

The evolution of influenza virus is a significant public health problem and necessitates the annual evaluation of influenza vaccine formulation to keep pace with viral escape from herd immunity. Influenza B virus is a serious health concern for children, in particular, yet remains understudied compared to influenza A virus. Influenza B virus evolves more slowly than influenza A virus, but the factors underlying this are not completely understood. We studied how the within-host diversity of influenza B virus relates to its global evolution by sequencing viruses from a community-based cohort. We found that influenza B virus populations have lower within-host genetic diversity than influenza A virus and experience a tight genetic bottleneck during transmission. Our work provides insights into the varying dynamics of influenza viruses in human infection.
KEYWORDS: influenza B virus, bottleneck, transmission, diversity, evolution, influenza virus
ABSTRACT
Influenza B virus (IBV) undergoes seasonal antigenic drift more slowly than influenza A virus, but the reasons for this difference are unclear. While the evolutionary dynamics of influenza viruses play out globally, they are fundamentally driven by mutation, reassortment, drift, and selection at the level of individual hosts. These processes have recently been described for influenza A virus, but little is known about the evolutionary dynamics of IBV during individual infections and transmission events. Here, we define the within-host evolutionary dynamics of IBV by sequencing virus populations from naturally infected individuals enrolled in a prospective, community-based cohort over 8,176 person-seasons of observation. Through analysis of high depth-of-coverage sequencing data from samples from 91 individuals with influenza B, we find that IBV accumulates lower genetic diversity than previously observed for influenza A virus during acute infections. Consistent with studies of influenza A viruses, the within-host evolution of IBVs is characterized by purifying selection and the general absence of widespread positive selection of within-host variants. Analysis of shared genetic diversity across 15 sequence-validated transmission pairs suggests that IBV experiences a tight transmission bottleneck similar to that of influenza A virus. These patterns of local-scale evolution are consistent with the lower global evolutionary rate of IBV.
IMPORTANCE The evolution of influenza virus is a significant public health problem and necessitates the annual evaluation of influenza vaccine formulation to keep pace with viral escape from herd immunity. Influenza B virus is a serious health concern for children, in particular, yet remains understudied compared to influenza A virus. Influenza B virus evolves more slowly than influenza A virus, but the factors underlying this are not completely understood. We studied how the within-host diversity of influenza B virus relates to its global evolution by sequencing viruses from a community-based cohort. We found that influenza B virus populations have lower within-host genetic diversity than influenza A virus and experience a tight genetic bottleneck during transmission. Our work provides insights into the varying dynamics of influenza viruses in human infection.
INTRODUCTION
Influenza viruses rapidly mutate and evolve through selection, genetic drift, and reassortment (1). On a global scale, influenza A virus (IAV) and influenza B virus (IBV) evolve under strong positive selection driven by pressure for escape from preexisting population immunity (2, 3). Selection of new antigenic variants contributes to reduced effectiveness of seasonal influenza vaccines, necessitating annual updates of vaccine strains (4). IAV and IBV both undergo seasonal antigenic drift and share a similar genomic architecture, but their ecology and evolution differ in important ways (5). While IBV accounts for roughly one-third of influenza’s burden of morbidity and mortality (6, 7), it circulates only in humans and seals and is considered to be a lower pandemic risk than IAV due to its limited animal reservoirs. Like IAV, there are cocirculating, antigenically distinct lineages of IBV that are included in the quadrivalent influenza vaccine. Two lineages of IBV diverged in the 1980s, B/Victoria/2/87-like and B/Yamagata/16/88-like, here referred to as B/Victoria and B/Yamagata, respectively (8).
IBV evolves more slowly than IAV on a global scale and has a lower rate of antigenic drift, but the reasons for this are poorly understood (5, 9). Similar evolutionary forces are involved in the antigenic evolution of IAV and IBV, generally characterized by nonsynonymous substitutions at antigenic sites in the surface hemagglutinin (HA) protein (10, 11) and reassortment within and between lineages (12–14). The IBV polymerase has a lower mutation rate than that of IAV (15). However, it is unclear whether the slower global evolution of IBV is driven by its lower mutation rate or other differences in selection at the global scale.
All new seasonal influenza variants are ultimately derived from de novo mutations within individual hosts (16). Therefore, understanding how new variants arise within individuals and transmit between them is essential to defining how novel viruses spread in host populations. For example, if the relative mutation rate is a major factor underlying the global evolutionary differences across IAV and IBV, we might also expect to see differences in their within-host dynamics. We and others have used next-generation sequencing (NGS) to investigate the within- and between-host evolutionary dynamics of IAV in humans (16–21). We have found that there is little accumulation of intrahost variants during acute infections of immunocompetent individuals (18, 19), and we have not found evidence of changes in intrahost diversity by vaccination status or other proxies for immunological history (19, 20, 22). The IAV transmission bottleneck is stringent (19), which generally means that few variants that arise within hosts are able to transmit. Together, these studies suggest that positive selection of novel variants is an inefficient process in IAV-infected hosts, contrasting with its patterns of significant positive selection at the global level (23). Despite the importance of intrahost processes to influenza virus evolution, these dynamics have not been systematically investigated in IBV.
Here, we use NGS to define the within-host diversity of IBV populations from individuals enrolled in the Household Influenza Vaccine Evaluation (HIVE) study, a community-based household cohort initiated in 2010. We apply a previously validated sequencing approach and bioinformatic pipeline (19, 20, 24) to identify intrahost single-nucleotide variants (iSNV) arising during infection with B/Victoria and B/Yamagata viruses. We find that IBV has significantly lower intrahost diversity than IAV, consistent with its lower mutation rate and lower rate of evolution. We analyze shared iSNV across 15 genetically validated household transmission pairs and find that like IAV, IBV is also subject to a tight genetic bottleneck at transmission. These data provide the first systematic evaluation of the genetic architecture of IBV populations during natural human infection and provide insights into the comparative epidemiology and evolution of influenza viruses.
(This article was submitted to an online preprint archive [25].)
RESULTS
We used high depth-of-coverage sequencing to define the intrahost genetic diversity in IBV-positive samples collected from individuals in the HIVE, a prospective, household cohort in southeastern Michigan that follows 200 to 350 households annually (Table 1). This cohort provides an opportunity to investigate natural infections and transmission events in a community context. Individuals that meet symptom-based criteria for an upper respiratory illness during the surveillance period undergo collection of nasal and throat swabs for molecular detection of respiratory viruses by reverse transcription-PCR (RT-PCR). Starting in 2014-2015, individuals also provided a sample collected at home prior to subsequent collection of a second specimen at the on-site clinic.
TABLE 1.
Influenza B viruses over seven seasons in a household cohort
| Parameter | Value for indicated seasone |
||||||
|---|---|---|---|---|---|---|---|
| 2010–2011 | 2011–2012 | 2012–2013 | 2013–2014 | 2014–2015 | 2015–2016 | 2016–2017 | |
| Households | 328 | 213 | 321 | 232 | 340 | 227 | 208 |
| Participants | 1,441 | 943 | 1,426 | 1,049 | 1,431 | 996 | 890 |
| Vaccinated [no. (%)]a | 934 (65) | 554 (59) | 942 (66) | 722 (69) | 992 (69) | 681 (68) | 611 (69) |
| IBV-positive individualsb | 45 | 7 | 49 | 4 | 44 | 11 | 30 |
| B/Yamagata | 1 | 3 | 38 | 4 | 34 | 5 | 26 |
| B/Victoria | 37 | 0 | 10 | 0 | 10 | 6 | 4 |
| IBV-positive householdsc | |||||||
| Two individuals | 10 | 2 | 5 | 0 | 11 | 2 | 4 |
| Three individuals | 0 | 1 | 1 | 0 | 1 | 0 | 2 |
| High-quality NGS datad | 13 | 2 | 20 | 1 | 32 | 11 | 20 |
Self-reported or confirmed receipt of vaccine prior to the specified season.
RT-PCR-confirmed infection. The lineage (e.g. B/Yamagata or B/Victoria) could not be determined on several samples.
Two individuals, households in which two individuals were positive within 7 days of each other; three individuals, the putative chains could have no pair with onset >7 days apart.
Samples with >103 genome copies per μl of transport medium, adequate amplification of all 8 genomic segments, and average sequencing coverage of >103 per nucleotide.
Values are numbers unless indicated otherwise.
Over seven seasons (2010-2011 through 2016-2017) and 8,176 person-seasons of observation, we identified 111 individuals infected with B/Yamagata and 67 infected with B/Victoria (Table 1). Several households had clusters of infections of two or three IBV-positive individuals within 7 days of each other, suggestive of within-household transmission. Because variant identification is sensitive to input viral titer (24), we first measured viral loads of all available IBV-positive samples by RT-quantitative PCR (RT-qPCR) (Fig. 1A). Any samples with a viral load below 103 copies/μl were not submitted for sequencing. For samples with a viral load in the range of 103 to 105 copies/μl, we performed two independent RT-PCRs and sequenced replicate libraries on separate sequencing runs. We sequenced samples with viral loads above 105 copies/μl of transport medium in a single replicate. From the available IBV-positive samples, we were able to obtain sequence data on 106 samples from 91 individuals, consisting of 35 individuals infected with B/Victoria and 56 infected with B/Yamagata (Table 1).
FIG 1.

Viral load and sequencing coverage. (A) Boxplot of viral load (genome copies per microliter of swab transport medium, y axis) by day of sampling relative to symptom onset (x axis). The boxes display median and 25th and 75th percentiles, with whiskers extending to the most extreme point within the range of the median ± 1.5 times the interquartile range. (B) Sequencing coverage is plotted with read depth on the y axis and location within a concatenated influenza B virus genome on the x axis. The mean coverage for each sample was calculated over a sliding window of size 200 and a step size of 100. The data are displayed for all samples at each window as a boxplot, showing the median and 25th and 75th percentiles, with whiskers extending to the most extreme point within the range of the median ± 1.5 times the interquartile range; all values outside this range are shown as individual points.
We identified intrahost single nucleotide variants (iSNV) using our previously validated bioinformatic pipeline. As in our previous work, we report iSNV at frequencies of 2% or above, for which we have well-defined sensitivity and specificity (19). We consider sites with >98% frequency to be essentially fixed, setting the frequency at those sites to 100% (see Materials and Methods). We achieved a mean coverage of 10,000× per sample across most genome segments, with generally lower coverage on segments encoding nucleoprotein (NP) and nonstructural protein (NS) (Fig. 1B). We restricted our analysis of iSNV to samples with an average genome coverage of greater than 1,000×, which includes 99 of the original 106 sequenced samples.
Within-host genetic diversity of IBV in natural infections.
All samples exhibited low genetic diversity. The vast majority had no iSNV above the 2% cutoff. Of the 99 samples with high-quality NGS data, 70 had no minority iSNV, 17 had one iSNV, 7 had two iSNV, and 3 samples had 3 iSNV (median, 0; interquartile range [IQR], 0 to 1) (Table 2). Two outliers had a large number of iSNV, with 8 and 20 iSNV. These two samples came from the same individual, with one collected at home and the second at the study clinic 2 days later. Most of the iSNV in these two samples were present at similar frequencies, 3 to 5% in the home sample and 17 to 23% in the clinic sample (Table 3), both of which were sequenced in duplicate on separate Illumina runs. The high number of mutations present at similar frequencies is suggestive of a mixed infection with distinct haplotypes or strains as opposed to de novo mutations arising on a single genetic background. The iSNV in the home-collected sample are all found in the subsequent clinic-collected sample, each with a similar change in frequency across the two samples. This further supports the conclusion that these mutations are on the same genome in a mixed infection with two distinct strains.
TABLE 2.
Identified iSNV, excluding samples from one putative mixed infection
| Enrollee | Specimen | Seasona | Lineage | Viral loadb | Gene | Nucleotidec | Amino acidd | Frequency | Coveragee | Mutation typef | Vaccinatedg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 50207 | MH15919 | 16/17 | B/Victoria | 3.50E+03 | M | A650G | N207S | 0.024 | 13,574 | NS | IIV4 |
| 331001 | MH2671 | 12/13 | B/Victoria | 3.20E+05 | NA | C276T | L73F | 0.086 | 6,107 | NS | LAIV3 |
| 331001 | MH2671 | 12/13 | B/Victoria | 3.20E+05 | PA | A2047G | K671R | 0.474 | 5,024 | NS | LAIV3 |
| 330171 | MH3227 | 12/13 | B/Victoria | 7.00E+06 | NA | A385C | N109T | 0.162 | 14,964 | NS | IIV3 |
| 330171 | MH3227 | 12/13 | B/Victoria | 7.00E+06 | PA | C1982T | A649A | 0.461 | 8,159 | S | IIV3 |
| 301587 | M53957 | 10/11 | B/Victoria | 3.30E+04 | HA | G1603A | G522R | 0.081 | 34,517 | NS | No |
| 301587 | M53957 | 10/11 | B/Victoria | 3.30E+04 | NA | A863C | T268T | 0.038 | 45,106 | S | No |
| 301202 | M54308 | 10/11 | B/Victoria | 4.40E+04 | PA | C1037T | N334N | 0.195 | 20,857 | S | No |
| 50003 | MH10403 | 14/15 | B/Victoria | 8.20E+04 | NS | A103C | T18T | 0.063 | 3,245 | S | No |
| 50004 | MH10404 | 14/15 | B/Victoria | 8.20E+04 | NP | A577G | N171S | 0.057 | 13,413 | NS | No |
| 50004 | MH10404 | 14/15 | B/Victoria | 8.20E+04 | NA | A1457G | L466L | 0.223 | 4,398 | S | No |
| 50004 | MH10404 | 14/15 | B/Victoria | 8.20E+04 | PA | G1617A | V528M | 0.497 | 17,758 | NS | No |
| 50424 | HS1876 | 14/15 | B/Victoria | 1.60E+03 | NP | G1191A | D376N | 0.034 | 1,073 | NS | IIV4 |
| 50051 | HS1909 | 14/15 | B/Victoria | 1.90E+03 | M | G709A | E227K | 0.343 | 2,138 | NS | Yes, Unk |
| 50004 | HS1788 | 14/15 | B/Victoria | 8.30E+05 | NP | A577G | N171S | 0.054 | 12,857 | NS | No |
| 50004 | HS1788 | 14/15 | B/Victoria | 8.30E+05 | PA | G1617A | V528M | 0.389 | 16,217 | NS | No |
| 50004 | HS1788 | 14/15 | B/Victoria | 8.30E+05 | NA | A1457G | L466L | 0.045 | 3,259 | S | No |
| 50312 | HS2019 | 15/16 | B/Victoria | 2.00E+05 | NP | G987A | V308I | 0.420 | 6,593 | NS | No |
| 50312 | HS2019 | 15/16 | B/Victoria | 2.00E+05 | PA | G1346A | E437E | 0.467 | 9,225 | S | No |
| 51123 | HS2680 | 15/16 | B/Victoria | 3.50E+05 | NP | G1511A | R482R | 0.344 | 7,393 | S | No |
| 320779 | MH0776 | 11/12 | B/Yamagata | 3.40E+05 | NP | A735G | S223S | 0.023 | 9,674 | S | IIV3 |
| 320779 | MH0776 | 11/12 | B/Yamagata | 3.40E+05 | PB2 | G661A | R211R | 0.222 | 14,675 | S | IIV3 |
| 51092 | MH10076 | 14/15 | B/Yamagata | 1.20E+04 | PB1 | A223G | I66V | 0.116 | 7,370 | NS | IIV4 |
| 50650 | MH16167 | 16/17 | B/Yamagata | 5.20E+04 | PA | T2019C | L662L | 0.373 | 5,397 | S | IIV4 |
| 50650 | MH16167 | 16/17 | B/Yamagata | 5.20E+04 | PB1 | C345T | A106A | 0.159 | 6,639 | S | IIV4 |
| 331060 | MH3065 | 12/13 | B/Yamagata | 3.70E+05 | PA | A1912G | K626R | 0.051 | 10,393 | NS | LAIV3 |
| 331397 | MH4247 | 12/13 | B/Yamagata | 2.40E+04 | PB2 | A676G | R216R | 0.370 | 20,110 | S | IIV3 |
| 330459 | MH4289 | 12/13 | B/Yamagata | 2.10E+05 | HA | G1102A | A355T | 0.024 | 14,033 | NS | IIV3 |
| 330460 | MH4364 | 12/13 | B/Yamagata | 2.10E+05 | PB2 | G520A | V164V | 0.032 | 7,919 | S | IIV3 |
| 50006 | MH16139 | 16/17 | B/Yamagata | 1.20E+05 | HA | T728C | F230S | 0.148 | 14,877 | NS | No |
| 331471 | MH2216 | 12/13 | B/Yamagata | 8.60E+04 | PB2 | G1936A | Q636Q | 0.024 | 30,012 | S | No |
| 331470 | MH2246 | 12/13 | B/Yamagata | 1.20E+04 | PA | G1535A | A500A | 0.029 | 9,483 | S | No |
| 331470 | MH2246 | 12/13 | B/Yamagata | 1.20E+04 | PB2 | A2253G | K742R | 0.124 | 4,478 | NS | No |
| 331470 | MH2246 | 12/13 | B/Yamagata | 1.20E+04 | PB2 | C769T | H247H | 0.023 | 12,295 | S | No |
| 331364 | MH4166 | 12/13 | B/Yamagata | 2.80E+04 | HA | C746T | T236I | 0.037 | 29,665 | NS | No |
| 331364 | MH4166 | 12/13 | B/Yamagata | 2.80E+04 | PA | G1298A | L421L | 0.093 | 15,898 | S | No |
| UM41536 | MH6592 | 13/14 | B/Yamagata | 2.00E+04 | PB1 | G1893A | R622R | 0.022 | 20,159 | S | No |
| 51093 | HS1747 | 14/15 | B/Yamagata | 3.90E+04 | PA | G1433A | L466L | 0.087 | 5,646 | S | IIV4 |
| 50419 | HS3214 | 16/17 | B/Yamagata | 5.40E+05 | PB1 | C345T | A106A | 0.046 | 7,545 | S | IIV4 |
| 51121 | HS3258 | 16/17 | B/Yamagata | 1.10E+04 | PB1 | A2079G | E684E | 0.022 | 17,567 | S | No |
Seasons are indicated as, e.g., 16/17 for 2016–2017.
Viral load measured by RT-qPCR, expressed in genome copies per microliter of transport medium.
Consensus nucleotide followed by position on reference genome and variant nucleotide.
Consensus amino acid followed by codon position on reference genome and variant amino acid.
Coverage expressed as the total sequencing read depth at the site of the identified variant.
Nonsynonymous (NS) or synonymous (S) variant relative to sample consensus.
Self-reported or confirmed receipt of vaccine prior to the specified season. IIV4, quadrivalent inactivated; LAIV3, trivalent live attenuated; IIV3, trivalent inactivated; Unk, vaccine product unknown.
TABLE 3.
Identified iSNV in one vaccinated individuala with a putative mixed infection during the 2014–2015 season
| Specimena | Viral loadb | Gene | Nucleotidec | Amino acidd | Frequency | Coveragee | Mutation typef |
|---|---|---|---|---|---|---|---|
| HS1875 | 2.90E+04 | HA | G1061A | R341K | 0.029 | 22,065 | NS |
| 2.90E+04 | NP | G1666A | G534D | 0.027 | 4,688 | NS | |
| 2.90E+04 | NA | G1210A | G384D | 0.054 | 20,907 | NS | |
| 2.90E+04 | NA | A798G | S247G | 0.052 | 25,817 | NS | |
| 2.90E+04 | NS | G1004A | V100V | 0.045 | 1,944 | S | |
| 2.90E+04 | PB2 | G817A | V263V | 0.036 | 24,333 | S | |
| 2.90E+04 | PB2 | A1231C | I401I | 0.032 | 26,905 | S | |
| 2.90E+04 | PB2 | A793G | E255E | 0.035 | 23,840 | S | |
| MH10536 | 3.80E+04 | HA | G1061A | R341K | 0.236 | 21,341 | NS |
| 3.80E+04 | HA | C366T | C109C | 0.213 | 17,059 | S | |
| 3.80E+04 | M | A114G | L28L | 0.189 | 2,792 | S | |
| 3.80E+04 | NP | G1666A | G534D | 0.193 | 3,039 | NS | |
| 3.80E+04 | NP | G1257A | R398R | 0.166 | 11,007 | S | |
| 3.80E+04 | NA | G1210A | G384D | 0.239 | 20,531 | NS | |
| 3.80E+04 | NA | C1286T | Y409Y | 0.218 | 16,887 | S | |
| 3.80E+04 | NA | C1319T | C420C | 0.217 | 12,891 | S | |
| 3.80E+04 | NA | G1148A | R363R | 0.225 | 23,087 | S | |
| 3.80E+04 | NA | A798G | S247G | 0.233 | 24,631 | NS | |
| 3.80E+04 | NA | T816C | F253L | 0.06 | 24,779 | NS | |
| 3.80E+04 | NS | G1004A | V100V | 0.185 | 1,310 | S | |
| 3.80E+04 | NS | G596A | V183I | 0.190 | 10,074 | NS | |
| 3.80E+04 | NS | T469A | V140V | 0.198 | 8,807 | S | |
| 3.80E+04 | NS | T66C | M6T | 0.173 | 1,986 | NS | |
| 3.80E+04 | PA | G1279A | S415N | 0.188 | 19,813 | NS | |
| 3.80E+04 | PB1 | T1932A | S635S | 0.214 | 19,999 | S | |
| 3.80E+04 | PB2 | G817A | V263V | 0.217 | 17,558 | S | |
| 3.80E+04 | PB2 | A1231C | I401I | 0.218 | 19,869 | S | |
| 3.80E+04 | PB2 | A793G | E255E | 0.221 | 17,325 | S |
Specimens from enrollee 50425. HS indicates home specimen and MH indicates clinic specimen, both from the same individual.
Viral load measured by RT-qPCR, expressed in genome copies per microliter of transport medium.
Consensus nucleotide followed by position on reference genome and variant nucleotide.
Consensus amino acid followed by codon position on reference genome and variant amino acid.
Coverage expressed as the total sequencing read depth at the site of the identified variant.
Nonsynonymous (NS) or synonymous (S) variant relative to sample consensus.
We examined how within-host diversity changes by day of sampling during IBV infections, as the virus population rapidly expands and contracts. As we have previously shown that specimen viral load can affect the sensitivity and specificity of variant identification (24), we sought to control for this variable in our analysis. Although viral load generally decreased with time after symptom onset (Fig. 1A), we found that within-host diversity as measured by number of identified minority iSNV did not vary with viral load (Fig. 2A) (P = 0.2996, adjusted r2 = 0.0009) or with day of infection (Fig. 2B) (P = 0.62, analysis of variance [ANOVA]). The frequencies of the identified iSNV were consistent across replicate libraries from the same samples, indicating that our measurements of iSNV frequency are precise (Fig. 2C).
FIG 2.

Intrahost minority SNV by day post-symptom onset and viral load. (A) The number of minority iSNV per sample is plotted on the y axis by day post-symptom onset on the x axis. Data are displayed as boxplots representing the median and 25th and 75th percentiles, with whiskers extending to the most extreme point within the range of the median ± 1.5 times the interquartile range. The raw data points are shown in black overlaid on top of the boxplots; points from mixed infection samples are shown in orange. (B) Scatterplot relating the number of minority iSNV per sample on the y axis to the log10 of viral load, in genome copies per microliter, on the x axis. Data points from the mixed infection are shown in orange. (C) Frequency of minority iSNV in samples sequenced in duplicate. Orange dots represent variants identified in samples with a viral load of 103 to 104 genome copies per microliter, and blue dots represent variants in samples with a viral load of 104 to 105 genome copies per microliter.
We detected minority iSNV across all eight genome segments (Fig. 3). The ratio of nonsynonymous to synonymous iSNV was 0.74, which given the excess of nonsynonymous sites across the genome suggests significant purifying selection. There was only one minority iSNV present in more than one individual; we identified a variant encoding a synonymous mutation in PB1 in two individuals from separate households infected with B/Yamagata in the 2016–2017 season. We did not identify any nonsynonymous minority iSNV in the known antigenic sites of IBV hemagglutinin, which suggests that positive selective pressure for variants that escape antibody-mediated immunity is not particularly strong within hosts. We found that there is no difference in distribution of the number of iSNV per sample between vaccinated and nonvaccinated individuals (Fig. 4A). During the first few seasons of the study, some individuals received trivalent vaccines, which contain only one of the two IBV lineages. We therefore repeated this analysis, excluding three individuals for whom we had no information about specific vaccine product and reclassifying six individuals who received trivalent vaccines and were infected with a lineage not included in that season’s trivalent formulation as “unvaccinated.” We again found no difference in the number of iSNV between groups (Mann-Whitney U [MWU] test, P = 0.9103). Together, these data indicate that vaccine-induced immunity is not a major diversifying force for IBV within hosts in our study population. This is consistent with our previous work on IAV in the HIVE as well as a randomized-controlled trial of vaccine efficacy (FLU-VACS), both of which showed no difference in intrahost diversity based on same-season vaccination status (19, 20). Intrahost diversity was similar between B/Victoria and B/Yamagata virus populations (Fig. 4B), consistent with our previous comparison of subtype A/H3N2 and A/H1N1 viruses (19).
FIG 3.

Intrahost SNV frequency by genome position and mutation type. All minority (<50%) iSNV from 99 samples are displayed with their frequency on the y axis and their position within a concatenated influenza B virus genome on the x axis. Synonymous mutations are shown in orange, and nonsynonymous mutations are shown in blue.
FIG 4.

Intrahost SNV by vaccination status and IBV lineage. (A) Numbers of minority iSNV per sample across all 99 samples are shown (y axis) by current-season vaccination status of the host (x axis). Samples from the mixed infection are shown in orange. (B) Numbers of minority iSNV per sample are shown (y axis) by IBV lineage (x axis). Samples from the mixed infection are shown in orange. (C) Pairwise nucleotide diversity (π, y axis) by influenza virus type (x axis), stratified by iSNV frequency cutoff (top). Medians are shown as red lines. Data for influenza A virus are from 243 samples described by McCrone et al. (18). Data on influenza B virus are from 97 high-quality samples in the present study. Samples from mixed infections in both studies are excluded. (D) Numbers of minority iSNV in 43 of the 99 high-quality samples (y axis), consisting of B/Yamagata from the 2014–2015 season, B/Victoria from the 2015–2016 season, and B/Yamagata from the 2016–2017 season based on alignments to the original references from the 2012–2013 season versus season-matched reference genomes (x axis).
We compared the within-host genetic diversity of IBV to our previously published data on IAV from the HIVE cohort (19). Here, IBV exhibits lower within-host diversity than IAV (Table 4) (P < 0.001, MWU test). IBV samples had a lower median number of minority iSNV (median, 0; IQR, 0 to 1) than IAV samples (median, 2; IQR, 1 to 3); 71% of IBV samples contained no minority iSNV, compared to 20% of IAV samples. The difference in within-host diversity was robust to relaxation of the iSNV frequency cutoff to 1% and 0.5%. IBV also exhibited a lower within-host diversity, as measured by nucleotide diversity (π), which takes both the number and the frequency of iSNV into account (Fig. 4C and Table 4). To ensure that our results were not an artifact of overly stringent quality thresholds, we also identified minority iSNV with less conservative read mapping quality (MapQ) and base quality (Phred) scores. We identified the same set of minority iSNV with a MapQ cutoff of 20 as with the original cutoff of 30. Similarly, reduction of the Phred base-quality cutoff to >25 in addition to a MapQ score cutoff of >20 resulted in only 20 more minority iSNV, 8 of which were found in the individual with a mixed infection. The other additional 12 minority iSNV were dispersed across specimens and did not significantly change the overall distribution of within-host diversity. We also examined whether our results were biased by use of a single B/Yamagata and B/Victoria reference for alignment and variant calling, which were both drawn from the 2012-2013 season (see Materials and Methods). We realigned sequence data from 43 of the original 99 samples to season-specific reference genomes isolated in southeastern Michigan. We found that the overall alignment rate for any given specimen was similar between the original reference and the new season-matched reference. Variant identification based on the new references and the original quality thresholds resulted in the same distribution of within-host diversity, although the identities of some iSNV were different (Fig. 4D).
TABLE 4.
Within-host diversity of IAV versus IBV
| Frequency cutoff (%) | Minority iSNV richness |
Nucleotide diversity (π) |
||||
|---|---|---|---|---|---|---|
| IAVa | IBVa | P value (MWU test) | IAVa | IBVa | P value (MWU test) | |
| 2 | 1 (1–3, 0.23) | 0 (0–1, 0.73) | <0.001 | 1.47e–05 (3.56e–06 to 3.28e–05, 0.23) | 0 (0 to 3.07e–06, 0.73) | <0.001 |
| 1 | 2 (1–3, 0.20) | 0 (0–1, 0.56) | <0.001 | 1.52e–05 (3.77e–06 to 3.37e–05, 0.20) | 0 (0 to 4.16e–06, 0.56) | <0.001 |
| 0.5 | 2 (1–4, 0.16) | 1 (0–2, 0.32) | <0.001 | 1.61e–05 (3.96e–06 to 3.46e–05, 0.16) | 1.73e–06 (0 to 6.4e–06, 0.32) | <0.001 |
Median (IQR, proportion at zero).
Together, these results indicate that our measurements of within-host diversity are robust to several technical aspects of variant identification, which are unlikely to account for the lower observed diversity of IBV. Because these data are from the same cohort and were generated using the same sequencing approach and analytic pipeline as our previous IAV data sets, the observed differences likely reflect true biological differences between IAV and IBV.
Identification of household transmission pairs.
We compared viral diversity across samples from individuals in the same household to investigate the genetic bottleneck that influenza B viruses experience during natural transmission. Over the seven influenza seasons, 39 households in the HIVE cohort had two or more individuals positive for the same IBV lineage within a 7-day interval (Table 1). This epidemiologic linkage is suggestive of transmission events but does not rule out coincident community-acquired infection (19). We identified 16 putative transmission pairs for which we sequenced at least one sample from each individual. In one of these pairs, the putative recipient was the individual with a mixed infection. The donor did not have evidence of a mixed infection based on number of iSNV, which would imply that the recipient may have been infected twice or that the second virus was lost from the donor by the time of sampling. This pair was excluded from the between-host analysis, leaving 15 putative transmission pairs for which we have high-quality sequencing data on both donor and recipient influenza populations.
We used our sequencing data to determine which of these epidemiologically linked household pairs were actual IBV transmission pairs. We generated maximum likelihood phylogenetic trees for samples from the two IBV lineages using the concatenated coding consensus sequences. Phylogenetic analysis provided genetic evidence that the 15 epidemiologically linked pairs were indeed true transmission pairs, as epidemiologically linked pairs were found nearest each other in each tree (Fig. 5A and B; vertical bars with household identifiers [ID]). We also validated these transmission pairs by analyzing the genetic distance across viral populations. True transmission pairs should have genetically similar populations exhibiting low genetic distance, while individuals with coincident community acquisition are more likely to have populations with a higher genetic distance. We compared the genetic distance between epidemiologically linked household pairs and random community pairs from the same season and infected with the same IBV lineage, using L1-norm as the measurement of genetic distance (Fig. 5C). The distribution of random community pairs functions as a null model of genetic distances among locally circulating strains. All of the 15 putative transmission pairs fell on the tail of this distribution, below the 5th percentile of the community pair L1-norm distribution, indicating that they are true transmission pairs (Fig. 5C). While the L1-norm is a function of both the consensus sequence and the iSNV, this signal was driven predominantly by consensus differences, as reflected in the phylogenetic analysis.
FIG 5.

Identification of household transmission pairs. Maximum likelihood phylogenetic tree of all B/Victoria (A) and B/Yamagata (B) samples from this study. Concatenated consensus coding sequences were aligned with MUSCLE, and phylogenetic trees were constructed with RAxML. Tip labels are denoted as enrollee ID, household ID, season, and lineage, separated by underscores; tip labels are colored by season. (C) Histogram of genetic distance, as measured by L1-norm, between household pairs and random community pairs from the same season and lineage. The bar heights for each group are normalized to the maximum for each group for comparison. Community pairs are shown in orange, and household pairs are shown in blue. The dotted red line indicates the 5th percentile of the community pair distribution.
Comparison of viral diversity across transmission pairs.
Transmission bottlenecks restrict the genetic diversity that is passed between hosts. With a loose transmission bottleneck, many unique genomes will be passed from donor to recipient. Because this will allow two variants at a given site to be transmitted, sites that are polymorphic in the donor are more likely to be polymorphic in the recipient. However, in the case of a tight or stringent bottleneck, sites that are polymorphic in the donor will likely be either fixed or absent in the recipient. We have previously demonstrated that influenza A virus experiences a tight transmission bottleneck of 1 to 2 unique genomes (19). Across our 15 IBV transmission pairs, we found no sites that were polymorphic in the donor and the recipient (Fig. 6). iSNV present in the donor were either fixed (100%) or absent (0%) in the recipient. These data suggest a stringent transmission bottleneck for influenza B virus, similar to that of influenza A virus. As there were fewer samples, transmission pairs, and iSNV in our IBV data set, we were unable to obtain a robust and precise estimate of bottleneck size.
FIG 6.
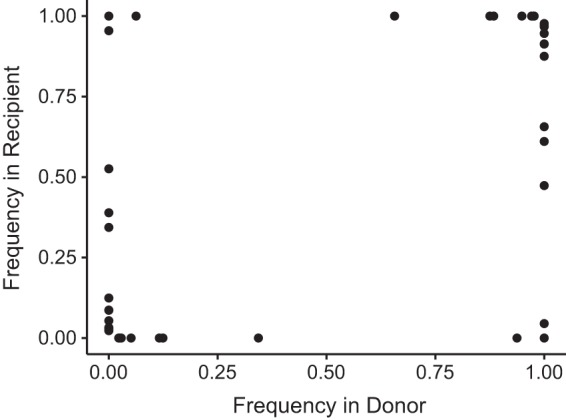
Shared diversity across household transmission pairs with influenza B virus. iSNV for 15 validated transmission pairs using samples closest to the time of transmission (inferred based on day of symptom onset) are shown. Each iSNV is plotted as a point, with its frequency in the recipient (y axis) versus its frequency in the donor (x axis).
DISCUSSION
Here, we define the within-host genetic diversity of IBV in natural infections by sequencing 106 samples collected over 8,176 person-seasons of observation in a household cohort. Because the HIVE study prospectively identifies individuals with acute respiratory illness regardless of severity, these samples capture IBV dynamics in a natural setting, reflective of infections occurring in the community. We show that within-host diversity of IBV is remarkably low, with most samples displaying no intrahost variants above our level of detection. We also find that IBV experiences a tight transmission bottleneck, limiting the diversity that is passed between hosts. IBV exhibits significantly lower within-host diversity than IAV. These findings reflect the lower relative evolutionary rate of IBV compared to IAV.
Our findings are largely consistent with what has been observed in IAV infections in humans (17–20). We found that only a minority of samples contain iSNV, the majority of which encode synonymous changes, consistent with a predominance of purifying selection within hosts. If immune-driven selective pressures were sufficiently strong to drive positive selection of antigenic variants at the individual level, we would expect to see enrichment of variants in antigenic regions. However, variants were no more common in the antigenic proteins hemagglutinin and neuraminidase, and we found no intrahost variants in known antigenic regions of hemagglutinin. We also found that the extent of within-host diversity did not vary with current-season vaccination status, further suggesting that immune selection is not particularly strong within hosts (19, 20, 22). Our data suggest that selective sweeps occur infrequently at the individual level, with selection evident only over a broader scale of time and space (19, 26). We recognize, however, that it is possible for individual level selective pressure to vary in magnitude by age, locale, influenza infection history, or immune status (27).
We do find that there are important differences in the within-host evolution of IAV and IBV. IBV displays significantly lower within-host diversity than IAV. Since measurements of within-host diversity can vary based on host population, sequencing approach, and variant calling algorithm (28), a strength of our study is that our comparison is based on samples from the same cohort with the same sequencing approach and analytic pipeline. In both of our studies, we have sequenced swab samples directly without prior culture, accounted for the confounding effect of viral load, and used a standardized, empirically validated analytic pipeline for variant identification (24). The only difference in methodology between these two studies is the multiplex amplification primers. In both viruses, these primers target the highly conserved ends of influenza virus genome segments, making it unlikely that this factor would drastically alter the amplification efficiency of within-host variants. Our analytic pipeline includes rigorous quality criteria to reduce false positives that can be introduced by amplification and Illumina sequencing. Importantly, these empirical quality criteria did not mask diversity actually present in these samples, strengthening the conclusion that IBV exhibits lower within-host diversity than IAV.
The most likely biological explanation for IBV’s lower within-host diversity is its de novo mutation rate, which is thought to be at least 2-fold lower than that of IAV (15). Viral mutation rates are critical to the diversification of rapidly evolving viruses within hosts. Under a neutral model, the number and frequency of minority variants are dependent on the mutation rate and demographics of the population (16). In such a model, the expected number of variants is highly sensitive to variation in the mutation rate across the range commonly estimated in RNA viruses. In light of our results, a more thorough comparison of mutation rates across influenza viruses is needed.
Another possible factor underlying IBV’s reduced diversity is the mutational robustness of the IBV genome relative to that of IAV. If IBV were less robust to mutation, stronger negative selection on multiple genes in IBV could result in more limited within-host diversity, perhaps located to certain regions of the genome. However, we found that the distributions of iSNV across IAV and IBV genomes are relatively similar. Furthermore, we have previously shown that the distribution of mutational fitness effects in influenza A/WSN/33/H1N1 matches that of other RNA and single-stranded DNA (ssDNA) viruses (29). Given that viruses across families with vastly different genomic architecture have similar mutational robustness, this is unlikely to account for the differences in within-host diversity between IAV and IBV.
We find that IBV experiences a stringent genetic bottleneck between hosts. A stringent transmission bottleneck places a constraint on the rate of adaptation of viral populations within and between individual hosts. Population bottlenecks reduce the effective population size, which increases random genetic drift and decreases the efficiency of selection (30). This results in a reduced ability of selection to fix beneficial mutations and to remove deleterious ones, which can decrease population fitness. However, there are potential evolutionary advantages to stringent bottlenecks, including removal of defective interfering particles (31, 32). While we were not able to estimate the size of the transmission bottleneck for IBV as precisely as for IAV, it is likely that the bottleneck sizes are comparable across the two viruses given the similarities in their transmission routes and ecology in the human population. Data from many more transmission pairs are necessary for a more robust estimate.
Together, our results are consistent with the lower rate of global evolution observed in IBV lineages than that in both seasonal A/H1N1 and A/H3N2 (10, 12, 14, 33). We suggest that a lower intrinsic mutation rate leads to reduced within-host diversity. With a comparably tight bottleneck, fewer de novo variants will rise to a level where they can be transmitted and spread through host populations. Combined with a lower incidence of IBV than of IAV, this would result in fewer variants that eventually spread and influence global dynamics. However, further investigation in larger populations are required to evaluate the within-host dynamics of both types of seasonal influenza viruses and how they contribute to larger-scale evolutionary patterns.
MATERIALS AND METHODS
Description of the HIVE cohort.
The HIVE study is a prospective, community-based household cohort in southeastern Michigan based at the University of Michigan School of Public Health (34–39). The cohort was initiated in 2010, with enrollment of households with children occurring on an annual basis and an active surveillance period lasting from October through May. In 2014, active surveillance was expanded to take place year-round. Participating adults provided informed consent for themselves and their children, and children ages 7 to 17 years provided oral assent. Individuals in each household were followed prospectively for acute respiratory illness, defined as two or more of the following: cough, fever or feverishness, nasal congestion, chills, headache, body aches, or sore throat. Study participants meeting the criteria for acute respiratory illness attended a study research clinic at the University of Michigan School of Public Health, where a combined throat and nasal swab, or a nasal swab only for children less than 3 years old, was collected by the study team. Beginning in the 2014-2015 season, study participants with acute respiratory illnesses took an additional nasal swab at home at the time of illness onset, collected either by themselves or by a parent. The study was approved by the Institutional Review Board of the University of Michigan Medical School.
Viral detection, lineage typing, and viral load quantification.
We processed upper respiratory specimens (combined nasal and throat swab or nasal swab) for confirmation of influenza virus infection by reverse transcription-PCR (RT-PCR). We extracted viral RNA with either QIAamp viral RNA mini kits (Qiagen) or PureLink Pro 96 viral RNA/DNA purification kits (Invitrogen) and tested samples using the SuperScript III Platinum one-step quantitative RT-PCR system with ROX (Invitrogen) and primers and probes for universal detection of influenza A and B viruses (CDC protocol, 28 April 2009). Specimens positive for influenza virus were tested using subtype/lineage primer and probe sets, which are designed to detect influenza A (H3N2), A (H1N1)pdm09, B (Yamagata), and B (Victoria) viruses. An RNAseP primer/probe set was run for each specimen to confirm specimen quality and successful RNA extraction.
We quantified the viral load in each sample by RT-qPCR using primers specific for the open reading frame of segment 8 (NS1/NEP): forward primer 5′-TCCTCAACTCACTCTTCGAGCG-3′, reverse primer 5′-CGGTGCTCTTGACCAAATTGG-3′, and probe 5′-(FAM)-CCAATTCGAGCAGCTGAAACTGCGGTG-(BHQ1)-3′. Each reaction mixture contained 5.4 μl of nuclease-free water, 0.5 μl of each primer at 50 μM, 0.1 μl of ROX dye, 0.5 μl SuperScript III RT/Platinum Taq enzyme mix, 0.5 μl of 10 μM probe, 12.5 μl of 2× PCR buffer master mix, and 5 μl of extracted viral RNA. To relate genome copy number to threshold cycle (CT) value, we used a standard curve based on serial dilutions of a plasmid control, run in duplicate on the same plate.
Amplification, library preparation, and sequencing.
We amplified viral cDNA from all eight genomic segments using the SuperScript III one-step RT-PCR Platinum Taq high-fidelity kit (Invitrogen). Each reaction mixture contained 5 μl of extracted viral RNA, 12.5 μl of 2× PCR buffer, 2 μl of primer cocktail, 0.5 μl of enzyme mix, and 5 μl of nuclease-free water. The primer cocktail was a mixture of B-PBs-UniF, B-PBs-UniR, B-PA-UniF, B-PA-UniR, B-HANA-UniF, B-HANA-UniR, B-NP-UniF, B-NP-UniR, B-M-Uni3F, B-Mg-Uni3F, B-M-Uni3R, B-NS-Uni3F, and B-NS-Uni3R (sequences and proportions are listed in reference 40). The thermocycler protocol was as follows: 45°C for 60 min, 55°C for 30 min, and 94°C for 2 min, followed by 5 cycles of 94°C for 20 s, 40°C for 30 s, and 68°C for 3 min 30 s, followed by 40 cycles of 94°C for 20 s, 58°C for 30 s, and 68°C for 3 min 30 s, and a final extension of 68°C for 10 min. We confirmed IBV genome amplification by gel electrophoresis. We sheared amplified cDNA (100 to 500 ng) on a Covaris ultrasonicator with the following settings: time, 80 s; duty cycle, 10%; intensity, 4; cycles per burst, 200. We prepared sequencing libraries with NEBNext Ultra DNA library prep kits (NEB) and sequenced them on an Illumina NextSeq with 2 × 150 paired end reads (mid-output run, v2 chemistry). To increase the specificity of variant identification, samples with a viral load between 103 and 105 genome copies/μl of transport medium were amplified and sequenced in duplicate. Samples amplified from B/Victoria and B/Yamagata plasmid clones were included on each sequencing run to account for sequencing errors. The plasmids used in the control reactions were generated by segment-specific RT-PCR from clinical samples of B/Victoria and B/Yamagata strains from the 2012-2013 season, followed by gel extraction and TOPO-TA cloning (Invitrogen). The sequence of each plasmid was determined by Sanger sequencing. We generated the plasmid control amplicons included on each Illumina sequencing run by using the same multiplex amplification protocol but with cloned plasmid DNA as the template.
Identification of iSNV.
Intrahost single-nucleotide variants (iSNV) were identified using a previously described analytic pipeline (24). We identified iSNV in samples that had an average genome coverage greater than 1,000× and a viral load greater than 103 genome copies/μl of transport medium in the original sample. Sequencing adapters were removed with Cutadapt (41), and reads were aligned to the sequences derived from the B/Victoria and B/Yamagata plasmid controls with Bowtie2 (42). Duplicate reads were marked and removed with Picard and samtools (43). Putative variants were identified with the R package deepSNV using data from the clonal plasmid controls of each sequencing run (44). Minority iSNV (<50% frequency) were identified using the following empirically derived criteria: deepSNV P value, <0.01; average mapping quality, >30; average Phred score, >35; and average read position, in the middle 50% (positions 37 and 113 for 150-bp reads). For samples processed in duplicate, we used only variants that were present in both replicates; the frequency of the variant in the replicate with greater coverage at that site was used. Lastly, variants with a frequency of <2%, which have a higher false-positive rate from RT-PCR and/or sequencing errors, were not included in downstream analyses.
In our previous work on IAV, we found that there were multiple sites with mutations that were essentially fixed (>0.95) relative to those of the plasmid control and in which the base in the plasmid control was therefore identified as a minority variant in the sample (19). At these sites, deepSNV is unable to estimate the base-specific error rate and cannot distinguish true minority iSNV; however, we found that we could accurately identify minority variants at these sites at a frequency of 2% or above (19). This frequency threshold was incorporated into the pipeline for iSNV identification at these sites. Therefore, we identify intrahost variants with frequencies between 2 and 98%. Minority iSNV are the subset of these variants with a frequency between 2 and 50% relative to the sample consensus, which we use as a metric of within-host diversity. For each minority iSNV, we identify the majority iSNV present at a frequency of 50 to 98%. Any sites that were monomorphic after application of quality filters were assigned a frequency of 100%. Nucleotide diversity (π) was calculated using identified iSNV in each sample by the formula described by Zhao and Illingworth (45).
Data and code availability.
Raw sequence data, with human content filtered out, are available at the NCBI Sequence Read Archive under BioProject accession number PRJNA561158. Code for the variant identification pipeline is available at http://github.com/lauringlab/variant_pipeline. Analysis code is available at http://github.com/lauringlab/Host_level_IBV_evolution.
ACKNOWLEDGMENTS
We acknowledge the individuals enrolled in the HIVE study for their participation. We thank Maria Virgilio for technical assistance.
This work was supported by grants R01 AI 118886 and NIH R21 AI 141832 from the NIH and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases award to A.S.L. The HIVE cohort was supported by grant U01 IP 001034 from the CDC to A.S.M. and E.T.M. and by grant U01 IP000474 from the CDC and grant R01 097150 from the NIH to A.S.M. A.L.V. was supported by grant T32 GM 007863 from the NIH.
A.S.M. reports personal fees from Sanofi and personal fees from Seqirus. E.T.M. reports consulting fees from Pfizer and research support from Merck. A.S.L. reports consulting fees from Sanofi and Roche. In all cases, these are outside the submitted work.
REFERENCES
- 1.Moya A, Holmes EC, González-Candelas F. 2004. The population genetics and evolutionary epidemiology of RNA viruses. Nat Rev Microbiol 2:279–288. doi: 10.1038/nrmicro863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. 2008. The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson MI, Holmes EC. 2007. The evolution of epidemic influenza. Nat Rev Genet 8:196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- 4.Yamayoshi S, Kawaoka Y. 2019. Current and future influenza vaccines. Nat Med 25:212–220. doi: 10.1038/s41591-018-0340-z. [DOI] [PubMed] [Google Scholar]
- 5.Petrova VN, Russell CA. 2018. The evolution of seasonal influenza viruses. Nat Rev Microbiol 16:47–60. doi: 10.1038/nrmicro.2017.118. [DOI] [PubMed] [Google Scholar]
- 6.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 7.Paul Glezen W, Schmier JK, Kuehn CM, Ryan KJ, Oxford J. 2013. The burden of influenza B: a structured literature review. Am J Public Health 103:e43–e51. doi: 10.2105/AJPH.2012.301137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175:59–68. doi: 10.1016/0042-6822(90)90186-u. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita M, Krystal M, Fitch WM, Palese P. 1988. Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology 163:112–122. doi: 10.1016/0042-6822(88)90238-3. [DOI] [PubMed] [Google Scholar]
- 10.Chen R, Holmes EC. 2008. The evolutionary dynamics of human influenza B virus. J Mol Evol 66:655. doi: 10.1007/s00239-008-9119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen J, Kirk BD, Ma J, Wang Q. 2009. Diversifying selective pressure on influenza B virus hemagglutinin. J Med Virol 81:114–124. doi: 10.1002/jmv.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langat P, Raghwani J, Dudas G, Bowden TA, Edwards S, Gall A, Bedford T, Rambaut A, Daniels RS, Russell CA, Pybus OG, McCauley J, Kellam P, Watson SJ. 2017. Genome-wide evolutionary dynamics of influenza B viruses on a global scale. PLoS Pathog 13:e1006749. doi: 10.1371/journal.ppat.1006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudas G, Bedford T, Lycett S, Rambaut A. 2015. Reassortment between influenza B lineages and the emergence of a coadapted PB1-PB2-HA gene complex. Mol Biol Evol 32:162–172. doi: 10.1093/molbev/msu287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijaykrishna D, Holmes EC, Joseph U, Fourment M, Su YC, Halpin R, Lee RT, Deng Y-M, Gunalan V, Lin X, Stockwell TB, Fedorova NB, Zhou B, Spirason N, Kühnert D, Bošková V, Stadler T, Costa A-M, Dwyer DE, Huang QS, Jennings LC, Rawlinson W, Sullivan SG, Hurt AC, Maurer-Stroh S, Wentworth DE, Smith GJ, Barr IG. 2015. The contrasting phylodynamics of human influenza B viruses. Elife 4:e05055. doi: 10.7554/eLife.05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nobusawa E, Sato K. 2006. Comparison of the mutation rates of human influenza A and B viruses. J Virol 80:3675–3678. doi: 10.1128/JVI.80.7.3675-3678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue KS, Moncla LH, Bedford T, Bloom JD. 2018. Within-host evolution of human influenza virus. Trends Microbiol 26:781–793. doi: 10.1016/j.tim.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinis JM, Florek NW, Fatola OO, Moncla LH, Mutschler JP, Charlier OK, Meece JK, Belongia EA, Friedrich TC. 2016. Deep sequencing reveals potential antigenic variants at low frequencies in influenza A virus-infected humans. J Virol 90:3355–3365. doi: 10.1128/JVI.03248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonard AS, McClain MT, Smith GJD, Wentworth DE, Halpin RA, Lin X, Ransier A, Stockwell TB, Das SR, Gilbert AS, Lambkin-Williams R, Ginsburg GS, Woods CW, Koelle K. 2016. Deep sequencing of influenza A virus from a human challenge study reveals a selective bottleneck and only limited intrahost genetic diversification. J Virol 90:11247–11258. doi: 10.1128/JVI.01657-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCrone JT, Woods RJ, Martin ET, Malosh RE, Monto AS, Lauring AS. 2018. Stochastic processes constrain the within and between host evolution of influenza virus. Elife 7:e35962. doi: 10.7554/eLife.35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debbink K, McCrone JT, Petrie JG, Truscon R, Johnson E, Mantlo EK, Monto AS, Lauring AS. 2017. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog 13:e1006194. doi: 10.1371/journal.ppat.1006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue KS, Bloom JD. 2019. Reconciling disparate estimates of viral genetic diversity during human influenza infections. Nat Genet 51:1298–1301. doi: 10.1038/s41588-019-0349-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han AX, Maurer-Stroh S, Russell CA. 2018. Individual immune selection pressure has limited impact on seasonal influenza virus evolution. Nat Ecol Evol 3:302–311. doi: 10.1038/s41559-018-0741-x. [DOI] [PubMed] [Google Scholar]
- 23.Xue KS, Bloom JD. 2019. Linking influenza virus evolution within and between human hosts. bioRxiv doi: 10.1101/812016. [DOI] [PMC free article] [PubMed]
- 24.McCrone JT, Lauring AS. 2016. Measurements of Intrahost viral diversity are extremely sensitive to systematic errors in variant calling. J Virol 90:6884–6895. doi: 10.1128/JVI.00667-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valesano AL, Fitzsimmons WS, McCrone JT, Petrie JG, Monto AS, Martin ET, Lauring AS. 2019. Influenza B viruses exhibit lower within-host diversity than influenza A viruses in human hosts. bioRxiv doi: 10.1101/791038. [DOI] [PMC free article] [PubMed]
- 26.Nelson MI, Simonsen L, Viboud C, Miller MA, Taylor J, George KS, Griesemer SB, Ghedin E, Ghedi E, Sengamalay NA, Spiro DJ, Volkov I, Grenfell BT, Lipman DJ, Taubenberger JK, Holmes EC. 2006. Stochastic processes are key determinants of short-term evolution in influenza A virus. PLoS Pathog 2:e125. doi: 10.1371/journal.ppat.0020125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JM, Eguia R, Zost SJ, Choudhary S, Wilson PC, Bedford T, Stevens-Ayers T, Boeckh M, Hurt AC, Lakdawala SS, Hensley SE, Bloom JD. 2019. Mapping person-to-person variation in viral mutations that escape polyclonal serum targeting influenza hemagglutinin. Elife 8:e49324. doi: 10.7554/eLife.49324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, Tan AL, Paul LM, Brackney DE, Grewal S, Gurfield N, Van Rompay KKA, Isern S, Michael SF, Coffey LL, Loman NJ, Andersen KG. 2019. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visher E, Whitefield SE, McCrone JT, Fitzsimmons W, Lauring AS. 2016. The mutational robustness of influenza A virus. PLoS Pathog 12:e1005856. doi: 10.1371/journal.ppat.1005856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCrone JT, Lauring AS. 2018. Genetic bottlenecks in intraspecies virus transmission. Curr Opin Virol 28:20–25. doi: 10.1016/j.coviro.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zwart MP, Elena SF. 2015. Matters of size: genetic bottlenecks in virus infection and their potential impact on evolution. Annu Rev Virol 2:161–179. doi: 10.1146/annurev-virology-100114-055135. [DOI] [PubMed] [Google Scholar]
- 32.Vignuzzi M, López CB. 2019. Defective viral genomes are key drivers of the virus-host interaction. Nat Microbiol 4:1075–1087. doi: 10.1038/s41564-019-0465-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bedford T, Riley S, Barr IG, Broor S, Chadha M, Cox NJ, Daniels RS, Gunasekaran CP, Hurt AC, Kelso A, Klimov A, Lewis NS, Li X, McCauley JW, Odagiri T, Potdar V, Rambaut A, Shu Y, Skepner E, Smith DJ, Suchard MA, Tashiro M, Wang D, Xu X, Lemey P, Russell CA. 2015. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 523:217–220. doi: 10.1038/nature14460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrie JG, Malosh RE, Cheng CK, Ohmit SE, Martin ET, Johnson E, Truscon R, Eichelberger MC, Gubareva LV, Fry AM, Monto AS. 2017. The household influenza vaccine effectiveness study: lack of antibody response and protection following receipt of 2014-2015 influenza vaccine. Clin Infect Dis 65:1644–1651. doi: 10.1093/cid/cix608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monto AS, Malosh RE, Petrie JG, Thompson MG, Ohmit SE. 2014. Frequency of acute respiratory illnesses and circulation of respiratory viruses in households with children over 3 surveillance seasons. J Infect Dis 210:1792–1799. doi: 10.1093/infdis/jiu327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohmit SE, Petrie JG, Malosh RE, Johnson E, Truscon R, Aaron B, Martens C, Cheng C, Fry AM, Monto AS. 2016. Substantial influenza vaccine effectiveness in households with children during the 2013-2014 influenza season, when 2009 pandemic influenza A(H1N1) virus predominated. J Infect Dis 213:1229–1236. doi: 10.1093/infdis/jiv563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohmit SE, Petrie JG, Malosh RE, Cowling BJ, Thompson MG, Shay DK, Monto AS. 2013. Influenza vaccine effectiveness in the community and the household. Clin Infect Dis 56:1363–1369. doi: 10.1093/cid/cit060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrie JG, Ohmit SE, Cowling BJ, Johnson E, Cross RT, Malosh RE, Thompson MG, Monto AS. 2013. Influenza transmission in a cohort of households with children: 2010-2011. PLoS One 8:e75339. doi: 10.1371/journal.pone.0075339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monto AS, Malosh RE, Evans R, Lauring AS, Gordon A, Thompson MG, Fry AM, Flannery B, Ohmit SE, Petrie JG, Martin ET, Aaron B, Callear AP, Truscon R, Johnson E, Cheng CK, Kaniclides A, Williams N, Martens C. 2019. Data resource profile: Household Influenza Vaccine Evaluation (HIVE) study. Int J Epidemiol 48:1040–1040g. doi: 10.1093/ije/dyz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, Barr IG, Wentworth DE. 2014. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol 52:1330–1337. doi: 10.1128/JCM.03265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 42.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, Beerenwinkel N. 2012. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 3:811. doi: 10.1038/ncomms1814. [DOI] [PubMed] [Google Scholar]
- 45.Zhao L, Illingworth C. 2019. Measurements of intrahost viral diversity require an unbiased diversity metric. Virus Evol 5:vey041. doi: 10.1093/ve/vey041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequence data, with human content filtered out, are available at the NCBI Sequence Read Archive under BioProject accession number PRJNA561158. Code for the variant identification pipeline is available at http://github.com/lauringlab/variant_pipeline. Analysis code is available at http://github.com/lauringlab/Host_level_IBV_evolution.