

Abstract
The Podostemaceae are ecologically and morphologically unusual aquatic angiosperms that survive only in rivers with pristine hydrology and high water quality and are at a relatively high risk of extinction. The taxonomic status of Podostemaceae has always been controversial. Here, we report the first high-quality genome assembly for Cladopus chinensis of Podostemaceae, obtained by incorporating Hi-C, Illumina and PacBio sequencing. We generated an 827.92 Mb genome with a contig N50 of 1.42 Mb and 27,370 annotated protein-coding genes. The assembled genome size was close to the estimated size, and 659.42 Mb of the assembly was assigned to 29 superscaffolds (scaffold N50 21.22 Mb). A total of 59.20% repetitive sequences were identified, among which long terminal repeats (LTRs) were the most abundant class (28.97% of the genome). Genome evolution analysis suggested that the divergence time of Cladopus chinensis (106 Mya) was earlier than that of Malpighiales (82 Mya) and that this taxon diverged into an independent branch of Podestemales. A recent whole-genome duplication (WGD) event occurred 4.43 million years ago. Comparative genomic analysis revealed that the expansion and contraction of oxidative phosphorylation, photosynthesis and isoflavonoid metabolism genes in Cladopus chinensis are probably related to the genomic characteristics of this growing submerged species. Transcriptome analysis revealed that upregulated genes in the shoot group compared to the root group were enriched in the NAC gene family and transcription factors associated with shoot development and defense responses, including WUSCHEL (WUS), ASYMMETRIC LEAVES (ASL), SHOOT MERISTEMLESS (STM), NAC2, NAC8, NAC29, NAC47, NAC73, NAC83 and NAC102. These findings provide new insights into the genomic diversity of unusual aquatic angiosperms and serve as a valuable reference for the taxonomic status and unusual shoot apical meristem of Podostemaceae.
Subject terms: High-throughput screening, Bioinformatics, Genomic analysis
Introduction
Podostemaceae are freshwater hydrophytes whose center of diversity is the Neotropics. In the Amazon, these species form subaquatic meadows producing autochthonous carbon, which serve as a source of food for the associated fauna1. The Amazon is the richest region of Podostemaceae occurrence2. These plants require rivers with pristine hydrology and good water quality, which are very important characteristics for their productivity and for maintaining the associated fauna, and their destruction can lead to ecological and economic losses as well as the loss of cultural and hedonic value3,4.
Podostemaceae is a morphologically and esthetically aquatic angiosperm family distributed in the rivers of tropical, subtropical and temperate regions5,6. Angiosperms normally exhibit a common system of gravitropism (the growth movement of organs in response to gravity). The roots grow downward into the soil, and the shoots upward in this gravitropism system. The plant meristems, including the root apical meristem (RAM) and shoot apical meristem (SAM), are unique structures of undifferentiated pluripotent stem cells7. However, Podostemaceae, Streptocarpus and Lemnaceae exhibit unusual modifications of the SAM that may reflect adaptation to new habitats and can subsequently diversify in the evolution of plants8.
Several studies have focused on the establishment of the SAM, including the auxin-related gene activities, transcription factors and morphological characteristics involved9–13. A low auxin level contributes to the initial establishment of the apical-basal axis that extends to form the apical meristems of the shoots9. The monopteros (MP) gene encodes an auxin response factor whose activity as a transcriptional activator facilitates auxin flow by promoting vascular development. MP expression is relatively weak in auxin-deficient conditions10. WUSCHEL (WUS) is a homeodomain transcription factor whose expression establishes a de novo stem cell population11. SHOOT MERISTEMLESS (STM) encodes a MEINOX/three-amino acid loop extension (TALE)-HD-type transcription factor required for the initiation and maintenance of the SAM12. Hamada et al. reported that mutations of the WUS gene in Arabidopsis thaliana halt the entire process of SAM formation and result in no production of juvenile leaves13. Based on these studies, the SAM is deemed vital for the vegetative growth of the stem and produces determinate lateral leaves with shoot branches at their axils, which is associated with the expression of key regulatory genes in the evolution of plant organization.
Podostemaceae species grow submerged on rock surfaces in rapids and waterfalls during the rainy season and bear flowers above the water during the dry season, when the water level decreases14. Studies involving large amounts of data have mainly been concentrated in certain geographical regions and focused on the molecular phylogenetic analysis of Podostemaceae among other angiosperms, including the evaluation of the phylogeny, classification, and biogeography of Podostemaceae as a whole15,16. However, little is known about the evolution of the distinct shoot and root systems of Podostemaceae. Katayama et al. examined the expression patterns of the STM, WUS and asymmetric leaves (ASL) genes related to shoot development in Tristichoideae and Podostemoideae8. The subfamily Tristichoideae shows typical shoot organogenesis with a tunica-corpus SAM that produced leaves, but Podostemoideae is devoid of SAM, and new leaves in these plants develop below the older leaves without a SAM structure. Katayama et al. found that the leaves or bracts of Podostemoideae were involved in SAM initiation and maintenance and differentiated into single apical leaves or bracts, resulting in the evolution of mixed shoot-leaf organs in Podostemaceae according to the analysis of phylogenetic and expression patterns8. However, questions remain regarding how the unusual modification of the SAM arose during the evolution of Podostemaceae and what key regulatory genes are involved in the evolution of adaptation to the environment. The reasons for these phenomena are not clear. Furthermore, genomic information for Podostemaceae species has not been previously reported.
Cladopus chinensis (C. chinensis) is a species of the genus Cladopus that exhibits flattened roots creeping on rock surfaces, and leaves and flowers are produced from the roots (Fig. 1). C. chinensis can be used as an excellent material for studying the evolution of angiosperm morphology due to its extraordinary aerial shoots and underground root system17. Because of the limited genomic information available, an in-depth investigation of the genetic basis of C. chinensis is still lacking.
Fig. 1. Cladopus chinensis.

a Flowering plants in the habitat of the species. b Roots. c Leaves on sterile stems. d Flowering plants. e Flowering shoots (an-anther; ov-ovary; st-style). Bar = 3 mm
In this study, to systematically understand the evolution of organogenesis in C. chinensis, we performed third-generation sequencing (TGS) on the PacBio SEQUEL platform and Hi-C technology to generate a high-quality genome assembly and annotation of C. chinensis. We also performed transcriptome sequencing to identify the genes that are involved in the diversity of SAM establishment. These results provide insight into the evolutionary model underlying shoot novelty in Podostemaceae.
Results
Genome sequencing and quality assembly
We estimated a genome size of 820 Mb for C. chinensis, with heterozygosity of 0.73% and a repeat content of 51.0% (Fig. S1). A total of 95.36 Gb of PacBio long reads (~105X coverage of the genome) and 106.2 Gb of Illumina clean reads (~118X coverage of the genome) were generated, resulting in approximately 224.2-fold coverage of the C. chinensis genome (Table 1). The total size of all reads assembled from the C. chinensis genome was 827.92 Mb, consisting of 5629 contigs. The contig N50 was 1.42 Mb, and the longest contig was 8.89 Mb (Fig. 2 and Table 2). The genome size was close to the results based on flow cytometry (Fig. S2) and genome surveys (Fig. S1). The assembly quality was assessed through BUSCO analysis and the alignment of Illumina short reads to the genome. The NGS short reads from the Illumina sequencing platform were mapped to the contigs by using Bowtie2 software, and approximately 99.78% of the Illumina resequencing reads were mapped to the assembly (Table S1). BUSCO analysis revealed that the assembly completeness was 90.7% in the C. chinensis genome (Table S2). Using the contact matrix and the agglomerative hierarchical clustering method in LACHESIS18, the 538 contigs were successfully clustered into 29 superscaffolds (Fig. 3; Table S3). The scaffold N50 reached 1.42 and 21.22 Mb (Table 2), providing the first high-quality genome assembly for C. chinensis. Moreover, a total of 836 syntenic blocks were detected in the C. chinensis genome, which involved 15,410 genes. Synteny analysis of the 29 superscaffolds of C. chinensis confirmed that superscaffold no. 4 shared the most syntenic blocks with superscaffold no. 6 (Fig. 2).
Table 1.
Sequencing data used for C. chinensis genome construction
| Library resource | Sequencing platform | Insert size (bp) | Clean data (Gb) | Sequence coverage (X) | Use of the data |
|---|---|---|---|---|---|
| Genome | Illumina HiSeq X Ten | 250 bp | 106.2 | 118 | Genome estimation and polishing |
| Genome | PacBio SEQUEL | 20 kb | 95.36 | 105 | Genome assembly |
| Hi-C | Illumina HiSeq X Ten | 250 bp | 96.4 | 107 | Chromosome construction |
| Transcriptome | PacBio SEQUEL | 0.6–3 kb | 77.7 | — | Difference analysis and annotation |
Note that the sequence coverage was calculated using the K-mer-based estimate of genome size
Fig. 2. Characterization of the elements in the superscaffold of the C. chinensis genome.

a The 29 assembled superscaffolds of the genome. b Distribution of GC content in the genome. c Distribution of gene density within sliding windows of 1 Mb. Higher density is shown in green. d Expression values of root- and shoot-expressed genes. e Percent coverage of TEs in nonoverlapping windows. f Schematic presentation of major interchromosomal relationships in the C. chinensis genome. Each line represents a syntenic block; block size = 3 kb
Table 2.
Assembly statistics for the C. chinensis genome
| Items | Canu | Hi-C | ||
|---|---|---|---|---|
| Contig_len(Mb) | Contig_number | Scaffold_len(Mb) | Scaffold_number | |
| Total | 827.92 | 5629 | 827.92 | 4929 |
| Max | 8.89 | — | 33.49 | — |
| Number ≥ 2 kb | — | 5387 | — | 4737 |
| N50 | 1.42 | 184 | 21.22 | 18 |
Fig. 3. C. chinensis genome-wide all-by-all Hi-C interaction heat map.

The map shows high-resolution individual superscaffolds, which were scaffolded and assembled independently
Gene prediction and functional annotation
A combination of reference plant protein homology support, transcriptome data, and ab initio gene prediction was used to generate all gene models. All gene models were merged, and redundancy was removed with MAKER, leading to a total of 27,370 protein-coding genes. The average transcript length was 1465 bp, and the average CDS length was 868 bp (Table 3). We functionally annotated 22,564, 14,193, 22,564 and 11,950 genes to the eggNOG, GO, COG and KEGG databases, respectively, leading to 23,163 (84.63% of the total) genes showing at least one hit to the public databases (Table 3). In total, 2156 transcription factors were identified in the C. chinensis genome, and these genes were classified into 28 families, including 705 protein kinase family, 459 PPR, 155 MYB and 123 bHLH superfamily proteins (Table S4).
Table 3.
Annotation statistics for the C. chinensis genome
| Annotation statistics for the genome | Number | Percent (%) |
|---|---|---|
| Total protein | 27,370 | |
| eggNOG | 22,564 | 82.44 |
| GO | 14,193 | 51.86 |
| COG | 22,564 | 82.44 |
| KEGG | 11,950 | 43.66 |
| In all databases | 8549 | 31.23 |
| In at least one database | 23,163 | 84.63 |
Annotation of noncoding RNAs (ncRNAs)
We identified snRNA, miRNA and rRNA genes in the C. chinensis genome from the Rfam database using BLASTN software (E-value ≤ 1e-5), and we used tRNAscan-SE and RNAmmer to predict tRNAs and rRNAs, resulting in a C. chinensis genome with 79 miRNAs, 1997 tRNAs, 397 rRNAs, 116 sRNAs, and 128 snRNAs (Table S5).
Repeat element (TE) annotation and burst analysis
The C. chinensis genome contained 490.13 Mb of repetitive sequences, accounting for 59.20% of the genome (Table S6). Long terminal repeat (LTR) retrotransposons accounted for 28.97% of the genome, with 22.30% Ty1/copia and 0.72% Ty3/gypsy sequences. Kimura distance analysis indicated an LTR burst (Fig. S3) involving the Ty1/copia and Ty3/gypsy superfamilies. Tandem Repeats Finder identified over 50,071 tandem repeats, accounting for 7.40% of the C. chinensis genome (Table S6). Telomeric and centromeric repeats were identified by searching the ends of contigs, and 8 telomeric repeat sequences and 572 centromeric repeat sequences were identified (Tables S7, S8).
Evolution of the C. chinensis genome
We collected the genome sequences of representative plant species and performed comparative genomic analysis with C. chinensis to reveal the genome evolution and divergence time of C. chinensis. The results suggested that gene family contractions outnumbered expansions in C. chinensis, R. communis, O. sativa, P. alba, B. oleracea, B. rapa, J. curcas, M. esculenta, A. thaliana, C. pepo and F. vesca, in contrast to the other seven species (Fig. 4a). The results support the view that Malpighiales and Rosales share a common Rosidae ancestor, which is the basal taxon of dicotyledons. The phylogenetic tree showed that C. chinensis phylogenetically diverged into an independent branch approximately 106 million years ago (Mya), without clustering into Malpighiales after the divergence of the A. thaliana (Brassicales) lineage 108 Mya. The data further confirmed that the divergence time of C. chinensis (106 Mya) was earlier than those of Malpighiales (82 Mya) and Rosales (102 Mya) and later than that of Brassicales (108 Mya). We also found that the internal branches separating Brassicales, Podestemales, Rosales, and Malpighiales were very short, as shown in Fig. 4a, suggesting the rapid diversification of these major clades. To obtain a highly reliable phylogenetic tree, we identified 91 single-copy homologous genes and performed coalescent analysis with RAxML and ASTRAL to estimate the species tree from gene trees. This result showed that C. chinensis phylogenetically clusters into Malpighiales, which is consistent with the results from NCBI Taxonomy (Fig. 4b). On the basis of the number of transversions at four-fold degenerate sites, we calculated an age distribution for all duplicate gene pairs. Using 8,074 paralogous gene pairs of similar ages and excluding tandem or local duplications, a large peak centered on a synonymous substitution rate (Ks) of approximately 0.14 was observed in the C. chinensis genome (Fig. 5), indicating only one recent whole-genome duplication (WGD) event and one ancient WGD that occurred 4.43 million years ago.
Fig. 4. Phylogenetic tree and evolution analysis of C. chinensis.

a Phylogenetic tree showing the number of gene families displaying expansion (green) and contraction (red) among 18 plant species. The pie charts show the proportions of expanded (green), contracted (red) and conserved (blue) gene family among all gene families. The estimated divergence time (million years ago) is displayed below the phylogenetic tree in black. *The time of the occurrence of a recent whole-genome duplication (WGD) event in C. chinensis. MRCA, most recent common ancestor. b The species tree of 18 species inferred from the complete dataset of single-copy homologous gene using concatenation (RAxML) and gene-tree-based coalescent (ASTRAL) methods
Fig. 5.
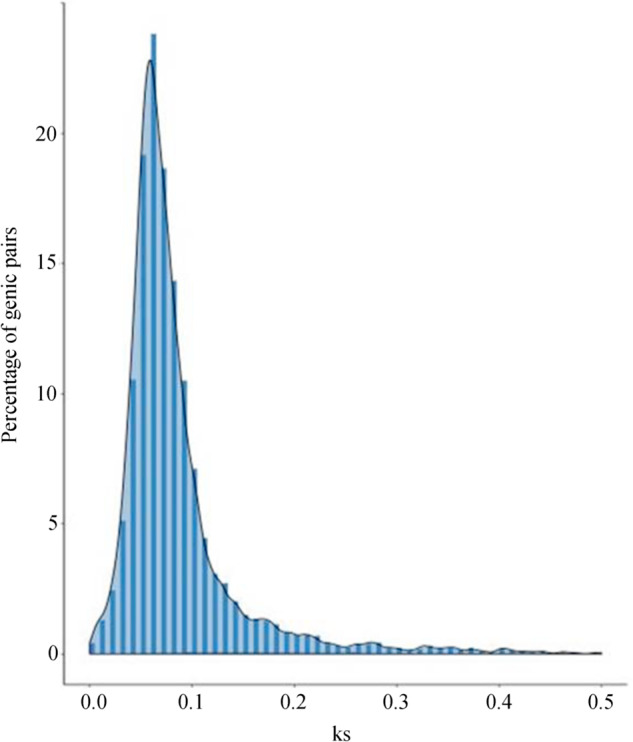
Ks distributions for duplicated gene pairs in C. chinensis
Gene family analysis
We performed comparative genomic analyses among 18 plant species and detected 81,585 families of homologous genes. A total of 17,964 gene families were identified in C. chinensis, among which 2164 and 14,356 gene families showed expansion and contraction, respectively (Fig. 4). A total of 2164 expanded gene families were annotated to KEGG pathways (Table S9) and GO terms (Table S10). KEGG analysis showed that most of the expanded genes were clustered in the categories of energy metabolism, signal transduction and aging. GO analysis showed that the expanded orthogroups were related to metabolic process and cellular component categories and the stimulus response, cysteine synthase, phototropism and sugar carrier terms. A total of 14,356 of the contracted gene families were involved in carbohydrate metabolism, signal transduction, secondary metabolism biosynthesis, amino acid metabolism, the nervous system, the metabolism of terpenoids and polyketides, the immune system, environmental adaptation and aging (Table S11). The GO terms of the contracted genes were related to the cellular component, flavonoid biosynthetic process, developmental process, stimulus response, catalytic activity, biological regulation, transcription regulator activity, immune system process, signaling, growth and reproduction process categories (Table S12). When we sought to investigate the environmental adaptation of C. chinensis, we found that a series of photosynthesis and energy metabolism-associated gene families exhibit significant expansion, including the NADPH oxidase (NDHF, NDHB1, GPSA), NADH dehydrogenase (NAD1, NDHD, NAD4L, NAD7, NDHG), chlorophyll a-b binding protein (LHCB) and cytochrome P450 (CYP86) families (Table S13). Notably, CYP450 genes are involved in the regulation of basic developmental processes such as cell differentiation and growth, indicating effects on the expression of developmental genes19. Additionally, approximately half of the encoded proteins catalyze specific biochemical reactions in various metabolic pathways. To obtain insights into the evolutionary relationships among C. chinensis CYP450 family proteins, we constructed a phylogenetic trees based on the sequences of full-length CYP450 proteins from 216 amino acid sequences of C. chinensis (121) and A. thaliana (95) using the neighbor-joining method. The numbers of most CYP450 types, including CYP71, CYP86 and CYP90, were higher than in A. thaliana indicating a close relationship of the development and metabolism of C. chinensis plants (Fig. S4).
The comparison of C. chinensis, A. thaliana, O. sativa, P. alba, M. esculenta, P. euphratica, J. curcas, R. communis, C. pepo and H. brasiliensis revealed that 4858 (31.37%) of the 15,485 C. chinensis gene families were shared by the other nine species, whereas 5,636 gene families were unique to C. chinensis (Fig. 6; Fig. S5). These 5,636 unique families have been specific to C. chinensis during its long history of evolution. Some of them may have been lost in other species, although we believe that some gene families originated de novo in Podostemaceae. Functional analysis performed via GO analysis revealed that these 5,636 unique families were enriched in suberin biosynthesis, cationic antimicrobial peptides, folate biosynthesis and ABC transporters (Table S14). KEGG analysis showed that most of the 5636 unique families were clustered in the signal transduction, lipid metabolism, energy metabolism and environmental adaptation categories (Table S15).
Fig. 6. UpSet plot of the intersection of gene families in C. chinensis, A. thaliana, O. sativa, P. alba, M. esculenta, P. euphratica, J. curcas, R. communis, C. pepo and H. brasiliensis.

The numbers of gene families (clusters) are indicated for each species and species intersection
Phylogenetic analysis of the WRKY, NAC and resistance gene families
A phylogenetic tree was constructed based on the WRKY gene family members from A. thaliana (72), P. trichocarpa (102) and C. chinensis (68) to examine the evolutionary relationships among them. According to our results, a total of 68 WRKY genes were identified in the C. chinensis genome and classified into 7 groups: I, IIa, IIb, IIc, IId, IIe and III. The subfamily III group was one of the most ancient WRKY types, while subfamily I had fewer members, with no more than six members in each species. The identified WRKY genes of C. chinensis were only classified into the IIc subfamily (Fig. 7a).
Fig. 7. The evolutionary tree of WRKY and NAC gene families in C. chinensis.

a The evolutionary tree and expression values of WRKY box genes in C. chinensis, P. trichocarpa and A. thaliana. b The evolutionary tree and expression values of NAC box genes in C. chinensis, P. trichocarpa and A. thaliana. Genes in the WRKY and NAC families were clustered separately using MEGA7 software via the neighbor-joining method
To obtain insights into the evolutionary relationships among C. chinensis NAC family proteins, phylogenetic trees were constructed based on the sequences of full-length NAC-box proteins from 350 amino acid sequences of C. chinensis (81), P. trichocarpa (165) and A. thaliana (104) using the neighbor-joining method. The 81 NAC genes of C. chinensis were classified into 18 categories, including NAC1, NAP, NAM and TIP subfamily genes. The number of NAM and NAP subfamily genes from the NAC family was higher than in A. thaliana and P. trichocarpa. We speculate that the members of the NAM and NAP subfamilies could play roles in the development and defense responses of C. chinensis (Fig. 7b).
Furthermore, we analyzed the evolutionary relationships among resistance (R) gene proteins from C. chinensis (26) and A. thaliana (423) via phylogenetic tree analysis. The results showed that the R gene proteins could be classified into three groups, including TNL, CNL and RLP/RLK subfamily genes. Interestingly, the number of R gene proteins from A. thaliana (423) was 16.26-fold higher than that in C. chinensis (26), and the RLP/RLK subfamily genes of C. chinensis were identified and clustered. It is speculated that the presence of most R gene proteins reduces resistance and environmental adaptability, resulting in the survival of C. chinensis only in good-quality water (Fig. S6).
Positively selected genes in C. chinensis
The Ka/Ks ratios of the single-copy genes were evaluated, and positively selected genes were identified in C. chinensis. A total of 490 candidate genes in C. chinensis underwent positive selection (P < 0.05). Most of which were enriched in GO terms related to the biological regulation, cellular process, developmental process, growth, metabolic process, stimulus response, catalytic activity, binding, positive regulation of biological process and cell part categories (Table S16; Fig. S7). KEGG enrichment analyses showed that the positively selected genes were enriched in the categories of environmental adaptation, endocrine system, metabolism of terpenoids and polyketides, lipid metabolism, energy metabolism, carbohydrate metabolism, biosynthesis of other secondary metabolites, amino acid metabolism, translation, signal transduction and cell growth (Table S17; Fig. S8).
Comparative analysis of gene expression
To obtain an overview of the transcriptome profile, principal component analysis (PCA) was performed according to normalized log10 (FPKM + 1) values. The first principal component (PC1), which explained 99.66% of the total variance, showed clearly different gene expression profiles between the root group and the shoot group (Fig. S9). We identified the specific genes responsible for shoot development and defense responses in C. chinensis. In comparison with the root group, we identified 3376 DEGs (log2 FC ≥ 2 and p-value ≤ 0.05) in the shoot group, including 2002 downregulated and 1374 upregulated genes (Table S18). Some of these DEGs were associated with shoot development, including WUS, ASL and STM. Among these genes, CcWUS10, CcASL4, CcASL9, CcSTM3 and CcSTM4 were expressed at low levels in the shoots, whereas CcWUS1, CcASL1, CcASL2 and CcASL7 were highly expressed in the shoots (Fig. 8a, Tables S18–S21). Podostemaceae species have extraordinary body plans showing unusual shoot organogenesis involving the modification of the SAM, making C. chinensis an ideal system for studying the development of roots and shoots in plants. To identify the potential key genes or transcription factors involved in shoot formation in C. chinensis, we identified 609 genes that were highly expressed in shoots (log2 FC ≥ 1.5 and p-value ≤ 0.01) (Table S22; Fig. S10), and these highly expressed genes were significantly enriched in the metabolic process, stimulus response, developmental process, environmental adaptation, signal transduction, energy metabolism, carbohydrate metabolism and secondary metabolite synthesis were identified according to KEGG and GO enrichment analyses (Figs. S11, S12; Tables S23, S24). Additionally, we found that the 609 highly expressed genes in the shoots included hormone biosynthesis and signaling genes, including 11 auxin, 1 cytokinin, 2 abscisic acid, 5 gibberellin and 1 brassinosteroid gene. The development of the plant shoot is dependent on the SAM, MYB and bHLH transcription factors, which could regulate meristem initiation, meristem function and leaf patterning. Notably, 107 transcription factors were identified from the 609 highly expressed genes in the shoots, including 11 MYB, 11 bHLH and HD-ZIP family proteins (Table S25). In addition, we observed that the CcASL1, CcASL2, CcASL7, CcASL8 and CcSTM1 genes were involved in shoot development and were significantly expressed in specific shoot tissues (Fig. S13). Furthermore, we identified 132 and 146 MYB proteins in C. chinensis and A. thaliana. These MYB genes are divided into 13 subfamilies, and the numbers of genes in subfamilies I, III, and X are greater than those in the other subfamilies of A. thaliana (Fig. 8b). MYB transcription factors are widely involved in the regulation of various physiological responses and phenylpropanoid metabolic pathways, indicating a close relationship with the growth of C. chinensis on submerged rock surfaces in rapids and waterfalls during the rainy season.
Fig. 8. Expression patterns of the root- and shoot-specific expression and evolutionary tree of MYB gene family in C. chinensis.

a Heatmap showing the root- and shoot-specific expression of the members of WUS, STM, ASL and NAC. Each box represents an individual gene, and the red and blue boxes represent relatively high levels and low levels of gene expression, respectively. b Evolutionary tree and expression values of MYB box genes in C. chinensis and A. thaliana. Genes in the MYB family were separately clustered using MEGA7 software via the neighbor-joining method. The genes of C. chinensis and A. thaliana are indicated by blue and purple, respectively
The expression of WUS, STM and ASL is involved in the initiation and maintenance of shoot development in Tristichoideae and Podostemoideae, respectively8. To confirm the accuracy of the obtained unigene expression levels, nine target genes (CcWUS1, CcWUS10, CcASL1, CcASL2, CcASL4, CcASL7, CcASL9, CcSTM3, CcSTM4) associated with shoot development were analyzed in the present study (Fig. S13). The relative levels of the amplified mRNAs were evaluated according to the 2−ΔΔCt method. Compared with the FPKM values, the relative expression levels of all nine unigenes determined by qRT-PCR were consistent with the RNA-seq data.
Discussion
The Podostemaceae family comprises ~300 species classified into ~50 genera, among which 3 genera, Cladopus, Terniopsis and Hydrobryum, have been confirmed from Fujian, Hainan, Guangdong and Yunnan in China (Fig. S14). Recently, a set of studies assessing the phylogeny, classification, biogeography and morphological evolution of the Podostemaceae family were reported. Katayama et al. examined the expression patterns of the STM, WUS and ARP genes during shoot development in Tristichoideae and Podostemoideae and tried to reveal the genetic basis for the evolution of shoots in Podostemaceae8. Mutations in the WUS gene of A. thaliana reported by Hamada et al. halted the entire process of SAM formation and resulted in the production of no juvenile leaves13. Here, we assembled the genome of C. chinensis to the superscaffold scale using the Hi-C technique. The C. chinensis genome assembly has been vastly improved by Hi-C analysis, producing an N50 scaffold size for the assembly of 21.22 Mb, compared to the N50 contig size of 1.42 Mb for the de novo-assembled genome obtained with Canu software. These results demonstrated that a high-quality C. chinensis genome was produced and suggested that the numbers of repetitive sequences and ncRNA sequences were relatively high in C. chinensis compared with other dicotyledon species. The present genome provides a good reference for understanding the distinct aerial shoot and underground root system traits of C. chinensis and related species.
According to the NCBI taxonomy database, C. chinensis is classified into Podostemonaceae of Malpighiales based on morphological characteristics and partial nucleotide alignment (rbc L, atpB, 18 S rRNA), which is consistent with the integrated system of the angiosperm phylogeny group (APG) classification (APG, 1998; APG II, 2003; APG III, 2009; APG IV, 2016)20. However, Podostemonaceae was removed from Malpighiales and classified into Podestemales in the integrated system of the classification of flowering plants by Cronquist21 and Wu22. Based on a concatenated sequence alignment of C. chinensis and 17 other plant species, a phylogenetic tree was reconstructed (Fig. 4a). The bootstrap values were >99% for all the relationships. P. alba, J. curcas, M. esculenta, R. communis, P. euphratica, H. brasiliensis and P. trichocarpa were grouped together in Malpighiales. C. chinensis phylogenetically diverged into an independent branch approximately 106 million years ago (Mya), after the divergence of the Brassicales lineage 108 Mya but before the divergence of Malpighiales (82 Mya) and Rosales (102 Mya). We found that the internal branches separating Brassicales, Podestemales, Rosales, and Malpighiales were very short, as shown in Fig. 4a, suggesting the rapid diversification of these major clades. We performed coalescent analysis with RAxML and ASTRAL to estimate the species tree from gene trees. This result showed that C. chinensis phylogenetically clustered into Malpighiales, which is inconsistent with the findings of Cronquist21 and Wu22 (Fig. 4b). Based on our data, we speculated that C. chinensis may belong to the order Malpighiales, which is consistent with the conclusions from the APG and NCBI Taxonomy.
KEGG and GO analyses showed that the cytochrome P450 (CYP) gene and serine/threonine protein kinase families were significantly contracted in the C. chinensis genome (Tables S10, S11). The cytochrome P450 gene family (mostly the CYP81, CYP90, CYP85, CYP4 and CYP7 families) is related to isoflavonoid metabolic processes that regulate secondary metabolite synthesis and affect broad environmental adaptability. Previous research suggests that the cytochrome P450 gene family is important for the regulation of basic developmental processes. For example, a previous study showed that high levels of CYP85 family gene expression alleviate the jasmonate response, resulting in longer primary roots and more lateral roots and enhanced drought tolerance in tobacco19. The ROTUNDIFOLIA3 (ROT3) locus of A. thaliana, which is involved in the regulation of leaf morphogenesis, harbors a CYP90 sequence23. Serine/threonine protein kinases are involved in the response to environmental stresses (high salt, drought and low temperature) and regulate multiple life processes in cells24. It is speculated that the contraction of these types of gene families affects the accumulation of secondary metabolites and reduces environmental adaptability, resulting in the survival of C. chinensis only in good-quality water.
ATP is an energy carrier and is conserved in the form of NADH, and FADH2 must be converted to ATP (oxidative phosphorylation). NADH and NADPH undergo spontaneous enzymatic reactions that participate in photosynthesis induced by light via the phytochrome system and respiratory metabolism25. The chlorophyll a-b-binding proteins are located in the chloroplast thylakoid membrane, where Chl a and Chl b bind to form protein complexes that transfer absorbed light energy to photosystem reactions26. KEGG and GO analysis showed that most of the expanded genes in the C. chinensis genome are involved in plant energy metabolism, especially oxidative phosphorylation (NADH and NADPH) and photosynthesis (chlorophyll a-b-binding proteins) (Tables S8, S9). Thus, it is likely that the significant expansion of energy metabolism-associated genes in C. chinensis could increase oxidative phosphorylation and photosynthesis, which would be beneficial for the growth of the plants on submerged rock surfaces and for bearing flowers exposed to the air when the water level decreases.
Katayama et al. analyzed the expression patterns of key regulatory genes related to shoot development (i.e., STM, WUS and ARP orthologs) in Tristichoideae and Podostemoideae. They found that STM and WUS were expressed in the floral meristem, similar to the pattern in model plants. Because of limited genome information, it was impossible for these authors to annotate all members of the STM, WUS and ASL gene families. The probes used for in situ hybridization in their studies could only indicate the expression profiles of these genes according to the coexpression of all gene family members8. Based on the available genome information and transcriptome data, we annotated all members of these gene families and analyzed their gene expression patterns. Additionally, we found that the expression levels of different members of these gene families were upregulated or downregulated to very different extents other in shoots. In this study, we performed transcriptome sequencing to identify the genes that are involved in the diversity of SAM establishment in whole shoots and roots. In future work, we will perform single-cell transcriptome analysis to evaluate the expression patterns of these genes in fine fractions of different development stages (e.g., meristem, young shoot, and mature shoot) by laser capture microdissection to reveal the roles of STM, WUS and ASL gene family members in the shoot development of C. chinensis.
The NAC proteins are one of the largest families of transcription factors in plants; these proteins regulate gene expression during the life of plants and exhibit important biological functions in response to both stress and plant growth27. We annotated 81 members of the NAC gene family in C. chinensis, and the numbers belonging to the NAM and NAP subfamilies were much higher than those of other subfamilies. NAC102 is an important gene of the NAM subfamily that is thought to participate in the regulation of seed germination in flooded environments27. Our transcriptome analysis showed that some genes exhibiting significant expression in shoots were members of the NAC gene family, including NAC2, NAC5, NAC8, NAC29, NAC47, NAC73, NAC83 and NAC102. We also found that among these highly expressed NAC genes, NAC29 (Cladopus_020264), NAC47 (Cladopus_004681) and NAC83 (Cladopus_004734) were included in the genes that were duplicated via a WGD event in C. chinensis superscaffold no. 4 and superscaffold no. 6 (Fig. S15). Additionally, the gene pairs (Cladopus_020264-Cladopus_004681, Cladopus_004734-Cladopus_025202) exhibited 97.8 and 97.0% identity scores, respectively (Table S26), suggesting that they were functionally redundant and were likely duplicated during the evolution of shoot development in C. chinensis. It is speculated that the presence and expression of these genes play roles in the shoot development and environmental adaptation of C. chinensis. However, which of these genes are beneficial to the environmental adaptation and shoot development of C. chinensis remains to be further studied and analyzed.
Materials and methods
Genome sequencing and de novo assembly
The root, leaf and shoot samples of C. chinensis (sample number CBH03085, Fig. S16) used for genome sequencing and assembly were collected from the Tingjiang River in Changting, Fujian, China (E116。25'10", N25。51'03", Alt. 281.3 m)28. The voucher specimens of C. chinensis were deposited in the Plant Herbaria of the College of Life Science, Fujian Normal University (collection number FNU0039809). To generate enough short and long reads for the genome assembly of C. chinensis, next-generation sequencing (NGS) on the Illumina HiSeq X Ten platform and third-generation sequencing (TGS) on the PacBio SEQUEL platform were applied for genome sequencing. Genomic DNA was produced via the CTAB method, and the integrity of the DNA was checked via agarose gel electrophoresis. The purity and concentration of the DNA were analyzed by using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). For Illumina X Ten sequencing, we constructed a paired-end library with 250 base pair (bp) sequences using the method indicated by the manufacturer. As a result, 125.95 Gb (~140X coverage of the estimated genome size, Table 1) of short reads were generated from the Illumina platform, which were further cleaned by using Trimmomatic (version 0.36) with the default parameters, resulting in 106.2 Gb (~118×, Table 1) of cleaned data for the following analysis. For PacBio library construction and sequencing, SMRTbell libraries (~20 Kbp) were obtained according to the PacBio protocol. After the removal of adapters, 95.36 Gb of subreads (~105×, Table 1) were corrected, trimmed and assembled using CANU (version 1.6) with the parameters corOutCoverage = 80 and corMinCoverage = 0. To improve accuracy, the primary contigs were further filtered with the Pilon program29 using 106.2 Gb (118×) of Illumina paired-end reads. We summed the statistics of the assemblies in Table 1. Genome completeness was assessed using the BUSCO databases30 with embryophyta_odb10 models.
Estimation of genome size and heterozygosity
Weused NGS short reads and a K-mer-based method to estimate the genome size, heterozygosity and repeat content of C. chinensis. Approximately 106.2 Gb of reads (250 bp) were generated and used to determine the abundance of 21-K-mers in the Illumina data. The frequency of 21-K-mers was counted with Jellyfish software31.
Root, leaf and shoot samples of C. chinensis were prepared according to a previously reported protocol32 and stained with propidium iodide (50 mg/ml). Quantification was performed using flow cytometry in a BD FACSCalibur cytometer (Becton Dickinson, San Jose, CA), and the results were calculated as the ratio of the mean fluorescence of C. chinensis to that of Zea mays B73. The estimated genome size of C. chinensis was 835 ± 5.52 Mb.
High-quality assembly using Hi-C technology
Samples were examined for the integrity of nuclei by DAPI staining to ensure the quality of the Hi-C library. Samples with confirmed high-quality nuclei were subjected to the Hi-C procedure33,34. Chromatin was digested with the restriction enzyme Mbo I or Hind III and ligated together in situ after biotinylation. DNA fragments were enriched via the interaction of biotin and blunt-end ligation and then subjected to HiSeq sequencing. From Hi-C library sequencing, approximately 96.4 Gb of data were generated (Table 1). The sequencing reads were mapped to the C. chinensis genome with Bowtie software. We applied an iterative alignment method to increase the read mapping ratio. We aligned the two read ends to the genome independently, and only the read pairs in which both ends were uniquely aligned to the reference genome were used for the detection and filtering of valid interaction products by using HiC-Pro (version 2.7.8)35. The order and direction of scaffolds/contigs were clustered into superscaffolds by using LACHESIS36 based on the relationships among valid reads.
RNA extraction and sequencing
Total RNA was prepared from C. chinensis roots and shoots (Fig. S17) using TRIzol reagent (Invitrogen, California, USA). A NanoDrop 2000 spectrophotometer (Waltham, MA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) were applied to check RNA quality; the absorbance at 260 nm/280 nm was 1.8, and the RIN value was 9.1. Equal amounts of RNA from each tissue were used for cDNA library construction. Approximately 77.7 Gb of transcript data were produced for C. chinensis from the Illumina HiSeq X Ten sequencing platform and processed using Trimmomatic (version 0.36) with the default parameters. Reads originating from RNA-seq were aligned to the reference genome using HISAT237. FPKM values and read counts were estimated using Stringtie38 and Ballgown39. The differential expression of genes was analyzed using edgeR40, for which the criteria were a log2-fold change (FC) ≥ 1 and a false discovery rate (FDR) ≤ 0.05.
Genome annotation
In the C. chinensis genome, de novo- and homology-based approaches were combined to search TEs and other repetitive sequences. We identified repeat sequences using Tandem Repeats Finder41, LTR_FINDER42, RepeatProteinMask and RepeatMasker43 and used Tandem Repeats Finder to search for tandem repeats in the genome assembly with the following parameters: Mismatch = 7, Match = 2, Delta = 7, PI = 10, Minscore = 50, PM = 100, and MaxPerid = 2,000. Using LTR_FINDER (version 1.0.6), a de novo repeat library was built. Subsequently, we aligned the genome sequences to Repbase TE (version 3.2.9)44 by using RepeatMasker for the searching of known repeat sequences and mapping onto the de novo repeat libraries to identify novel types of repeat sequences. tRNAscan-SE (version 2.0.3)45 was used to detect reliable tRNA positions, and noncoding RNA (ncRNA) was predicted by searching the RFAM (version 12.0)46 databases by using Infernal software (version 1.0)47 with the default parameters. Centromere and telomere repeats were identified with Tandem Repeats Finder (version 4.07b)41. We transformed the resulting ‘dat_dir file’ into a GFF3 file, which was used to identify centromeric and telomeric repeats.
The C. chinensis genome assembly was annotated via the following approaches: homology-based, transcriptome-based, and ab initio annotation. Thirteen representative species were selected to perform homology annotation, including Manihot esculenta (M. esculenta)48, Populus euphratica (P. euphratica)49, Populus alba (P. alba)50, Jatropha curcas (J. curcas)51, Ricinus communis (R. communis)52, Hevea brasiliensis (H. brasiliensis)53, Arabidopsis thaliana (A. thaliana)54, Cucurbita pepo (C. pepo)55, Fragaria vesca (F. vesca)56, Malus domestica (M. domestica)57, Populus trichocarpa (P. trichocarpa)52, Pyrus x bretschneideri (P. bretschneideri)58 and Rosa chinensis (R. chinensis)59. The protein sequences of these species were aligned to C. chinensis genome sequences using TBLASTN software60 with an E-value ≤ 1e-5. Genewise (version 2.2.0)61 was utilized to predict the exact gene structures based on all TBLASTN results. We used Cufflinks (version 2.2.1)62 to preliminarily identify gene structures. Augustus63 was used for ab initio annotation with the repeat-masked genome sequences. We integrated all genes predicted from the three annotation procedures with MAKER software64.
Functional annotation of the protein-coding genes was carried out by using BLASTP with an E-value ≤ 1e-5 in four integrated protein sequence databases: eggNOG, GO, COG and KEGG. We used InterProScan (version 4.8)65 and HMMER (version 3.1)66 to annotate protein domains by searching the INTERPRO (version 32.0)67 and Pfam (version 27.0)68 databases. Gene Ontology (GO) terms were produced from the InterPro or Pfam entry69. The pathways were assigned through BLAST searches in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (release 53) with an E-value ≤ 1e-570. The functions of the genes were predicted and classified using the Clusters of Orthologous Groups (COG) database71.
Phylogenetic analysis and divergence time estimation
We investigated the relationships of C. chinensis with 17 other species and performed a phylogenetic analysis based on protein-coding genes from the C. chinensis genome and 17 other species. We extracted and downloaded the protein sequences of single-copy genes from 18 species from the NCBI database, including M. esculenta, P. euphratica, P. alba, J. curcas, R. communis, H. brasiliensis, A. thaliana, C. pepo, F. vesca, M. domestica, P. trichocarpa, P. bretschneideri, R. chinensis, Brassica oleracea (B. oleracea), Brassica rapa (B. rapa), Raphanus sativus (R. sativus) and Oryza sativa (O. sativa). The similarities among the proteins from all species were searched in an all-to-all manner by using BLASTP software with an E-value ≤ 1e-5. By using OrthoFinder software (version 2.27)72, we generated multiple sequence alignments for the protein sequences in each single-copy family with the default parameters as well as for phylogenetic tree construction. We designated O. sativa as the outgroup of the phylogenetic tree. The phylogenetic relationships were constructed through the superalignment of the coding DNA sequences (CDSs) using the maximum likelihood (ML) method, and the divergence time between species was estimated using the MCMCtree program of PAML (http://abacus.gene.ucl.ac.uk/software/paml.html). The TimeTree database (http://www.time.org/) was used for the divergence time recalibration of these plant species. The CDSs were aligned with the guidance of the protein alignments and then concatenated into the superalignment matrix of each family. We compared the cluster size differences between the ancestor and each species and analyzed the expansion and contraction of the gene families by using CAFE software (version 2.1)73. For coalescent analysis, the concatenated alignment of single-copy homologous genes for 18 species was used as the input for ML inference with RAxML (version 7.2.8)74. The alignment was partitioned by the genes, and ProtTest75 was used to select the appropriate model of amino acid substitution for each partition. We used the -f a option of RAxML to generate 200 rapid bootstrap replicates, followed by a search for the best-scoring ML tree, and then used ASTRAL76 to estimate the species tree from the gene trees.
Synteny analysis
The conserved paralogs of the protein sequences of C. chinensis were obtained with BLASTP (E-value ≤ 1E-5). By using MCScanX (http://chibba.pgml.uga.edu/mcscab2), we identified collinearity blocks in the genome. The Circos tool (http://www.circos.ca) was used to map gene density, GC content and repeat content as well as gene synteny on individual pseudochromosomes.
Whole-genome duplication
We took advantage of the high-quality genome of C. chinensis, analyzed WGD events and determined the source of the high number of genes in C. chinensis. First, the protein sequences from C. chinensis were searched to identify syntenic blocks by using BLASTP with an E-value ≤ 1e-5. We identified gene synteny and collinearity by using MCScanX software77 and calculated the synonymous substitution rate (Ks) for syntenic gene pairs using KaKs_Calculator software78 and the Nei-Gojobori method79.
Quantitative real-time PCR (qRT-PCR) validation
Total RNA was extracted from the three replicates using the TransZol Up Plus RNA Kit (Transgen Biotech, Beijing). Primers were designed using Primer Premier 5.0, and the sequences are listed in Table S27. qRT-PCR was performed using the ABI 7300 Real-time PCR System (Framingham, MA, USA) with SYBR Green PCR Master Mix (TaKaRa) following procedures described previously80. All PCR assays were performed in triplicate. The reference gene was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The relative expression level was quantified via the 2−ΔΔCt method81.
Supplementary information
Table S25 Transcription factors identified from 609 specific expressed genes in shoots
Table S14 Statistics of GO enrichment of the 5636 gene families in the C. chinensis
Table S15 Statistics of KEGG enrichment of the 5636 gene families in the C. chinensis
Table S8 Size and location of centromere satellite repeat arrays
Table S9. KEGG enrichment of the expansion familes genes identified in the C. chinensis
Table S10. GO enrichment of the expansion familes genes identified in the C. chinensis
Table S11. KEGG enrichment of the contraction familes genes identified in the C. chinensis
Table S12. GO enrichment of the contraction familes genes identified in the C. chinensis
Table S16 GO enrichment of the positively genes identified in the C. chinensis
Table S16 GO enrichment of the positively genes identified in the C. chinensis
Table S18 my.gene.counts.matrix.root_vs_shoot.DESeq2
Table S22 609 specific expressed genes in shoots
Table S23 GO enrichment of the 609 specific expressed genes in the shoot of C. chinensis
Table S24 KEGG enrichment of the 609 specific expressed genes in the shoot of C. chinensis
Acknowledgements
This manuscript was edited for proper English language by the highly qualified native English-speaking editors at American Journal Experts. This work was supported by the Natural Science Foundation of Fujian Province, China (grant number 2017J01622), and the Sugar Crop Research System (grant number CARS-170501).
Author contributions
Y.Z., J.Z., X.Z., and Y.C. designed and coordinated the entire project. T.X., X.Z., D.C., B.C., and Y.C. led and performed the entire project together. L.L., J.C., W.C., and B.C. performed the collection and processing of samples. T.X., X.Z., D.C., N.C., L.L., S.C., Z.H., W.H., J.C., W.C., B.C., and X.Z. performed the analyses of genome evolution and gene families. T.X., X.Z., D.C., and Y.C. participated in manuscript writing and revision. All authors read and approved the final manuscript.
Data availability
The genome sequence data that support the findings of this study have been deposited in the BIG Sub system under BioProject accession number CRA002215 (http://bigd.big.ac.cn/gsa/s/a118ohEn). Raw sequencing data for RNA-Seq were used for annotation and biological analyses and have been deposited in the BIG Sub system under BioProject accession number CRA002218 (http://bigd.big.ac.cn/gsa/s/0VZ87KD1).
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Binghua Chen, Email: bhchen@fjnu.edu.cn.
Xingtan Zhang, Email: tanger.zhang@gmail.com.
Youqiang Chen, Email: yqchen@fjnu.edu.cn.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-0269-5).
References
- 1.Vinicius NL, et al. High importance of autochthonous basal food source for the food web of a brazilian tropical stream regardless of shading. Int. Rev. Hydrobiol. 2016;101:132–142. doi: 10.1002/iroh.201601851. [DOI] [Google Scholar]
- 2.Sobral-Leite M, et al. Anthecology and reproductive system of mourera fluviatilis (podostemaceae): pollination by bees and xenogamy in a predominantly anemophilous and autogamous family? Aquat. Bot. 2011;95:0–87. doi: 10.1016/j.aquabot.2011.03.010. [DOI] [Google Scholar]
- 3.Ram HYM, Sehgal A. In vitro studies on developmental morphology of indian podostemaceae. Aquat. Bot. 1997;57:0–132. doi: 10.1016/S0304-3770(96)01124-2. [DOI] [Google Scholar]
- 4.Cruden, Robert W. Pollen-ovule ratios: a conservative indicator of breeding systems in flowering plants. Evolution. 1977;31:32–46. doi: 10.1111/j.1558-5646.1977.tb00979.x. [DOI] [PubMed] [Google Scholar]
- 5.Koi S, Kato M. Paracladopus chanthaburiensis, a new species of Podostemaceae from thailand, with notes on its morphology, phylogeny and distribution. Taxon. 2008;57:201–210. [Google Scholar]
- 6.Katayama N, Kato M, Imaichi R. Habitat specificity enhances genetic differentiation in two species of aquatic Podostemaceae in Japan. Am. J. Bot. 2016;103:317–324. doi: 10.3732/ajb.1500385. [DOI] [PubMed] [Google Scholar]
- 7.Koi S, Imaichi R, Kato M. Endogenous leaf initiation in the apical‐meristemless shoot ofcladopus queenslandicus (Podostemaceae) and implications for evolution of shoot morphology. Int. J. Plant. Sci. 2005;166:199–206. doi: 10.1086/427482. [DOI] [Google Scholar]
- 8.Katayama N, Koi S, Kato M. Expression of shoot meristemless, wuschel, and asymmetric leaves1 homologs in the shoots of Podostemaceae: implications for the evolution of novel shoot organogenesis. Plant Cell. 2010;22:2131–2140. doi: 10.1105/tpc.109.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shani E, Yanai O, Ori N. The role of hormones in shoot apical meristem function. Curr. Opin. Plant. Biol. 2006;9:484–489. doi: 10.1016/j.pbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Luo L, et al. A molecular framework for auxin-controlled homeostasis of shoot stem cells in Arabidopsis. Mol. Plant. 2018;7:899–913. doi: 10.1016/j.molp.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Landau U, et al. The erecta, clavata and class iii hd-zip pathways display synergistic interactions in regulating floral meristem activities. PLoS ONE. 2015;10:e0125408. doi: 10.1371/journal.pone.0125408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cole M. Nuclear import of the transcription factor shoot meristemless depends on heterodimerization with blh proteins expressed in discrete sub-domains of the shoot apical meristem of arabidopsis thaliana. Nucleic Acids Res. 2006;34:1281–1292. doi: 10.1093/nar/gkl016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamada S, et al. Mutations in the WUSCHEL gene of Arabidopsis thaliana result in the development of shoots without juvenile leaves. Plant J. 2000;24:91–101. doi: 10.1046/j.1365-313x.2000.00858.x. [DOI] [PubMed] [Google Scholar]
- 14.Rutishauser R. Evolution of unusual morphologies in lentibulariaceae (bladderworts and allies) and podostemaceae (river-weeds): a pictorial report at the interface of developmental biology and morphological diversification. Ann. Bot. 2016;117:811–832. doi: 10.1093/aob/mcv172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koi S, et al. Molecular phylogenetic analysis of Podostemaceae: implications for taxonomy of major groups. Bot. J. Lin. Soc. 2012;169:461–492. doi: 10.1111/j.1095-8339.2012.01258.x. [DOI] [Google Scholar]
- 16.Kato M, Wong MCK, Lo JP, Satoshi K. A cladopus species (Podostemaceae) rediscovered from Hong Kong. Acta Phytotax. 2017;68:17–22. [Google Scholar]
- 17.Chao HC. Teratological variations of stamen in Lawiella chinensis Chao (Podostemonaceae) in relation to the taxonomic position of the genus. J. Univ. Chin. Aca. Sci. 1951;1:343–359. [Google Scholar]
- 18.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fangmeng D, et al. Overexpression of socyp85a1, a spinach cytochrome p450 gene in transgenic tobacco enhances root development and drought stress tolerance. Front. Plant. Sci. 2017;8:1909. doi: 10.3389/fpls.2017.01909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chase MW, et al. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016;181:1–20. doi: 10.1111/boj.12385. [DOI] [Google Scholar]
- 21.Cronquist A. Angiosperm orders and families. (Book Reviews: An Integrated System of Classification of Flowering Plants) Science. 1981;216:1217–1218. [Google Scholar]
- 22.Wu, Z. Y. et al. (eds) The families and genera of angiosperms in China (Science Press, 2003).
- 23.Kim TG, et al. Changes in the shapes of leaves and flowers upon overexpression of cytochrome P450 in Arabidopsis. Proc. Natl Acad. Sci. USA. 1999;96:9433–9437. doi: 10.1073/pnas.96.16.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhaskar G, et al. Spermidine-mediated in vitro phosphorylation of transcriptional regulator osbz8 by snf1-type serine/threonine protein kinase sapk4 homolog in indica rice. Acta Physiol. Plant. 2012;34:1321–1336. doi: 10.1007/s11738-012-0929-7. [DOI] [Google Scholar]
- 25.Zhang J, Wang CX, Ying WH. Sirt2 and akt mediate nad+-induced and nadh-induced increases in the intracellular atp levels of bv2 microglia under basal conditions. Neuroreport. 2018;29:65. doi: 10.1097/WNR.0000000000000876. [DOI] [PubMed] [Google Scholar]
- 26.Krol M. Chlorophyll a/b-binding proteins, pigment conversions, and early light- induced proteins in a chlorophyll b-less barley mutant. Plant Physiol. 1995;107:873–883. doi: 10.1104/pp.107.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SY, et al. Exploring membrane-associated nac transcription factors in arabidopsis: implications for membrane biology in genome regulation. Nucleic Acids Res. 2007;35:203–213. doi: 10.1093/nar/gkl1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo LQ, et al. Study on the biological characteristics of Cladopus chinensis (Podostemaceae) Bull. Biol. 2014;49:15–16. [Google Scholar]
- 29.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 31.Guillaume C, et al. A model-based method for investigating bioenergetic processes in autotrophically growing eukaryotic microalgae: application to the green algae Chlamydomonas reinhardtii. Biotechnol. Progre. 2011;27:631–640. doi: 10.1002/btpr.596. [DOI] [PubMed] [Google Scholar]
- 32.Doležel J. Flow cytometric analysis of nuclear DNA content in higher plants. Phytochem. Anal. 1991;2:143–154. doi: 10.1002/pca.2800020402. [DOI] [Google Scholar]
- 33.Chen P, et al. The effect of environment on the microbiome associated with the roots of a native woody plant under different climate types in China. Appl. Microbiol. Biot. 2019;12:661–677. doi: 10.1007/s00253-019-09747-6. [DOI] [PubMed] [Google Scholar]
- 34.Yang XL, et al. The chromosome-level quality genome provides insights into the evolution of the biosynthesis genes for aroma compounds of Osmanthus fragrans. Hortic. Res. 2018;5:72. doi: 10.1038/s41438-018-0108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keel BN, Snelling WM. Comparison of burrows-wheeler transform-based mapping algorithms used in high-throughput whole-genome sequencing: application to illumina data for livestock genomes. Front. Genet. 2018;9:35. doi: 10.3389/fgene.2018.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pertea M, et al. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016;11:1650. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikolayeva O, Robinson MD. Edger for differential rna-seq and chip-seq analysis: an application to stem cell biology. Methods Mol. Biol. 2014;1150:45–79. doi: 10.1007/978-1-4939-0512-6_3. [DOI] [PubMed] [Google Scholar]
- 41.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences (Calgary Univ. Press, 2004). [DOI] [PubMed]
- 44.Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 45.Lowe TM, Chan PP. Trnascan-se on-line: integrating search and context for analysis of transfer rna genes. Nucleic Acids Res. 2016;44:54–57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardner PP, et al. Rfam: updates to the rna families database. Nucleic Acids Res. 2009;37:136–140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daniell H, et al. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpf in Malpighiales: rna editing and multiple losses of a group ii intron. Theor. Appl. Genet. 2008;116:723–737. doi: 10.1007/s00122-007-0706-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao G, Zhang X, Tengfei YU, Liu B. Comparison of three evapotranspiration models with eddy covariance measurements for a Populus euphratica in an arid region of northwestern China. J. Arid Land. 2016;8:146–156. doi: 10.1007/s40333-015-0017-0. [DOI] [Google Scholar]
- 50.Liu YJ, Wang XR, Zeng QY. De novo assembly of white poplar genome and genetic diversity of White poplar population in irtysh river basin in china. Sci. China. 2019;62:3–12. doi: 10.1007/s11426-018-9345-5. [DOI] [PubMed] [Google Scholar]
- 51.Kancharla N, et al. De novo sequencing and hybrid assembly of the biofuel crop Jatropha curcas L.: identification of quantitative trait loci for geminivirus resistance. Genes. 2019;10:1. doi: 10.3390/genes10010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeng L, et al. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 2017;214:1338–1354. doi: 10.1111/nph.14503. [DOI] [PubMed] [Google Scholar]
- 53.Tang CR, et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants. 2016;23:73–78. doi: 10.1038/nplants.2016.73. [DOI] [PubMed] [Google Scholar]
- 54.Zapata L, et al. Chromosome-level assembly of Arabidopsis thaliana reveals the extent of translocation and inversion polymorphisms. Proc. Natl Acad. Sci. USA. 2016;113:52–57. doi: 10.1073/pnas.1607532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monteropau J, et al. De novo assembly of the zucchini genome reveals a whole genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 2018;16:1161–1171. doi: 10.1111/pbi.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shulaev V, et al. The genome of woodland strawberry (Fragaria vesca) Nat. Genet. 2011;43:109–116. doi: 10.1038/ng.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daccord N, et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017;49:1099–1106. doi: 10.1038/ng.3886. [DOI] [PubMed] [Google Scholar]
- 58.Wu J, et al. The genome of the pear (Pyrus bretschneideri rehd) Genome Res. 2013;23:396–408. doi: 10.1101/gr.144311.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raymond O, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018;50:772–777. doi: 10.1038/s41588-018-0110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams TH. Computer software design for pediatric practice. Clin. Pediatr. 1984;23:5–11. doi: 10.1177/000992288402300101. [DOI] [PubMed] [Google Scholar]
- 61.Birney E, Durbin R. Using genewise in the drosophila annotation experiment. Genome Res. 2000;10:547–548. doi: 10.1101/gr.10.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C, et al. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stanke M, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:435–439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.None. Molecular biology: Protein maker and gene regulator. Nature. 2011;473:127–127. [Google Scholar]
- 65.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klingenberg H, et al. Protein signature-based estimation of metagenomic abundances including all domains of life and viruses. Bioinformatics. 2013;29:973–980. doi: 10.1093/bioinformatics/btt077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Daly TK, Sutherland-Smith AJ, David P. In silico resurrection of the major vault protein suggests it is ancestral in modern Eukaryotes. Genome Biol. Evol. 2013;5:1567–1583. doi: 10.1093/gbe/evt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashburner M, et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanehisa M, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tatusov RL, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu Y, et al. Removal behavior research of orthophosphate by CaFe-layered double hydroxides. Desalin. Water Treat. 2016;57:7918–7925. doi: 10.1080/19443994.2015.1039602. [DOI] [Google Scholar]
- 74.Stamatakis A. Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abascal F, et al. Prottest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 76.Chao Z, et al. Astral-iii: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018;19:153. doi: 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Z, et al. KaKs_calculator: calculating ka and ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006;4:259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 80.Wang HY, et al. Use of hTERT and HPV E6/E7 mRNA RT-qPCR TaqMan assays in combination for diagnosing high-grade cervical lesions and malignant tumors. Am. J. Clin. Pathol. 2015;143:344. doi: 10.1309/AJCPF2XGZ2XIQYQX. [DOI] [PubMed] [Google Scholar]
- 81.Wang X, et al. Expression analysis of KAP9.2 and Hoxc13 genes during different cashmere growth stages by qRT-PCR method. Mol. Biol. Rep. 2014;41:5665–5668. doi: 10.1007/s11033-014-3435-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S25 Transcription factors identified from 609 specific expressed genes in shoots
Table S14 Statistics of GO enrichment of the 5636 gene families in the C. chinensis
Table S15 Statistics of KEGG enrichment of the 5636 gene families in the C. chinensis
Table S8 Size and location of centromere satellite repeat arrays
Table S9. KEGG enrichment of the expansion familes genes identified in the C. chinensis
Table S10. GO enrichment of the expansion familes genes identified in the C. chinensis
Table S11. KEGG enrichment of the contraction familes genes identified in the C. chinensis
Table S12. GO enrichment of the contraction familes genes identified in the C. chinensis
Table S16 GO enrichment of the positively genes identified in the C. chinensis
Table S16 GO enrichment of the positively genes identified in the C. chinensis
Table S18 my.gene.counts.matrix.root_vs_shoot.DESeq2
Table S22 609 specific expressed genes in shoots
Table S23 GO enrichment of the 609 specific expressed genes in the shoot of C. chinensis
Table S24 KEGG enrichment of the 609 specific expressed genes in the shoot of C. chinensis
Data Availability Statement
The genome sequence data that support the findings of this study have been deposited in the BIG Sub system under BioProject accession number CRA002215 (http://bigd.big.ac.cn/gsa/s/a118ohEn). Raw sequencing data for RNA-Seq were used for annotation and biological analyses and have been deposited in the BIG Sub system under BioProject accession number CRA002218 (http://bigd.big.ac.cn/gsa/s/0VZ87KD1).