

Abstract
Germline mutation rates in humans have been estimated for a variety of mutation types, including single-nucleotide and large structural variants. Here, we directly measure the germline retrotransposition rate for the three active retrotransposon elements: L1, Alu, and SVA. We used three tools for calling mobile element insertions (MEIs) (MELT, RUFUS, and TranSurVeyor) on blood-derived whole-genome sequence (WGS) data from 599 CEPH individuals, comprising 33 three-generation pedigrees. We identified 26 de novo MEIs in 437 births. The retrotransposition rate estimates for Alu elements, one in 40 births, is roughly half the rate estimated using phylogenetic analyses, a difference in magnitude similar to that observed for single-nucleotide variants. The L1 retrotransposition rate is one in 63 births and is within range of previous estimates (1:20–1:200 births). The SVA retrotransposition rate, one in 63 births, is much higher than the previous estimate of one in 900 births. Our large, three-generation pedigrees allowed us to assess parent-of-origin effects and the timing of insertion events in either gametogenesis or early embryonic development. We find a statistically significant paternal bias in Alu retrotransposition. Our study represents the first in-depth analysis of the rate and dynamics of human retrotransposition from WGS data in three-generation human pedigrees.
Non–long terminal repeat (non-LTR) retrotransposons have played a large role in shaping the human genome by creating structural variation and influencing gene expression (Elbarbary et al. 2016; Bourque et al. 2018). In addition, there are at least 130 documented instances of retrotransposition events associated with human disease (Hancks and Kazazian 2016; Kazazian and Moran 2017). These retrotransposons mobilize via a “copy-and-paste” mechanism using an mRNA intermediate that is reverse-transcribed into the genome. There are three currently active non-LTR retrotransposons in humans: the autonomous long interspersed element 1 (L1); and two nonautonomous elements, the Alu short interspersed elements (SINE), and the composite element SINE-R-VNTR-Alu (SVA). These three retrotransposon families alone account for >25% of the human genome, and younger copies are polymorphic for their presence or absence in humans (Cordaux and Batzer 2009). There are more than 1.5 million non-LTR retrotransposons in the human genome (Cordaux and Batzer 2009), and a small fraction of them are active and still capable of creating new mobile element insertions (MEIs) in germline and somatic tissue. L1 elements, for example, are active and have been extensively studied in the human brain (for review, see Faulkner and Billon 2018) and in tumors (for review, see Burns 2017).
Inherited retrotransposition events occur either in the parental gametes or in early embryogenesis of the individual, with the latter leading to mosaicism of the element. Studies have suggested that the majority of inherited MEIs originate in the male germline (Nellåker et al. 2012), and likely in individuals with compromised control of retrotransposition (Newkirk et al. 2017). A few de novo Alu and L1 elements in humans have been tracked to either the germline (Kazazian et al. 1988; Wallace et al. 1991; Richardson et al. 2017) or early embryogenesis (van den Hurk et al. 2007). L1 retrotransposition studies in mice indicate that retrotransposition mainly occurs in early embryogenesis (Kano et al. 2009; Richardson et al. 2017; for review, see Richardson and Faulkner 2018). The timing of Alu and SVA element insertions remains largely unknown.
Alu, L1, and SVA germline retrotransposition rates have been estimated through phylogenetic and disease-based studies. It is estimated that one de novo Alu insertion occurs in about every 20 births and a de novo L1 insertion event occurs once in about every 150 live human births (Deininger and Batzer 1999; Kazazian 1999; Li et al. 2001; Cordaux et al. 2006; Xing et al. 2009b; Ewing and Kazazian 2010; Huang et al. 2010; Hormozdiari et al. 2011; Hancks and Kazazian 2012). There are only a few thousand SVA elements in the human genome, and the current estimate for the rate of new SVA insertion events is one in roughly every 900 live human births (Xing et al. 2009b). Although previous studies have identified de novo Alu, L1, and SVA insertions in large cohorts using whole-genome sequencing (WGS) (Werling et al. 2018) and whole-exome sequencing (WES) (Gardner et al. 2018), there has not yet been a rigorous empirical study of heritable retrotransposition and retrotranspositional timing in multigenerational pedigrees. Moreover, it is unknown whether human germline retrotransposition is affected by the parent's age or sex, or whether retrotransposition rates differ among pedigrees.
We undertook WGS of 599 members of 33 three-generation Utah Centre d'Etude du Polymorphisme Humain (CEPH) pedigrees (Dausset et al. 1990) because of the historical significance of this cohort in human genetic research and because of the unique research opportunities offered by these large multigenerational pedigrees. The Utah CEPH pedigrees were used to help establish the human linkage map (White et al. 1985), and trios from these pedigrees (CEPH from Utah [CEU]) were an important component of the International HapMap Project (The International HapMap Consortium 2003, 2007) and the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010). The pedigrees were drawn from a population of primarily northern European descent which has experienced very low consanguinity (Jorde 1989), no evidence of founder effect (McLellan et al. 1984), and heterozygosity similar to that of other populations of European ancestry (Xing et al. 2009a). A previous study identified several related pairs of individuals in the Utah and non-Utah CEPH pedigrees (Stevens et al. 2012), but only one mating pair used in our study had detectable consanguinity, with a coefficient of relationship of 0.001. Here, we present our findings of de novo L1, SVA, and Alu retrotransposition events in these pedigrees using three MEI-calling tools: MELT (Gardner et al. 2017), RUFUS (https://github.com/jandrewrfarrell/RUFUS) (Ostrander et al. 2018), and TranSurVeyor (Rajaby and Sung 2018).
Results
Analysis of de novo MEIs in three-generation pedigrees
Blood-derived DNA samples from 599 individuals in 33 three-generation pedigrees were whole-genome sequenced at an average depth of ∼30× using Illumina paired-end technology (Supplemental Table S1). In these pedigrees, we designate the grandparents as generation 1, their offspring as generation 2, and their grandchildren as generation 3. A separate study (Sasani et al. 2019) presents an analysis of single-nucleotide variants (SNVs) and small indels in these pedigrees.
To maximize sensitivity (at the expense of specificity), we used liberal criteria for initial MEI detection in the three MEI-calling tools. This resulted in a large number of false-positive cases that were subsequently identified by Integrative Genomics Viewer (IGV) evaluation (Robinson et al. 2011; Thorvaldsdóttir et al. 2013). MELT identified 907 candidate de novo loci from 12,594 called Alu, SVA, and L1 loci. These candidates were evaluated in IGV for characteristic signatures of MEIs, including a target site duplication (TSD), a poly(A) tail, and split/discordant reads with pairs that mapped to a retrotransposon family (Methods; Supplemental Data S1). Nineteen loci met these criteria and were absent in the parents, and all were validated via PCR and Sanger sequencing (Supplemental Figs. S1 and S2). TranSurVeyor identified 21 de novo loci from 86,649 breakpoints, including 14 of the 19 identified by MELT and an additional six loci not found by MELT (Supplemental Data S2). The RUFUS algorithm called 23 de novo loci from 44,190 breakpoints (Supplemental Data S3), including 22 called by MELT or TranSurVeyor, and one additional de novo locus. In total, we identified and PCR-validated 26 de novo MEIs, including eight L1, seven SVA, and 11 Alu insertions in 16 of 33 CEPH pedigrees (Table 1; Fig. 1; Supplemental Table S2; Supplemental Figs. S1, S2). PCR validation showed that every locus with preliminary evidence of a MEI event was a true-positive de novo insertion.
Table 1.
Characteristics of 26 de novo MEIs identified in 437 births

Figure 1.

Distribution of de novo MEIs throughout the genome. (A) Genomic map of de novo MEIs using HumanIdiogramLibrary (https://zenodo.org/record/1210245#.XVhePuhKiUk). The numbers to the right of the triangles indicate the ID number of each element listed in Table 1. (B) RepeatMasker (UCSC Genome Browser) context of de novo MEIs (Kent et al. 2002). (C) Genic context of de novo MEIs (UCSC Genome Browser) (Kent et al. 2002). The genomic context in B and C was determined using the TSD region of each locus.
Twenty-four of 26 de novo MEIs contain all of the hallmarks of L1-mediated retrotransposition: a poly(A) tail, a TSD, and the endonuclease cleavage site motif (5′-TTTT/AA-3′) (for review, see Cordaux and Batzer 2009; Hancks and Kazazian 2016). The insertion sites of the remaining two loci, Alu #4 and L1 #1, do not fit the canonical pattern. Alu #4 is full-length but has a 1.7-kb deletion at its 5′ flanking region, which may have occurred during the insertion event, and thus does not have a TSD. Alu #4 is de novo in individual 8327 (NA07355) but is also present at low levels by IGV and PCR in sibling 8439 (NA07351) (Supplemental Fig. S1). Amplification of a nearby SNP indicates that there is low-level sample contamination of 8327 (NA07355) in 8439 (NA07351), but this had no effect on the results in this study (Supplemental Fig. S1). L1 #1 is 5′ and 3′ truncated, does not have hallmarks of retrotransposition, and contains a deletion of an “A” at the insertion site. This indicates a nonclassical L1 insertion event, which is hypothesized to play a role in double-stranded break repair (Morrish et al. 2002; Sen et al. 2007). Because L1 #1 was not inserted through retrotransposition, we excluded it from the retrotransposition rate estimates. With 437 trios in this data set, we estimate retrotransposition rates of one Alu event in 39.7 births (95% CI 22.2–79.4), 1 L1 in 62.5 births (95% CI 30.6–153.8), and 1 SVA in 62.5 births (95% CI 30.6–153.8) (Methods).
The genomic context of the 26 de novo MEIs is shown in Figure 1. Detailed information on each breakpoint is provided in Supplemental Figure S2. The MEIs are randomly distributed across the genome (Fig. 1A). Forty-two percent of the loci inserted outside of repetitive DNA regions (Fig. 1B). Nearly all of the MEIs inserted in intergenic or intronic regions (Fig. 1C). L1 #7 inserted 25 bp away from exon 4 in PM20D2 (Supplemental Fig. S2). L1 # 5 inserted within the 3′ UTR of PGRMC2 and created a 628-bp TSD (Supplemental Fig. S2). As expected for a nondisease cohort and this number of MEIs, we did not find any de novo MEIs in exons.
Subfamily analysis of the de novo MEIs
We performed subfamily characterization for the de novo MEIs using MELT's CALU tool and Repbase (Bao et al. 2015; Gardner et al. 2017). The 11 Alu elements belong to seven subfamilies. Alu elements #1 and #5 are exact matches to the Yb8 subfamily, whereas Alu #8 and #9 belong to the Ya5 subfamily. Alu #10 is truncated by >250 bp and could possibly belong to many Y (or the older S) subfamilies (Kryatova et al. 2017). Sequence alignment and FASTA files for the 11 Alu elements are presented in Supplemental Figure S3 and Supplemental Data S4. We matched the full-length L1 #2 to the young L1Ta1d subfamily. We did not get sequence information for the other full-length L1 (L1 #7), and the other six elements are too truncated for classification. SVA #3–5 and #7 contain part of the 5′ transduction of MAST2 exon 1 and therefore belong to the SVA_F1 subfamily (Bantysh and Buzdin 2009; Damert et al. 2009; Hancks et al. 2009). SVA #4 also contains a 3′ transduction of an AluSp, which is present in the SVA_F1 master element H10_1 (Damert et al. 2009; Hancks et al. 2009). The sequences of the SINE-R regions for SVA #1–2 and #6 align to the other known active subfamilies, D-F. The subfamily assignment for each element is in Supplemental Table S2.
Several de novo MEIs have hallmarks of retrotransposition activity
To determine whether any of the de novo Alu elements are capable of further retrotransposition, we examined each element for its potential capacity for retrotransposition activity. Hallmarks of active Alu elements include intact box A and B internal RNA polymerase III (Pol III) promoters (Mills et al. 2007; Bennett et al. 2008; Comeaux et al. 2009), intact SRP9/14 sites, an uninterrupted poly(A) tail at least 20 bases long (Dewannieux and Heidmann 2005), and a Pol III termination sequence, TTTT, preferably within 15 bp of the TSD downstream from the poly(A) tail (Comeaux et al. 2009). In addition, there are 124 conserved nucleotides in active Alu elements, and multiple mutations in these nucleotides may affect retrotransposition efficiency (Bennett et al. 2008). Alu elements #1 and #8 contain all of these hallmarks and therefore may be active (Supplemental Data S4).
To identify potentially active L1/SVA elements, we focused on the full-length de novo elements in our data set. L1 #2 is potentially active because it is not truncated relative to its source element and has two intact open reading frames (ORFs 1 and 2) as determined by L1Base2 (Penzkofer et al. 2017). L1 #7 is full length, but we were unable to sequence the ORFs to determine activity potential. The other six L1 elements are 5′ truncated and therefore not active. SVA #2 is the only element with the CCCTCT hexamer promoter and may be active, although we were unable to sequence through the VNTR region. SVA #5 and #7 are de novo SVA_F1 elements with the full MAST2 promoter and therefore could be active. The other SVA elements do not contain the CCCTCT hexamer but may be transcribed if they inserted downstream from a promoter.
Identification of source elements
We used the human reference genome (hg19) and reconstructed FASTA files from the MELT output to identify potential source elements of the de novo MEIs (Methods; Fig. 1). Alu #2 and #4 each had a unique match to a reference Alu element (hg19 Chr 3: 190,156,698–190,156,966 and Chr 1: 246,470,713–246,471,020, respectively). Alu #3 is 40 bp truncated but uniquely matches a full-length polymorphic Alu element identified by MELT that was paternally transmitted (hg19 Chr 2: 185,125,618). SVA #1 is identical to a reference SVA_D element (hg19 Chr 17: 42,314,401–42,316,970) except for a 725-bp deletion region as a result of splicing (Supplemental Fig. S2). SVA #5 contains a 22-bp deletion within the MAST2 promoter, which is unique to a reference SVA_F1 element (hg19 Chr 3: 48,251,893–48,254,907) (Damert et al. 2009). There were too many potential source elements to pinpoint the candidate source element for the remaining eight Alu and five SVA elements.
We identified the unique source element for the three L1 elements with 3′ transductions (Figs. 1, 2). L1 #2 contains an 82-bp 3′ transduction that maps to an active L1 on Chr4q25. This source element was paternally transmitted. L1 #4 contains an 846-bp 3′ transduction from a L1 on Chr5q22 that was maternally transmitted. L1 #6 is a 497-bp orphan 3′ transduction (i.e., the entire L1 was 5′ truncated) that maps to the 3′ end of a ∼2 kb 3′ transduction from Chr13q21.2. We identified four additional 3′ transduction events from these source elements in our data set by examining the source loci in IGV (Fig. 2A; Supplemental Table S3). Two loci were present in a single grandparent, one locus was polymorphic in a pedigree, and the other locus was polymorphic in 17 pedigrees (Supplemental Table S3). All three source elements are nonreference insertions that are polymorphic across nearly all of the major population groups in the Simons Genome Diversity Project (Fig. 2B; Supplemental Fig. S4; Mallick et al. 2016). These three source elements have also been previously found to produce somatic 3′ transductions in cancer genomes (Tubio et al. 2014).
Figure 2.

Three source L1 elements identified by 3′ transductions. (A) Circlize plot of L1 elements to identified offspring elements in the CEPH data set (Gu et al. 2014). Source elements are highlighted with a star. (B) Minor allele frequency (MAF) of the three source elements in the Simons Genome Diversity Project (Mallick et al. 2016). Genotypes were manually typed from IGV screenshots (Supplemental Table S4).
Estimation of parental origin of MEIs
We used the three-generation pedigree structure to infer the stage at which the retrotransposition event occurred during development for all de novo MEIs in generation 2 (three Alu, one L1, and two SVA). These six second generation MEIs were all found in females, which is statistically significant (exact binomial test P-value <0.0313), but this pattern was not seen in the 21 third generation MEIs (n = 10/18 exact binomial test P-value >0.814). Using the haplotype of the children that inherited the de novo MEI, all six second generation insertions were phased to the maternal grandfather's chromosome (Fig. 3; Supplemental Tables S5–S10). We reason that de novo MEIs that are inherited in Mendelian ratios (50:50) in generation 3 and are cotransmitted with the grandfathers’ haplotype likely arose in the grandfathers’ germline. In contrast, de novo MEIs that are inconsistently associated with the grandfathers’ haplotype provide evidence that the MEIs arose during early embryogenesis in the mother, making her cells mosaic for the MEI.
Figure 3.

Tracking de novo retrotransposition in multigenerational pedigrees. The maternal grandfather's haplotype is shown in light blue, and the maternal grandmother's haplotype is shown in light red. An individual with the de novo MEI is in black.
The three Alu elements that arose in generation 2 were transmitted to generation 3 at Mendelian ratios (χ2 test with one degree of freedom, two-tailed P-value >0.05), and the Alu insertions were always cotransmitted with the maternal grandfathers’ haplotype (Fig. 3). This suggests that the Alu elements originated during the development of the maternal grandfathers’ germline, rather than in early embryogenesis in the mothers. In contrast, L1 #4 and SVA #2 and #5 are not transmitted at the expected ratios (χ2 test with one degree of freedom, two-tailed P-value <0.02). These MEIs were only transmitted to one offspring each, and there were multiple offspring in each pedigree who inherited the maternal grandfathers’ haplotypes but not the MEIs (Fig. 3). Further, because the source element for L1 #4 was maternally transmitted but L1 #4 inserted on the paternal chromosome, L1 #4 was inserted post-zygotically. Both the transmission frequency and haplotype inconsistencies indicate that the three L1/SVA insertions are somatic/germline (gonosomal) mosaic in the second generation.
Another approach for determining whether each de novo MEI is mosaic or nonmosaic in an individual is to calculate the breakpoint allele frequency (BAF), which is the percentage of reads that support the MEI breakpoint (Supplemental Figs. S5, S6). We chose the highest BAF of the two breakpoints for each locus, but this may still be a slight underestimate (i.e., split reads may have mapped elsewhere). BAFs for the 22 third generation individuals who inherited a de novo MEI ranged from 25% to 58%. Therefore, we used a threshold of 25% to estimate heterozygosity. BAFs of the gonosomal mosaic generation 2 L1 and SVA elements ranged from 12% to 21%. This is in the reported range of allelic imbalances of SNV/SV gonosomal mosaicism in parents (Campbell et al. 2014; Acuna-Hidalgo et al. 2015; Rahbari et al. 2016; Jónsson et al. 2018). In contrast, BAFs for the three second generation individuals who have a de novo Alu element were 38%–50%, which is within the range of the inherited de novo MEIs and likely reflects retrotransposition in the parental germline. BAFs of all of the third generation Alu elements were within the range of inherited MEIs, although Alu #7–8 were the lowest at 25.8% and 31%. L1 #2–3 and SVA #3 were the only L1/SVA elements to have BAFs indicating potential heterozygosity (41%–58%). The hemizygous L1 #6 and #8 had BAFs of 100%. These results support the hypothesis indicated by the multigenerational analysis that Alu retrotransposition generally occurs in the germline.
We identified the parental origin of the chromosome for half of the de novo generation 3 MEIs using sex chromosome hemizygosity and SNP-based phasing approaches including two SVA, two L1, and six Alu insertions (Methods; Supplemental Table S2). SVA #4 inserted on the maternal chromosome, and SVA #1 inserted on the paternal chromosome, although these insertions are likely mosaic in the individuals indicated by their BAFs. The hemizygous L1 #6 and L1 #8 in two male individuals inserted on the maternal (Chr X) and paternal (Chr Y) chromosomes. We identified the parental chromosome for six of the eight third generation de novo Alu elements, except for Alu #6–7. Including both second and third generation Alu elements, we found that eight Alu elements were transmitted on the paternal chromosome, and one element was transmitted on the maternal chromosome (exact binomial test, P-value <0.04). We conclude that assuming these elements occurred during gametogenesis, there is a statistically significant paternal sex bias with respect to Alu retrotransposition, whereas L1/SVA retrotransposition appears to generally occur post-zygotically. We did not find statistical support for a paternal age effect on Alu retrotransposition (P-value = 0.26), although the sample size is small (Supplemental Fig. S7).
Evaluation and comparison of the three MEI-calling tools
We used three tools with different approaches to identify MEIs to maximize the likelihood of finding all de novo MEIs. MELT uses a transposon reference file to identify and characterize nonreference MEIs for each transposon family (Gardner et al. 2017). In contrast, RUFUS and the recently published TranSurVeyor identify breakpoints regardless of the transposon family, each producing tens of thousands of false-positive breakpoints (https://github.com/jandrewrfarrell/RUFUS) (Ostrander et al. 2018; Rajaby and Sung 2018). MELT missed the orphan transduction (L1 #6) as well as six other MEIs. MELT preliminarily identified SVA #6 in the individual but then misgenotyped it as homozygous reference (BAF 9.4%). We hypothesize that the other MEIs were missed by not aligning to the transposon family by either having too many differences or a lack of split reads outside of the poly(A) tail. RUFUS missed the two SVA elements in the parents and SVA #6, which may be attributable to the low BAFs in these MEIs. TranSurveyor did not detect five MEIs, but we could not detect a pattern that explained why these were missed. Our results show the importance of utilizing different tools for MEI detection (Ewing 2015; Rishishwar et al. 2016; Goerner-Potvin and Bourque 2018), because only half of the validated de novo MEIs were detected by all three tools, and 12% of the de novo MEIs were detected by a single tool (Supplemental Table S2).
With our three-generation pedigrees, we were able to identify obligate carriers of a MEI in generation 2 as individuals whose parent (generation 1) carried the MEI and whose offspring (generation 3) inherited the MEI (Supplemental Fig. S8). This allowed us to estimate MELT's sensitivity. MELT's unfiltered call set has a sensitivity of 68% for all MEIs, whereas MELT's standard call set has a sensitivity of 94%, because it excluded many incorrect calls. For our identified de novo MEIs, we estimate sensitivity for MELT, RUFUS, and TranSurVeyor calls as ∼73%, 88%, and 77%, respectively. Using only loci that passed MELT's filters (i.e., “PASS”) would have reduced the de novo candidate list from 907 to 217 loci, but 42% (8 of 19) of the de novo loci would have been undetected. Nevertheless, even using three tools, we may have missed additional de novo MEIs because of low sequencing depth or their location in regions with high repeat content. Therefore, our retrotransposition rate estimates should be regarded as lower bounds.
Discussion
With rapid advances in high-throughput sequencing technology, a large number of human pedigrees have been sequenced, and many studies have directly estimated the single-nucleotide de novo mutation rate (Roach et al. 2010; Jónsson et al. 2017). New technology also affords an opportunity to estimate the rate of de novo retrotransposition, which generates genomic variation through an entirely different mutation mechanism. From 437 births, we estimate an Alu retrotransposition rate of about 1:39.7 births (95% CI 22.4–79.4), a SVA rate of about 1:62.5 births (95% CI 30.6–153.8), and a L1 rate of about 1:62.5 births (95% CI 30.6–153.8) (Fig. 4). MELT was used previously to identify de novo MEI transmission in 519 quartets in the Simons Simplex Collection (SSC) (Werling et al. 2018). Using these published data, we estimated comparative retrotransposition rates for Alu, L1, and SVA elements (Fig. 4). The Alu retrotransposition rate in SSC is nearly identical to the estimate in this study, but our L1 and SVA retrotransposition rates are 2.4× and 5.5× higher but do not differ significantly (Fig. 4, 95% CIs; Werling et al. 2018). The latter differences reflect in part our use of multiple MEI-calling tools, which showed that MELT detects 91% of de novo Alu elements detected by TranSurVeyor and RUFUS, but only 75% of the L1 and 43% of the SVA elements detected by the latter tools (Supplemental Table S2). These two data sets both estimate an Alu retrotransposition rate that is twofold lower than previous phylogenetic and disease-based estimates. Given MELT's high sensitivity for Alu detection (Gardner et al. 2017) as well as the use of multiple MEI-calling tools, it is unlikely that our lower rate is caused by false-negative calls, although we could be missing calls in highly repetitive regions. Instead, it is likely that the phylogenetically estimated rate is affected by assumptions about the divergence time of humans and chimpanzees, the effective population size of the human-chimpanzee ancestral population, and retrotransposition rate variation over time (Cordaux and Batzer 2009; Roach et al. 2010; Campbell and Eichler 2013; Ségurel et al. 2014).
Figure 4.
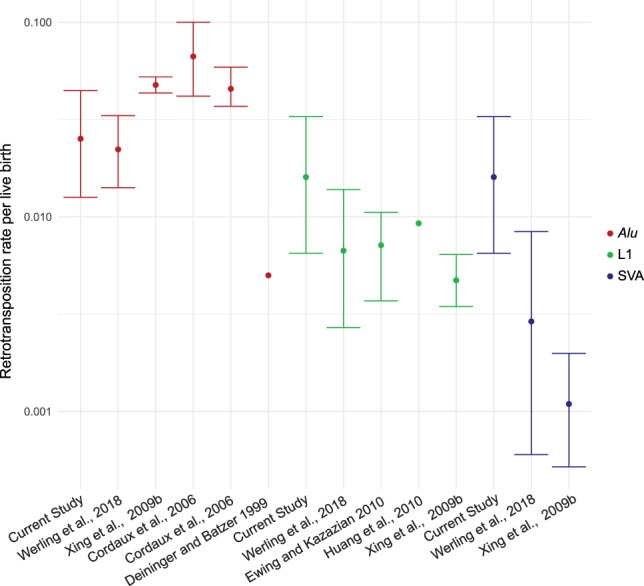
Estimated retrotransposition rates. Estimated retrotransposition rates for previous studies are listed (Deininger and Batzer 1999; Cordaux et al. 2006; Xing et al. 2009b; Ewing and Kazazian 2010; Huang et al. 2010). Confidence intervals are shown if available from the study. Rates and binomial 95% CI were determined for Werling et al. (2018) and this study. Alu element rates are shown in red, L1 in green, and SVA in blue.
Although preliminary, our results suggest there may be differences in retrotransposition timing among the non-LTR retrotransposon families. All of the de novo Alu elements appear heterozygous in WGS, and all three Alu elements in generation 2 conform to Mendelian expectations and cosegregate with the paternal grandfather's chromosome, indicating retrotransposition in the germline. Further, there appears to be a paternal sex bias in de novo Alu retrotransposition, which is similar to the paternal transmission bias seen in SNVs and short tandem repeats (Jónsson et al. 2017; Willems et al. 2017). We found evidence of L1 retrotransposition events in both the germline (the hemizygous elements, L1 #6 and #8) and early embryogenesis (L1 #4) (Fig. 3), which corroborates previous findings (van den Hurk et al. 2007; Richardson et al. 2017). The two second generation SVA elements appear to be mosaic in the germline and somatic tissue in the mother and likely arose during early embryogenesis. Inheritance of gonosomal mosaic L1 and SVA elements in large pedigree analyses has thus far only been seen in females: three in this study and four in a recent mouse study (Richardson et al. 2017). The observation of likely post-zygotic SVA element insertions suggests that SVA elements may be underreported in studies of somatic or tumor cells.
Our data allow us to identify the subfamily distribution of active mobile element subfamilies. Yb8 and Ya5 subfamilies accounted for 72% of 322 polymorphic Alu elements in a previous study (Konkel et al. 2015), yet only 36% of de novo Alu elements identified here belong to the Ya5 or Yb8 subfamilies (Fisher's exact test, P-value <0.02). Our identification of only two Yb8 elements corroborates our pilot ME-Scan study of Yb8/9 elements in the CEPH data set, in which we initially discovered Alu #1 (the individual with Alu #5 was not included in the study) (Supplemental Methods; Supplemental Tables S11, S12). Indeed, the variety of de novo Alu sequences detected here corroborates the “stealth model” hypothesis of Alu amplification, in which there are multiple active subfamilies that proliferate, rather than one large, active subfamily/locus (Deininger et al. 1992; Deininger and Batzer 1999; Han et al. 2005; Konkel et al. 2015). We detected insertions of all active SVA subfamilies, with the youngest SVA_F1 subfamily (Bantysh and Buzdin 2009; Damert et al. 2009; Hancks et al. 2009) accounting for 57% of the de novo SVA elements. Our data show that there are many active AluY subfamilies, and the youngest SVA subfamily, SVA_F1, may be currently one of the most active SVA subfamilies.
In addition to the three non-LTR retrotransposon families, there are other substrates of retrotransposition in the human genome. Processed pseudogene insertions occur when processed mRNA is inserted into the genome using the L1 machinery (Esnault et al. 2000; Abyzov et al. 2013; Ewing et al. 2013; Schrider et al. 2013). There are also several polymorphic HERV-K (HML-2) elements in humans, including at least one potentially active insertion (Wildschutte et al. 2016). We searched for HERV-K (HML-2) elements using MELT and did not identify any candidate de novo loci (Methods; Supplemental Data S1). RUFUS and TranSurVeyor did not detect any de novo pseudogene or HERV-K (HML-2) insertions. Processed pseudogene retrotransposition events are rare (Ewing et al. 2013; Gardner et al. 2018), and tools specific to identifying these events in WGS would allow for retrotransposition rate estimates in pedigrees.
Retrotranspositional activity may differ across pedigrees and populations (Chaisson et al. 2019), similar to how polymorphic PRDM9 variants affect recombination hotspot activity (Baudat et al. 2010; Kong et al. 2010). It is predicted that every human contains 80–100 active L1 elements (Brouha et al. 2003), and this may influence variation in retrotransposition activity among humans. The three source L1 elements in this study are present in all major regional groups in the SGDP (Mallick et al. 2016), which suggests that these elements may also be active in non-European populations (Fig. 2; Supplemental Fig. S4). However, we did not investigate any polymorphic internal variants that may affect the “hotness” of the source element (Seleme et al. 2006). We identified an overabundance (six) of de novo MEIs in pedigree 1331; siblings 8549 (NA07033) and 8310 (NA07023) were also the only individuals with more than one de novo MEI in the data set (Alu #1 and SVA #4 in 8549 [NA07033], and L1 #5 and SVA #1 in 8310 [NA07023]). Preliminary analysis of the pedigree did not reveal any pathogenic SNPs in a gene list of proteins that restrict retrotransposition activity (Goodier 2016). Future studies of retrotransposition in large pedigree-based cohorts may help to elucidate variants and genetic factors involved in the regulation of L1-mediated retrotransposition activity.
Methods
CEPH individuals
Blood-derived DNA samples from 599 individuals, including 454 trios within larger pedigrees, were collected from either the original CEPH cohort (Dausset et al. 1990) or the Utah Genetic Reference Project (Prescott et al. 2008). These samples were whole-genome sequenced at ∼30× coverage (Supplemental Methods) and aligned to the GRCh37 reference genome using BWA-MEM v0.7.15 (Li and Durbin 2009). BAMs were not realigned to the updated GRCh38 because de novo MEIs are by definition not found in the reference sequence. SAMBLASTER was used to de-duplicate the aligned BAM files (Faust and Hall 2014). GATK v3.50 was used to realign regions containing potential short insertions and deletions and base quality score recalibration (DePristo et al. 2011). Alignment quality metrics for the BAM files were calculated by running samtools stats and flagstats (Li et al. 2009). Approximate coverage estimates for each BAM file were calculated using the covstats tool (goleft v0.1.17; https://github.com/brentp/goleft) (Supplemental Table S1). Box plots for coverage of each pedigree are shown in Supplemental Figure S9. Evaluation by peddy identified nine individuals with a het_ratio > 0.2 who were also declared duplicates, indicating potential sample contamination before sequencing (Pedersen and Quinlan 2017). All 17 trios with these individuals were removed from the rate estimate post-IGV evaluation. Therefore, 437 births were used in the rate estimates. All sampled individuals provided informed consent. All ascertainment was performed under University of Utah institutional review board approvals.
Identification of MEIs in the CEPH data set
We used three complementary approaches to identify de novo MEIs in this data set. All 599 individuals were joint-called with the MELT-Split protocol in MELT (v2.14) for detection of Alu, L1, SVA, and HERV-K (HML-2) elements using the consensus transposon files provided by MELT (Gardner et al. 2017). Coverage estimates for each BAM file were rounded down for the IndivAnalysis step. To increase sensitivity, loci were not filtered using the filtering criteria provided by MELT. To identify de novo MEIs in generations 2 and 3 simultaneously, the Genotype Query Tools (GQT) package (Layer et al. 2016) was used to identify loci that were restricted to a unique CEPH pedigree and homozygous reference in generation 1.
All 454 trios were processed through RUFUS, and all structural variant breakpoints were extracted for detection of L1-associated retrotransposition events (https://github.com/jandrewrfarrell/RUFUS) (Ostrander et al. 2018). RUFUS was unable to process trios 1788, 2020, and 4877 successfully. These trios were not among the 17 removed after peddy analysis.
Each sample was individually processed through TranSurVeyor, and unfiltered breakpoints with fewer than four discordant reads of support were removed (Rajaby and Sung 2018). Then, we merged overlapping breakpoints in each individual using the BEDtools merge command (Quinlan and Hall 2010) and merged samples into three BED files: children, parents, and grandparents. We next used BEDtools intersect to identify MEIs that are present in parents and absent in the grandparents, and children MEIs that are absent in the parent and grandparent BED files.
We created a BED file that contained each candidate locus and included the sample BAM ID in the fourth column. This was processed through a custom Python script that generated an IGV batch script. Scripts to generate IGV images in one individual and a trio are available (https://github.com/julieefeusier/IGV-Batch-Script-Generator-for-bed-files) (Supplemental Code S1). Each candidate locus was visualized as a trio in IGV to identify candidate de novo MEIs (Robinson et al. 2011; Thorvaldsdóttir et al. 2013). Any image with evidence of a structural variant (but not small indels) or MEI was flagged for further investigation. These criteria include the presence of one or more features: discordant read pairs, split reads, clear breakpoints, TSDs, and poly(A) tails. Breakpoints were then further investigated in IGV and in BLAT to rule out non-MEI SVs. Candidate loci that passed these initial steps were then locally reassembled for PCR validation. TranSurVeyor took 4.5 h on average per individual. For RUFUS, k-mer counting took on average 2 h per sample, and each trio run took 6 h using 40 cores. MELT took about 1 wk per mobile element family with 10 threads for the individual steps.
Local reassembly of candidate MEIs for primer design
After IGV evaluation, the de novo TE insertion breakpoints provided by the three tools were further analyzed by extracting the reads mapped to a 250-bp region flanking the breakpoint in each individual. Discordant reads mapped to that 500-bp window were identified, and mates of those discordant reads mapped elsewhere in the BAM file were collected (http://broadinstitute.github.io/picard/) (Li et al. 2009). A local de novo assembly of all the extracted reads was performed (Huang and Madan 1999) for each breakpoint in each individual. The assembled contigs were further probed for the presence of TEs. These steps were performed using a custom Perl script (https://github.com/jainy/local_assembly_nonreferenceTE) (Supplemental Code S2).
PCR/Sanger validation of de novo MEIs
PCR amplifications of about 10–25 ng of template DNA (blood-derived or transformed lymphoblast DNA) were performed in 25-µL reactions according to the Phusion Hot Start Flex DNA Polymerase protocol (using 5× GC buffer) and Q5 Hot Start DNA Polymerase (using GC Enhancer). The thermocycler conditions were initial denaturation for 30 sec at 98°C, 40 cycles of denaturation for 10 sec at 98°C, for 30 sec at optimal annealing temperature (58°C–68°C), a 30 sec–3 min extension at 72°C, and a final extension for 5 min at 72°C. Every primer set reaction was performed on the pedigree with the candidate de novo MEI, a positive control, and sterile water. PCR amplicons were run on a 1%–2% gel containing 0.12 mg/mL ethidium bromide for 75–90 min at 120 V. Gels were imaged using a Fotodyne Analyst Investigator Eclipse machine. Bands were cut out and purified for Sanger sequencing using the Qiagen QIAquick Gel Extraction Kit. Primer sets for Alu elements are in Supplemental Table S13, and primers sets for L1/SVA elements are in Supplemental Table S14.
L1 and SVA elements were amplified using the Thermo Fisher Scientific Platinum SuperFi DNA polymerase and cloned using Thermo Fisher Scientific Zero Blunt Topo II/4 kits. We followed the Platinum SuperFi PCR setup for 25-µL reactions using 2 µL starting DNA (∼5 ng/µL). For the PCR procedure, each reaction was denatured for 30 sec at 98°C, and then amplified for 35 cycles (for 10 sec at 98°C, annealing for 10 sec, an extension for 30 sec or longer at 72°C based on amplicon size). Annealing temperatures were estimated for each primer pair based on Thermo Fisher Scientific calculations. A final extension was performed for 5 min at 72°C. The Invitrogen PureLink Quick Plasmid Miniprep Kit was used to extract DNA from the clones. Clones were Sanger sequenced through the whole length of the fragment (Supplemental Table S15). We used several internal primers from previous studies (Scott et al. 2016; Feusier et al. 2017). The three generation 2 L1/SVA elements were analyzed in generation 3 because of the availability of DNA.
Retrotransposition rate estimates
The retrotransposition rate and 95% confidence intervals were calculated using an exact binomial confidence interval estimate with x = number of de novo Alu, L1, or SVA elements and N = 437 births. We dropped L1 #1 from the number of L1 elements because this insertion did not likely occur by retrotransposition. This rate was also calculated for the SSC data set using the identified 23 Alu, seven L1, and three SVA elements in 1038 births (Werling et al. 2018). We included all listed MEIs in the rate estimates, including one SVA element that did not have orthogonal support (PCR, Microarray, or liWGS) (Werling et al. 2018). The estimates and confidence intervals are listed in Supplemental Table S16.
Investigation of source elements
MELT lists differences from the consensus for each MEI locus in the DIFF section of the INFO column (Gardner et al. 2017). These differences were extracted and converted to FASTA format using the MELT consensus transposon FASTA file as the reference. A custom Python script was used for this step (https://github.com/julieefeusier/MEI-VCF-to-FASTA) (Supplemental Code S3). Each de novo MEI sequence was compared to the MELT FASTA file using the “grep” command to identify potential source elements. The de novo MEIs were also compared to the hg19 reference genome using BLAT (Kent 2002; Kent et al. 2002).
Source elements in Simons Genome Diversity Project and CEPH
Paired-end BAM DNA sequences (hg19) for 279 individuals from the Simons Genome Diversity Project (Mallick et al. 2016) were downloaded from the European Nucleotide Archive at the European Bioinformatics Institute (PRJEB9586). DNA samples were remapped to hg38. The locations of the three source elements were converted to hg38 using liftOver in the UCSC Genome Browser (Kent et al. 2002). Individuals were genotyped from IGV screenshots for each of the three source elements (Supplemental Table S4).
We used IGV of the source element to identify additional 3′ transduction events in CEPH. IGV screenshots of the source element and 2 kb downstream from the MEI were generated in each CEPH individual for the three source elements. These 3′ transduction events were discovered by identifying one breakpoint downstream whose mate pair mapped to a retrotransposition event elsewhere in the genome.
Parental origin analysis
For haplotype phasing of de novo MEIs in the parents (second generation), we extracted SNPs in a 200-kb window surrounding the MEI's position. We filtered on SNPs that were heterozygous in the parent and the parental grandparents and absent in the other parent (Supplemental Tables S5–S10). The children were assigned a grandparent haplotype based on the transmission of the SNPs from the grandparent individuals. Then, the transmission of the de novo MEI was placed on the children's grandparent haplotype to determine the parental origin. There was no evidence of recombination between the de novo MEI and the markers on the grandparent haplotype.
Parental origin of the chromosome of de novo MEIs in generation 3 was analyzed using sex chromosome hemizygous status and SNP phasing (Supplemental Table S2). We considered a hemizygous insertion on a sex chromosome to be a retrotransposition event on the parental chromosome in the germline. For SNP phasing, we considered informative SNPs to be either heterozygous to one parent and the child, or heterozygous in the child and homozygous ref/alt in the parents. Paired-end reads that connected the MEI and a nearby informative SNP confirmed parental origin. For Alu elements with SNPs <5 kb away, we designed primers that amplified the Alu-SNP region and confirmed the SNPs of the children and their parents via Sanger sequencing (primers listed in Supplemental Table S13).
Estimating BAF in de novo MEIs
We manually estimated BAF in each individual with a de novo MEI, including the children (third generation) with the inherited de novo MEIs. In IGV, we counted the number of split/discordant reads supporting the MEI at the position that was 1 bp outside of the TSD. Hard-clipped reads were counted as supporting evidence of the breakpoint. We summed the total reads at that position and excluded any reads that could not reliably distinguish the MEI from the reference sequence. We performed these steps for both breakpoints of the TSD. Alu #4, and L1 #1 did not have TSDs, and the L1 #5 TSD was 628 bp, so the BAFs for these loci were calculated using the two positions 1 bp before the start of the breakpoints. Then, the number of reads supporting the MEI was divided by the total number of reads at the position for each breakpoint. We used the highest BAF of the two breakpoints for each MEI. The BAFs and the average BAF are included in Supplemental Table S17.
MELT sensitivity analysis
We used the MELT genotype output to estimate its sensitivity in the three-generation CEPH cohort. Each transposon family was analyzed separately. For each pedigree, we used GQT (Layer et al. 2016) to extract loci that were present in at least one grandparent and at least two grandchildren to identify all of the inherited loci. Then we extracted loci that were present in at least one grandparent, at least two grandchildren, and absent in both parents (generation 2). These were deemed false-negative calls. The false-negative rate was calculated by dividing the total false-negative calls by the total inherited loci. We also calculated the false-negative/sensitivity rates for filtered loci by extracting only loci with MELT's “PASS” filter after identifying loci in GQT. These results are in Supplemental Figure S7.
Data access
Whole-genome sequencing data for the human samples from this study have been submitted to the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs001872.v1.p1.
Supplementary Material
Acknowledgments
Funding for this project was provided by the Utah Genome Project, the George S. and Dolores Doré Eccles Foundation, and National Institutes of Health grants GM118335 and GM059290 (to L.B.J.) and R00HG005846 (to H.H. and J.X.). We thank Aaron Quinlan, Brent Pedersen, Ryan Layer, and Thomas Sasani for helpful discussions. Sanger sequencing was performed at the DNA Sequencing Core Facility, University of Utah. We thank William Richards and Matt Velinder for their help with running and troubleshooting RUFUS. Finally, we wish to thank the investigators who organized the original Utah Centre d'Etude du Polymorphisme Humain collection, in particular, Ray White and Mark Leppert, as well as all the families who generously participated in the project.
Footnotes
[Supplemental material is available for this article.]
Article, supplemental material, and publication date are at http://www .genome.org/cgi/doi/10.1101/gr.247965.118.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Iskow R, Gokcumen O, Radke DW, Balasubramanian S, Pei B, Habegger L, The 1000 Genomes Project Consortium, Lee C, Gerstein M. 2013. Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division. Genome Res 23: 2042–2052. 10.1101/gr.154625.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, Hoischen A, Vissers LE, Gilissen C. 2015. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet 97: 67–74. 10.1016/j.ajhg.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantysh OB, Buzdin AA. 2009. Novel family of human transposable elements formed due to fusion of the first exon of gene MAST2 with retrotransposon SVA. Biochemistry 74: 1393–1399. 10.1134/S0006297909120153 [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, De Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. 10.1126/science.1183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EA, Keller H, Mills RE, Schmidt S, Moran JV, Weichenrieder O, Devine SE. 2008. Active Alu retrotransposons in the human genome. Genome Res 18: 1875–1883. 10.1101/gr.081737.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, et al. 2018. Ten things you should know about transposable elements. Genome Biol 19: 199 10.1186/s13059-018-1577-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr. 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100: 5280–5285. 10.1073/pnas.0831042100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KH. 2017. Transposable elements in cancer. Nat Rev Cancer 17: 415–424. 10.1038/nrc.2017.35 [DOI] [PubMed] [Google Scholar]
- Campbell CD, Eichler EE. 2013. Properties and rates of germline mutations in humans. Trends Genet 29: 575–584. 10.1016/j.tig.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wiśniowiecka-Kowalnik B, et al. 2014. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet 95: 173–182. 10.1016/j.ajhg.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 1784 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeaux MS, Roy-Engel AM, Hedges DJ, Deininger PL. 2009. Diverse cis factors controlling Alu retrotransposition: What causes Alu elements to die? Genome Res 19: 545–555. 10.1101/gr.089789.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703. 10.1038/nrg2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Hedges DJ, Herke SW, Batzer MA. 2006. Estimating the retrotransposition rate of human Alu elements. Gene 373: 134–137. 10.1016/j.gene.2006.01.019 [DOI] [PubMed] [Google Scholar]
- Damert A, Raiz J, Horn AV, Löwer J, Wang H, Xing J, Batzer MA, Löwer R, Schumann GG. 2009. 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res 19: 1992–2008. 10.1101/gr.093435.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R. 1990. Centre d'Etude du polymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics 6: 575–577. 10.1016/0888-7543(90)90491-C [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. 1999. Alu repeats and human disease. Mol Genet Metab 67: 183–193. 10.1006/mgme.1999.2864 [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA, Hutchison CA III, Edgell MH. 1992. Master genes in mammalian repetitive DNA amplification. Trends Genet 8: 307–311. 10.1016/0168-9525(92)90139-U [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewannieux M, Heidmann T. 2005. Role of poly(A) tail length in Alu retrotransposition. Genomics 86: 378–381. 10.1016/j.ygeno.2005.05.009 [DOI] [PubMed] [Google Scholar]
- Elbarbary RA, Lucas BA, Maquat LE. 2016. Retrotransposons as regulators of gene expression. Science 351: aac7247 10.1126/science.aac7247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. 2000. Human LINE retrotransposons generate processed pseudogenes. Nat Genet 24: 363–367. 10.1038/74184 [DOI] [PubMed] [Google Scholar]
- Ewing AD. 2015. Transposable element detection from whole genome sequence data. Mob DNA 6: 24 10.1186/s13100-015-0055-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH Jr. 2010. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20: 1262–1270. 10.1101/gr.106419.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Ballinger TJ, Earl D, Broad Institute Genome Sequencing and Analysis Program and Platform, Harris CC, Ding L, Wilson RK, Haussler D. 2013. Retrotransposition of gene transcripts leads to structural variation in mammalian genomes. Genome Biol 14: R22 10.1186/gb-2013-14-3-r22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, Billon V. 2018. L1 retrotransposition in the soma: a field jumping ahead. Mob DNA 9: 22 10.1186/s13100-018-0128-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30: 2503–2505. 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feusier J, Witherspoon DJ, Scott Watkins W, Goubert C, Sasani TA, Jorde LB. 2017. Discovery of rare, diagnostic AluYb8/9 elements in diverse human populations. Mob DNA 8: 9 10.1186/s13100-017-0093-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, Mills RE, The 1000 Genomes Project Consortium, Devine SE. 2017. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res 27: 1916–1929. 10.1101/gr.218032.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EJ, Prigmore E, Gallone G, Short PJ, Sifrim A, Singh T, Chandler KE, Clement E, Lachlan KL, Prescott K, et al. 2018. Contribution of retrotransposition to developmental disorders. bioRxiv 10.1101/471375 [DOI] [PMC free article] [PubMed]
- Goerner-Potvin P, Bourque G. 2018. Computational tools to unmask transposable elements. Nat Rev Genet 19: 688–704. 10.1038/s41576-018-0050-x [DOI] [PubMed] [Google Scholar]
- Goodier JL. 2016. Restricting retrotransposons: a review. Mob DNA 7: 16 10.1186/s13100-016-0070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, Brors B. 2014. Circlize implements and enhances circular visualization in R. Bioinformatics 30: 2811–2812. 10.1093/bioinformatics/btu393 [DOI] [PubMed] [Google Scholar]
- Han K, Xing J, Wang H, Hedges DJ, Garber RK, Cordaux R, Batzer MA. 2005. Under the genomic radar: the stealth model of Alu amplification. Genome Res 15: 655–664. 10.1101/gr.3492605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22: 191–203. 10.1016/j.gde.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH. 2016. Roles for retrotransposon insertions in human disease. Mob DNA 7: 9 10.1186/s13100-016-0065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Ewing AD, Chen JE, Tokunaga K, Kazazian HH Jr. 2009. Exon-trapping mediated by the human retrotransposon SVA. Genome Res 19: 1983–1991. 10.1101/gr.093153.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, Yorukoglu D, Dao P, Bakhshi M, Sahinalp SC, et al. 2011. Alu repeat discovery and characterization within human genomes. Genome Res 21: 840–849. 10.1101/gr.115956.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. 1999. CAP3: a DNA sequence assembly program. Genome Res 9: 868–877. 10.1101/gr.9.9.868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, et al. 2010. Mobile interspersed repeats are major structural variants in the human genome. Cell 141: 1171–1182. 10.1016/j.cell.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2003. The International HapMap Project. Nature 426: 789–796. 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. 10.1038/nature06258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, et al. 2017. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549: 519–522. 10.1038/nature24018 [DOI] [PubMed] [Google Scholar]
- Jónsson H, Sulem P, Arnadottir GA, Pálsson G, Eggertsson HP, Kristmundsdottir S, Zink F, Kehr B, Hjorleifsson KE, Jensson BÖ, et al. 2018. Multiple transmissions of de novo mutations in families. Nat Genet 50: 1674–1680. 10.1038/s41588-018-0259-9 [DOI] [PubMed] [Google Scholar]
- Jorde LB. 1989. Inbreeding in the Utah Mormons: an evaluation of estimates based on pedigrees, isonymy, and migration matrices. Ann Hum Genet 53: 339–355. 10.1111/j.1469-1809.1989.tb01803.x [DOI] [PubMed] [Google Scholar]
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH Jr. 2009. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23: 1303–1312. 10.1101/gad.1803909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH. 1999. An estimated frequency of endogenous insertional mutations in humans. Nat Genet 22: 130 10.1038/9638 [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Moran JV. 2017. Mobile DNA in health and disease. N Engl J Med 377: 361–370. 10.1056/NEJMra1510092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. 1988. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332: 164–166. 10.1038/332164a0 [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-Like Alignment Tool. Genome Res 12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The Human Genome Browser at UCSC. Genome Res 12: 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103. 10.1038/nature09525 [DOI] [PubMed] [Google Scholar]
- Konkel MK, Walker JA, Hotard AB, Ranck MC, Fontenot CC, Storer J, Stewart C, Marth GT, The 1000 Genomes Consortium, Batzer MA. 2015. Sequence analysis and characterization of active human Alu subfamilies based on the 1000 Genomes pilot project. Genome Biol Evol 7: 2608–2622. 10.1093/gbe/evv167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryatova MS, Steranka JP, Burns KH, Payer LM. 2017. Insertion and deletion polymorphisms of the ancient AluS family in the human genome. Mob DNA 8: 6 10.1186/s13100-017-0089-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Kindlon N, Karczewski KJ; Exome Aggregation Consortium, Quinlan AR. 2016. Efficient genotype compression and analysis of large genetic-variation data sets. Nat Methods 13: 63–65. 10.1038/nmeth.3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Scaringe WA, Hill KA, Roberts S, Mengos A, Careri D, Pinto MT, Kasper CK, Sommer SS. 2001. Frequency of recent retrotransposition events in the human factor IX gene. Hum Mutat 17: 511–519. 10.1002/humu.1134 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, et al. 2016. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538: 201–206. 10.1038/nature18964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan T, Jorde LB, Skolnick MH. 1984. Genetic distances between the Utah Mormons and related populations. Am J Hum Genet 36: 836–857. [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, Devine SE. 2007. Which transposable elements are active in the human genome? Trends Genet 23: 183–191. 10.1016/j.tig.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV. 2002. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet 31: 159–165. 10.1038/ng898 [DOI] [PubMed] [Google Scholar]
- Nellåker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, Flint J, Adams DJ, Frankel WN, Ponting CP. 2012. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol 13: R45 10.1186/gb-2012-13-6-r45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newkirk SJ, Lee S, Grandi FC, Gaysinskaya V, Rosser JM, Vanden Berg N, Hogarth CA, Marchetto MCN, Muotri AR, Griswold MD, et al. 2017. Intact piRNA pathway prevents L1 mobilization in male meiosis. Proc Natl Acad Sci 114: E5635–E5644. 10.1073/pnas.1701069114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander BEP, Butterfield RJ, Pedersen BS, Farrell AJ, Layer RM, Ward A, Miller C, DiSera T, Filloux FM, Candee MS, et al. 2018. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom Med 3: 22 10.1038/s41525-018-0061-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Quinlan AR. 2017. Who's who? Detecting and resolving sample anomalies in human DNA sequencing studies with Peddy. Am J Hum Genet 100: 406–413. 10.1016/j.ajhg.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzkofer T, Jäger M, Figlerowicz M, Badge R, Mundlos S, Robinson PN, Zemojtel T. 2017. L1Base 2: more retrotransposition-active LINE-1s, more mammalian genomes. Nucleic Acids Res 45: D68–D73. 10.1093/nar/gkw925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SM, Lalouel JM, Leppert M. 2008. From linkage maps to quantitative trait loci: the history and science of the Utah genetic reference project. Annu Rev Genomics Hum Genet 9: 347–358. 10.1146/annurev.genom.9.081307.164441 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. 2016. Timing, rates and spectra of human germline mutation. Nat Genet 48: 126–133. 10.1038/ng.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaby R, Sung WK. 2018. TranSurVeyor: an improved database-free algorithm for finding non-reference transpositions in high-throughput sequencing data. Nucleic Acids Res 46: e122 10.1093/nar/gky685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Faulkner GJ. 2018. Heritable L1 retrotransposition events during development: understanding their origins. Bioessays 40: 1700189 10.1002/bies.201700189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Gerdes P, Gerhardt DJ, Sanchez-Luque FJ, Bodea GO, Muñoz-Lopez M, Jesuadian JS, Kempen MHC, Carreira PE, Jeddeloh JA, et al. 2017. Heritable L1 retrotransposition in the mouse primordial germline and early embryo. Genome Res 27: 1395–1405. 10.1101/gr.219022.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishishwar L, Mariño-Ramírez L, Jordan IK. 2016. Benchmarking computational tools for polymorphic transposable element detection. Brief Bioinform 18: 908–918. 10.1093/bib/bbw072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639. 10.1126/science.1186802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasani TA, Pedersen BS, Gao Z, Baird L, Przeworski M, Jorde LB, Quinlan AR. 2019. Large, three-generation CEPH families reveal post -zygotic mosaicism and variability in germline mutation accumulation. bioRxiv 10.1101/552117 [DOI] [PMC free article] [PubMed]
- Schrider DR, Navarro FC, Galante PA, Parmigiani RB, Camargo AA, Hahn MW, de Souza SJ. 2013. Gene copy-number polymorphism caused by retrotransposition in humans. PLoS Genet 9: e1003242 10.1371/journal.pgen.1003242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EC, Gardner EJ, Masood A, Chuang NT, Vertino PM, Devine SE. 2016. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res 26: 745–755. 10.1101/gr.201814.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségurel L, Wyman MJ, Przeworski M. 2014. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet 15: 47–70. 10.1146/annurev-genom-031714-125740 [DOI] [PubMed] [Google Scholar]
- Seleme MC, Vetter MR, Cordaux R, Bastone L, Batzer MA, Kazazian HH Jr. 2006. Extensive individual variation in L1 retrotransposition capability contributes to human genetic diversity. Proc Natl Acad Sci 103: 6611–6616. 10.1073/pnas.0601324103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen SK, Huang CT, Han K, Batzer MA. 2007. Endonuclease-independent insertion provides an alternative pathway for L1 retrotransposition in the human genome. Nucleic Acids Res 35: 3741–3751. 10.1093/nar/gkm317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens EL, Heckenberg G, Baugher JD, Roberson ED, Downey TJ, Pevsner J. 2012. Consanguinity in Centre d’Étude du Polymorphisme Humain (CEPH) pedigrees. Eur J Hum Genet 20: 657–667. 10.1038/ejhg.2011.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. 2014. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345: 1251343 10.1126/science.1251343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, et al. 2007. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet 16: 1587–1592. 10.1093/hmg/ddm108 [DOI] [PubMed] [Google Scholar]
- Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS. 1991. A de novo Alu insertion results in neurofibromatosis type 1. Nature 353: 864–866. 10.1038/353864a0 [DOI] [PubMed] [Google Scholar]
- Werling DM, Brand H, An J, Stone MR, Zhu L, Glessner JT, Collins RL, Dong S, Layer RM, Markenscoff-Papadimitriou E, et al. 2018. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet 50: 727–736. 10.1038/s41588-018-0107-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Leppert M, Bishop DT, Barker D, Berkowitz J, Brown C, Callahan P, Holm T, Jerominski L. 1985. Construction of linkage maps with DNA markers for human chromosomes. Nature 313: 101–105. 10.1038/313101a0 [DOI] [PubMed] [Google Scholar]
- Wildschutte JH, Williams ZH, Montesion M, Subramanian RP, Kidd JM, Coffin JM. 2016. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci 113: E2326–E2334. 10.1073/pnas.1602336113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems T, Zielinski D, Yuan J, Gordon A, Gymrek M, Erlich Y. 2017. Genome-wide profiling of heritable and de novo STR variations. Nat Methods 14: 590–592. 10.1038/nmeth.4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Watkins WS, Witherspoon DJ, Zhang Y, Guthery SL, Thara R, Mowry BJ, Bulayeva K, Weiss RB, Jorde LB. 2009a. Fine-scaled human genetic structure revealed by SNP microarrays. Genome Res 19: 815–825. 10.1101/gr.085589.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, et al. 2009b. Mobile elements create structural variation: analysis of a complete human genome. Genome Res 19: 1516–1526. 10.1101/gr.091827.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.