also done with leaky cells (■), i.e. in the throughout presence of 0.01 mg/ml of the pore-forming agent filipin. N = 3.
Finally, by retaining dissociated ligands to a compartment with a small volume such as a cell plasma membrane could favor their propensity to rebind/re-associate to the same or to neighboring receptor molecules. Such compartments could also include synapses (Coombs and Goldstein, 2004), tortuous paths with blind pockets are tissues such as the brain and even spaces between cells in a monolayer culture (Hrabetová and Nicholson, 2004, Spivak et al., 2006, Vargová and Syková, 2008). In radioligand dissociation experiments, this “rebinding” phenomenon leads to an apparent decrease in the radioligand's dissociation rate in wash-out medium only (i.e. when radioligand rebinding is possible) when compared to similar wash-out experiments in the presence of an unlabelled competing ligand at high concentration; i.e. when radioligand rebinding is effectively prevented (Vauquelin and Szczuka, 2007). Based on intact cell experiments, rebinding was recently shown to be so extreme as to approach virtual irreversibility in the case of the lipophilic D2-type dopamine receptor antagonist spiperone (Packeu et al., 2008) and the CB1-type cannabinoid receptor inverse agonist taranabant (Szczuka et al., 2009). These experiments are compatible with the ability of cell membrane confinement to exacerbate rebinding phenomena with lipophilic drugs and, as suggested by recent simulation studies (Szczuka et al., 2009), rebinding could explain the long-lasting effect of salmeterol as pertinently as the exosite model. In the same line, rebinding could also explain the need for the simultaneous presence of receptors and their lipid environment for the wash-resistant xanomeline binding to take place (Jakubík et al., 2004).
Finally, while tortuosities in extracellular spaces in tissues could already limit the diffusion of molecules in general, the repeated partitioning in cell membranes is likely to further exacerbate this process in the case of hydrophobic ligands (Lovich et al., 2001, Hrabetová and Nicholson, 2004). By retaining a hydrophobic ligand in its target tissue, repeated partitioning could greatly contribute to the often-observed long-lasting effect of such molecules in vivo as well as in in vitro experiments with intact tissues.
7. Concluding remarks
The so-called non-specific partitioning of amphiphilic/hydrophobic ligands in plasma membranes may exert control on their receptor binding characteristics in many ways such as by altering the time they need to reach a receptor molecule, their conformation and the receptor-domain towards which the final approach takes place. These effects, along with the accumulation of such ligands in a small volume around the receptors may affect their association rate, their affinity as well as their in vivo residence time at the receptor. However, general rules are hard to formulate, as membrane partitioning does not always exert a positive influence on the ligand's receptor binding characteristics. Novel membrane-connected concepts, including exosite binding and ligand translocation between the receptor's α-helical transmembrane domains have been advanced as well but, by lack of tangible proof, the physical reality of these concepts is likely to remain matter of debate for some time to come. In addition, new questions recently emerged such as why some antagonists display distinct receptor-binding kinetics in intact cell systems when compared to leaky cells and membrane preparations thereof (Hara et al., 1998, Fierens et al., 2002, Smith et al., 2006). An additional example thereof is shown in Fig. 8B. Further research in these areas (and about hydrophobic/amphiphilic ligand–receptor interactions in general) is highly desirable, especially because of the increasing awareness that receptor-binding kinetics may influence the in vivo effectiveness and duration of ligand action and, accordingly, that kinetic issues should also be addressed in screening studies.
References
- Adam G., Delbrück M. Reduction of dimensionality in biological diffusion processes. In: Rich A., Davidson N., Freeman W.H., editors. Structural Chemistry and Molecular Biology. W.H. Freeman & Co.; San Francisco: 1968. pp. 198–215. [Google Scholar]
- Aiello M., Moran O., Pisciotta M., Gambale F. Interaction between dihydropyridines and phospholipid bilayers: a molecular dynamics simulation. Eur. Biophys. J. 1998;27:211–218. doi: 10.1007/s002490050127. [DOI] [PubMed] [Google Scholar]
- Anderson G.P. In: Anderson G.P., Chapman I.D., Morley J., editors. vol. 34. Birkhauser Verlag; 1991. pp. 97–115. (In New Drugs for Asthma Therapy. Agents and Actions Supplement). [Google Scholar]
- Anderson G.P. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. doi: 10.1016/0024-3205(93)90729-m. [DOI] [PubMed] [Google Scholar]
- Anderson G.P., Lindén A., Rabe K.F. Why are long-acting beta-adrenoceptor agonists long-acting? Eur. Respir. J. 1994;7:569–578. doi: 10.1183/09031936.94.07030569. [DOI] [PubMed] [Google Scholar]
- Austin R.P., Barton P., Bonnert R.V., Brown R.C., Cage P.A., Cheshire D.R., Davis A.M., Dougall I.G., Ince F., Pairaudeau G., Young A. QSAR and the rational design of long-acting dual D2-receptor/beta2-adrenoceptor agonists. J. Med. Chem. 2003;46:3210–3320. doi: 10.1021/jm020886c. [DOI] [PubMed] [Google Scholar]
- Axelrod D. New dimensions in two dimensions. Biophys. J. 1994;67:1799–1800. doi: 10.1016/S0006-3495(94)80660-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D., Wang M.D. Reduction-of-dimensionality kinetics at reaction-limited cell surface receptors. Biophys. J. 1994;66:588–600. doi: 10.1016/s0006-3495(94)80834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader R., Bettio A., Beck-Sickinger A.G., Zerbe O. Structure and dynamics of micelle-bound neuropeptide Y: comparison with unligated NPY and implications for receptor selection. J. Mol. Biol. 2001;305:307–329. doi: 10.1006/jmbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- Ball D.I., Brittain R.T., Coleman R.A., Denyer L.H., Jack D., Johnson M., Lunts L.H., Nials A.T., Sheldrick K.E., Skidmore I.F. Salmeterol, a novel, long-acting beta 2-adrenoceptor agonist: characterization of pharmacological activity in vitro and in vivo. Br. J. Pharmacol. 1991;104:665–671. doi: 10.1111/j.1476-5381.1991.tb12486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangalore R., Baindur N., Rutledge A., Triggle D.J., Kass R.S. L-type calcium channels: asymmetrical intramembrane binding domain revealed by variable length, permanently charged 1,4-dihydropyridines. Mol. Pharmacol. 1994;46:660–666. [PubMed] [Google Scholar]
- Barnett-Norris J., Lynch D., Reggio P.H. Lipids, lipid rafts and caveolae: their importance for GPCR signaling and their centrality to the endocannabinoid system. Life Sci. 2005;77:1625–1639. doi: 10.1016/j.lfs.2005.05.040. [DOI] [PubMed] [Google Scholar]
- Berg H.C., Purcell E.M. Physics of chemoreception. Biophys. J. 1977;20:193–219. doi: 10.1016/S0006-3495(77)85544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergendal A., Lindén A., Skoogh B.E., Gerspacher M., Anderson G.P., Lolfdahl C.G. Salmeterol mediated reassertion of relaxation persists in guinea-pig trachea pretreated with aliphatic side chain structural analogues. Br. J. Pharmacol. 1996;117:1009–1015. doi: 10.1111/j.1476-5381.1996.tb16690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist M., Lindstrom F., Watts A., Grobner G. Two types of Alzheimer's beta-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- Brown A.M., Kunze D.L., Yatani A. The agonist effect of dihydropyridines on Ca channels. Nature. 1984;311:570–572. doi: 10.1038/311570a0. [DOI] [PubMed] [Google Scholar]
- Castanho M.A.R.B., Fernandes M.X. Lipid membrane-induced optimization for ligand–receptor docking: recent tools and insights for the “membrane catalysis” model. Eur. Biophys. J. 2006;35:92–103. doi: 10.1007/s00249-005-0007-9. [DOI] [PubMed] [Google Scholar]
- Chester D.W., Herbette L.G., Mason R.P., Joslyn A.F., Triggle D.J., Koppel D.E. Diffusion of dihydropyridine calcium channel antagonists in cardiac sarcolemmal lipid multibilayers. Biophys. J. 1987;52:1021–1030. doi: 10.1016/S0006-3495(87)83295-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A., Pierce T.L., Sorman J.L., El-Fakahany E.E. On the unique binding and activating properties of xanomeline at the M1 muscarinic acetylcholine receptor. Mol. Pharmacol. 1998;53:1120–1130. [PubMed] [Google Scholar]
- Clark R.B., Allal C., Friedman J., Johnson M., Barber R. Stable activation and desensitization of beta 2-adrenergic receptor simulation of adenylyl cyclase by salmeterol: evidence for quasi-irreversible binding to an exosite. Mol. Pharmacol. 1996;49:182–189. [PubMed] [Google Scholar]
- Coleman R.A., Johnson M., Nials A.T., Vardey C.J. Exosites: their current status and their relevance to the duration of action of long-acting β2-adrenoceptor agonists. Trends Pharmacol. Res. 1996;17:324–330. [PubMed] [Google Scholar]
- Contreras L.L., de Almeida R.F.M., Villalaín J., Fedorov A., Prieto M. Interaction of α-melanocyte stimulating hormone with binary phospholipid membranes: structural changes and relevance of phase behavior. Biophys. J. 2001;80:2273–2283. doi: 10.1016/S0006-3495(01)76199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs D., Goldstein B. Effects of the geometry of the immunological synapse on the delivery of effector molecules. Biophys. J. 2004;87:2215–2220. doi: 10.1529/biophysj.104.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R.A., Pompliano D.L., Meek T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- DeLisi C. The effect of cell size and receptor density on ligand–receptor reaction rate constants. Mol. Immunol. 1981;18:507–511. doi: 10.1016/0161-5890(81)90128-0. [DOI] [PubMed] [Google Scholar]
- DeLisi C., Wiegel F.W. Effect of nonspecific forces and finite receptor number on rate constants of ligand-cell bound-receptor interactions. Proc. Natl. Acad. Sci. 1981;78:5569–5572. doi: 10.1073/pnas.78.9.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M. Diffusion control in biochemical reactions. In: Mintz S.L., Widmayer S.N., editors. Quantum Statistical Mechanics in the Natural Sciences. Plenum Press; New York: 1974. pp. 37–61. [Google Scholar]
- Erne D., Rolka K., Schwyzer R. Membrane structure of substance P. III. Secondary structure of substance P in 2,2,2-trifluoroethanol, methanol, and on flat lipid membranes studied by infrared spectroscopy. Helv. Chim. Acta. 1986;69:1807–1816. [Google Scholar]
- Fierens F.L., Vanderheyden P.M., Roggeman C., Vande Gucht P., De Backer J.P., Vauquelin G. Distinct binding properties of the AT1 receptor antagonist [3H]candesartan to intact cells and membrane preparations. Biochem. Pharmacol. 2002;63:1273–1279. doi: 10.1016/s0006-2952(02)00859-6. [DOI] [PubMed] [Google Scholar]
- Green S.A., Spasoff A.P., Coleman R.A., Johnson M., Liggett S.B. Sustained activation of a G protein-coupled receptor via “anchored” agonist binding. Molecular localization of the salmeterol exosite within the β2-adrenergic receptor. J. Biol. Chem. 1996;271:24029–24035. doi: 10.1074/jbc.271.39.24029. [DOI] [PubMed] [Google Scholar]
- Greff D., Thoma F., Fermandjian S., Löw M., Kisfaludy L. Conformational studies of corticotropin1-32 and constitutive peptides by circular dichroism. Biochim. Biophys. Acta. 1976;439:219–231. doi: 10.1016/0005-2795(76)90177-x. [DOI] [PubMed] [Google Scholar]
- Gremlich H.U., Fringeli U.P., Schwyzer R. Conformational changes of adrenocorticotropin peptides upon interaction with lipid membranes revealed by infrared attenuated total reflection spectroscopy. Biochemistry. 1983;22:4257–4264. doi: 10.1021/bi00287a015. [DOI] [PubMed] [Google Scholar]
- Hara M., Tozawa F., Itazaki K., Mihara S., Fujimoto M. Endothelin ET(B) receptors show different binding profiles in intact cells and cell membrane preparations. Eur. J. Pharmacol. 1998;345:339–342. doi: 10.1016/s0014-2999(98)00016-8. [DOI] [PubMed] [Google Scholar]
- Herbette L.G., Trumbore M., Chester D.W., Katz A.M. Possible molecular basis for the pharmacokinetics and pharmacodynamics of three membrane-active drugs: propranolol, nimodipine and amiodarone. J. Mol. Cell. Cardiol. 1988;20:373–378. doi: 10.1016/s0022-2828(88)80128-7. [DOI] [PubMed] [Google Scholar]
- Herbette L.G. Membrane pathways for drug/ion channel interactions: molecular basis for pharmacokinetic properties. Drug Dev. Res. 1994;33:214–224. [Google Scholar]
- Hrabetová S., Nicholson C. Contribution of dead-space microdomains to tortuosity of brain extracellular space. Neurochem. Int. 2004;45:467–477. doi: 10.1016/j.neuint.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Jakubík J., Tucek S., El-Fakahany E.E. Allosteric modulation by persistent binding of xanomeline of the interaction of competitive ligands with the M1 muscarinic acetylcholine receptor. J. Pharmacol. Exp. Ther. 2002;301:1033–1041. doi: 10.1124/jpet.301.3.1033. [DOI] [PubMed] [Google Scholar]
- Jakubík J., Tucek S., El-Fakahany E.E. Role of receptor protein and membrane lipids in xanomeline wash-resistant binding to muscarinic M1 receptors. J. Pharmacol. Exp. Ther. 2004;308:105–110. doi: 10.1124/jpet.103.058594. [DOI] [PubMed] [Google Scholar]
- Johnson M., Butchers P.R., Coleman R.A., Nials A.T., Strong P., Sumner M.J., Vardey C.J., Whelan C.J. The pharmacology of salmeterol. Life Sci. 1993;52:2131–2143. doi: 10.1016/0024-3205(93)90728-l. [DOI] [PubMed] [Google Scholar]
- Johnson M., Coleman R.A. In: Mechanisms of Action of β2-Adrenoceptor Agonists. Busse W.W., Holgate S.T., editors. Asthma & Rhinitis; Cambridge, Blackwell: 1995. pp. 1278–1295. [Google Scholar]
- Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J. Allergy Clin. Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Kaiser E.T., Krzdy F.J. Amphiphilic secondary structure: design of peptide hormones. Science (Wash., DC) 1984;223:249–255. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- Kapur A., Hurst D.P., Fleischer D., Whitnell R., Thakur G.A., Makriyannis A., Reggio P.H., Abood M.E. Mutation studies of Ser7.39 and Ser2. 60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol. 2007;71:1512–1524. doi: 10.1124/mol.107.034645. [DOI] [PubMed] [Google Scholar]
- Kokubun S., Reuter H. Dihydropyridine derivatives prolong the open state of Ca channels in cultured cardiac cells. Proc. Natl. Acad. Sci. U.S.A. 1984;81:4824–4827. doi: 10.1073/pnas.81.15.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindén A., Bergendal A., Ullman A., Skoogh B.E., Löfdahl C.G. High concentration of formoterol and salmeterol in the isolated guinae-pig trachea: reassertion of smooth muscle relaxation after beta blockade followed by wash-out. Am. Rev. Respir. Dis. 1991;143(4(2)):A749. [Google Scholar]
- Lovich M.A., Creel C., Hong K., Hwang C.W., Edelman E.R. Carrier proteins determine local pharmacokinetics and arterial distribution of paclitaxel. J. Pharm. Sci. 2001;90:1324–1335. doi: 10.1002/jps.1085. [DOI] [PubMed] [Google Scholar]
- Lutz J., Romano-Götsch R., Escrieut C., Fourmy D., Mathä B., Müller G., Kessler H., Moroder L. Mapping of ligand binding sites of the cholecystokinin-B/gastrin receptor with lipo-gastrin peptides and molecular modeling. Biopolymers. 1997;41:799–817. doi: 10.1002/(SICI)1097-0282(199706)41:7<799::AID-BIP8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Lynch D.L., Reggio P.H. Molecular dynamics simulations of the endocannabinoid N-arachidonoylethanolamine (anandamide) in a phospholipid bilayer: probing structure and dynamics. J. Med. Chem. 2005;48:4824–4833. doi: 10.1021/jm058185d. [DOI] [PubMed] [Google Scholar]
- Lynch D.L., Reggio P.H. Cannabinoid CB1 receptor recognition of endocannabinoids via the lipid bilayer: molecular dynamics simulations of CB1 transmembrane helix 6 and anandamide in a phospholipid bilayer. J. Comput. Aided Mol. Des. 2006;20:495–509. doi: 10.1007/s10822-006-9068-9. [DOI] [PubMed] [Google Scholar]
- Mason R.P., Moring J., Herbette L.G. A molecular model involving the membrane bilayer in the binding of lipid soluble drugs to their receptors in heart and brain. Int. J. Rad. Appl. Instrum. B. 1990;17:13–33. doi: 10.1016/0883-2897(90)90004-k. [DOI] [PubMed] [Google Scholar]
- Mason R.P., Rhodes D.G., Herbette L.G. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J. Med. Chem. 1991;34:869–877. doi: 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- Makriyannis A., Tian X., Guo J. How lipophilic cannabinergic ligands reach their receptor sites. Prostaglandins Other Lipid Mediat. 2005;77:210–218. doi: 10.1016/j.prostaglandins.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Mavromoustakos T., Zoumpoulakis P., Kyrikou I., Zoga A., Siapi E., Zervou M., Daliani I., Dimitriou D., Pitsas A., Kamoutsis C., Laggner P. Efforts to understand the molecular basis of hypertension through drug:membrane interactions. Curr. Top. Med. Chem. 2004;4:445–459. doi: 10.2174/1568026043451339. [DOI] [PubMed] [Google Scholar]
- McCloskey M.A., Poo M.M. Rates of membrane-associated reactions: reduction of dimensionality revisited. J. Cell. Biol. 1986;102:88–96. doi: 10.1083/jcb.102.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroder L., Romano R., Guba W., Mierke D.F., Kessler H., Delporte C., Winand J., Christophe J. New Evidence for membrane-bound pathway in hormone receptor binding. Biochemistry. 1993;32:13551–13559. doi: 10.1021/bi00212a022. [DOI] [PubMed] [Google Scholar]
- Moroder L., Romano R. Synthesis, conformational and biological properties of lipophilic derivatives of gastrin and cholecystokinin peptides. Pure Appl. Chem. 1994;66:2111–2114. [Google Scholar]
- Moroder L. On the mechanism of hormone recognition and binding by the CCK-B/gastrin receptor. J. Pept. Sci. 1997;3:1–14. doi: 10.1002/(SICI)1099-1387(199701)3:1%3C1::AID-PSC85%3E3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Murphy J.W., Kendall D.A. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55, 940 but not SR 141716A. Biochem. Pharmacol. 2003;65:1623–1631. doi: 10.1016/s0006-2952(03)00155-2. [DOI] [PubMed] [Google Scholar]
- Nials A.T., Ball D.I., Butchers P.R., Coleman R.A., Humbles A.A., Johnson M., Vardey C.J. Formoterol on airway smooth muscle and human lung mast cells: a comparison with salbutamol and salmeterol. Eur. J. Pharmacol. 1994;251:127–135. doi: 10.1016/0014-2999(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Oliveira C.R., Lima M.C.P., Carvalho C.A.M., Leysen J.E., Carvalho A.P. Partition coefficients of dopamine antagonists in brain membranes and liposomes. Biochem. Pharmacol. 1989;38:2113–2120. doi: 10.1016/0006-2952(89)90065-8. [DOI] [PubMed] [Google Scholar]
- Packeu A., De Backer J.P., Van Liefde I., Vanderheyden P., Vauquelin G. Antagonist-dopamine D2L-receptor interactions in intact cells. Biochem. Pharmacol. 2008;75:2192–2203. doi: 10.1016/j.bcp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Peterson B.Z., Catterall W.A. Allosteric interactions required for high-affinity binding of dihydropyridine antagonists to Ca(V)1.1 channels are modulated by calcium in the pore. Mol. Pharmacol. 2006;70:667–675. doi: 10.1124/mol.105.020644. [DOI] [PubMed] [Google Scholar]
- Price M.R., Baillie G.L., Thomas A., Stevenson L.A., Easson M., Goodwin R., McLean A., McIntosh L., Goodwin G., Walker G., Westwood P., Marrs J., Thomson F., Cowley P., Christopoulos A., Pertwee R.G., Ross R.A. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Rhodes D.G., Sarmiento J.G., Herbette L.G. Kinetics of binding of membrane-active drugs to receptor sites. Diffusion-limited rates for a membrane bilayer approach of 1,4-dihydropyridine calcium channel antagonists to their active site. Mol. Pharmacol. 1985;27:612–623. [PubMed] [Google Scholar]
- Rhodes D.G., Newton R., Butler R., Herbette L. Equilibrium and kinetic studies of the interactions of salmeterol with membrane bilayers. Mol. Pharmacol. 1992;42:596–602. [PubMed] [Google Scholar]
- Rocha e Silva A thermodynamic approach to problems of drug antagonism. I. The “Charnière theory”. Eur. J. Pharmacol. 1969;6:294–302. doi: 10.1016/0014-2999(69)90188-5. [DOI] [PubMed] [Google Scholar]
- Romano R., Musiol H.J., Weyher E., Dufresne M., Moroder L. Peptide hormone-membrane interactions: the aggregational and conformational state of lipo-gastrin derivatives and their receptor binding affinity. Biopolymers. 1992;32:1545–1558. doi: 10.1002/bip.360321112. [DOI] [PubMed] [Google Scholar]
- Sargent D.F., Schwyzer R. Membrane lipid phase as catalyst for peptide–receptor interactions. Proc. Natl. Acad. Sci. U.S.A. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent D.F., Bean J.W., Schwyzer R. Conformation and orientation of regulatory peptides on lipid membranes. Key to the molecular mechanism of receptor selection. Biophys. Chem. 1988;31:183–193. doi: 10.1016/0301-4622(88)80024-3. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. 100 years lock-and-key concept: are peptide keys shaped and guided to their receptors by the target cell membrane? Biopolymers. 1995;37:5–16. doi: 10.1002/bip.360370104. [DOI] [PubMed] [Google Scholar]
- Szczuka, A., Wennerberg, M., Packeu, A., Vauquelin, G., 2009. Molecular mechanism of the persistent bronchodilatory effect of the partial β2-adrenoceptor agonist salmeterol. Br. J. Pharmacol., in press. [DOI] [PMC free article] [PubMed]
- Smith C., Rahman T., Toohey N., Mazurkiewicz J., Herrick-Davis K., Teitler M. Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor. Mol. Pharmacol. 2006;70:1264–1270. doi: 10.1124/mol.106.024612. [DOI] [PubMed] [Google Scholar]
- Spivak C.E., Oz M., Beglan C.L., Shrager R.I. Diffusion delays and unstirred layer effects at monolayer cultures of Chinese hamster ovary cells: radioligand binding, confocal microscopy, and mathematical simulations. Cell. Biochem. Biophys. 2006;45:43–58. doi: 10.1385/CBB:45:1:43. [DOI] [PubMed] [Google Scholar]
- Stone S.R., Mierke J.F., Jackson G.E. Evidence for a C-terminal structural motif in gastrin and its bioactive fragments in membrane mimetic media. Peptides. 2007;28:1561–1571. doi: 10.1016/j.peptides.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Teschemacher A., Lemoine H. Kinetic analysis of drug-receptor interactions of long-acting beta2 sympathomimetics in isolated receptor membranes: evidence against prolonged effects of salmeterol and formoterol on receptor-coupled adenylyl cyclase. J. Pharmacol. Exp. Ther. 1999;288:1084–1092. [PubMed] [Google Scholar]
- Theodoropoulou E., Marsh D. Interactions of angiotensin II non-peptide antagonist losartan with phospholipid membranes by combined use of differential scanning calorimetry and electron spin resonance spectroscopy. Biochim. Biophys. Acta. 1999;1461:135–146. doi: 10.1016/s0005-2736(99)00155-8. [DOI] [PubMed] [Google Scholar]
- Tian X., Guo J., Yao F., Yang D.P., Makriyannis A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J. Biol. Chem. 2005;280:29788–29795. doi: 10.1074/jbc.M502925200. [DOI] [PubMed] [Google Scholar]
- Tummino P.J., Copeland R.A. Residence time of receptor–ligand complexes and its effect on biological function. Biochemistry. 2008;47:8465. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Vargová L., Syková E. Extracellular space diffusion and extrasynaptic transmission. Physiol. Res. 2008;57:S89–S99. doi: 10.33549/physiolres.931603. [DOI] [PubMed] [Google Scholar]
- Vauquelin G., Van Liefde I. Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Trends Pharmacol. Sci. 2006;51:254–260. doi: 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Vauquelin G., Szczuka A. Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem. Int. 2007;51:254–260. doi: 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Voss, H.P., 1994. Long-acting β2-adrenoceptor agonists in asthma: molecular pharmacological aspects. Thesis, Vrije Universiteit, Amsterdam.
- Wang D., Gou S.Y., Axelrod D. Reaction rate enhancement by surface diffusion of adsorbates. Biophys. Chem. 1992;43:117–137. doi: 10.1016/0301-4622(92)80027-3. [DOI] [PubMed] [Google Scholar]
- Xie X.Q., Melvin L.S., Makriyannis A. The conformational properties of the highly selective cannabinoid receptor ligand CP-55,940. J. Biol. Chem. 1996;271:10640–10647. doi: 10.1074/jbc.271.18.10640. [DOI] [PubMed] [Google Scholar]
- Zoumpoulakis P., Daliani I., Zervou M., Kyrikou I., Siapi E., Lamprinidis G., Mikros E., Mavromoustakos T. Losartan's molecular basis of interaction with membranes and AT1 receptor. Chem. Phys. Lipids. 2003;125:13–25. doi: 10.1016/s0009-3084(03)00053-7. [DOI] [PubMed] [Google Scholar]