

Abstract
Protein kinases are essential mediators of cellular signal transduction and are often dysregulated in disease. Among these, protein tyrosine kinases (PTKs) have received specific interest due to their common roles in various diseases including cancer, and emerging observations indicating that PTK signalling pathways are susceptible to regulation by reactive oxygen species (ROS), which are also frequently implicated in disease pathology. While it is well recognized that ROS can impact on tyrosine kinase signalling by inhibiting tyrosine phosphatases, more recent studies highlight additional modes of redox-based regulation of tyrosine kinase signalling by direct redox modification of non-catalytic cysteines within tyrosine kinases or other protein components of this signalling pathway. In this review, we will present recent advancements with respect to redox-based mechanisms in regulating PTK signalling, with a specific focus on recent studies demonstrating direct redox regulation of Src-family kinases and epidermal growth factor receptor kinases. Importantly, redox-based modulation of tyrosine kinases may be relevant for many other kinases and has implications for current approaches to develop pharmacological inhibitors for these proteins.
Keywords: Redox, cysteine, Src; EGFR; NOX
Cellular signal transduction is regulated by reversible post-translational modifications, allowing cells to respond to external stimuli thereby translating them into intracellular biochemical responses. Reversible protein phosphorylation by protein phosphotransferase enzymes, otherwise known as kinases, represents a major mechanism by which cellular processes such as cell survival, proliferation, differentiation and migration, are regulated. Protein kinases have also received much attention as drug targets in the treatment of various diseases. Protein kinases found in eukaryotes are divided into ∼428 serine/threonine kinases and ∼90 tyrosine kinases, based on their substrate target residues. All kinase enzymes catalyse the transfer of the γ-phosphate from ATP to the hydroxyl group of a protein side chain (Fig. 1) (1). The installation of a phosphate comprises a mechanistic switch that serves to modulate target protein activity and alters protein binding affinities by recognition and binding of specialized protein domains (e.g. SH2 domains binding phosphotyrosine) (2). Protein kinases can be activated by diverse external stimuli, which commonly involves phosphorylation modifications within the kinase enzyme, thus promoting signal transduction in a phosphorylation cascade. Protein phosphorylation is reversed by the action of phosphatase enzymes, which dephosphorylate proteins via phospho-amino acid hydrolysis (Fig. 1) (3). Since phosphatases can in turn also be regulated by phosphorylation, this overall signalling machinery can create a rich array of regulatory processes with both negative and positive feedback mechanisms.
Fig. 1.
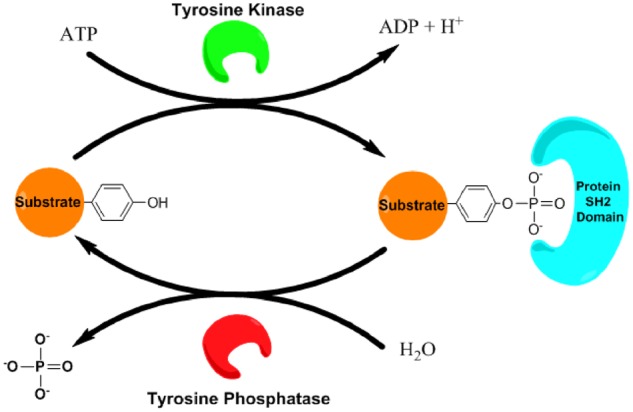
Basic components of tyrosine kinase signalling. Tyrosine kinase signalling involves the phosphorylation of a substrate protein via the addition of a phosphate to a substrate Tyr residue, catalysed by a kinase, leading to activation or inactivation of this target protein. Phosphorylated Tyr residues can act as a recognition site for SH2 domain-containing proteins that ‘read’ and transmit the signal. Protein tyrosine phosphorylation can be reversed through hydrolysis by a Tyr phosphatase.
Among the protein kinase families, protein tyrosine kinases (PTKs) appear to be present primarily in multicellular organisms, which evolved during episodes of increasing atmospheric oxygen concentrations. As such, PTKs are particularly relevant in processes related to cell differentiation and specialized cell functions, and have become recognized as important clinically responsive targets for tyrosine kinase inhibitors (TKI) in treatment of, e.g. various cancers (4). The evolution of multicellular organisms has largely relied on the use of oxygen in mitochondrial respiration, which results in the generation of reactive metabolites from molecular O2, termed reactive oxygen species (ROS). Multicellular organisms also contain multiple isoforms of NADPH oxidases (NOX), which produce ROS in a regulated fashion for specific purposes (5). Primary ROS include superoxide anion (O2−) or hydrogen peroxide (H2O2), but other ROS can be generated via interactions with transition metal ions (e.g. hydroxyl radical, OH•) or nitric oxide (NO•; to form, e.g. peroxynitrite, ONOO−) (5). Although elevated ROS production is often referred to as ‘oxidative stress’ and associated with systemic cellular distress (6), emerging evidence has led to the widely embraced concept of ‘redox-based signalling’, involving reversible oxidative post-translational modifications (PTMs) as an important biological mechanism of protein regulation (7). Indeed, accumulating evidence has indicated that PTK signalling is subject to regulation by oxidant-dependent mechanisms in an orthogonal fashion, largely through reversible modification of redox-active protein cysteine residues (8). As will be discussed in this review, such redox-based mechanisms can regulate protein tyrosine phosphorylation at various levels, and have important implications for ongoing approaches to therapeutically target this signalling pathway and for our overall understanding of oxidant-driven processes in health and disease.
Redox-Based Signalling: General Concepts
From a chemical perspective, protein redox regulation most commonly involves oxidation of cysteine residues on their thiol side chains (9). Cysteines (Cys) are statistically under-utilized in the proteome, yet are often highly conserved when present in primary gene sequences, illustrating their importance in protein structure and function. Reaction of ROS (typically H2O2) with Cys generally proceeds through reaction with its reactive thiol (P-SH), typically as a deprotonated thiolate (P-S−), and initially forms a sulphenic acid (P-SOH), which often possesses limited stability and undergoes further reactions with other thiols or ROS (9). Indeed, P-SOH is known to rapidly react with local reduced thiols to form intra- or intermolecular disulphide bonds or alternative redox-based PTMs with glutathione or free Cys, resulting in S-glutathionylation or S-cysteinylation, respectively (9). In some cases, P-SOH reacts with the protein main chain amide to form an intramolecular sulphenyl amide, which can induce structural alterations and thereby modulate protein function (9). Each of these modifications can be reversed by endogenous cellular reducing systems, such as thioredoxins and glutaredoxins (9). Additionally, overoxidation can occur when P-SOH is further oxidized to corresponding sulphinic or sulphonic acids (P-SO2H and P-SO3H, respectively), which are generally irreversible (9).
Protein cysteine thiols can also be modified by NO•-derived products resulting in S-nitrosylation (P-SNO) (9). Additionally, cysteine thiols can be converted to persulphides or polysulphides (P-SnSH), through the action of hydrogen sulphide or related reactive sulphur species, which forms an area of rapidly growing interest (10). Since P-SnSH are typically more nucleophilic than the corresponding P-SH, they are also subject to additional oxidative modifications (11). Protein Cys residues are also subject to alternative biological modifications, such as alkylation by electrophilic metabolites formed during lipid oxidation (e.g. acrolein, 4-hydroxynonenal, nitro-lipids) (12, 13), or acylation (e.g. palmitoylation) regulated by enzymatic actions of acyl transferase enzymes and acyl-protein thioesterases (14). Although these latter modifications are technically not the result of ROS chemistry, they have the potential to interfere with redox-based thiol modifications and can thereby impact on redox signalling pathways. Moreover, each of these modifications can be reversed by cellular reducing systems, although cysteine alkylation is generally thought to be relatively irreversible.
With considerable emphasis on Cys residues as redox-sensitive protein targets, it should be noted that other amino acids are also redox active and therefore subject to redox-based modifications. Selenocysteine (Sec), a rare amino acid in a limited number of proteins, is highly susceptible to redox modification due to the similar electrochemical properties of selenium and sulphur (15). Another sulphur-containing amino acid, methionine (Met), is also susceptible to oxidation by ROS and can be oxidized to methionine sulphoxide and/or sulphone, and the existence of methionine sulphoxide reductase enzymes suggests the biological importance of Met oxidation in regulating protein function (16). Indeed, emerging evidence from bioinformatics studies suggests that methionine oxidation may also be important in controlling protein phosphorylation (17). Finally, oxidation of other amino acids (histidine, tyrosine, tryptophan) has been demonstrated in response to various ROS, although these are generally considered irreversible modifications and their relevance for biological protein regulation is unclear (18).
Although biological protein oxidation is clearly highly diverse and multifactorial, the functional relevance of such oxidative modifications can be categorized in two general classes: (i) enzymatic loss-of-function due to modifications of catalytic amino acids (e.g. cysteines or selenocysteines) and (ii) structural changes upon modification of non-catalytic amino acids that affect enzyme activity indirectly or impact on subcellular protein targeting, protein–protein interactions, or protein stability. Especially in these latter cases, our understanding of how diverse cysteine modifications control protein structure and/or function is still limited, and much remains to be learned about the molecular mechanisms by which these specific oxidative modifications affect protein function. The next sections will highlight growing evidence indicating that protein tyrosine phosphorylation is strongly impacted by redox-based cysteine modifications at various levels, involving both catalytic and non-catalytic cysteines in diverse protein components that comprise this signalling pathway.
Redox-Dependent Inactivation of Protein Tyrosine Phosphatases
The first reported examples of redox-based regulation of protein tyrosine phosphorylation involved oxidant-mediated inactivation of a protein tyrosine phosphatase (PTP), thereby indirectly prolonging PTK activation (19). All PTPs contain an active site Cys residue located in a (I/V)HCXAGXGR(S/T) active site motif that is critical for catalytic function, as this cysteine is responsible for nucleophilic attack on the phosphotyrosine substrate to generate an intermediate phospho-cysteine, after which the phosphate group is eliminated by hydrolysis via a conserved Asp residue with in their WDP loop to reconstitute the native enzyme (20). Since this catalytic cysteine typically has a low pKa to promote its function, it is also particularly susceptible to oxidation (21). PTP oxidation can occur by elevated ROS concentrations during NOX activation or mitochondrial dysfunction, and allow for prolonged PTK activity (5, 21). The best studied PTP with respect to redox signalling is protein tyrosine phosphatase 1B (PTP1B), and its inactivation by, e.g. H2O2 proceeds through oxidation of its active site Cys residue to a sulphenic acid (P-SOH) (22). Structural elucidation by X-ray crystallography of oxidant-inactivated PTP1B revealed a sulphenyl amide modification within the catalytic site (23), but in the cellular context the main inactive form of PTP1B appears to exist as an S-glutathionylated protein, due to reaction of P-SOH of the sulphenyl amide with GSH (24). PTP1B can also be reversibly inactivated by nitric oxide-derived oxidants via S-nitrosylation (25). Other PTPs can be similarly inactivated, such as SHP-1/2 (Src homology region 2 domain-containing phosphatase-1/2) (26), PTEN (phosphatase and tensin homolog) and KIM (kinase interaction motif) family PTPs (27, 28), and in the latter cases the active site cysteine becomes engaged in intra- or intermolecular disulphide bonds with other protein cysteines. The diverse nature of these various modifications is likely not relevant for regulating enzymatic function, as each of these would inactivate the catalytic cysteine. Instead, the various modes of cysteine modification in PTPs are likely more critical for regulating the duration of inactivation, as distinct reduction mechanisms are involved in their reversal (29). In some instances, redox-dependent modification can also enhance phosphatase function, as has been reported for some dual-specificity phosphatases in which oxidation of Cys residues in Zn-binding domains enhances phosphatase activity through disulphide bonding and associated structural changes (30, 31).
Direct Redox Regulation of Tyrosine Kinases
The catalytic activity of PTKs is exquisitely controlled through protein structural changes (32). All tyrosine kinases contain a catalytic domain that consists of two structured lobes comprising a catalytic cleft where ATP binds and is hydrolysed to ADP along with installation of phosphate on the protein target (33). Catalytic activity is critically dependent on structural changes of the N-lobe αC-helix (red in Fig. 2) and C-lobe activation loop (A-loop, orange in Fig. 2). In the inactive state, the A-loop is folded and prevents the binding of protein substrates, stabilizing the αC-helix in the ‘outward’ position (33). Activation of tyrosine kinases occurs by diverse actions, but generally involves ‘inward’ rotation of the αC-helix, enabling the engagement of a conserved glutamic acid within the ATP binding site, as well as unfolding of the A-loop allowing for substrate access to the kinase active site (34). Diverse cellular and biochemical mechanisms universally influence these PTK structural elements to directly modulate the activity of PTKs, and these often involve protein (auto)phosphorylation (e.g. within the A-loop) to promote or sustain kinase activation.
Fig. 2.
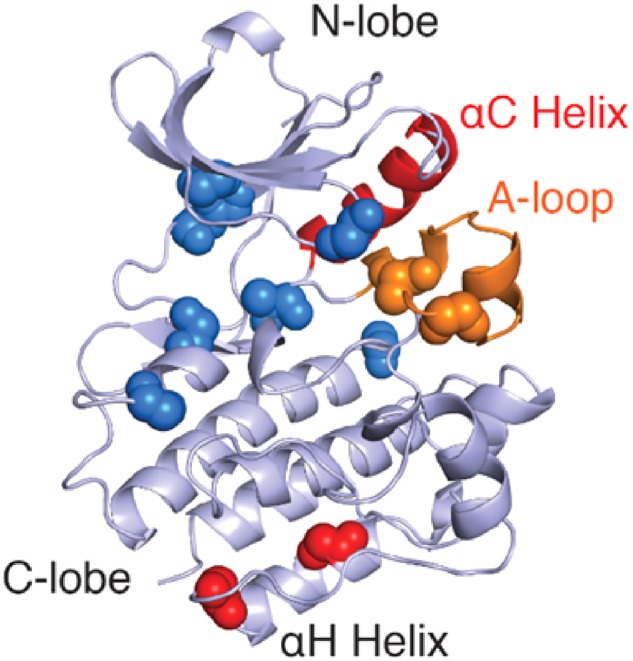
Conservation of Cys residues in SFKs. Architecture of the c-Src kinase domain with regulatory αC-helix (red cartoon) and A-loop (orange cartoon) (PDB: 2SRC). Main chain spheres represent the composite locations of conserved cysteine residues across the kinome within the kinase domain (blue), the activation loop (orange) and within a conserved cysteine cluster distant from the ATP binding site (red).
While various post-translational modifications (including phosphorylation events) can regulate PTK function, recent evidence indicates that redox-dependent cysteine modifications can also represent an important regulatory mechanism for PTK function. Over 80% of all PTKs contain highly conserved cysteine residues within a CXXXXXXXMXXCW motif found in their C-lobe αH-helix (Fig. 2, red spheres), which is in a structured region and is speculated to be important for activity and/or protein stability (35–37). In addition, tyrosine kinases, as well as other protein kinase families, frequently contain cysteines within their ATP binding regions (Fig. 2). The function of these cysteines is typically unknown and they are often considered appealing targets for irreversible covalent kinase inhibitors. Indeed, bioinformatics analysis has suggested that >150 protein kinases (among which several PTKs) contain suitable cysteines for such covalent targeting (38). However, a number of studies over the past decade have indicated that several of these conserved cysteine residues in PTKs are susceptible to redox-based modification, with variable impact on kinase function (8, 39). Table I summarizes some of the major studies to date that have addressed the role(s) of specific cysteine modifications in regulating PTKs, but the molecular basis is still only superficially understood. Intriguingly, in some cases such redox-dependent activation of kinase function involves cysteines that are also targets for irreversible inhibitors of these kinases (e.g. C797 in EGFR and C277 in Src) (44, 59, 60). In the following sections, we will discuss recent advances with respect to the molecular mechanism by which reversible cysteine oxidation regulates PTK function in some detail, focussing primarily on Src family kinases (SFKs) and the epidermal growth factor receptor as prototypical examples of non-receptor tyrosine kinases (NRTKs) and receptor tyrosine kinases (RTKs), respectively.
Table I.
Direct regulation of PTKs by oxidant signalling
| Kinase | Involved residue(s) | Modification | Functional outcome | Reference |
|---|---|---|---|---|
| Non-receptor PTKs | ||||
| Src | ND | ND | Activation | (40) |
| ND | Sulphenylation, glutathionylation | Activation | (41, 42) | |
| C185 | Disulphide | Cortactin binding | (43) | |
| C185, C277 | Sulphenic acid | Activation | (44) | |
| C245, C487 | Proposed disulphide | Activation | (45) | |
| C245, C277, C487, C498 | ND | Activation | (46) | |
| C277 | Disulphide | Inactivation | (47) | |
| C487 | ND | Activation | (48) | |
| C498 | S-Nitrosylation | Activation | (49) | |
| C483, C487, C496, C498 | ND | Activation | (50) | |
| Yes | C491, C506 | ND | Activation | (51) |
| Fyn | C488 | ND | Activation | (52) |
| Lyn | C466 | ND | Activation | (53) |
| C479 | ND | Activation | (51) | |
| Lck | C464, C475 | ND | Activation | (36) |
| Hck | ND | ND | Activation | (54) |
| JAK2 | C866, C917 | Disulphide | Inactivation | (55) |
| C1094, C1105 | ND | Inactivation | (55) | |
| CSK | C122, C164 | Disulphide | Activation | (56) |
| SYK | C206 | Inhibitor adduction | Inactivation | (57) |
| ZAP-70 | C39 | Inhibitor adduction | Inactivation | (57) |
| Receptor PTKs | ||||
| FGFR | C488 | Disulphide | Inactivation | (47) |
| EGFR | C797 | Sulphenylation | Activation | (59) |
| ND | S-Glutathionylation | Nuclear trafficking | (41, 42) | |
| C166, C305 | S-Nitrosylation | Inhibition | (58) | |
| C797 | Acrolein adduction | Inhibition | (61) | |
| C1025, C1122 | Palmitoylation | Receptor trafficking | (62) | |
| IGF-1R | ND | Nitrosylation | Inactivation | (63) |
| RET | C376 | Potential disulphide | UV activation | (37) |
| VEGFR | C1199, C1206 | Disulphide | Inhibition | (64) |
‘ND’ indicates that the published study did not specifically detect this property. Underlined residues indicate Cys found in the CXXXXXXXMXXCW motif.
Non-receptor tyrosine kinases
Src family kinases
The Src family kinases (SFK) represent one of the most widely studied families of NRTKs (65). Structurally, SFKs consist of an N-terminal Src homology (SH) 4 domain that contains lipid modification sites, a poorly conserved ‘unique’ domain, an SH3 domain that binds to specific proline-rich sequences, an SH2 domain that can bind to specific tyrosine phosphorylated proteins, and a C-terminal tyrosine kinase domain followed by an unstructured negative regulatory C-terminal peptide tail. In their inactive state, SFKs are held in a ‘clamped’ autoinhibited state via intramolecular binding of a C-terminal phosphotyrosine (pY527; based on chicken c-Src numbering) to their SH2 domains, as well as binding of their SH3 domains to a polyproline sequence within the kinase linker domain (66). Activation of SFKs occurs in a mechanism initiated by dephosphorylation of pY527 (or its homolog) and results in detachment of the SH2/SH3 domains from the back of the kinase domain, thereby inducing ‘unclamping’ and allowing for recognition of these SH2/SH3 domains by target proteins (66). In a concerted fashion, SH2/SH3 domain detachment ‘unclamps’ the kinase domain in an allosteric mechanism leading to unfolding of the activation loop, exposing a conserved A-loop tyrosine (Y416 in Src) for phosphorylation, and inward rotation of the αC-helix. PTP-dependent dephosphorylation of Y416 and phosphorylation of Y527 by, e.g. C-terminal Src kinase (CSK) can reverse this activation and restore the inactive state (67).
In addition to these classical SFK regulating mechanisms, recent discoveries highlight the impact of redox regulation on the inactive/active state equilibrium of these kinases through direct Cys oxidation, although these mechanisms vary between the different family members based on the evolutionary conservation of Cys residues (50) (Fig. 3A). The Src protein contains 9 Cys residues that are distributed across its SH2 and kinase domains (Fig. 3B). None of these cysteines are involved in structural disulphide bonding, and several are contained within the conserved CXXXXXXXMXXCW motif. The first indication that Src is redox regulated came from studies showing that various Cys-SH targeting alkylating agents can abrogate Src activity (68). Later, it was observed that exposure of fibroblasts or immunoprecipitated c-Src to NO-generating molecules could in fact promote Src activation, independent of its Y527 phosphorylation status, and this was found to be associated with redox-dependent formation of putative Src multimers through disulphide formation, although for such disulphide formation was not directly demonstrated (69). Follow up studies by the same group suggested a role for the four clustered C-terminal Cys residues (including the CXXXXXXXMXXCW motif C487 and 498) in Src activation induced by HgCl2 or P-SNO (49–51). These and subsequent studies suggested that increased cellular NO• production in, e.g. breast cancers could promote cell invasion and oncogenesis by S-nitrosylation of Src and/or associated disulphide formation (49, 70).
Fig. 3.

(A) The nine Cys residues found in the c-Src kinase and their conservation within the SFKs. The Cys residues contained within the CXXXXXXXMXXCW domain conserved within Tyr kinases are indicated. (B) Crystal structure of c-Src (PDB: 2SRC) indicating the location of the nine Cys residues, as well as the regulatory Y416 and Y527 residues. CXXXXXXXMXXCW domain Cys residues are highlighted in red. Modified from (44).
Giannoni et al. highlighted an alternative mechanism of Src redox regulation by cellular ROS in the context of anchorage-dependent growth. They found that Src activation was associated with cysteine oxidation within the Src protein following adhesion-induced activation of 5-lipoxygenase via integrin signalling, and mutation studies implicated C245 within the SH2 domain and C487 within the CXXXXXXXMXXCW motif in such oxidative regulation (45). Subsequent studies by the same authors also implicated these Cys residues in redox-dependent Src activation in increased anoikis resistance via EGFR-dependent mechanisms (71, 72). The authors postulated formation of a putative intramolecular disulphide bond between these residues in such oxidative Src activation (45), although this would seem unlikely from a structural perspective since these Cys are quite distant from each other in the inactive Src structure (e.g.Fig. 2B). However, since Src activation was recently shown to involve the formation of asymmetric dimers (73), is it plausible that intermolecular disulphide bonds between two Src monomers may be involved. Other studies have highlighted a role for C487 in redox-dependent Src activation in angiotensin II-mediated cellular hypertrophy or in IGF-1 signalling during hyperglycaemia, which was linked to cellular H2O2 production by the NADPH oxidase isoform NOX4 (40, 48). NOX activation was also associated with Src activation in the context of TGF-β signalling, and mutagenesis studies suggested the involvement of C245, C277, C487 and C498 (46). Although none of these studies actually demonstrated the ability of H2O2 to directly enhance Src activity, this was demonstrated more recently in studies with immunoprecipitated Src (74).
Most of the Cys residues highlighted above are conserved among all SFKs, and other SFKs have also been shown to be regulated by ROS. For example, Fyn and Yes represent ubiquitously expressed SFK members that also contain a CXXXXXXXMXXCW motif (Fig. 3), and activation of Fyn in keratinocytes in response to solar UV-light exposure was found to involve oxidation of C488 (homologous to Src C487), which was thought to represent a potential redox sensing mechanism to induce protective cellular apoptosis and prevent melanoma (52). In a model of zebrafish tail fin regeneration, haematopoietic cell-specific Lyn was found to be a critical mediator of neutrophil recruitment to areas of high ROS production at injury sites, which was dependent on oxidation of the kinase CXXXXXXXMXXCW motif residue C466 (homologous to Src C487) (53). Similar cysteine residues were also implicated in HgCl2-induced activation of Yes (C491/C506; homologous to Src C483/C498) and Lyn (C479; homologous to Src C498) (51) and in redox-dependent activation of the lymphocyte-specific SFK Lck (at residues C464 and C475, homologous to Src C487 and C498, respectively) (36). Finally, Hck was also found to be activated by the oxidant peroxynitrite (ONOO−), although no specific Cys residues were implicated (54).
In aggregate, these various studies strongly implicate the importance of CXXXXXXXMXXCW motif cysteines within αH-helix of SFK kinase domains, perhaps in combination with other Cys residues, in redox-mediated activation of several SFKs. However, the mechanisms by which modification of these cysteines control kinase function are still not understood, largely because the relevant cysteine modifications have not been identified. No structural studies exist, to our knowledge, to directly link oxidation of CXXXXXXXMXXCW motif cysteines to SFK activation, and this region is typically thought to be rather inconsequential for kinase activation based on available structural studies (66). Indeed, recent molecular dynamics (MD) simulations of Src C498 oxidation (to its primary oxidation product P-SOH) did not reveal significant structural alterations that would explain increases in kinase function (44). We therefore speculate that redox-dependent modification of these cysteines may involve altered interactions with other associated proteins (such as other SFK monomers), potentially via disulphide-mediated interactions, but this has to date not been addressed directly.
Unique cysteine modifications in redox-dependent activation of Src
Our research group recently demonstrated the importance of the NOX isozyme DUOX1 in airway epithelial wound responses via ATP-dependent stimulation of purinergic P2YR2 receptors and redox-dependent activation of Src (75). Src activation by P2YR2 stimulation was found to involve recruitment of Src to the P2YR2 via its PXXP domain, and also appears to involve recruitment of DUOX1. We attempted to associate Src activation with specific redox-based modifications within the Src protein (P-SOH and P-SSG), which suggested that Src activation corresponded most closely with formation of P-SOH (41). Studies with recombinant Src protein indicated that H2O2 was capable of enhancing Src kinase activity, whereas formation of P-SSG by addition of GSSG was ineffective (41). Mass spectrometry analysis using P-SOH-specific probes, to identify the main redox-sensitive cysteines within recombinant Src protein, highlighted prominent sulphenylation of two cysteines that are rather unique within Src compared to other SFK members, namely C185 within the SH2 domain and C277 within the ATP binding region of the kinase domain (Figs 2 and 3). Mutation of either cysteine significantly attenuated H2O2-dependent Src activation, both in studies with recombinant proteins in vitro and in a cellular context (44). Moreover, MD simulations demonstrated that introduction of sulphenylated forms (P-SOH) of either C185 or C277 within the autoinhibited Src crystal structure (PDB 2SRC) promoted electrostatic and structural changes that are consistent with Src activation mechanisms (44). Indeed, modelling of C277-SOH indicated that this modification directly influences the A-loop, promoting energetically favourable unfolding and solvent exposure of Y416. Inclusion of C185-SOH altered interactions with the inhibitory pY527, destabilizing its binding affinity to the SH2 domain and enhancing its solvent accessibility and potential susceptibility to dephosphorylation (44). Since both cysteine residues appeared to contribute to ROS-dependent Src activation in a cellular context, we proposed a stepwise redox-based mechanism of Src activation, involving the successive oxidation of both C277 and C185 to P-SOH (44). Subsequent S-glutathionylation of either residue would be expected to lock the Src kinase into its active ‘unclamped’ state until these modifications are reversed by cellular reductases (41).
The most intriguing aspect of our recent studies relates to the fact that it concerns relatively unique cysteines that are not present in most other SFK members, although similar cysteines are found in other PTKs. C277 is conserved in only two other SFK members and its function is largely unknown, and has therefore been targeted by newly developed covalent Src inhibitors (38). Studies with recombinant Src proteins indicated that removal of DTT in the kinase buffer causes loss of kinase activity, and this was attributed to formation of disulphide-linked homodimers via C277 (47), but the significance of such a disulphide-based mechanisms of Src inactivation has to date not been demonstrated in a cellular context. The importance of C185 in the SH2-domain of Src is also poorly understood, and all other SFKs contain a serine in the corresponding position (Fig. 3A), although similar cysteines can be found in other SH2 domains (43). Mutation studies with recombinant Src proteins suggested that the presence of a cysteine in this position may weaken binding affinity to phosphotyrosine-containing peptides that mimic the autoinhibitory pY527-containing domain, and that Src may thus be more readily activated compared to other SFKs (76). A more recent study implicated Src C185 in recognition of substrates through formation of an intermolecular disulphide, as was demonstrated for the Src substrate cortactin (43).
Overall, it follows that redox mechanisms of SFK regulation are exceedingly complex and may involve intrinsic mechanisms that induce structural alterations that alter enzyme function, but may also involve redox modifications that impact on interactions with substrates (43) or potentially affect asymmetric Src dimerization as a critical mechanism in promoting autophosphorylation and Src activation (73). Some SFKs, but not Src itself, also contain additional Cys residues within their N-terminal SH4 domains that are modified by S-acylation (palmitoylation) to regulate SFK trafficking and membrane targeting (77). It is not known to date whether these cysteines are also subject to redox regulation.
Other NRTKs
Redox-based mechanisms have also been implicated in regulation of other NRTKs, such as the Janus Kinases (JAK), which are predominantly involved in cytokine signalling and activation of STAT family of transcription factors (78). JAK proteins also contain a CXXXXXXXMXXCW motif (including C1094 and C1105) but, in contrast to SFKs, are generally found to be inactivated by oxidative mechanisms through oxidation of unique cysteines (79). Four distinct Cys residues (C866, C917, C1094 and C1105) in JAK2 were found to be independently responsible for redox-dependent suppression of kinase activity, with C866 and C917 postulated to form a disulphide bond based on their spatial proximity (55). The PTK CSK, which negatively regulates Src by mediating Y527 phosphorylation (80), is redox regulated by formation of an intramolecular disulphide bond within its SH2 domain (between its C122 and C164 residues), and this inhibits kinase activity compared to the fully reduced enzyme (56). Other uniquely regulated non-receptor kinases include the spleen tyrosine kinase (SYK) and its related family members, which are predominantly involved in T-cell activation and adaptive immune signalling (81). SYK, as well as its related family member ZAP-70, were found to be susceptible to Cys-targeted covalent inhibitors as well as H2O2, which target Cys residues within their dual SH2 domain motif and thereby prevent these kinases from binding substrate peptides (57). These examples indicate that cysteine oxidation can regulate tyrosine kinase signalling by multiple distinct processes, not only by directly affecting kinase activity but also by regulating kinase signalling more indirectly, e.g. by interfering with phosphopeptide substrate binding.
Receptor tyrosine kinases
Epidermal growth factor receptor
RTKs comprise several families of high-affinity cell surface receptors for various growth factors and hormones (82). Among these, the epidermal growth factor receptor (EGFR), a member of the ErbB family, has been extensively studied because of its broad roles in various cellular processes (e.g. proliferation, migration, cell survival, immune signalling) and the prevalence of diverse activating mutations within this protein in various cancers (83). Similar to other RTKs, EGFR is a transmembrane receptor expressed on the cell surface that is activated by one of its seven cognate ligands (e.g. EGF) through homodimerization or heterodimerization with other ErbB family members. Dimerization of the extracellular ectodomain leads to kinase activation through the formation of an asymmetric kinase dimer complex comprising an activator and receiver kinase pair (Fig. 4) followed by EGFR tail (auto)phosphorylation on several tyrosine residues (84). EGFR can also be activated by transactivation processes, in which other kinases (such as SFKs) phosphorylate unique tyrosines within the EGFR kinase domain to enhance kinase function (75). The full-length EGFR protein contains 60 Cys residues, most of which are present in the extracellular domain and are involved in structurally important disulphide bonds (85). However, the intracellular kinase domain also contains six cysteine residues (and some others are also present in the C-terminal phosphorylation tail), including cysteines contained within the previously mentioned CXXXXXXXMXXCW motif within the kinase αH-helix. Since this αH-helix plays a critical role in the activation of EGFR through direct coil–coil interaction with the αC-helix of the receiver kinase in the asymmetric dimer (Fig. 4), it is conceivable that redox-modification of these cysteines could affect such dimer interactions. However, in contrast to studies with SFKs discussed above, no studies to date have addressed the specific function of these CXXXXXXXMXXCW motif cysteines within EGFR.
Fig. 4.

Active asymmetric EGFR kinase dimer model showing the positions of the activator αH helix, containing the conserved CXXXXXXXMXXCW motif (cysteines of this motif shown as red spheres), and αC-helix (red) inward positioning in the active receiver kinase. αH and αC helix of the receiver and activator kinases, respectively, are labelled for clarity.
It has been recognized for over 20 years that EGFR activation can promote production of cellular ROS (e.g. by NOX activation) and that this promotes EGFR-dependent signalling (86, 87). While this was largely attributed to redox-dependent inactivation of PTPs (as described earlier), recent pioneering studies by Kate Carroll et al. have highlighted the ability of ROS to directly regulate EGFR. They illustrated that H2O2 can directly activate EGFR by oxidation of a conserved cysteine (C797) within the ATP binding region (59, 60). Among the six cysteines within the EGFR kinase domain, C797 is the most solvent accessible and redox-sensitive, due to its relatively low pKa (60). Indeed, studies with recombinant EGFR kinase domains showed that, although C797 is not critical for intrinsic kinase activity, the ability of H2O2 to enhance kinase activity relies fully on the presence of C797 (59, 60). Redox-dependent EGFR activation by ligand stimulation was found to involve activation of the NOX isoform NOX2, which is recruited to the EGFR signalling complex following EGF stimulation (41, 59). Our research group recently highlighted activation of EGFR in airway epithelial cells in the context of ATP-dependent wound signalling, and this was found to involve activation of a different NOX isoform, DUOX1, which promotes EGFR transactivation by initial redox-dependent activation of Src associated with cysteine oxidation within Src as well as EGFR (41, 75). DUOX1-dependent EGFR activation was also demonstrated to contribute to innate airway responses to allergens and to features of allergic asthma (88, 89). Evaluation of distinct cysteine modifications indicated that H2O2-induced EGFR activation is mediated specifically by formation of P-SOH, whereas other modifications (P-SSG, P-SO2/3H) did not appear to alter intrinsic kinase activity (41, 90). MD simulations indicated that formation of C797-SOH can alter local electrostatic interactions due to hydrogen bonding between C797-SOH and a critical R841 residue within the ATP binding region, thereby potentially aiding in coordinating ATP binding (60). Interestingly, C797 within the EGFR kinase domain is also the target for covalent EGFR TKI inhibitors (91, 92) and for inhibition by other thiol-reactive electrophiles such as acrolein (61). Hence, oxidation of EGFR C797 would be expected to negatively impact on efficacy of irreversible kinase inhibitors, as was indeed observed in several studies (60, 90).
The importance of cysteine oxidation in EGFR has been implicated in EGFR-dependent functions in normal epithelial biology (41, 75), but may be dysregulated in the context of lung cancer, especially those involving oncogenic EGFR mutants. For example, the oncogenic L858R/T790M double mutant, which inserts an Arg residue onto the A-loop opposite the active site in addition to the Met gatekeeper mutation, was found to display increased sulphenylation over a higher range of EGF concentrations in comparison to other oncogenic cell lines (60). Alternatively, redox events may also regulate subcellular EGFR trafficking as was illustrated by increased caveolin-dependent perinuclear trafficking in cancer cells under conditions of oxidative stress (93). More recent findings indicated that the absence of DUOX1 in lung cancers can promote EGFR internalization and nuclear localization, and this was associated with altered redox modification(s) within EGFR (42). In this regard, it is important to consider that EGFR is also regulated by palmitoylation on several cysteine residues, including C797, with apparent consequences for EGFR membrane interactions and dimerization, and such palmitoylation has been implicated in development of tumour resistance against EGFR kinase inhibitors (94). In addition to C797, cysteines within the C-terminal tail of EGFR (C1025 and C1122), have also been identified as sites for palmitoylation, regulating receptor turnover and recycling (62). Inhibition of EGFR palmitoylation by inhibiting the palmitoyltransferase DHHC20 was found to result in sustained EGFR activation, and to sensitise cancer cells to EGFR TKI inhibitors (62). Redox modification of C797 or these C-terminal cysteines might therefore indirectly affect EGFR function by interfering with EGFR palmitoylation. Intriguingly, recent studies highlighted development of tumour resistance against covalent EGFR inhibitors that target C797, which was found to involve acquired mutation of this Cys to a Ser (95), which would also suggest that EGFR signalling is in these cases refractory to redox regulation or palmitoylation at this cysteine.
Other RTKs
Other RTKs may be similarly redox regulated based on the presence of conserved cysteines. For example, other ErbB family members such as ErbB2/HER2 or ErbB4/HER4 contain homologous cysteines to C797 in EGFR (35, 39), as do some other RTKs (96), but the importance of redox regulation of these cysteines has not yet been addressed. The insulin-like growth factor 1 receptor (IGF-1R) was recently found to be subject to S-nitrosylation, which was reasoned to be disrupting IGF-1R activation and downstream signaling (63). Fibroblast growth factor receptors (FGFR) contain a cysteine within their ATP binding regions that is homologous to C277 in Src, and studies with recombinant FGFR1 protein showed loss of kinase activity under oxidizing conditions (in the absence of DTT) corresponding to formation of disulphide-based homodimers similar to Src (47), although the relevance for potential redox regulation in a cellular context is unclear. Exposure of vascular endothelial cells to H2O2 was found to be capable of promoting intramolecular disulphide bonds within the VEGF receptor, between C1199 and C1206 within its cytoplasmic domain, which resulted in its activation (64). Finally, disulphide-linked dimers were also implicated in the activation of the RET kinase due to UV-mediated oxidation, which was observed to involve C376 within its kinase domain (37).
Alternative Redox-Dependent Regulation of PTK Signalling
In addition to the aforementioned redox regulation of EGFR kinase domains, some studies have indicated that NO• is capable of inhibiting EGFR activation and tumour cell proliferation, which was associated with S-nitrosylation of extracellular C166 and C305 residues within EGFR (58). However, contrasting reports demonstrated that NO donors can enhance tumour cell proliferation and pointed to P-SNO associated EGFR activation involving Src and β-catenin (70), although specific cysteine residues were not identified in this case. Another potential redox modification involves the rapidly emerging aspect of hydrogen sulphide and related persulphides/polysulphides, which can modify various proteins by formation of persulphides (P-SSH). Indeed, recent studies by our group indicated that persulphides exist within EGFR as well as Src and may also be subject to oxidation, leading to perthiosulphenylation (P-SSOH) (11), but the functional importance of these modifications is unknown.
In addition to redox regulation of PTPs and PTKs, cysteine oxidation within SH2 domains could also affect phosphotyrosine signalling more indirectly by impacting on phospho-Tyr binding, as was shown recently with respect to C185 within the SH2 domain of Src and its involvement in intermolecular disulphide cross-linking with the Src substrate protein cortactin (43). An estimated one quarter of all SH2 domains contain cysteine residues within a similar phosphotyrosine binding region (43), and such redox-based regulation of SH2 domains and their phosphoprotein substrate recognition might apply more broadly. Indeed, modification of similar SH2 domain cysteines within zeta-chain associated protein of 70 kDa (ZAP-70) or SYK by thiol-reactive small molecules or by H2O2 has been found to inhibit phosphopeptide binding (57). As another example, formation of a disulphide within the SH2 domain of the CSK, a constitutively active kinase that phosphorylates and inhibits Src, attenuates CSK function as an indirect redox-based mechanism involved in Src activation (56).
Summary and Future Perspectives
The prevailing literature highlighting redox-based regulation of tyrosine kinase signalling has attributed such oxidant sensitivity to the inhibition of PTPs, since they are typically considered as highly redox sensitive and oxidation of their catalytic cysteines invariably inactivates their phosphatase function. However, it should be apparent from this review that recent studies over the past decade have dramatically expanded the redox-based mechanisms that may control tyrosine kinase signalling, which includes oxidation of various non-catalytic cysteines within PTKs, thereby regulating intrinsic tyrosine kinase activity, as well as oxidation of cysteines with in SH2 domains to affect interactions with phosphotyrosine-containing substrates (Fig. 5). This diversity in redox-based mechanisms may be important in fine-tuning the overall impact on tyrosine kinase signalling, which may highly depend on specific subcellular mechanisms of ROS generation (e.g. NOX enzymes) in relation to these kinase signalling pathways. In some cases, the various actions may act in concert (e.g. direct activation of PTKs as well as inactivation of PTPs), but such redox modifications may also affect this signalling pathway at different levels, e.g. by controlling specificity in tyrosine kinase signalling via altering interactions with specific tyrosine kinase substrates, or by regulating subcellular localization (42, 62, 74). Clearly, many questions still remain, and our understanding is still very limited with respect to the molecular mechanisms by which specific redox modifications, which can be highly diverse (97, 98), affect intrinsic PTK function or their interactions with other regulatory proteins or substrates. For example, we do not know in which state (active or inactive) PTKs are most susceptible to cysteine oxidation. While PTPs are most likely subject to cysteine oxidation in their active state (leading to loss of activity), this is much less clear for PTKs such as Src that undergo significant conformational changes during their activation. In fact, studies with recombinant PTKs (e.g. Src or EGFR) indicate that their intrinsic activity can be enhanced by cysteine oxidation (41, 44). This could mean that overall activity is increased by oxidation of initially inactive kinase, but it is also possible that cysteine oxidation further enhances initially active kinase, as was suggested previously (45). Further work is needed to specifically address this question.
Fig. 5.
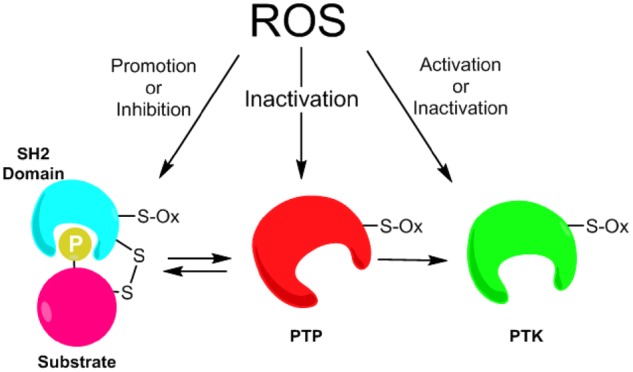
Diverse mechanisms of redox regulation of tyrosine kinase signalling. Cellular ROS (typically H2O2 but also including various RNS) can modify cysteines within PTPs, thereby inhibiting their activity and alleviating their inhibitory effects on PTK function or substrate phosphorylation (middle), leading to higher levels of activation. Alternatively, ROS can either positively or negatively regulate PTK function by cysteine oxidation (right), depending on the nature or site of cysteine modification. Additionally, ROS can modify cysteines in SH2 domain regions to impact on interaction with tyrosine phosphorylated proteins (left) or promote activation of some PTPs. The recognition of these domains can be influenced by PTP redox modification as SH2 domains require phospho-Tyr for target binding, but may also be influenced by redox on PTPs that have SH2 domains themselves. See manuscript text for further details.
One major problem in this field is the overall lack in available approaches to specifically detect these diverse site-specific modifications in selected proteins of interest in relation to their spatiotemporal activation in real time, although some recent studies (99) have begun to address this. For example, while we have access to many diverse phosphospecific antibodies to assess specific tyrosine phosphorylation events in various biological settings, we do not yet have similar tools to assess specific cysteine oxidations, and we have to rely largely on more indirect approaches. Also, much of our understanding with respect to redox regulation of kinases depends on mutation studies, which may induce unforeseen effects on protein stability or subcellular localization, or on studies with recombinant (or purified) kinases, which do not provide insights into potential impact on interactions with other proteins in a more relevant cellular context. Further development of novel approaches that would allow for more direct analysis of specific redox modifications in biological contexts would help address these questions. In any case, it should hopefully be clear from this review that tyrosine kinase signalling is closely interrelated with redox-based signalling, with redox-based mechanisms impacting on tyrosine kinase signalling at various levels, but also by regulatory effects of phosphorylation events on cellular ROS production by actions of mitochondrial function (100) or NOX activation (101). In light of the wide interest in tyrosine kinases as therapeutic targets in various diseases, such increased understanding of redox-based mechanisms may also aid in development of improved inhibitors.
Acknowledgement
The authors gratefully acknowledge research support from NIH (R01 HL085646, R01 HL138178, and F31 HL142221).
Conflict of Interest
None declared.
Glossary
Abbreviations
- CSK
C-terminal Src kinase
- EGFR
epidermal growth factor receptor
- FGFR
fibroblast growth factor receptors
- H2O2
hydrogen peroxide
- IGF
insulin growth factor
- JAK
Janus kinase
- MD
molecular dynamics
- NO
nitric oxide
- NOX
NADPH oxidases
- PTK
protein tyrosine kinase
- PTM
post-translational modification
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- Sec
selenocysteine
- SYK
spleen tyrosine kinase
- TKI
tyrosine kinase inhibitor
References
- 1. Endicott J.A., Noble M.E., Johnson L.N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 2. Pawson T., Gish G.D., Nash P. (2001) SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 11, 504–511 [DOI] [PubMed] [Google Scholar]
- 3. Barford D., Das A.K., Egloff M.P. (1998) The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27, 133–164 [DOI] [PubMed] [Google Scholar]
- 4. Zhang J., Yang P.L., Gray N.S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PubMed] [Google Scholar]
- 5. Holmstrom K.M., Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 [DOI] [PubMed] [Google Scholar]
- 6. Schieber M., Chandel N.S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones D.P., Sies H. (2015) The redox code. Antioxid. Redox Signal. 23, 734–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corcoran A., Cotter T.G. (2013) Redox regulation of protein kinases. FEBS J. 280, 1944–1965 [DOI] [PubMed] [Google Scholar]
- 9. Janssen-Heininger Y.M., Mossman B.T., Heintz N.H., Forman H.J., Kalyanaraman B., Finkel T., Stamler J.S., Rhee S.G., van der Vliet A. (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Filipovic M.R., Zivanovic J., Alvarez B., Banerjee R. (2018) Chemical biology of H2S signaling through persulfidation. Chem. Rev. 118, 1253–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heppner D.E., Hristova M., Ida T., Mijuskovic A., Dustin C.M., Bogdandi V., Fukuto J.M., Dick T.P., Nagy P., Li J., Akaike T., van der Vliet A. (2018) Cysteine perthiosulfenic acid (Cys-SSOH): a novel intermediate in thiol-based redox signaling? Redox Biol. 14, 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai J., Bhatnagar A., Pierce W.M. Jr. (2009) Protein modification by acrolein: formation and stability of cysteine adducts. Chem. Res. Toxicol. 22, 708–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schopfer F.J., Cipollina C., Freeman B.A. (2011) Formation and signaling actions of electrophilic lipids. Chem. Rev. 111, 5997–6021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang H., Zhang X., Chen X., Aramsangtienchai P., Tong Z., Lin H. (2018) Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem. Rev. 118, 919–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reich H.J., Hondal R.J. (2016) Why nature chose selenium. ACS Chem. Biol. 11, 821–841 [DOI] [PubMed] [Google Scholar]
- 16. Hoshi T., Heinemann S. (2001) Regulation of cell function by methionine oxidation and reduction. J. Physiol. 531, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao R., Xu D., Thelen J.J., Miernyk J.A. (2013) Circles within circles: crosstalk between protein Ser/Thr/Tyr-phosphorylation and Met oxidation. BMC Bioinformatics 14, S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Vliet A., Janssen-Heininger Y.M.W., Anathy V. (2018) Oxidative stress in chronic lung disease: from mitochondrial dysfunction to dysregulated redox signalling. Mol. Aspects Med. 63, 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finkel T. (1998) Oxygen radicals and signalling. Curr. Opin. Cell Biol. 10, 248–253 [DOI] [PubMed] [Google Scholar]
- 20. Stone R.L., Dixon J.E. (1994) Protein-tyrosine phosphatases. J. Biol. Chem. 269, 31323–31326 [PubMed] [Google Scholar]
- 21. Tonks N.K. (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121, 667–670 [DOI] [PubMed] [Google Scholar]
- 22. Denu J.M., Tanner K.G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 23. Salmeen A., Andersen J.N., Myers M.P., Meng T.C., Hinks J.A., Tonks N.K., Barford D. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 24. Barrett W.C., DeGnore J.P., Konig S., Fales H.M., Keng Y.F., Zhang Z.Y., Yim M.B., Chock P.B. (1999) Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38, 6699–6705 [DOI] [PubMed] [Google Scholar]
- 25. Chen Y.Y., Chu H.M., Pan K.T., Teng C.H., Wang D.L., Wang A.H., Khoo K.H., Meng T.C. (2008) Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 283, 35265–35272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weibrecht I., Bohmer S.A., Dagnell M., Kappert K., Ostman A., Bohmer F.D. (2007) Oxidation sensitivity of the catalytic cysteine of the protein-tyrosine phosphatases SHP-1 and SHP-2. Free Radic. Biol. Med. 43, 100–110 [DOI] [PubMed] [Google Scholar]
- 27. Lee S.R., Yang K.S., Kwon J., Lee C., Jeong W., Rhee S.G. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342 [DOI] [PubMed] [Google Scholar]
- 28. Machado L., Shen T.L., Page R., Peti W. (2017) The KIM-family protein-tyrosine phosphatases use distinct reversible oxidation intermediates: intramolecular or intermolecular disulfide bond formation. J. Biol. Chem. 292, 8786–8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frijhoff J., Dagnell M., Godfrey R., Ostman A. (2014) Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid. Redox Signal. 20, 1994–2010 [DOI] [PubMed] [Google Scholar]
- 30. Fox G.C., Shafiq M., Briggs D.C., Knowles P.P., Collister M., Didmon M.J., Makrantoni V., Dickinson R.J., Hanrahan S., Totty N., Stark M.J., Keyse S.M., McDonald N.Q. (2007) Redox-mediated substrate recognition by Sdp1 defines a new group of tyrosine phosphatases. Nature 447, 487–492 [DOI] [PubMed] [Google Scholar]
- 31. Bonham C.A., Vacratsis P.O. (2009) Redox regulation of the human dual specificity phosphatase YVH1 through disulfide bond formation. J. Biol. Chem. 284, 22853–22864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson L.N., Noble M.E., Owen D.J. (1996) Active and inactive protein kinases: structural basis for regulation. Cell 85, 149–158 [DOI] [PubMed] [Google Scholar]
- 33. Taylor S.S., Radzio-Andzelm E. (1994) Three protein kinase structures define a common motif. Structure 2, 345–355 [DOI] [PubMed] [Google Scholar]
- 34. Huse M., Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 35. Nakashima I., Takeda K., Kawamoto Y., Okuno Y., Kato M., Suzuki H. (2005) Redox control of catalytic activities of membrane-associated protein tyrosine kinases. Arch. Biochem. Biophys. 434, 3–10 [DOI] [PubMed] [Google Scholar]
- 36. Veillette A., Dumont S., Fournel M. (1993) Conserved cysteine residues are critical for the enzymatic function of the lymphocyte-specific tyrosine protein kinase p56lck. J. Biol. Chem. 268, 17547–17553 [PubMed] [Google Scholar]
- 37. Kato M., Iwashita T., Takeda K., Akhand A.A., Liu W., Yoshihara M., Asai N., Suzuki H., Takahashi M., Nakashima I. (2000) Ultraviolet light induces redox reaction-mediated dimerization and superactivation of oncogenic Ret tyrosine kinases. Mboc. 11, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rao S., Gurbani D., Du G., Everley R.A., Browne C.M., Chaikuad A., Tan L., Schroder M., Gondi S., Ficarro S.B., Sim T., Kim N.D., Berberich M.J., Knapp S., Marto J.A., Westover K.D., Sorger P.K., Gray N.S. (2019) Leveraging compound promiscuity to identify targetable cysteines within the kinome. Cell Chem. Biol. 26, 818–829 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Truong T.H., Carroll K.S. (2013) Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 48, 332–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xi G., Shen X., Maile L.A., Wai C., Gollahon K., Clemmons D.R. (2012) Hyperglycemia enhances IGF-I-stimulated Src activation via increasing Nox4-derived reactive oxygen species in a PKCzeta-dependent manner in vascular smooth muscle cells. Diabetes 61, 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heppner D.E., Hristova M., Dustin C.M., Danyal K., Habibovic A., van der Vliet A. (2016) The NADPH oxidases DUOX1 and NOX2 play distinct roles in redox regulation of epidermal growth factor receptor signalling. J. Biol. Chem. 291, 23282–23293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Little A.C., Hristova M., van Lith L., Schiffers C., Dustin C.M., Habibovic A., Danyal K., Heppner D.E., Lin M.J., van der Velden J., Janssen-Heininger Y.M., van der Vliet A. (2019) Dysregulated redox regulation contributes to nuclear EGFR localization and pathogenicity in lung cancer. Sci. Rep. 9, 4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Evans J.V., Ammer A.G., Jett J.E., Bolcato C.A., Breaux J.C., Martin K.H., Culp M.V., Gannett P.M., Weed S.A. (2012) Src binds cortactin through an SH2 domain cystine-mediated linkage. J. Cell Sci. 125, 6185–6197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heppner D.E., Dustin C.M., Liao C., Hristova M., Veith C., Little A.C., Ahlers B.A., White S.L., Deng B., Lam Y.W., Li J., van der Vliet A. (2018) Direct cysteine sulfenylation drives activation of the Src kinase. Nat. Commun. 9, 4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giannoni E., Buricchi F., Raugei G., Ramponi G., Chiarugi P. (2005) Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25, 6391–6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang H., Davies K.J., Forman H.J. (2015) TGFbeta1 rapidly activates Src through a non-canonical redox signaling mechanism. Arch. Biochem. Biophys. 568, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kemble D.J., Sun G. (2009) Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. U.S.A. 106, 5070–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Block K., Eid A., Griendling K.K., Lee D.Y., Wittrant Y., Gorin Y. (2008) Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 283, 24061–24076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rahman M.A., Senga T., Ito S., Hyodo T., Hasegawa H., Hamaguchi M. (2010) S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 285, 3806–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oo M.L., Senga T., Thant A.A., Amin A.R., Huang P., Mon N.N., Hamaguchi M. (2003) Cysteine residues in the C-terminal lobe of Src: their role in the suppression of the Src kinase. Oncogene 22, 1411–1417 [DOI] [PubMed] [Google Scholar]
- 51. Senga T., Hasegawa H., Tanaka M., Rahman M.A., Ito S., Hamaguchi M. (2008) The cysteine-cluster motif of c-Src: its role for the heavy metal-mediated activation of kinase. Cancer Sci. 99, 571–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim J.E., Roh E., Lee M.H., Yu D.H., Kim D.J., Lim T.G., Jung S.K., Peng C., Cho Y.Y., Dickinson S., Alberts D., Bowden G.T., Einspahr J., Stratton S.P., Curiel-Lewandrowski C., Bode A.M., Lee K.W., Dong Z. (2016) Fyn is a redox sensor involved in solar ultraviolet light-induced signal transduction in skin carcinogenesis. Oncogene 35, 4091–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoo S.K., Starnes T.W., Deng Q., Huttenlocher A. (2011) Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 480, 109–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mallozzi C., Di Stasi M.A., Minetti M. (2001) Peroxynitrite-dependent activation of src tyrosine kinases lyn and hck in erythrocytes is under mechanistically different pathways of redox control. Free Radic. Biol. Med. 30, 1108–1117 [DOI] [PubMed] [Google Scholar]
- 55. Smith J.K., Patil C.N., Patlolla S., Gunter B.W., Booz G.W., Duhe R.J. (2012) Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic. Biol. Med. 52, 1101–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mills J.E., Whitford P.C., Shaffer J., Onuchic J.N., Adams J.A., Jennings P.A. (2007) A novel disulfide bond in the SH2 Domain of the C-terminal Src kinase controls catalytic activity. J. Mol. Biol. 365, 1460–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Visperas P.R., Winger J.A., Horton T.M., Shah N.H., Aum D.J., Tao A., Barros T., Yan Q., Wilson C.G., Arkin M.R., Weiss A., Kuriyan J. (2015) Modification by covalent reaction or oxidation of cysteine residues in the tandem-SH2 domains of ZAP-70 and Syk can block phosphopeptide binding. Biochem. J. 465, 149–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murillo-Carretero M., Torroglosa A., Castro C., Villalobo A., Estrada C. (2009) S-Nitrosylation of the epidermal growth factor receptor: a regulatory mechanism of receptor tyrosine kinase activity. Free Radic. Biol. Med. 46, 471–479 [DOI] [PubMed] [Google Scholar]
- 59. Paulsen C.E., Truong T.H., Garcia F.J., Homann A., Gupta V., Leonard S.E., Carroll K.S. (2012) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Truong T.H., Ung P.M., Palde P.B., Paulsen C.E., Schlessinger A., Carroll K.S. (2016) Molecular basis for redox activation of epidermal growth factor receptor kinase. Cell Chem. Biol. 23, 837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Danyal K., de Jong W., O'Brien E., Bauer R.A., Heppner D.E., Little A.C., Hristova M., Habibovic A., van der Vliet A. (2016) Acrolein and thiol-reactive electrophiles suppress allergen-induced innate airway epithelial responses by inhibition of DUOX1 and EGFR. Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L913–L923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Runkle K.B., Kharbanda A., Stypulkowski E., Cao X.J., Wang W., Garcia B.A., Witze E.S. (2016) Inhibition of DHHC20-mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol. Cell 62, 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Okada K., Zhu B.T. (2017) S-nitrosylation of the IGF-1 receptor disrupts the cell proliferative action of IGF-1. Biochem. Biophys. Res. Commun. 491, 870–875 [DOI] [PubMed] [Google Scholar]
- 64. Kang D.H., Lee D.J., Lee K.W., Park Y.S., Lee J.Y., Lee S.H., Koh Y.J., Koh G.Y., Choi C., Yu D.Y., Kim J., Kang S.W. (2011) Peroxiredoxin II is an essential antioxidant enzyme that prevents the oxidative inactivation of VEGF receptor-2 in vascular endothelial cells. Mol. Cell 44, 545–558 [DOI] [PubMed] [Google Scholar]
- 65. Parsons S.J., Parsons J.T. (2004) Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909 [DOI] [PubMed] [Google Scholar]
- 66. Xu W., Doshi A., Lei M., Eck M.J., Harrison S.C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638 [DOI] [PubMed] [Google Scholar]
- 67. Roskoski R., Jr. (2005) Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 331, 1–14 [DOI] [PubMed] [Google Scholar]
- 68. Fukazawa H., Mizuno S., Uehara Y. (1990) Effects of herbimycin-a and various Sh-reagents on P60v-Src kinase-activity in vitro. Biochem. Biophys. Res. Commun. 173, 276–282 [DOI] [PubMed] [Google Scholar]
- 69. Akhand A.A., Pu M., Senga T., Kato M., Suzuki H., Miyata T., Hamaguchi M., Nakashima I. (1999) Nitric oxide controls src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J. Biol. Chem. 274, 25821–25826 [DOI] [PubMed] [Google Scholar]
- 70. Switzer C.H., Glynn S.A., Cheng R.Y., Ridnour L.A., Green J.E., Ambs S., Wink D.A. (2012) S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 10, 1203–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Giannoni E., Fiaschi T., Ramponi G., Chiarugi P. (2009) Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene 28, 2074–2086 [DOI] [PubMed] [Google Scholar]
- 72. Giannoni E., Buricchi F., Grimaldi G., Parri M., Cialdai F., Taddei M.L., Raugei G., Ramponi G., Chiarugi P. (2008) Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 15, 867–878 [DOI] [PubMed] [Google Scholar]
- 73. Spassov D.S., Ruiz-Saenz A., Piple A., Moasser M.M. (2018) A dimerization function in the intrinsically disordered N-terminal region of Src. Cell Rep. 25, 449–463 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krasnowska E.K., Pittaluga E., Brunati A.M., Brunelli R., Costa G., De Spirito M., Serafino A., Ursini F., Parasassi T. (2008) N-acetyl-l-cysteine fosters inactivation and transfer to endolysosomes of c-Src. Free Radic. Biol. Med. 45, 1566–1572 [DOI] [PubMed] [Google Scholar]
- 75. Sham D., Wesley U.V., Hristova M., van der Vliet A. (2013) ATP-mediated transactivation of the epidermal growth factor receptor in airway epithelial cells involves DUOX1-dependent oxidation of Src and ADAM17. PLoS One 8, e54391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bradshaw J.M., Mitaxov V., Waksman G. (1999) Investigation of phosphotyrosine recognition by the SH2 domain of the Src kinase. J. Mol. Biol. 293, 971–985 [DOI] [PubMed] [Google Scholar]
- 77. Sato I., Obata Y., Kasahara K., Nakayama Y., Fukumoto Y., Yamasaki T., Yokoyama K.K., Saito T., Yamaguchi N. (2009) Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 122, 965–975 [DOI] [PubMed] [Google Scholar]
- 78. Yamaoka K., Saharinen P., Pesu M., Holt V.E. 3rd, Silvennoinen O., O'Shea J.J. (2004) The Janus kinases (Jaks). Genome Biol. 5, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Duhe R.J. (2013) Redox regulation of Janus kinase: the elephant in the room. JAKSTAT 2, e26141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Okada M., Nada S., Yamanashi Y., Yamamoto T., Nakagawa H. (1991) CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 266, 24249–24252 [PubMed] [Google Scholar]
- 81. Mocsai A., Ruland J., Tybulewicz V.L. (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ullrich A., Schlessinger J. (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212 [DOI] [PubMed] [Google Scholar]
- 83. Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M.R., Carotenuto A., De Feo G., Caponigro F., Salomon D.S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 [DOI] [PubMed] [Google Scholar]
- 84. Zhang X., Gureasko J., Shen K., Cole P.A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 85. Abe Y., Odaka M., Inagaki F., Lax I., Schlessinger J., Kohda D. (1998) Disulfide bond structure of human epidermal growth factor receptor. J. Biol. Chem. 273, 11150–11157 [DOI] [PubMed] [Google Scholar]
- 86. Gamou S., Shimizu N. (1995) Hydrogen peroxide preferentially enhances the tyrosine phosphorylation of epidermal growth factor receptor. FEBS Lett. 357, 161–164 [DOI] [PubMed] [Google Scholar]
- 87. Goldkorn T., Balaban N., Matsukuma K., Chea V., Gould R., Last J., Chan C., Chavez C. (1998) EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am. J. Respir. Cell Mol. Biol. 19, 786–798 [DOI] [PubMed] [Google Scholar]
- 88. Habibovic A., Hristova M., Heppner D.E., Danyal K., Ather J.L., Janssen-Heininger Y.M., Irvin C.G., Poynter M.E., Lundblad L.K., Dixon A.E., Geiszt M., van der Vliet A. (2016) DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma. JCI Insight 1, e88811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hristova M., Habibovic A., Veith C., Janssen-Heininger Y.M., Dixon A.E., Geiszt M., van der Vliet A. (2016) Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J. Allergy Clin. Immunol. 137, 1545–1556 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schwartz P.A., Kuzmic P., Solowiej J., Bergqvist S., Bolanos B., Almaden C., Nagata A., Ryan K., Feng J., Dalvie D., Kath J.C., Xu M., Wani R., Murray B.W. (2014) Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cross D.A., Ashton S.E., Ghiorghiu S., Eberlein C., Nebhan C.A., Spitzler P.J., Orme J.P., Finlay M.R., Ward R.A., Mellor M.J., Hughes G., Rahi A., Jacobs V.N., Red Brewer M., Ichihara E., Sun J., Jin H., Ballard P., Al-Kadhimi K., Rowlinson R., Klinowska T., Richmond G.H., Cantarini M., Kim D.W., Ranson M.R., Pao W. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Singh J., Petter R.C., Baillie T.A., Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 [DOI] [PubMed] [Google Scholar]
- 93. Khan E.M., Heidinger J.M., Levy M., Lisanti M.P., Ravid T., Goldkorn T. (2006) Epidermal growth factor receptor exposed to oxidative stress undergoes Src- and caveolin-1-dependent perinuclear trafficking. J. Biol. Chem. 281, 14486–14493 [DOI] [PubMed] [Google Scholar]
- 94. Thomas R., Srivastava S., Katreddy R.R., Sobieski J., Weihua Z. (2019) Kinase-inactivated EGFR is required for the survival of wild-type EGFR-expressing cancer cells treated with tyrosine kinase inhibitors. Int. J. Mol. Sci. 20, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Thress K.S., Paweletz C.P., Felip E., Cho B.C., Stetson D., Dougherty B., Lai Z., Markovets A., Vivancos A., Kuang Y., Ercan D., Matthews S.E., Cantarini M., Barrett J.C., Janne P.A., Oxnard G.R. (2015) Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Truong T.H., Carroll K.S. (2012) Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 51, 9954–9965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Forman H.J., Davies M.J., Kramer A.C., Miotto G., Zaccarin M., Zhang H., Ursini F. (2017) Protein cysteine oxidation in redox signaling: caveats on sulfenic acid detection and quantification. Arch. Biochem. Biophys. 617, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Heppner D.E., Janssen-Heininger Y.M.W., van der Vliet A. (2017) The role of sulfenic acids in cellular redox signaling: reconciling chemical kinetics and molecular detection strategies. Arch. Biochem. Biophys. 616, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tsutsumi R., Harizanova J., Stockert R., Schroder K., Bastiaens P.I.H., Neel B.G. (2017) Assay to visualize specific protein oxidation reveals spatio-temporal regulation of SHP2. Nat. Commun. 8, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pagliarini D.J., Dixon J.E. (2006) Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem. Sci. 31, 26–34 [DOI] [PubMed] [Google Scholar]
- 101. Matsushima S., Kuroda J., Zhai P., Liu T., Ikeda S., Nagarajan N., Oka S., Yokota T., Kinugawa S., Hsu C.P., Li H., Tsutsui H., Sadoshima J. (2016) Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Invest. 126, 3403–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]