

Abstract
Recent advances have highlighted the ability of hematopoietic stem and progenitor cells in the bone marrow to sense peripheral inflammation or infection and adapt through increased proliferation and skewing toward the myeloid lineage. Such adaptations can meet the increased demand for innate immune cells and can be beneficial in response to infection or myeloablation. However, the inflammation-induced adaptation of hematopoietic and myeloid progenitor cells towards enhanced myelopoiesis might also perpetuate inflammation in chronic inflammatory or cardio-metabolic diseases by generating a feed-forward loop between inflammation-adapted hematopoietic progenitor cells and the inflammatory disorder. Sustained adaptive responses of progenitor cells in the bone marrow can also contribute to trained immunity, a non-specific memory of earlier encounters that in turn facilitates their heightened response, as well as that of their progeny, to future challenges. Here, we discuss the mechanisms that govern the adaptation of hematopoietic progenitor cells to inflammation and its sequelae in the pathogenesis of human disease.
Hematopoietic stem cells (HSCs) and hematopoietic progenitor cells in the bone marrow (BM) are responsible for the maintenance of steady-state and stress-adapted hematopoiesis1–3. HSCs (also named long-term HSC (LT-HSCs)) reside at the top of the hematopoietic hierarchy, have self-renewal and multi-lineage differentiation capacity and give rise to all mature blood cells. LT-HSCs can differentiate into short-term HSCs (ST-HSCs) and multi-potent progenitors (MPPs), which are cells with relatively restricted differentiation potential; LT-HSCs, ST-HSCs and MPPs are collectively known as hematopoietic stem and progenitor cells (HSPCs)1,4. HSPCs are enriched within the lineage−Sca-1+c-Kit+ (LSK) population, wherein LT-HSCs and MPPs can be differentiated with distinct markers, for instance SLAM family markers1,4. MPPs differentiate to oligo-potent and uni-potent progenitor cells, such as common myeloid progenitors (CMPs), granulocyte-monocyte progenitors (GMPs) and others, which give rise to mature blood cells2,3.
HSC maintenance is facilitated by the highly specialized niche micro-environment, comprising different cell types, such as mesenchymal stromal cells (MSC), endothelial cells (EC) and megakaryocytes5. The HSC niche fosters a variety of juxtracrine (cell-cell or cell-matrix) and paracrine (via cytokines, chemokines or growth factors) interactions involving the HSCs, thereby regulating the self-renewal of HSCs and instructing the differentiation of hematopoietic progenitor cells5,6. As an adaptation to the hypoxic niche micro-environment, HSCs preferentially engage aerobic glycolysis for energy production rather than oxidative phosphorylation; consistently, the glycolytic metabolism of HSCs promotes the maintenance of their stemness7.
The traditional tree-like hierarchical model of hematopoiesis1, in which lineage decision takes place at the MPP stage, is constantly redrawn. The use of new technologies, such as single-cell analysis8–12, has resulted in alternative models of hematopoiesis that suggest a greater functional heterogeneity of HSCs (reviewed in ref.3). Additionally, several surface markers have been identified to characterize lineage-biased HSCs and MPPs13,14. For instance, based on the expression of Flt3 and CD150, MPPs may form three distinct subpopulations, which are primed towards erythro-megakaryocytic (Flt3−CD150+MPPs or MPP2), myeloid (Flt3−CD150−MPPs or MPP3) or lymphoid (Flt3+CD150−MPPs or MPP4) lineage14. Another example is the identification of CD41 as a marker of myeloid-biased HSCs13. Moreover, HSC clones primed for generation of megakaryocytes and platelets have been also reported8,9.
In both humans and mice, early lineage priming of HSCs is controlled by distinct gene expression modules, which are regulated by lineage-specific transcription factors 11,15. For example, the C/EBP family of transcription factors and PU.1 regulate the entry into myeloid lineage15–17. As outlined below in more detail, activation of such lineage-specific transcription factors is also critical for the adaptation of hematopoietic progenitors to inflammatory stimuli18. Further differentiation decisions in myelopoiesis, a process leading to the generation of specific myeloid cell subsets, granulocytes, monocytes and dendritic cells19,20, occur at the level of myeloid progenitors21 and are fine-tuned by additional transcription factors and epigenetic mechanisms22–24.
Although it is debated whether HSCs contribute to unperturbed hematopoiesis25,26, HSCs are critical for hematopoiesis reconstitution upon myeloablation. In this setting, activation of HSCs results in massively increased myelopoiesis as compared to lymphopoiesis25. Upon transplantation-associated hematopoietic stress, HSCs can bypass several steps of the traditional tree-like hematopoietic hierarchy via direct differentiation into myeloid progenitors through asymmetric division12, a process giving rise to progeny cells with distinct fates, e.g., a stem cell and a lineage-committed cell. Hence, upon demand (e.g., following myeloablation or inflammation), HSCs rapidly respond to replenish myeloid cells through a process designated as emergency myelopoiesis20. Furthermore, the rapid adaptation of hematopoietic progenitors to severe bacterial infections leading to peripheral blood neutrophilia is designated as emergency granulopoiesis2. Although meeting the increased demand for immune cell generation, such adaptations of hematopoietic progenitors may contribute to chronicity of inflammatory diseases27. Herein, we review emerging evidence that the adaptation of hematopoietic progenitor cells to inflammation acts as a central hub of the host response to infectious and inflammatory challenges.
Adaptation of hematopoietic progenitor cells to inflammation
Similar to the mature immune cells and committed myeloid progenitor cells in the BM, HSPCs can also become directly responsive to acute infection or chronic inflammatory conditions1,2,27. This HSPC adaptation is tightly orchestrated by a combination of cell-intrinsic (transcriptional, epigenetic and metabolic) and cell-extrinsic (soluble growth factors, cytokines, microbial ligands and adhesive interactions) mechanisms2,28. HSPCs are more sensitive to exogenous stimuli than traditionally thought, owing to their expression of receptors for microbial products, such as the Toll-like receptors (TLRs) and receptors for inflammatory cytokines and growth factors, such as IL-1β, IL-6, M-CSF, type 1 and 2 interferons (IFN), the abundance of which massively increases upon systemic infection, thereby enabling HSPCs to sense a systemic infectious or inflammatory challenge directly or indirectly1,2. While such HSPC responses can be beneficial in promoting elimination of an infection, they may also cause impairment of HSPC function and exhaustion if HSPC activation is chronically sustained2 or may contribute to chronicity of inflammatory pathologies27. The underlying mechanisms and functional outcomes of HSPC adaptation to inflammatory stimuli have only now begun to be understood. Below we discuss the complex regulatory interactions at the interface between inflammation and HSCs.
Effects of TLR signaling on HSC fate decision and regulation of myelopoiesis
The seminal observation that hematopoietic progenitors express TLRs and that TLR ligation induces myeloid differentiation in a manner dependent on the adaptor MyD88 provided new insight in the role of HSPCs in the host defense against pathogens29. Subsequent studies have identified indirect, cytokine-mediated, and direct effects of TLR ligands on HSPCs (Fig.1). Upon activation of the TLRs, ST-HSCs and MPPs produce substantial amounts of cytokines in an manner dependent on the transcription factor NF-κB30. These cytokines, particularly IL-6, can promote, in a paracrine fashion, the proliferation and myeloid differentiation of HSPCs30. Additionally, systemic LPS administration stimulates enhanced HSC proliferation31, while chronic low-dose LPS treatment results in expansion of HSCs and their myeloid skewing in a cell-intrinsic, TLR4-dependent manner, as shown in BM chimera experiments using cells from TLR4-deficient and -proficient mice32. However, chronic low-dose murine endotoxemia induces the functional impairment of HSCs and loss of quiescence, as shown by their reduced repopulation capacity in serial transplantation experiments33. Moreover, high-dose LPS results in increased BM cell death and dysfunction of HSCs, despite their initial expansion34.
Figure 1. Inflammatory adaptation of hematopoietic progenitors.
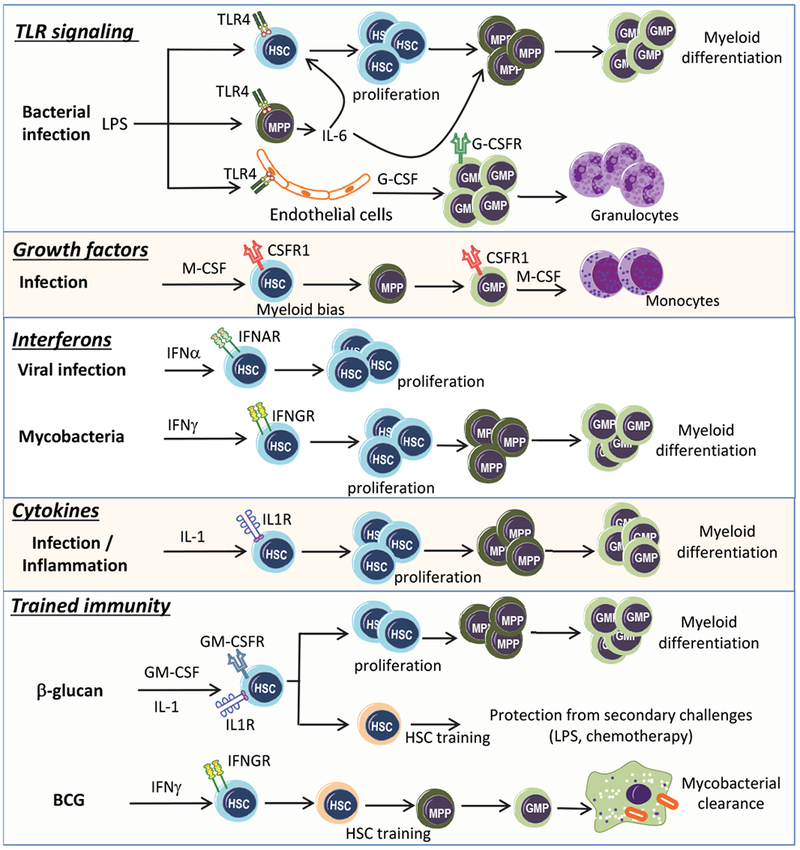
Hematopoietic stem cells (HSC) and multipotent progenitors (MPP) express Toll-like receptors (TLR) sensing directly pathogen-derived products, such as LPS, which can drive HSC proliferation31. LPS also stimulates release of cytokines, such as IL-6, from MPPs; IL-6 acts in a paracrine manner to promote HSC proliferation and enhanced myelopoiesis30. In parallel, LPS stimulates release of G-CSF from endothelial cells38. G-CSF acts on myeloid progenitors, especially on granulocyte macrophage progenitors (GMP) driving their differentiation towards granulocytes. In the course of infection, M-CSF can act direcly on HSCs, promoting their myeloid differentiation53, and on GMPs to promote monocyte generation51. IFN-α, produced in response to viral infections, induces HSC cell-cycle entry at the expense of their self-renewal potential55. IFN-γ, produced in the course of mycobacterial infection, results in HSC proliferation and their instruction towards the myeloid lineage64. Release of IL-1 during infection or inflammation drives proliferation and myeloid differentiation of HSCs18. Trained immunity induced by β-glucan drives the proliferation and sustained myeloid bias in HSCs through IL-1 and GM-CSF. Trained immunity mediates a beneficial response of HSCs to secondary challenges, such as chemotherapy and LPS administration98. Additionally, trained immunity induced by BCG reprograms HSCs resulting in the generation of macrophages with enhanced anti-mycobacterial properties68.
Hence, TLR4 activation on HSCs stimulates their proliferation, but may exert several detrimental actions on their function due to proliferative stress in HSCs upon TLR4 ligation33,35. Specifically, TLR4 activation stimulates the proliferation of dormant HSCs, while diminishing their self-renewal and repopulation capacity36. These effects are mediated by signaling dependent on the adaptor TRIF and subsequent downstream production of reactive oxygen species (ROS) and activation of the mitogen-activated protein kinase p38, leading to replication stress and activation of the DNA repair machinery36. Scavenging ROS or blockade of p38 prevents this endotoxemia-related HSC exhaustion36. Consistently, TRIF mediates the expansion and functional exhaustion of HSPCs in sepsis35.
Different TLR ligands may lead to different emergency myelopoiesis responses by engaging distinct pathways leading to the generation of Ly6Chi monocytes37. Specifically, stimulation of TLR4 by LPS causes an increase in neutrophils and neutrophil-like monocytes derived from GMPs, while stimulation of TLR9 by CpG results in expansion of dendritic cells and of a functionally distinct monocyte subtype derived from monocyte-dendritic cell progenitors37. Besides the direct effects of TLR agonists on hematopoietic progenitors, endotoxemia also induces the production of myeloid-lineage growth factors (discussed in detail below), thereby supporting the replenishment of blood neutrophils38. Importantly, the TLR4-MyD88 signaling pathway also mediates LPS-related emergency granulopoiesis through a paracrine pathway, which does not involve direct effects on HSPCs, but requires the production of the growth factor G-CSF by endothelial cells38 (Fig.1), as discussed below.
Effect of growth factors on the regulation of myelopoiesis
G-CSF is a major driver of granulopoiesis under both steady-state and emergency conditions, such as infection, by regulating the expression of myeloid lineage-specific transcription factors and of receptors for myeloid lineage-specific growth factors2,39. Other growth factors contributing to emergency myelopoiesis are M-CSF and GM-CSF2. However, mice deficient in these factors (G-MCF, GM-CSF and M-CSF) can still generate modest numbers of myeloid cells, at least in response to sterile peritoneal inflammation, highlighting a functional redundancy in myelopoiesis40. GM-CSF may act alone or together with other cytokines, such as IL-3, in the regulation of myelopoiesis, specifically during chronic inflammation or bacterial infection2,41.
During emergency granulopoiesis, G-CSF induces proliferative and lineage-specification signals by acting primarily on CMPs and GMPs, which expand and form well-defined clusters that differentiate into granulocytes39,42,43. Besides affecting the differentiation of myeloid progenitor cells, G-CSF may also induce the proliferation of HSCs44. Moreover, G-CSF stimulates the mobilization of HSCs from the BM in an indirect fashion, by acting on monocytes in the HSC niche45,46. G-CSF-induced activation of its receptor, G-CSFR, leads to signaling through the transcription factor STAT3, which directly stimulates the expression of the master transcriptional regulator of emergency granulopoiesis, C/EBPβ39,42; contrastingly, C/EBPα is the major driver of steady-state granulopoiesis2. The effects of STAT3 are counteracted by the regulator of cytokine signal transduction SOCS3, which downregulates G-CSF-induced emergency granulopoiesis; indeed, hematopoietic cell-specific deficiency of SOCS3 results in sustained G-CSF-induced activation of STAT3, enhanced neutrophilia and mobilization of hematopoietic progenitor cells47. Consistently, the epigenetic modifier TET2, which is mutated in patients with myeloid malignancies48, promotes infection-induced emergency myelopoiesis by suppressing the expression of SOCS349.
The generation of monocytes in the BM occurs in a manner dependent on the receptor CSFR1, which binds M-CSF50. M-CSF acts directly on hematopoietic and myeloid progenitor cells and induces their differentiation into monocytes (Fig.1). In vitro treatment of GMPs with M-CSF promotes their differentiation to mature monocytes51. The M-CSF-instructed monocyte lineage choice in GMPs is mediated by Src family kinase signaling52. M-CSF can instruct myeloid lineage identity in single HSCs, independently of HSC survival or proliferation, by inducing a PU.1-dependent molecular signature53. This mechanism of M-CSF-dependent myelopoiesis may protect against opportunistic infections post-HSC transplantation54.
Effect of IFN on HSCs fate decision and the regulation of myelopoiesis
Type 1 IFN, especially IFN-α, and type 2 IFN (IFN-γ) are central cytokines in the adaptation of HSPCs to inflammation. The type 1 IFN inducer polyinosinic:polycytidylic acid (polyI:C), or IFN-α itself, can drive proliferation of dormant HSCs (Fig.1) through a pathway dependent on the IFN-α/β receptor IFNAR and STAT1; however, chronic administration of IFN-α results in impaired HSC repopulation capacity55. The transcription factor Irf2, a negative regulator of type 1 IFN signaling, counteracts the effects of type 1 IFN on HSCs and protects quiescent HSCs from type 1 IFN-induced proliferation-associated exhaustion56. This type 1 IFN-dependent functional impairment and attrition of HSCs is due to induction of DNA damage in HSCs entering the cell cycle, which is associated with enhanced mitochondrial membrane potential and mitochondrial production of ROS57. However, type 1 IFN-triggered proliferation of HSCs may be brief, due to the transient reduction in the expression of genes enforcing quiescence (e.g., p27, p57, Foxo1, Foxo3a or Pten), while reestablishment of HSC quiescence can protect them from the pro-apoptotic effects of IFN-α58. Hematopoietic cells may engage additional homeostatic protective mechanisms against IFN-α-induced dysfunction. For example, all-trans retinoic acid-induced signaling protects dormant HSCs against polyI:C-induced replicative stress59. The cyclic GMP-AMP (cGAMP) synthase cGAS is an intracellular sensor of pathogen-derived DNAs, mostly from viruses. Upon DNA binding, cGAS generates cGAMP, which – together with the adapter STING – stimulates the production of type 1 IFN60. Interestingly, a circular RNA antagonizing cGAS synthase activity, named cia-cGAS, is highly enriched in the nucleus of LT-HSCs and promotes maintenance of their dormancy by protecting HSCs against cGAS-type 1 IFN-mediated exhaustion61. The complexity of the IFN-α effects on the BM is further exemplified by the observations that acute IFN-α-mediated inflammation acts on HSC-niche endothelial cells, affecting vascularity and vessel leakage in the BM62, whereas type 1 IFN can additionally stimulate proliferation and post-trancriptional protein synthesis in a primed subpopulation of stem cell-like megakaryocyte-committed progenitor cells phenotypically residing within the HSCs, leading to rapid platelet replenishment in acute inflammation63.
IFN-γ also modulates the maintenance and proliferation of HSCs. Infection with Mycobacterium avium triggers the proliferation of HSCs in an IFN-γ, rather than IFN-α-dependent manner64 (Fig.1). Consistently, HSC proliferation is abrogated in mice with genetic deficiency of either the receptor for IFN-γ or its downstream signal transducer STAT164, whereas administration of recombinant IFN-γ reverses the effect, suggesting that IFN-γ stimulates HSC cell cycle entry64. However, the effects of IFN-γ on HSCs are context-dependent, as IFN-γ inhibits the proliferation of HSCs, impairs their maintenance and negatively affects their recovery during infection with the lymphocytic choriomeningitis virus (LCMV)65. In the context of LCMV infection, IFN-γ induces myeloid differentiation by acting either directly on myeloid-biased HSC66 or indirectly; specifically, IFN-γ from cytotoxic T cells stimulates a paracrine pathway leading to enhanced production of IL-6 from BM-MSCs, resulting in heightened proliferation and myelopoiesis of MPPs67. Additionally, IFN-γ is responsible for the attrition of HSCs due to their differentiation into myeloid cells and loss of their self-renewal capacity during chronic mycobacterial infection in mice66. Furthermore, intravenously administered mycobacterial Bacille Calmette-Guérin (BCG) triggers the IFN-γ-dependent expansion of HSPCs and their myeloid differentiation through a myelopoiesis-related transcriptional program68 (Fig.1).
IL-1β and other cytokines effects on HSCs fate decision
IL-1β, a central mediator in innate immunity69, acts directly on HSCs in vitro, promoting their proliferation and myeloid differentiation through activation of the transcription factor PU.118 (Fig.1). Administration of IL-1β to mice results in increased numbers of myeloid-biased HSPCs, while BM recovery after chemotherapeutic injury is delayed in IL-1 receptor-deficient mice18. Chronic administration of IL-1β diminishes the self-renewal capacity of HSCs, although this effect is reversed upon IL-1β withdrawal18. IL-1β, in fact, may account for the hematologic abnormalities in patients with X-linked chronic granulomatous disease (X-CGD) and in the respective mouse model, both of which are characterized by IL-1β-associated hyperinflammation70. Patients with X-CGD have decreased HSC counts, whereas mice with targeted disruption in the X-linked gene gp91phox have increased HSC proliferation and impaired reconstitution capacity, as well as boosted myelopoiesis, as assessed by enhanced numbers of GMPs70. An IL-1 receptor antagonist partially restores hematopoiesis in mice with X-CGD70, suggesting IL-1 signaling is in part responsible for this phenotype. Short-term or low-dose administration of IL-1α or IL-1β prevents chemotherapy-induced myelosuppression71,72 and protects cyclophosphamide-induced neutropenic mice against sepsis72, indicating timing and dosing may be critical regulators of the beneficial or detrimental effects of IL-1 on HSCs. While IL-1 may promote restoration of myelopoiesis, persistent IL-1-mediated inflammation negatively impacts HSC functionality.
Tumor necrosis factor (TNF) induces proliferation and instructs myeloid lineage decisions in HSCs and may thus compromise their repopulation capacity73. TNF directly upregulates PU.1 in vitro and mediates its activation in LPS-administered mice, because PU.1 is not affected by LPS in TNF-deficient mice74. IL-3, derived from innate response activator B cells, amplifies myelopoiesis and inflammation in sepsis. Specifically, the expansion of HSPC and myeloid progenitor cells is hindered by IL-3 deficiency in murine sepsis, while high concentrations of IL-3 in plasma correlate with higher mortality in human sepsis75. IL-6 regulates HSPC proliferation and differentiation during emergency myelopoiesis2,30. In this context, IL-6 may be derived either from the HSPCs following TLR stimulation30 (Fig.1) or from BM-MSCs upon stimulation with cytotoxic T cell-derived IFN-γ67. IL-6, through activation of signaling though SHP2 and STAT3, can also support survival of TET2-deficient HSPCs and myeloid cells76; as TET2 mutations are linked with clonal hematopoiesis and a higher risk for leukemia development, these observations may indicate how inflammation may promote survival of preleukemic HSPCs76. IL-27, a member of the IL-6-IL-12 cytokine family, also acts on HSPCs and promotes emergency myelopoiesis77. In a model of malarial infection, deletion of the IL-27R subunit WSX-1 suppressed the expansion of progenitor cells and their differentiation into myeloid cells, which resulted in increased parasitemia77.
Modulation of inflammation-induced HSPC adaptation by the BM niche
Emergency myelopoiesis is also regulated by the HSC niche microenvironment in the BM. The niche modulates HSC quiescence, self-renewal, proliferation and differentiation into committed progenitor cells5,28,78,79 (Fig.2). Niche cells such as endothelial cells, nestin+ MSCs, perivascular stromal cells, including CXCL12-abundant reticular (CAR) cells, and osteolineage cells regulate HSC function through paracrine signals (growth factors or chemokines) or through adhesive interactions that may also involve extracellular matrix proteins5,28,78,79. DEL-1, an adhesive protein secreted by niche endothelial cells, CAR cells and osteolineage cells in the BM interacts with β3 integrin on HSCs and enhances the proliferation of LT-HSCs and their myeloid differentiation, predominantly in the context of LPS- or G-CSF-induced stress myelopoiesis80. Another matrix-associated protein, osteopontin, restrains emergency myelopoiesis in favor of lymphopoiesis through a dual mechanism mediated by two distinct isoforms: intracellular osteopontin is pro-apoptotic in myeloid progenitors, whereas secreted osteopontin is anti-apoptotic in differentiated lymphoid cells81. Furthermore, BM innervation and sympathoadrenergic activity are important HSC niche regulators and control the circadian HSC release into the circulation; conversely, loss of sympathoadrenergic nerves promotes aging of the niche and impairs regeneration of hematopoiesis82–84.
Figure 2. Regulation of HSCs by their progeny in the BM niche.
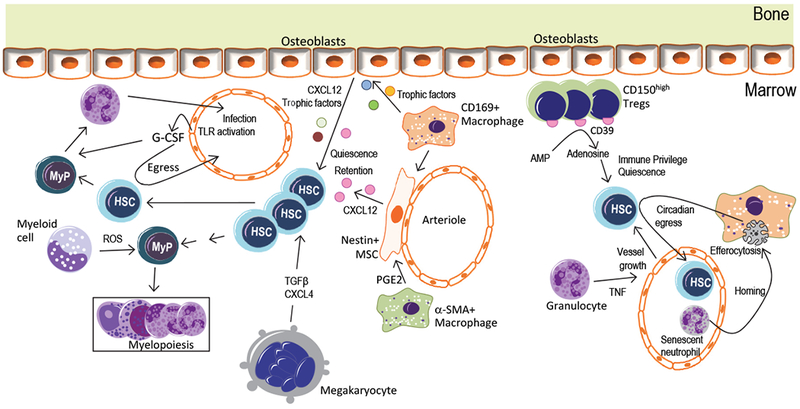
Innate and adaptive immune cells in the BM, as well as other HSC progeny, such as megakaryocytes, contribute to HSPC adaptation to inflammatory stimuli derived from systemic infection or inflammation. G-CSF, produced by endothelial cells in response to LPS or systemic infection, acts on myeloid progenitors (MyP) stimulating emergency granulopoiesis38,39 and promotes the egress of HSPCs from the BM45. CD169+ macrophages in the BM niche interact with osteoblasts or nestin+ perivascular cells and control their expression of HSC-trophic factors, including CXCL12, which contributes to HSC retention in the niche85,86. CD169+ macrophages phagocytose apoptotic neutrophils which, upon senescence, home back to the BM; this process promotes the circadian egress of hematopoietic progenitors into the circulation88. α-SMA+ macrophages secrete PGE2, which acts on nestin+ cells, which in turn secrete CXCL12, thereby contributing to HSC quiescence and retention87. In response to systemic infection or inflammation, Gr1+ myeloid cells in the BM release ROS, which stimulates the expansion and differentiation of MyPs, thus contributing to demand-adapted myelopoiesis89. Moreover, under inflammatory conditions, granulocytes in the BM can secrete TNF, which stimulates vessel growth and thus indirectly promotes HSPC regeneration90. CD4+CD25+FoxP3+ Treg cells accumulate on the endosteal BM niche and confer immune privilege to the HSC niche. CD150hi Treg cells support HSC quiescence and engraftment via ectoenzyme CD39-generated adenosine95. Megakaryocytes control HSC quiescence through TGFβ1 or CXCL4 signaling91,92.
The progeny of HSCs in the BM may be important mediators for the adaptation of HSPC to inflammatory signals. Monocytes are a major cellular target of G-CSF-induced egress of HSPCs from the BM niche and emergency granulopoiesis46. G-CSF-induced mobilization of HSPCs is linked with depletion of trophic endosteal macrophages in the BM85. CD169+ macrophages in the BM may interact with osteoblasts or nestin+ cells in the niche to control expression of trophic factors such as angiopoietin-1, CXCL12 and Kit ligand on HSC and retention of HSPC in the niche85,86. Consistently, depletion of BM macrophages is sufficient to induce HSPC mobilization85. α-SMA+ monocytes and macrophages may protect HSPCs from exhaustion in stress conditions such as sublethal irradiation, in part because PGE2 produced from these macrophages upregulates nestin+ stromal cell-derived CXCL12, which is critical for stem-cell quiescence87. BM macrophages can also regulate the HSC niche through the phagocytosis of apoptotic cells (known as efferocytosis). Senescent neutrophils in the circulation upregulate their expression of CXCR4 and home back to the BM, where they are efferocytosed by BM-resident CD169+ macrophages, a process that in turn promotes the circadian egress of hematopoietic progenitor cells into the circulation88 (Fig.2).
Neutrophils and their precursors in the BM also contribute to demand-adapted myelopoiesis. In response to systemic inflammation, Gr1+ myeloid cells in the BM release ROS, which stimulates the proliferation and differentiation of myeloid progenitor cells, thus enhancing myeloid cell output89. Granulocyte-derived TNF promotes vessel growth and thereby HSPC regeneration90. Megakaryocytes regulate HSC quiescence through TGFβ1 or CXCL4 and may promote post-injury regeneration of HSCs through FGF-191,92. Megakaryocytes preferentially regulate the proliferation of vWF+ platelet- and myeloid-biased HSCs93. In vivo high-resolution imaging revealed the co-localization of HSPCs with CD4+CD25+FoxP3+ regulatory T cells (Treg cells) that accumulate on the endosteal BM niche94. These Treg cells may confer immune privilege to the HSPC niche, thus preventing allogeneic rejection of transplanted HSPCs or protecting endogenous HSPCs from excessive inflammation. In support of this notion, a niche-specific subpopulation of CD150hi Treg cells promotes HSC quiescence and engraftment of HSCs through a mechanism dependent on adenosine generated by the ectoenzyme CD39, which protects HSCs from oxidative stress95 (Fig.2). Together, different cells in the HSC niche contribute to context-dependent HSPC adaptation to diverse inflammatory challenges.
The effect of trained immunity on HSPCs
Trained immunity is a form of adaptation that enhances the response of innate immune cells to secondary challenges independently of adaptive immunity96,97. Trained immunity can be triggered by certain microbial components, such as the fungal cell-wall constituent β-glucan or vaccines like BCG, and involves immunometabolic and epigenetic alterations in target cells96,97. Trained innate immunity operates in hematopoietic progenitor cells68,98, resolving an earlier paradox regarding the long-term effect of trained immunity in myeloid cells with a relatively short lifespan. Trained immunity induced by injection of β-glucan in mice leads to the expansion of myeloid-biased HSPCs, such as CD41+LT-HSCs and MPP3 cells, in the BM98. β-glucan-induced myelopoiesis requires signaling through IL-1β and is associated with enhanced glycolysis and cholesterol biosynthesis in HSPCs. Increased cholesterol amounts in HSPCs from β-glucan-trained mice results in enhanced signaling via CD131, the common β-subunit of the IL-3/GM-CSF receptor (IL-3Rβ), in LT-HSCs and MPPs98. Consistently, elevated expression of IL-3Rβ and enhanced myeloid bias are observed in HSPCs upon blockade of cholesterol efflux99. Importantly, β-glucan-induced training of HSPCs and enhancement of myelopoiesis confers a favorable outcome to secondary systemic LPS challenge and protection against chemotherapy-induced myelosuppression98 (Fig.1). BCG administration in mice also stimulates the expansion of HSPCs and promotes myelopoiesis in a manner requiring IFN-γ signaling. Importantly, macrophages derived from BCG-trained progenitor cells mediate host protection against Mycobacterium tuberculosis infection independently of adaptive immunity68 (Fig.1). Hence, innate immune training of hematopoietic progenitor cells68,98 shares common features with emergency myelopoiesis in response to systemic infection or inflammation2.
HSC expansion due to inflammatory stimuli such as LPS or IFN-α leads to exhaustion of HSCs, which is associated with compromised self-renewal and capacity for competitive repopulation and is mediated by DNA damage associated with replication stress35,36,57,100. Interestingly, β-glucan-triggered training of HSPCs is associated with attenuated LPS- or chemotherapy-induced replication stress in HSCs98. The beneficial effect of trained immunity in this case may lie in changes in cellular metabolism. While mitochrondrial ROS and activation of oxidative phosphorylation are associated with DNA damage in HSCs and lead to their functional impairment, LT-HSC maintenance depends on glycolysis7,57,101,102. HSPCs in β-glucan-trained mice retain a glycolytic signature even after chemotherapy, which may be linked with resistance to replication stress-related DNA damage98. This could suggest that certain agonists of trained immunity may act favorably on HSCs and myelopoiesis by counteracting inflammation-dependent stress and impaired function in HSC98. Genetic targeting of the pathways implicated in trained immunity, such as IL-1 signalling or cell metabolism in hematopoietic progenitor cells, mature myeloid cells and components of the HSC niche are required to clarify the mechanisms underlying the modulation of myelopoiesis by trained immunity.
Adaptation of hematopoietic progenitors in inflammatory diseases
There is increasing evidence that chronic metabolic-inflammatory conditions, such as obesity and diabetes, or cardiovascular-inflammatory conditions, such as atherosclerosis can alter hematopoiesis. Reciprocally, adaptation of hematopoietic progenitor cells leading to chronic leukocytosis may contribute to the perpetuation and chronicity of these disorders27,103,104 (Fig.3). Diabetes-associated hyperglycemia and obesity result in monocytosis and neutrophilia in mice105,106. In hyperglycemia, the neutrophil-derived damage-associated molecular pattern S100A8/A9 interacts with the receptor RAGE on CMPs and triggers the production of M-CSF and GM-CSF, which stimulate the proliferation of GMPs and in turn, monocytosis, neutrophilia and atherosclerosis105. Similarly, genetic or diet-induced obesity in mice stimulates the proliferation of myeloid progenitor cells in the BM, in a manner dependent on the NLRP3-inflammasome-dependent production of IL-1β in adipose tissue (AT) macrophages106 (Fig.3). Weight loss in humans following bariatric surgery significantly decreases the number of circulating monocytes and neutrophils106. The myeloid bias of HSPCs from mice with diet-induced obesity is maintained upon serial transplantation to recipient mice, and the obesity-related myeloid reprogramming is associated with increased numbers of LT-HSCs and MPPs in the BM107,108. The myeloid-biased reprogramming of HSPCs requires signaling through TLR4, MyD88 and TRIF and contributes to enhanced generation of proinflammatory macrophages in the AT, thereby exacerbating obesity-related AT inflammation and dysfunction107,108. Experiments with germ-free mice have also indicated an important role of the gut microbiota in myelopoiesis109. In this context, altered gut microbiota in mice due to high-fat diet promotes HSPC differentiation toward myelopoiesis at the expense of lymphopoiesis through modulation of niche MSCs110.
Figure 3: Adaptation of hematopoietic progenitors in cardio-metabolic disease.
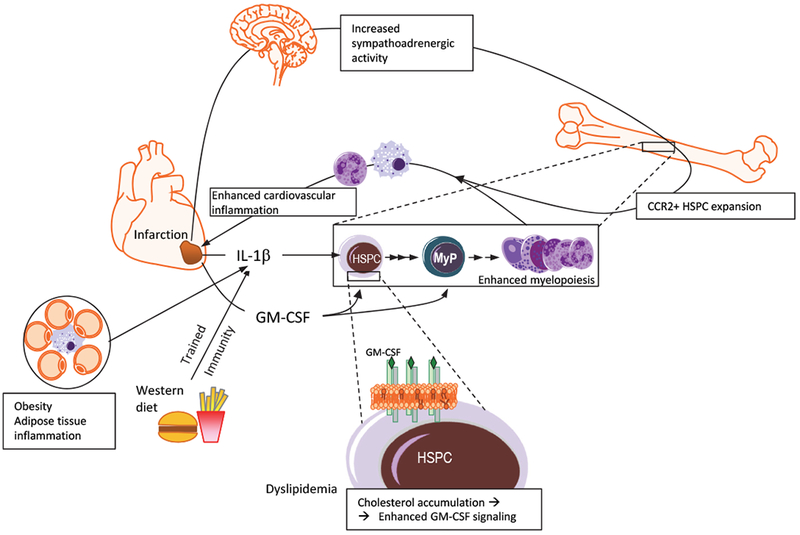
IL-1β can act directly on hematopoietic stem and progenitor cells (HSPCs) and is a crucial mediator promoting myelopoiesis under different cardio-metabolic settings like post-myocardial infarction MI, obesity and western-type diet106,115–117. Moreover, in MI, increased sympathoadrenergic activity (e.g., due to pain or anxiety) causes expansion and enhanced egress from the BM of HSPCs, including a subset of CCR2+ HSPCs, further contributing to enhanced myeloid cell output111,112. This enhanced generation of inflammatory myeloid cells may aggravate inflammation and interfere with tissue healing in the case of MI and perhaps in other inflammatory conditions. GM-CSF produced post-MI (or under other inflammatory disorders) promotes myelopoiesis by acting on HSPCs or myeloid progenitors (MyP)41,118. Cholesterol accumulation in HSPCs (e.g., due to dyslipidemia or hypercholesterolemia) causes membrane changes associated with enhanced IL-3 or GM-CSF-dependent signaling that induces myelopoiesis99,124. Such immunometabolic changes resulting in GM-CSF-dependent enhanced myelopoiesis are also associated with trained immunity-induced IL-1β98 and may, at least in part, explain why innate immune training aggravates cardiovascular inflammation117.
Altered hematopoiesis is also integral to atherosclerosis and cardiovascular disease27,103. Myocardial infarction (MI) results in enhanced myelopoiesis, which may involve several complementary and likely synergistic mechanisms27 (Fig.3). For instance, a myeloid-biased subset of HSPCs (CCR2+CD150+CD48−LSKs) with a higher proliferative and lower self-renewal capacity than HSCs contributes to the enhanced myelopoiesis post-MI111. Increased sympathoadrenergic activity causes enhanced mobilization of HSPCs from the BM to the spleen, which becomes a site for increased extramedullary myelopoiesis112. Interestingly, enhanced sympathoadrenergic activity mediates the increased, stress-induced, proliferation of LT-HSCs and myelopoiesis, thus likely explaining how psychosocial stress promotes atherosclerosis113. In a similar context, a disturbed sleep pattern contributes to atherosclerosis by dysregulating a neuro-immune axis linking hypothalamic hypocretin to regulation of myelopoiesis114. IL-1β is a central player in enhanced myelopoiesis post-MI, as it can promote HSPC proliferation in a direct or indirect, niche cell-dependent fashion, as well as stimulate splenic monocytopoiesis115,116. Detrimental effects of innate immune training may also contribute to western diet-associated atherosclerosis117. Western diet induces NLRP3- and IL-1-dependent transcriptomic and epigenetic reprogramming in GMPs in atherosclerosis-prone LDL-receptor (Ldlr)-deficient mice, driving the GMPs towards enhanced proliferation and inflammatory responses; consistent with the long-term effects of trained immunity, this progenitor cell reprogramming is sustained even after mice are switched back to chow diet117. Moreover, GM-CSF from the infarcted myocardium stimulates expansion of myeloid-biased CD131+ MPP3 in the BM118.
Cholesterol biosynthesis and/or mevalonate, the first metabolite of the cholesterol biosynthetic pathway, are important mediators of innate immune training in mature myeloid cells or hematopoietic progenitor cells98,119,120. The cholesterol-dependent immunometabolic crosstalk during trained immunity may also be intimately linked with the dyslipidemia- or hypercholesterolemia-associated altered hematopoiesis that leads to enhanced myelopoiesis during atherosclerosis97,103. Disrupted cholesterol efflux pathways and enhanced cholesterol accumulation in HSPCs drive myelopoiesis in a cell-intrinsic fashion99,121. Major players in the cellular cholesterol efflux are apolipoprotein E (ApoE) and the ATP-binding cassette transporters ABCA1 and ABCG1121. The latter are upregulated in pro-resolving macrophages upon efferocytosis dependent on transcription factor LXR and contribute to inflammation resolution programs122,123. Cholesterol accumulation in HSPCs due to double deficiency of ABCA1 and ABCG1, or due to ApoE deficiency, results in HSPC expansion and myeloid differentiation, and hence peripheral monocytosis and neutrophilia99,124. The underlying mechanism involves membrane alterations associated with enhanced intracellular cholesterol and elevated cell-surface expression of the lipid raft-associated IL-3Rβ (CD131), which facilitates enhanced IL-3- or GM-CSF-dependent signaling for inducing myelopoiesis99,124 (Fig.3). A similar mechanism, involving enhanced cholesterol accumulation and lipidomic membrane remodeling that augment CD131-dependent signaling in HSPCs leading to their myeloid differentiation operates in β-glucan-induced trained immunity in HSPCs98. Together, immunometabolic adaptations of hematopoietic progenitor cells to inflammatory signals may be the common denominator between the modes of action in trained innate immunity and cardio-metabolic disease. In addition, (mal)adaptive training of hematopoietic progenitor cells may provide a mechanistic basis for comorbidities, such as the enhanced risk of cardiovascular complications in patients with rheumatoid arthritis, periodontitis or other autoimmune or inflammatory disorders125,126.
The age-associated impairment in HSC function is associated with replication stress100, while decreased DNA damage response and repair pathways in aged HSCs may promote accumulation of mutations leading to hematological malignancies127. Autophagy counteracts the age-associated dysfunction of HSCs by clearing mitochondria, thereby facilitating maintenance of HSC quiescence and regenerative capacity128. Aged HSCs display diminished self-renewal and a myeloid bias129–131. CD61hi LT-HSCs are highly responsive to inflammatory stimuli and predominate amongst aged LT-HSCs as a myeloid-biased subset regulated by the transcription factors KLF5, IKZF1 and STAT3131.
The propensity of aged HSCs to accrue mutations and have a myeloid bias and enhanced responsiveness to inflammation may provide a mechanism for clonal hematopoiesis (also known as clonal hematopoiesis of indeterminate potential)132,133, whose prevalence strongly increases with age and is defined by mutations associated with myeloid malignancies, such as mutations in TET2, DNMT3A and ASXL1, with normal blood counts. People with clonal hematopoiesis have higher risk for hematologic malignancies and cardiovascular disease132–134. Consistently, murine deficiency of the epigenetic regulator TET2 results in the expansion of HSPCs with myeloid bias, leading to myeloproliferation135. Chimeric Ldlr-deficient mice partially or completely reconstituted with TET2-deficient BM cells have exacerbated atherosclerosis typified by the presence of pro-inflammatory macrophages with enhanced NLRP3-dependent production of IL-1β133,134. Clonal hematopoiesis may also accelerate heart failure in an IL-1β-dependent manner136. The concept of clonal hematopoiesis could, therefore, integrate aging- and inflammation-related adaptations in myelopoiesis progenitor cells with cardiometabolic inflammation. NLRP3 inflammasome activation is linked to clonal HSPC expansion and progression of the myelodysplastic syndrome (MDS)137, while IL-6-related inflammation promotes survival of TET2-deficient HSPCs76, implicating inflammation as a driver of clonal hematopoiesis and MDS.
IL-1β appears as the common denominator of innate immune training in hematopoietic progenitor cells, enhanced myelopoiesis in cardiometabolic disease and clonal hematopoiesis-related exacerbated vascular inflammation. The success of IL-1β blockade (CANTOS trial) in atherosclerosis138 further supports a major role of IL-1β in these conditions. Moreover, inflammation induced by TLR2 agonists or disseminated bacteria resulting from a disrupted intestinal barrier may promote the progression to pre-leukemic myeloproliferation in TET2-deficiency139. Further investigations could elucidate the potential link(s) amongst the aforementioned inflammatory (IL-1β-related) processes, especially trained innate immunity and clonal hematopoiesis, as they appear to participate in a self-sustained vicious cycle, wherein inflammatory disease and inflammatory adaptations of hematopoietic progenitors engage in reciprocally reinforced interactions.
Conclusions
The ability of hematopoietic progenitor cells to sense and adapt to inflammatory stimuli renders them central players in the context of acute peripheral inflammation or infection1,2. During infection, HSPCs react to TLR ligands, inflammatory cytokines, including IL-1 or IFNs, and growth factors. Such inflammatory signaling in HSPCs can drive their expansion and myelopoiesis, but may also cause HSPC exhaustion, if chronically sustained2. With the exception of chronic cardio-metabolic diseases27,103, the role of hematopoietic progenitors cells as central determinants of the host response in chronic diseases has been underappreciated. In cardio-metabolic disease (and possibly other chronic disorders), the adaptation to inflammation that drives myelopoiesis in HSPCs generates a destructive feed-forward loop (Fig.4), in which increased numbers of inflammatory myeloid cells enhance inflammation, which in turn perpetuates HSPC-mediated myelopoiesis27,103,111,112,116,121. Importantly, previous infectious or inflammatory challenges can epigenetically imprint an “inflammatory memory” in hematopoietic progenitor cells97,98,117, thereby contributing to trained immunity. As innate immune training facilitates cardiovascular inflammation117, it is intriguing to hypothesize that the development of clonal hematopoiesis, which is associated with higher cardio-metabolic inflammation132–134, might be linked with selective pressure for inflammatory memory96–98. A major question is whether adaptation of hematopoietic progenitors to inflammation can provide a unifying concept where inflammatory stimuli (e.g., infections) and multiple risk factors (e.g., obesity, dyslipidemia, psychosocial stress) can be integrated in a meaningful manner to better understand the pathogenesis of different forms of chronic inflammatory diseases or even malignant disorders. This challenging concept needs to be addressed by interdisciplinary studies bringing together experts in immunology, hematopoiesis, metabolism, cardiovascular medicine, as well as mathematical modeling, which could potentially lead to new risk assessment scores for chronic inflammatory diseases.
Figure 4. Detrimental feed-forward loop linking HSPC inflammatory adaptation to chronic inflammatory disease.
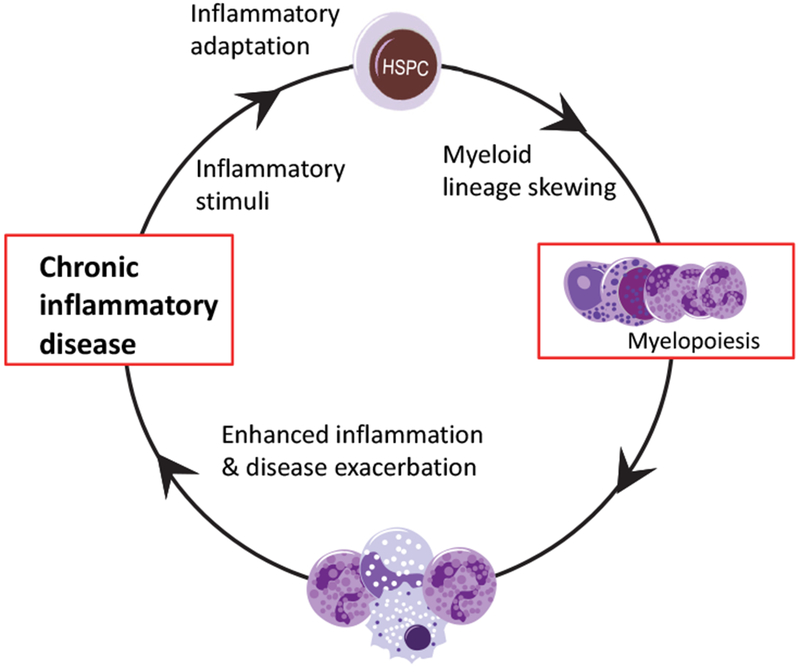
The ability of HSPCs to sense and adapt to inflammatory stimuli may have detrimental consequences in the setting of chronic inflammatory diseases. According to this hypothesis, the adaptation of HSPCs to inflammatory signals derived from on-going chronic inflammatory disorders (e.g., cardio-metabolic disease) promotes myelopoiesis and output of inflammatory myeloid cells, which in turn further enhance inflammation. This not only can exacerbate the disease but also perpetuate HSPC-mediated myelopoiesis. Thus, a feed-forward loop between inflammation-adapted hematopoietic progenitors and the inflammatory disorder is generated that may contribute to or underlie the chronicity of the disorder.
Acknowledgements:
The authors are supported by the NIH (DE015254, DE024153, DE024716 to GH and DE026152 to GH and TC), the European Research Council (DEMETINL-683145 to TC) and the Deutsche Forschungsgemeinschaft (SFB/TRR 205 to TC). IM is supported by the National Center for Tumor Diseases, Partner Site Dresden.
References
- 1.King KY & Goodell MA Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol 11, 685–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manz MG & Boettcher S Emergency granulopoiesis. Nat Rev Immunol 14, 302–314 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Jacobsen SEW & Nerlov C Haematopoiesis in the era of advanced single-cell technologies. Nat Cell Biol 21, 2–8 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Oguro H, Ding L & Morrison SJ SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei Q & Frenette PS Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 48, 632–648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cordeiro Gomes A et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 45, 1219–1231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suda T, Takubo K & Semenza GL Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Carrelha J et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 554, 106–111 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Fraticelli AE et al. Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu VWC et al. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 167, 1310–1322 e1317 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Velten L et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 19, 271–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto R et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Gekas C & Graf T CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121, 4463–4472 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Pietras EM et al. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giladi A et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol 20, 836–846 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Avellino R et al. An autonomous CEBPA enhancer specific for myeloid-lineage priming and neutrophilic differentiation. Blood 127, 2991–3003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh-Nakadai A et al. A Bach2-Cebp Gene Regulatory Network for the Commitment of Multipotent Hematopoietic Progenitors. Cell Rep 18, 2401–2414 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Pietras EM et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18, 607–618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that IL-1β is a major regulator of hematopoiesis by acting directly on HSCs and promoting their proliferation and myeloid differentiation. Although this mechanism is crucial for rapid myeloid recovery after acute BM injury, chronic IL-1β diminishes the self-renewal capacity of HSCs.
- 19.Ginhoux F & Jung S Monocytes and macrophages: developmental pathways and tissue homeostasis. Nature Reviews Immunology 14, 392 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Schultze JL, Mass E & Schlitzer A Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity 50, 288–301 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Geissmann F et al. Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Álvarez-Errico D, Vento-Tormo R, Sieweke M & Ballestar E Epigenetic control of myeloid cell differentiation, identity and function. Nature Reviews Immunology 15, 7 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Evrard M et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 48, 364–379 e368 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Paul F et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Busch K et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Sawai CM et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 45, 597–609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nahrendorf M Myeloid cell contributions to cardiovascular health and disease. Nat Med 24, 711–720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitroulis I, Kalafati L, Hajishengallis G & Chavakis T Myelopoiesis in the Context of Innate Immunity. J Innate Immun 10, 365–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagai Y et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao JL et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 14, 445–459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S & Manz MG Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med 208, 273–284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu A et al. Cutting Edge: Hematopoietic Stem Cell Expansion and Common Lymphoid Progenitor Depletion Require Hematopoietic-Derived, Cell-Autonomous TLR4 in a Model of Chronic Endotoxin. J Immunol 195, 2524–2528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esplin BL et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol 186, 5367–5375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Liu Y, Liu Y & Zheng P Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest 120, 4091–4101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H et al. Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88. Stem Cell Reports 6, 940–956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takizawa H et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 21, 225–240 e225 (2017). [DOI] [PubMed] [Google Scholar]; This study has demonstrated that direct TLR4 activation on HSCs stimulates, via TRIF-dependent signaling, their proliferation, while diminishing their self-renewal and repopulation capacity.
- 37.Yáñez A et al. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 47, 890–902.e894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boettcher S et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 124, 1393–1403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirai H et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol 7, 732–739 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Hibbs ML et al. Mice lacking three myeloid colony-stimulating factors (G-CSF, GM-CSF, and M-CSF) still produce macrophages and granulocytes and mount an inflammatory response in a sterile model of peritonitis. J Immunol 178, 6435–6443 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Becher B, Tugues S & Greter M GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 45, 963–973 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Zhang H et al. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 116, 2462–2471 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herault A et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544, 53–58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson A et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Greenbaum AM & Link DC Mechanisms of G-CSF-mediated hematopoietic stem and progenitor mobilization. Leukemia 25, 211–217 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Christopher MJ, Rao M, Liu F, Woloszynek JR & Link DC Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med 208, 251–260 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croker BA et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 20, 153–165 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Delhommeau F et al. Mutation in TET2 in myeloid cancers. N Engl J Med 360, 2289–2301 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Shen Q et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554, 123–127 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Auffray C, Sieweke MH & Geissmann F Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol 27, 669–692 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC & Schroeder T Hematopoietic cytokines can instruct lineage choice. Science 325, 217–218 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Endele M et al. CSF-1-induced Src signaling can instruct monocytic lineage choice. Blood 129, 1691–1701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mossadegh-Keller N et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 497, 239–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This pivotal study showed that M-CSF promotes myeloid differentiation in single HSCs, independently of their survival or proliferation, by inducing the myeloid-lineage master regulator PU.1.
- 54.Kandalla PK et al. M-CSF improves protection against bacterial and fungal infections after hematopoietic stem/progenitor cell transplantation. J Exp Med 213, 2269–2279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Essers MA et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908 (2009). [DOI] [PubMed] [Google Scholar]; This seminal report has shown that IFN-α drives proliferation of dormant HSCs, although, in a chronic setting, this mechanism impairs the self-renewal potential of HSCs.
- 56.Sato T et al. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med 15, 696–700 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Walter D et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 (2015). [DOI] [PubMed] [Google Scholar]; This study identified the mechanism whereby IFN-α causes impairment and attrition of HSCs. Specifically, the authors demonstrated IFN-α-induced DNA damage in LT-HSCs upon entering the cell cycle, associated with increased mitochondrial membrane potential and ROS production.
- 58.Pietras EM et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 211, 245–262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cabezas-Wallscheid N et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823 e819 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Roers A, Hiller B & Hornung V Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 44, 739–754 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Xia P et al. A Circular RNA Protects Dormant Hematopoietic Stem Cells from DNA Sensor cGAS-Mediated Exhaustion. Immunity 48, 688–701 e687 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Prendergast AM et al. IFNalpha-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica 102, 445–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haas S et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 17, 422–434 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Baldridge MT, King KY, Boles NC, Weksberg DC & Goodell MA Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Bruin AM, Demirel O, Hooibrink B, Brandts CH & Nolte MA Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood 121, 3578–3585 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Matatall KA et al. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep 17, 2584–2595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schurch CM, Riether C & Ochsenbein AF Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 14, 460–472 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Kaufmann E et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190 e119 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Garlanda C, Dinarello CA & Mantovani A The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weisser M et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J Allergy Clin Immunol 138, 219–228 e219 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Damia G et al. Prevention of acute chemotherapy-induced death in mice by recombinant human interleukin 1: protection from hematological and nonhematological toxicities. Cancer Res 52, 4082–4089 (1992). [PubMed] [Google Scholar]
- 72.van der Meer JW, Barza M, Wolff SM & Dinarello CA A low dose of recombinant interleukin 1 protects granulocytopenic mice from lethal gram-negative infection. Proc Natl Acad Sci U S A 85, 1620–1623 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pronk CJ, Veiby OP, Bryder D & Jacobsen SE Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med 208, 1563–1570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Etzrodt M et al. Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood (2018). [DOI] [PubMed] [Google Scholar]
- 75.Weber GF et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 347, 1260–1265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai Z et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 23, 833–849.e835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Furusawa J et al. Promotion of Expansion and Differentiation of Hematopoietic Stem Cells by Interleukin-27 into Myeloid Progenitors to Control Infection in Emergency Myelopoiesis. PLoS Pathog 12, e1005507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding L, Saunders TL, Enikolopov G & Morrison SJ Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morrison SJ & Scadden DT The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mitroulis I et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J Clin Invest 127, 3624–3639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kanayama M et al. Skewing of the population balance of lymphoid and myeloid cells by secreted and intracellular osteopontin. Nat Immunol 18, 973–984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lucas D et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nature Medicine 19, 695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maryanovich M et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nature Medicine 24, 782–791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Méndez-Ferrer S, Lucas D, Battista M & Frenette PS Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Winkler IG et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116, 4815–4828 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Chow A et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208, 261–271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ludin A et al. Monocytes-macrophages that express alpha-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol 13, 1072–1082 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Casanova-Acebes M et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153, 1025–1035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kwak HJ et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 42, 159–171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowers E et al. Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med 24, 95–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao M et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med 20, 1321–1326 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Bruns I et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med 20, 1315–1320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pinho S et al. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev Cell 44, 634–641 e634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fujisaki J et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474, 216–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirata Y et al. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 22, 445–453 e445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Netea MG et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penkov S, Mitroulis I, Hajishengallis G & Chavakis T Immunometabolic Crosstalk: An Ancestral Principle of Trained Immunity? Trends Immunol 40, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mitroulis I et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161 e112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper resolved the paradox regarding the long-term effects of trained immunity in mature myeloid cells despite their relatively short life span in the circulation. The paper demonstrated that metabolic and transcriptional adaptations in HSPCs leading to enhanced myelopoiesis are an integral component of trained immunity.
- 99.Yvan-Charvet L et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study has shown that cholesterol accumulation in HSPCs due to deficiency of cholesterol efflux mechanisms results in HSPC expansion and myeloid differentiation associated with elevated IL-3Rβ expression. These alterations can lead to leukocytosis and accelerate atherosclerosis.
- 100.Flach J et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Simsek T et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Takubo K et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Murphy AJ & Tall AR Disordered haematopoiesis and athero-thrombosis. Eur Heart J 37, 1113–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barrett TJ, Murphy AJ, Goldberg IJ & Fisher EA Diabetes-mediated myelopoiesis and the relationship to cardiovascular risk. Ann N Y Acad Sci 1402, 31–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nagareddy PR et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 17, 695–708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagareddy PR et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19, 821–835 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Griffin C et al. TLR4, TRIF, and MyD88 are essential for myelopoiesis and CD11c(+) adipose tissue macrophage production in obese mice. J Biol Chem 293, 8775–8786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singer K et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol Metab 3, 664–675 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khosravi A et al. Gut Microbiota Promote Hematopoiesis to Control Bacterial Infection. Cell Host & Microbe 15, 374–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luo Y et al. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metabolism 22, 886–894 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Dutta P et al. Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 16, 477–487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified CCR2+CD150+CD48− LSK cells as a myeloid-biased HSPC subset that exhibits higher proliferative and reduced self-renewal capacity than HSCs and contributes to post-MI enhanced myelopoiesis.
- 112.Dutta P et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Heidt T et al. Chronic variable stress activates hematopoietic stem cells. Nat Med 20, 754–758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McAlpine CS et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report has identified a neuro-immune mechanism that links sleep to regulation of hematopoiesis. Specifically, the authors showed that fragmented sleep in mice reduces the production of hypocretin from hypothalamus, in turn leading to enhanced M-CSF-dependent monocytosis and accelerated atherosclerosis.
- 115.Sager HB et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 132, 1880–1890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leuschner F et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209, 123–137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Christ A et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study has causally linked western diet to atherosclerosis through induction of innate immune training of myeloid progenitors leading to their enhanced proliferation and inflammatory responses. The mechanism mediated by western diet involves NLRP3 inflammasome- and IL-1-dependent transcriptomic and epigenetic reprogramming of the progenitors.
- 118.Anzai A et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med 214, 3293–3310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bekkering S et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172, 135–146 e139 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Arts RJ et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab 24, 807–819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tall AR & Yvan-Charvet L Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15, 104–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kourtzelis I et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol 20, 40–49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sallam T et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med 24, 304–312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murphy AJ et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest 121, 4138–4149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dragoljevic D et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur Heart J 39, 2158–2167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hajishengallis G Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15, 30–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gutierrez-Martinez P et al. Diminished apoptotic priming and ATM signalling confer a survival advantage onto aged haematopoietic stem cells in response to DNA damage. Nat Cell Biol 20, 413–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ho TT et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dykstra B, Olthof S, Schreuder J, Ritsema M & de Haan G Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 208, 2691–2703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yamamoto R et al. Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell Stem Cell 22, 600–607.e604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mann M et al. Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep 25, 2992–3005.e2995 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jaiswal S et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study linked aging-related clonal hematopoiesis with increased risk for coronary heart disease and showed that TET2 deficiency promotes atherosclerosis.
- 134.Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Consistent with the findings of ref. 133, this report has shown that deficiency in the epigenetic regulator TET2 is associated with HSPC expansion and a myeloid-bias that enhances atherosclerosis in low-density lipoprotein receptor-deficient mice.
- 135.Moran-Crusio K et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sano S et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol 71, 875–886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Basiorka AA et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 128, 2960–2975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Meisel M et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]