

Abstract
Intracellular vesicular trafficking and membrane fusion are important processes for nervous system development and for the function of neural circuits. Synaptosomal-associated protein 25 kDa (SNAP-25) is a component of neural soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) core complexes that mediate the exocytotic release of neurotransmitters at chemical synapses. Previous results from mouse mutant models and pharmacological/neurotoxin blockades have demonstrated a critical role for SNAP-25-containing SNARE complexes in action potential (AP)-dependent release at cholinergic and glutamatergic synapses and for calcium-triggered catecholamine release from chromaffin cells. To examine whether SNAP-25 participates in the evoked release of other neurotransmitters, we investigated the expression and function of SNAP-25 in GABAergic terminals. Patch-clamp recordings in fetal Snap25-null mutant cortex demonstrated that ablation of SNAP-25 eliminated evoked GABAA receptor-mediated postsynaptic responses while leaving a low level of spontaneous AP-independent events intact, supporting the involvement of SNAP-25 in the regulated synaptic transmission of early developing GABAergic neurons. In hippocampal cell cultures of wild-type mice, punctate staining of SNAP-25 colocalized with both GABAergic and glutamatergic synaptic markers, whereas stimulus-evoked vesicular recycling was abolished at terminals of both transmitter phenotypes in Snap25−/− neurons. Moreover, immunohistochemistry and fluorescence in situ hybridization revealed coexpression of SNAP-25, VGAT (vesicular GABA transporter), and GAD65/67 (glutamic acid decarboxylase 65/67) in interneurons within several regions of the adult brain. Our results thus provide evidence that SNAP-25 is critical for evoked GABA release during development and is expressed in the presynaptic terminals of mature GABAergic neurons, consistent with its function as a component of a fundamental core SNARE complex required for stimulus-driven neurotransmission.
Keywords: SNARE proteins, GABAergic neuron, inhibitory neurotransmission, SNAP-25, neuroexocytosis, CNS development, GABA transporter
Introduction
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein complexes are essential in promoting vesicular fusion for neurotransmission (Sudhof, 2004). Although the precise mechanisms of SNARE function remain unidentified, it is recognized that this family of proteins (Fasshauer et al., 1998; Bock et al., 2001) interact through amphipathic SNARE domains to create a four-helix bundle that is capable of bridging apposing membranes before fusion (Sutton et al., 1998). Different SNARE complexes appear to be involved in virtually all elements of vesicular trafficking, including intracellular transport, constitutive and stimulus-driven delivery of plasma membrane proteins, as well as regulated neuroexocytosis (Jahn et al., 2003; Hong, 2005). In neurons, SNARE complex composition may help distinguish process outgrowth during development from neurotransmitter release (Bark and Wilson, 1994; Martinez-Arca et al., 2001; Washbourne et al., 2002). In addition to acting directly in membrane fusion, SNARE complexes may also provide a scaffold to recruit accessory proteins that contribute distinct physiological properties required for various fusion events (Melia et al., 2002; Nagy et al., 2005).
The predominant neural SNARE complex is composed of plasma membrane-associated synaptosomal-associated protein 25 kDa (SNAP-25) and syntaxin 1a, and vesicular membrane-associated synaptobrevin (Sollner et al., 1993). The involvement of these SNARE proteins in neuroexocytosis has been addressed using the proteolytic tetanus and botulinum neurotoxins (BoNTs) that block neurotransmission (for review, see Montecucco et al., 2005). In particular, BoNT/A and /E target SNAP-25, disrupting activity-dependent release of various transmitters and peptides (Sadoul et al., 1995; Graham et al., 2002). Recently, we provided genetic evidence that SNAP-25 is critical for evoked glutamatergic and cholinergic transmission in central neurons and at neuromuscular junctions (Washbourne et al., 2002), and for fast calcium-triggered catecholamine release from adrenal chromaffin cells (Sørensen et al., 2003). Nevertheless, Snap25-null mutants retained stimulus-independent transmitter secretion (Washbourne et al., 2002; Sørensen et al., 2003), suggesting that alternative SNARE constituents could promote spontaneous vesicle fusion and transmitter release. However, it has yet to be established what importance SNAP-25-independent transmission might have in neural communication (for example, during brain development) (Molnar et al., 2002).
Recent evidence suggests that the presynaptic proteins Munc13-1, RIM1α (Rab3-interacting molecule 1α), and synapsin exert different modulatory affects on glutamate and GABA neurotransmission (Augustin et al., 1999; Schoch et al., 2002; Gitler et al., 2004) and that these accessory proteins may contribute to shaping physiological parameters that govern synaptic activity. Although SNAP-25 appears to be required for glycine release from spinal interneurons (Keller et al., 2004), it has been suggested that SNAP-25 is not expressed by central GABAergic neurons and that inhibitory transmission may occur without this neural SNARE protein (Verderio et al., 2004; Frassoni et al., 2005). Given the implication that calcium-triggered inhibitory and excitatory neurotransmission could be distinguished presynaptically by interchanging core components of the SNARE complex, we sought to examine whether SNAP-25 is required for GABA release in Snap25-null mutant fetal brain and whether it is expressed in developing and adult GABAergic neurons. Our findings indicate that SNAP-25 is critical for GABAergic transmission and support its role as a primary constituent of SNARE complexes required for stimulus-evoked neuroexocytosis.
Materials and Methods
Electrophysiology.
Snap25-null mutant mice were collected through a timed pregnancy protocol from heterozygous matings wherein pregnant animals (plug date, d 0) were killed by rapid cervical dislocation and decapitation. Fetuses were removed sequentially from the uterus, and tested for movement by a brief pinch to the hindlimb. Because Snap25-null homozygote mutant animals lack evoked neuromuscular transmission (Washbourne et al., 2002), fetuses were initially categorized based on the presence of movement, with subsequent PCR genotyping to confirm knock-out animals, and to distinguish between heterozygote and homozygote wild-type mice that served as control littermates. Pups were quickly decapitated and their brains were removed and placed in ice-cold PBS. All procedures were performed in accordance with guidelines of the University of New Mexico Health Sciences Center Laboratory Animal Care and Use Committee, and the National Institutes of Health.
Coronal brain slices (350–400 μm) were prepared from wild-type and mutant mice at embryonic day 17.5 (E17.5) as described previously (Carta et al., 2004), except that ketamine was not used. After a recovery period of ≥80 min, slices were transferred to a chamber perfused at rate of 2 ml/min with artificial CSF (ACSF) equilibrated with 95% O2/5% CO2 and containing in (mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2 and 10 glucose. When indicated, 20 μm bicuculline methiodide was added to the ACSF to block GABAA receptors. Whole-cell patch-clamp electrophysiological recordings from visualized cortical neurons were performed under infrared-differential interference contrast microscopy at 32°C, with an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Patch electrodes had a resistance of 3–5 MΩ. For voltage-clamp experiments, patch pipettes were filled with an internal solution containing the following (in mm): 140 CsCl, 2 MgCl2, 1 CaCl2, 2 Na2-ATP, 10 EGTA, and 10 HEPES, pH 7.3. Current-clamp experiments (Iholding = 0) were conducted with patch pipettes filled with an internal solution containing the following (in mm): 135 K-gluconate, 10 MgCl2, 0.1 CaCl2, 2 Na2-ATP, 1 EGTA, and 10 HEPES, pH 7.3. Access resistance was between 10 and 30 MΩ; if access resistance changed >20%, the recording was discarded. Spontaneous postsynaptic currents (sPSCs) were recorded at a holding potential of −65 mV in the presence of 10 μm 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2,3-dione (NBQX) (Axxorra, San Diego, CA), and 100 μm d,l-APV. Miniature PSCs (mPSCs) were recorded in presence of 500 nm tetrodotoxin (Calbiochem, La Jolla, CA), 10 μm NBQX, and 100 μm d,l-APV. Evoked PSCs (ePSCs) were recorded in presence of NBQX and d,l-APV with 4 mm QX-314 [(2,6-dimethylphenyl)carbamoylmethyl-triethyl-azanium] in the internal pipette solution and were elicited with a concentric bipolar stimulating electrode placed 100–200 μm from the patched cell. Data were acquired and analyzed with pClamp-9 (Molecular Devices). Minis Analysis program (Synaptosoft, Decatur, GA) was used to analyze mPSCs. Statistical analyses of pooled data were performed by unpaired Student’s t test (Prism 4; GraphPad Software, San Diego, CA). All values were expressed as mean ± SEM.
Protein analysis.
For protein analysis, E17.5 mouse brains were removed and immediately immersed in ice-cold PBS. Hippocampi and cortices were then dissected over ice, homogenized in 0.32 m sucrose, 20 mm Tris, pH 7.4, 1 mm DTT, 1% Nonidet P-40, and protease inhibitors (Complete Minitablet; Boehringer Mannheim, Indianapolis, IN), and processed through differential centrifugation to obtain LP2 protein fractions as described previously (Huttner et al., 1983). Protein concentrations of the protein samples were determined with Micro BCA assay kit (Pierce, Rockford, IL), and equal amounts of protein were loaded on 12% SDS-PAGE polyacrylamide gels with subsequent transfer to polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA). Blots were then probed with the antibodies to the following proteins: synaptophysin (1:1500; Invitrogen, Carlsbad, CA), vesicular glutamate transporter 1 (VGLUT1) (1:500; Synaptic Systems, Göttingen, Germany), and vesicular GABA transporter (VGAT) (1:200; Synaptic Systems). Primary antibodies were detected with species-appropriate HRP-conjugated secondary IgGs followed by chemiluminescence assay (ECL-Plus; Amersham Biosciences, Piscataway, NJ) and quantitated on a STORM PhosphoImager system (Molecular Dynamics, Sunnyvale, CA). One-way ANOVA analysis on the data were performed with Bonferroni’s comparisons post hoc test (Prism 4). All values are expressed as mean ± SEM.
FM 1-43 FX vesicular recycling assay.
Hippocampal neurons were isolated from individual E17.5 fetal mice, genotyped as detailed above, and grown as dispersed mixed cell cultures as described previously (Washbourne et al., 2002). Briefly, hippocampi were microdissected in ice-cold PBS containing 27 mm glucose, 17.5 mm sucrose, and 15 mm HEPES with a final pH of 7.4. After incubation with 0.05% trypsin-EDTA (Invitrogen) for 10 min at 37°C, the hippocampi were then transferred to Neurobasal A media (Invitrogen) supplemented with 10% FBS, 25 μm glutamate, 0.5 mm l-glutamine, and 1% penicillin/streptomycin, and gently triturated with flame-polished Pasteur pipettes. The dispersed cells were plated on poly-l-lysine/collagen-coated 12 mm coverslips (four coverslips per animal; ∼50,000 cells/coverslip). For the first 24 h, cultures were grown in the supplemented medium described above. The medium was then replaced with Neurobasal A medium containing B27 supplement (Invitrogen) instead of FBS, and after 5 d in vitro (DIV 5), glutamate was removed entirely from the culture medium. Every third day beyond this point, one-third of the medium was removed and replaced with Neurobasal A medium containing B27 supplement without glutamate.
To assay uptake of the fixable analog of N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl)pyridinium dibromide (FM1-43FX) dye, coverslips of DIV 12 neurons were briefly washed in PBS (37°C), and then incubated for 15 min in modified Tyrode’s solution (150 mm NaCl, 4 mm KCl, 2 mm MgCl, 10 mm glucose, 10 mm HEPES, and 2 mm CaCl2, pH 7.4). During this incubation and throughout the duration of the experiment, all buffers were used at 37°C and contained 10 μm CNQX, 25 μm APV, 20 μm bicuculline, and 0.6 μm tetrodotoxin (TTX) to prevent spontaneous action potential (AP)-dependent neuronal activity. Synaptic vesicles were loaded in the presence of 15 μm FM1-43FX dye (Invitrogen) after evoking exocytosis with either high K+ (64 mm NaCl, 90 mm KCl, 2 mm MgCl, 10 mm glucose, 10 mm HEPES, and 2 mm CaCl2, pH 7.4) for 90 s or hypertonic sucrose (500 mOsm added to modified Tyrode’s solution) for 30 s (Sara et al., 2002). Cells were then immediately washed in modified Tyrode’s solution for 15 min and either fixed with 4% paraformaldehyde (PFA)/4% sucrose or destained in high K+ solution for 2 min in the absence of dye before washing and fixation.
Fixed cultures were immunostained with either VGAT or VGLUT1 monoclonal antibodies (1:1000; Synaptic Systems) followed by Alexa 647-conjugated anti-mouse secondary antibody (Invitrogen), and then visualized with a Zeiss (Oberkochen, Germany) LSM 510 META/Axiovert 100M laser confocal microscope using a 63× oil differential interference contrast (DIC) objective [numerical aperture (NA), 1.4]. META photodetectors were configured to recognize fluorescent emissions within the spectral range of 556–716 nm, and the peak emissions of FM1-43FX (598 nm) and Alexa 647 (663 nm) were captured at 585–609 and 652–673 nm, respectively. FM1-43FX fluorescence intensity that colocalized within the immunoreactive punctate staining of either VGAT or VGLUT1 was measured by using the mean region of interest (ROI) intensity function of the LSM 510 bundled software. A ROI corresponds to a single point of five pixels that overlays the majority of area within each puncta. Areas of low neurite density were chosen to reduce the chance of incidental colocalization of the punctate vesicular transporter stain with nonsynaptic FM1-43FX membrane staining distributed along neurite fibers. Ten puncta present on no less than three neurites were measured per field, averaged, and used as a single value for each animal (n = 5 animals per genotype and experimental condition). After subtraction of background fluorescence, data were analyzed using one-way ANOVA with Bonferroni’s post hoc comparisons (Prism 4). All values are expressed as mean ± SEM. Although brightness and contrast was adjusted for clarity in images used in figures (Adobe Photoshop 7.0 software; Adobe Systems, San Jose, CA), all quantitative analysis was done on raw data before any manipulation of the image.
Fluorescent immunocytochemistry.
After 9–21 d in culture, E17.5 hippocampal neurons were washed in PBS at 37°C followed by a 5 min fixation in 4% PFA/sucrose (37°C) in 0.1 m phosphate buffer (PB) at 37°C. A second round of washing in warm PBS was followed by a 5 min incubation of the coverslips at room temperature in a quenching solution (0.38% glycine, 0.27% NH4Cl). Finally, the coverslips were stored at 4°C in PBS containing 0.02% sodium azide.
For tissue collection, adult mice [postnatal day >50 (>P50)] were anesthetized with pentobarbital (50 μg/g) and perfused transcardially with 30 ml of 0.1 m PB followed by 30 ml of 4% PFA in 0.1 m PB. Brains were dissected and then postfixed in 4% PFA/0.1 m PB overnight at 4°C, followed by immersion in 30% sucrose for 48 h before cryosectioning. Tissue was then frozen in 30% sucrose/0.1 m PB solution before sectioning. Thirty-micrometer coronal sections were cut using a sliding microtome.
Before applying antibodies, both cultured cells and brain sections were permeabilized and nonspecific antibody interactions were blocked by incubating in 1% donkey serum, 1% Tween 20, in TBS. Primary antibodies were used at the following dilutions: glutamic acid decarboxylase 65/67 (GAD65/67) rabbit polyclonal antibody (1:200; a generous gift from Michele Solimena, Dresden University of Technology, Dresden, Germany), and both mouse monoclonal and rabbit polyclonal antibodies to VGLUT1 and VGAT (1:1000; C1 317D5 and C1 117G4, respectively; Synaptic Systems). For the experiments described, we used the SNAP-25 monoclonal antibody SMI 81 (1:1000; Sternberger Monoclonals, Lutherville, MD). The specificity of this antibody was demonstrated by its robust reactivity to a single 25 kDa protein band in wild-type brain, but also through the absence of detectable immunoreactivity either by Western blot or immunofluorescence in SNAP-25 knock-out mutant neurons (Fig. 1). These criteria, in our hands, were not satisfactorily fulfilled by all SNAP-25 antibodies (data not shown). Coverslips and sections of fixed culture and tissue samples were then incubated with Cy3-conjugated affinipure donkey anti-rabbit IgG (H+L) (1:1000; Jackson ImmunoResearch Laboratories, West Grove, PA) and Alexa Fluor 488 donkey anti-mouse IgG antibodies (1:200; Invitrogen) as secondary antibodies for species-appropriate detection of primary antibodies. Finally, To-Pro-3 iodide (Invitrogen) was used for nuclear counterstaining.
Figure 1.
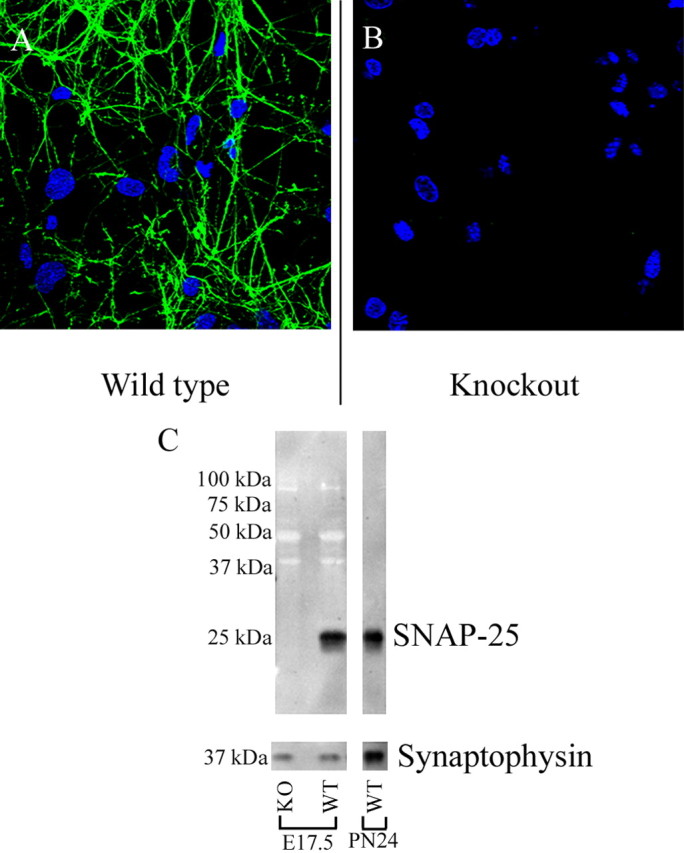
SMI81 monoclonal antibody shows robust and specific immunoreactivity in both cultured neurons and Western blots. A, Wild-type E17.5 cultured hippocampal neurons immunostained at 12 DIV using SMI81 monoclonal antibody and viewed by confocal microscopy with a 63× objective (NA, 1.4; optical slice, 0.81 μm). Probing for SNAP-25 (green; Alexa 488) resulted in staining that was both continuous throughout the neurites with regular punctate accumulations, but not appreciably within the soma or around the nucleus (blue; ToPro3). B, SMI81 showed no apparent immunoreactivity in Snap25-null mutant neuronal cultures. C, Protein fractions prepared from the cortices of Snap25−/− and wild-type animals at E17.5 (mutant, lane 1; wild type, lane 2; 30 μg) and P24 (wild type, lane 3; 1.0 μg) fractionated on a 12% SDS-PAGE gel, blotted, and probed for SNAP-25. SMI81 immunoreactivity was evident in wild-type lanes as a single band at ∼25 kDa with no cross-reactivity in the mutant lane. The synaptic vesicle protein synaptophysin was used as a loading control.
Images of the immunofluorescent staining were obtained using a Bio-Rad 2100 Radiance confocal microscope using a 63× oil immersion DIC objective (NA, 1.4) where x, y pixel corresponds to 0.2 μm per pixel with a resolution of 1024 × 1024. Under these conditions, our z-axis, or optical slice, was 0.8 μm. Before viewing double-stained material, we calibrated the appropriate laser intensity, amplifier gain, and signal offset settings for each marker with single-stained specimens to prevent saturation of their respective signals. Our confocal settings were optimized to acquire the linear range of fluorescence signal in our desired regions of interest, which in some cases may have resulted in slight pixel saturation in neighboring regions containing higher levels of synaptic density. In addition, our data were quantitated and collected before any manipulation of brightness or contrast that would compromise the integrity of pixel intensity. Punctate colocalization of separate color channels was then isolated and quantitated using comparable threshold-level adjustments (MetaMorph 6.1 software; Universal Imaging, Downingtown, PA). In short, this analysis did not count individual puncta but, rather, compared the total pixel number of each stain and calculated the percentage or degree of their overlap. Because of differing sizes of punctate fluorescence between stains, not all pixels overlapped, resulting in percentages <100%. It is important to note the percentages, therefore, do not reflect the percentage of synapses that are double positive, but correspond to the total pixel overlap across each field. Each colocalization value was determined using the average of three fields per animal (n = 3 animals). Statistical analysis of colocalization was performed using one-way ANOVA with Bonferroni’s post hoc comparisons using Prism 4 software. All values were expressed as mean ± SEM.
Fluorescence in situ hybridization.
Adult mice (>P50) were killed with isoflurane and decapitated. Brains were quickly removed and quick-frozen in a beaker of isopentane equilibrated in a dry ice/ethanol slurry and stored at −70°C until additional processing. Coronal brain sections (20 μm) were prepared using a cryostat and arranged on slides (Superfrost Plus; VWR Scientific, West Chester, PA) that were air-dried and stored frozen at −20°C until use.
Double-labeled fluorescence in situ hybridization (FISH) for SNAP-25 and GAD65/67 mRNA was performed as described in detail previously (Guzowski et al., 1999; Vazdarjanova et al., 2002; Vazdarjanova and Guzowski, 2004). Briefly, digoxigenin-labeled SNAP-25 and fluorescein-labeled GAD65/67 antisense riboprobes were generated from cDNA plasmids using a commercial transcription kit (MaxiScript; Ambion, Austin, TX) and premixed RNA labeling nucleotide mixes containing either digoxigenin- or fluorescein-labeled UTP (Roche Molecular Biochemicals, Palo Alto, CA). Hybridization with the tissue was performed overnight at 56°C. Then, the digoxigenin-labeled SNAP-25 riboprobe was detected with anti-digoxigenin-HRP conjugate (Roche Applied Science, Indianapolis, IN), amplified with a TSA-biotin kit (PerkinElmer Life Sciences, Emeryville, CA), and detected with streptavidin-cyanine-3 (Jackson ImmunoResearch Laboratories). Subsequently, the slides were then treated with 2% H2O2 to quench any residual HRP activity, and the fluorescein-labeled probe GAD65/67 was detected with an anti-fluorescein-HRP conjugate (Roche Applied Science) and a TSA-FITC substrate kit (FITC Direct FISH; PerkinElmer Life Sciences). Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen).
Images were acquired with a Nikon (Melville, NY) TE2000U epifluorescence microscope with a 20× dry objective (NA, 0.75) and captured using a CoolSNAP-Hq CCD Camera (Roper Scientific, Tucson, AZ). Images of DAPI (cell nuclei), CY3 (SNAP-25), and FITC (GAD65/67) were acquired and color-combined using MetaMorph software (Universal Imaging). The images were analyzed by counting the total number of GAD65/67-positive neurons and then determining the percentage of those that were double positive for SNAP-25 fluorescence using Image J software (made available as freeware through the National Institutes of Health). Statistical analysis was performed through one-way ANOVA with Bonferroni’s comparison using Prism 4 software. All values were expressed as mean percentage ± SEM per region.
Results
SNAP-25 is crucial for GABAergic transmission in fetal neurons
To assess whether SNAP-25 plays a role in GABAergic neurotransmission in fetal brain slices, we conducted whole-cell patch-clamp recording experiments in cortical slices from Snap25-null mutants (Washbourne et al., 2002) and control E17.5 fetal mice. This homozygous null mouse mutant does not possess a functional Snap25 gene and allows us to investigate the specific role that SNAP-25 may play in neurotransmission. Although mice homozygous for this mutation develop normally during gestation, these mutants die at birth at least in part because of a lack of cholinergic transmission to the diaphragm, resulting in respiratory failure.
Cortical pyramidal neurons were visualized with infrared microscopy, and then randomly patched and recorded in the presence of the NMDA and AMPA receptor antagonists (100 μm d,l-APV and 10 μm NBQX, respectively) to isolate GABAergic currents. In these whole-cell, patch-clamp experiments, the observed ePSCs were obtained at a holding potential of −65 mV evoked by stimulation of 100–150 pA for 100 μs with a bipolar electrode positioned near the target cell. As previously, recordings from wild-type and heterozygote Snap25-null mutant littermates were indistinguishable and thus used as controls for all electrophysiological experiments (Washbourne et al., 2002; Sørensen et al., 2003). As illustrated in Figure 2A, ePSCs were repeatedly and consistently triggered in control (n = 9) neurons after stimulation, whereas mutant (n = 8) cells showed an absence of evoked transmission. Treatment with the selective GABAA receptor antagonist bicuculline (20 μm) (Fig. 2A, dotted trace) abolished the ePSCs in control slices, confirming that these events are mediated by GABAA receptors.
Figure 2.
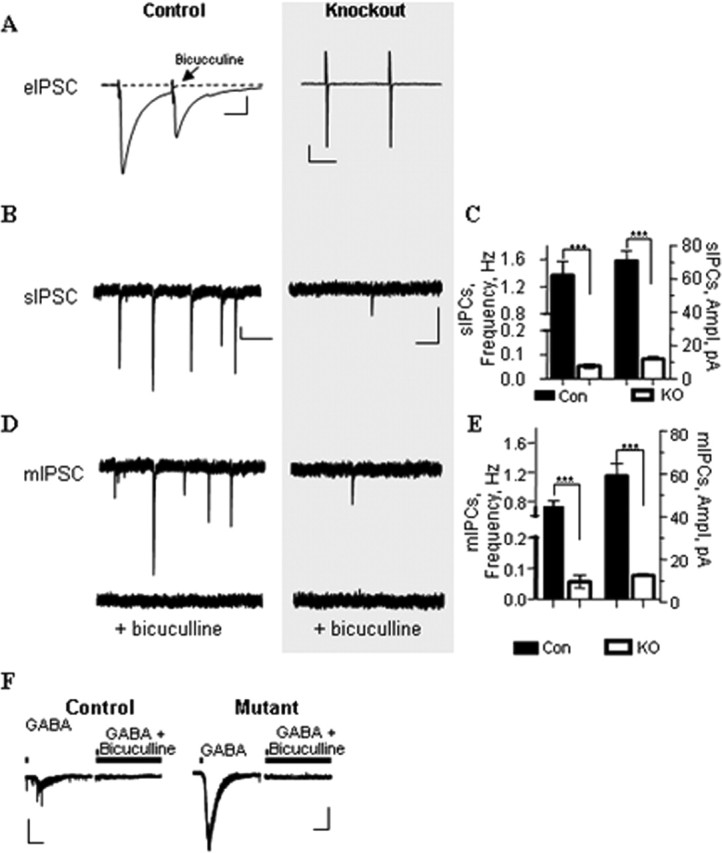
GABAergic transmission could not be evoked in SNAP-25-deficient mutant neurons, although spontaneous currents persist. A, Whole-cell patch-clamp recordings of field stimulation ePSCs obtained from cortical slices of E17.5 control and Snap25-null mutant fetuses. GABAergic responses were isolated by recording in the presence of NBQX (10 μm) plus APV (100 μm) to block glutamatergic transmission. Representative tracings are presented on the left with respective calibrations. The trace (dotted) obtained after application of 20 μm bicuculline to control slices is superimposed over the recording in the absence of GABAA receptor inhibition and indicates GABAergic origin of the response. Mutant slices did not show any detectable response to stimulation. B, Recordings of sPSCs in the absence of TTX. C, sPSCs from Snap25-null mutant slices (n = 9) were decreased in frequency (26.1-fold) and amplitude (6.0-fold) compared with controls (n = 5; ***p < 0.001). Error bars represent SEM. D, mPSCs recorded in the presence of TTX (0.5 μm). E, Both the amplitude (5.2-fold) and frequency (12.9-fold) of TTX-resistant mPSCs were decreased in Snap25-null mutants (n = 3) compared with controls (n = 3; ***p < 0.001). TTX treatment, however, did not significantly reduce the frequency or amplitude of mPSCs recorded from SNAP-25-deficient slices (see Results) indicating that action potential-dependent responses that contribute to the sPSCs of control slices are completely absent in Snap25-null mutants. Error bars represent SEM. F, Response of Snap25-null mutant (n = 5) and control slices (n = 5) to bath application of GABA (50 μm). Note that the response to exogenous GABA in Snap25-null mutant slices was more robust than control (8.4-fold; p < 0.001; n = 8), suggesting that the decreased amplitude for mIPSCs recorded from mutant slices was not attributable to inherent receptor defects and that GABAA receptors may be upregulated in SNAP-25-deficient fetal brain.
We next examined spontaneous GABAergic transmission by recording transient inward currents at a holding potential of −65 mV both with and without the administration of TTX. As shown in Figure 2, B and C, sPSCs observed in SNAP-25-deficient slices were significantly reduced in both frequency and amplitude when compared with controls. The frequency was reduced 26-fold from 1.36 ± 0.46 Hz for control neurons to 0.05 ± 0.03 Hz for mutant neurons. The amplitude of sPSCs was reduced sixfold from 71.0 ± 12.0 pA for control neurons to 11.8 ± 2.96 pA for mutant neurons. Recordings were then made after application of TTX (0.5 μm) to block action potential propagation and isolate mPSCs. As depicted in Figure 2, D and E, Snap25 mutant neurons still exhibited detectable levels of TTX-resistant GABAergic mPSCs. Similar to recordings of sPSCs, there was a significant reduction in the frequency and amplitude of mPSCs (0.06 ± 0.03 Hz; 11.2 ± 1.59 pA) from mutant neurons compared with those recorded from control neurons (0.73 ± 0.13 Hz; 57.7 ± 10.8 pA). Interestingly, TTX treatment did not significantly reduce the amplitude or frequency of either sPSC or mPSC events in Snap25-null mutants, whereas neurons from control slices showed a reduction in both amplitude (18.7%) and frequency (46%) of spontaneous activity when action potential propagation is inhibited. This is consistent with the complete loss of AP-dependent, evoked GABAergic transmission in the absence of SNAP-25, as reported for glutamatergic and cholinergic synapses (Washbourne et al., 2002). Treatment with bicuculline blocked both sPSC and mPSC activity in control and mutant slices, demonstrating that GABAA receptor activation mediated these spontaneous events.
The reduced level of GABAergic transmission in SNAP-25-deficient neurons may have been attributable to decreased postsynaptic GABA responsiveness, possibly by the downregulation of GABAA receptors. To examine GABAAR receptor function in Snap25−/− neurons, we recorded inward currents at a holding potential of −65 mV evoked by exogenous application of GABA (50 μm) in the presence of TTX (0.5 mm). Snap25-null mutant slices displayed a robust response that exceeded that of the control by over eightfold (−10.96 ± 1.43 and −1.30 ± 0.45 pA/pF, respectively) (Fig. 2F), consistent with a potential upregulation of the total available GABAA receptors. These results suggest that, despite the lack of evoked GABA release in SNAP-25-deficient mutants, postsynaptic GABAA receptors are present and capable of responding to GABA transmission in SNAP-25-deficient fetal brain and that the decreased amplitude of mPSCs was, therefore, not attributable to overall decreased GABAA receptor expression.
To examine whether the decrease in evoked and spontaneous GABAergic activity in Snap25-null mutants resulted from a comparable deficit of transmitter-specific synaptic vesicles, we performed Western blot analysis to obtain a quantitative comparison of the relative levels of vesicular glutamate and GABA transport proteins within the synapse. Synaptosome-containing LP2 fractions prepared from the cortex and hippocampus of individual fetal (E17.5) brains were therefore probed for VGAT, VGLUT1, and, as a control, the common protein constituent of synaptic vesicles, synaptophysin. As shown in Figure 3A, the intensity of the signals for VGAT and VGLUT1 transporters was comparable between the LP2 fraction of Snap25-null mutant mice and control extracts. After normalizing the intensities of VGAT and VGLUT1 to synaptophysin in the individual samples, no significant difference was found for the relative signal of VGAT or VGLUT1 between mutant and control LP2 fractions (p > 0.46; one-way ANOVA), indicating that the expression of GABA transporters is not specifically affected in SNAP-25 knock-out mutant synapses (Fig. 3B). Together, these findings suggest that the absence of detectable evoked GABAergic transmission, as well as the sixfold decrease in the amplitude of GABAergic sPSCs, in fetal SNAP-25-deficient cortex likely result from the inability to form AP-dependent neuroexocytotic machinery rather than the depletion of GABA-containing synaptic vesicles or a differential expression or absence of vesicular GABA transport proteins.
Figure 3.
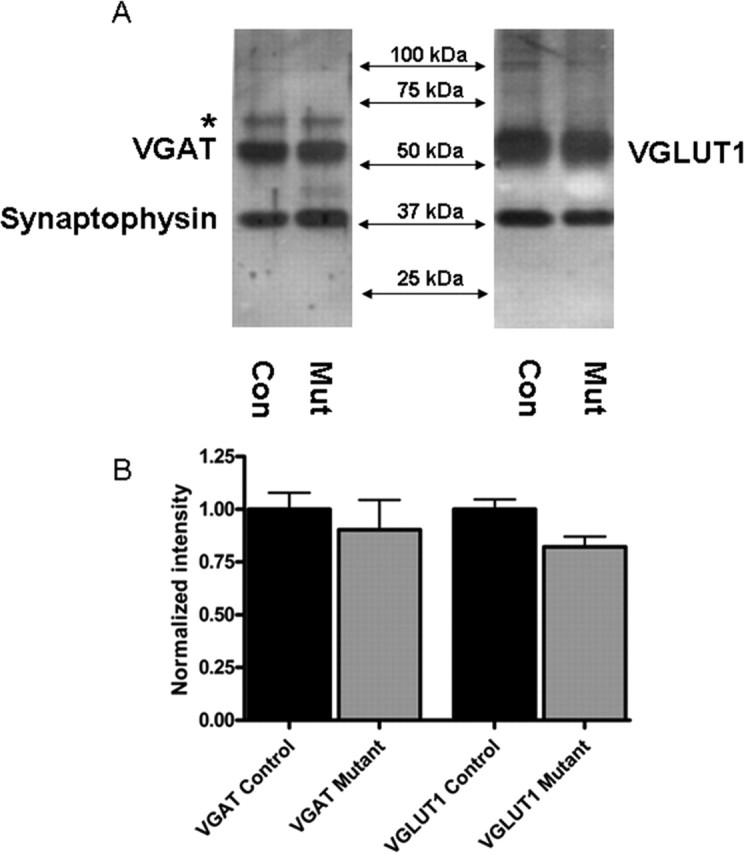
Similar levels of synaptic vesicle proteins expressed in SNAP-25-deficient and control mice. A, Crude synaptic vesicle fraction (LP2; 2.5 μg) prepared from cortex and hippocampus of E17.5 Snap25−/− and control littermates were fractionated on a 10% SDS-PAGE gel, blotted, and probed with antibodies to synaptophysin and VGAT, or VGLUT1, as indicated. B, Three animals per genotype were assayed in duplicate with the mean of each animal’s repeated values being normalized to the average synaptophysin levels of the control group. Error bars indicate SEM. One-way ANOVA analysis showed no difference between levels of immunoreactive VGAT, VGLUT1, and synaptophysin in mutant and control fractions, indicating that neither the total vesicular content nor specific GABA- or glutamate-containing vesicles are significantly decreased in SNAP-25-deficient neurons. The slower migrating species (indicated by an asterisk) recognized by VGAT antibodies likely reflects phosphorylated VGAT (see Results).
The VGAT antibody used in these Western blots detected a higher molecular weight species that has been reported to reflect a phosphorylated form of the transporter (Bedet et al., 2000). Compared with controls, there was a 55% reduction of this immunoreactive protein species (Fig. 3A, left panel, asterisk). Although phosphorylation of VGAT does not appear to affect the vesicular packaging of GABA, this modification may be involved in the trafficking of vesicles and/or the turnover of the transporter protein (Bedet et al., 2000). This suggests that a decrease in expression of phospho-VGAT in mutant GABAergic synapses could reflect changes in the availability of the readily releasable pool of GABAergic vesicles in SNAP-25-deficient mutants.
Stimulus-evoked vesicular recycling is blocked in both glutamatergic and GABAergic synapses of SNAP-25-deficient neurons
Because differences in amplitude and frequency of mPSCs between mutant and control brains could reflect the vesicular content or release of transmitter rather than synaptic vesicle fusion per se, we examined neuroexocytosis more directly by measuring vesicular recycling using FM1-43 uptake (Brumback et al., 2004). Affects on stimulus-driven endocytotic uptake of the styryl dye would provide additional evidence that the absence of AP-dependent release in SNAP-25-deficient neurons resulted from the loss of evoked neuroexocytosis and was not attributable to fusion of empty vesicles. For these experiments, we used the aldehyde fixable analog FM1-43FX to stain dispersed neuronal cultures and assess synaptic vesicle recycling at individual glutamatergic and GABAergic synapses that were subsequently identified by immunocytochemistry with antibodies to the respective vesicular transporters, VGLUT1 and VGAT. Although three isoforms of the vesicular glutamate transporter have been identified, only VGLUT1 and VGLUT2 are expressed exclusively in glutamatergic neurons throughout development (Herzog et al., 2001). Although VGLUT2 expression occurs at high levels in glutamatergic neurons during the first week after birth, VGLUT1 that is expressed initially at low levels in the first week postnatally becomes the predominant form in the postnatal brain (Nakamura et al., 2005). In contrast, VGLUT3 is expressed transiently during development in neurons with various neurotransmitter phenotypes (Gras et al., 2005). Therefore, we selected an antibody raised against VGLUT1 that does not cross-react with the two other isoforms, but allowed us to consistently track glutamatergic cells beyond DIV 9–21 for these and the following experiments described below.
Hippocampal neurons derived from Snap25−/− mutant and wild-type E17.5 fetuses were loaded either by depolarization through application of 90 mm KCl (90 s) or by exposure to hypertonic sucrose (30 s) to induce calcium-independent exocytosis of the readily releasable pool of synaptic vesicles (Rosenmund and Stevens, 1996). After washing away excess dye, neurons were fixed with 4% paraformaldehyde and immunostained with antibodies to either VGLUT1 or VGAT to assess the specific uptake in GABAergic and glutamatergic neurons. As shown in Figure 4, wild-type cultures showed robust punctate FM dye fluorescent staining after depolarization that colocalized with the punctate staining pattern for VGLUT1 (A, B1–B3) and VGAT (D, F1–F3). In contrast, no FM dye uptake was detectable in SNAP-25 knock-out neurons at either glutamatergic or GABAergic immunolabeled synapses (Fig. 4C,G; D1–D3,H1–H3, respectively). Similar images were obtained for wild-type and mutant neurons after hypertonic sucrose (data not shown). Quantitation of the fluorescence intensity of FM1-43 dye that colocalized to VGLUT1- and VGAT-positive terminals in response to either depolarization (Fig. 4I) or hypertonic sucrose (J) confirmed that both glutamatergic and GABAergic synapses in wild-type neurons readily endocytosed FM1-43 dye, which could be effectively unloaded after a second round of depolarization-triggered exocytosis (e.g., destain in I and J). Importantly, neither VGLUT1- nor VGAT-containing synapses of SNAP-25-deficient neurons showed significant uptake above background fluorescence after high K+ or sucrose, demonstrating a complete lack of stimulus driven-endocytosis and therefore highly compromised neuroexocytosis from VGAT-, as well as VGLUT1-containing synapses. Together, these results suggest that the lack of evoked PSCs and the decrease in mPSC amplitude seen in electrophysiology recordings reflects a defect in vesicular fusion in Snap25−/− neurons, and is not attributable to an alternative mechanism, such as the recycling of transmitter-depleted synaptic vesicles.
Figure 4.
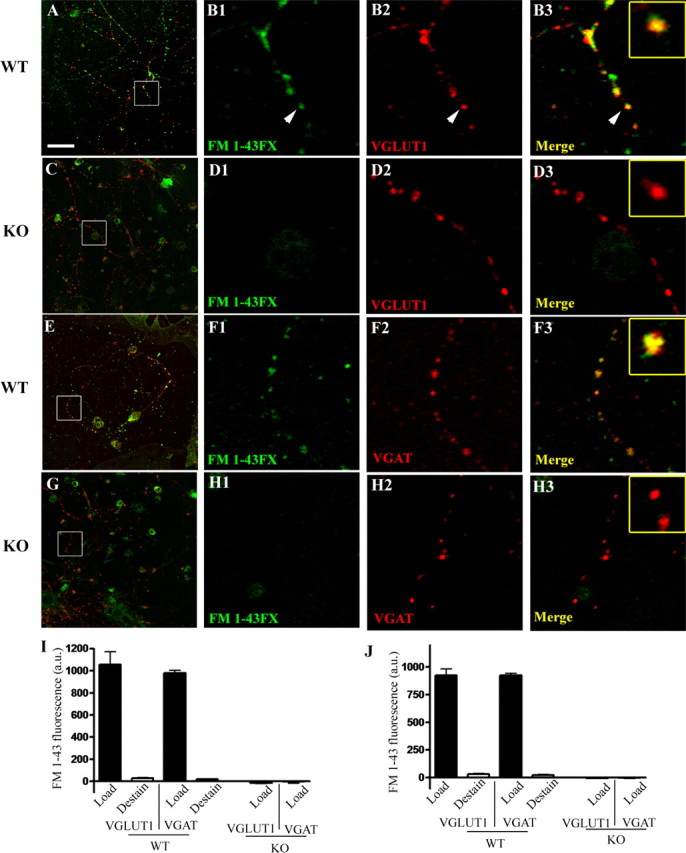
Vesicular recycling within both glutamatergic and GABAergic terminals requires SNAP-25. Hippocampal neurons prepared from both Snap25−/− (KO) and control (WT) E17.5 mice grown for 12 d in culture (DIV 12) were loaded with in the presence of 15 μm FM1-43FX by application of either high K+ or hypertonic sucrose buffer as described in Materials and Methods; control wild-type neurons were also destained by applying a subsequent 90 s exposure to 90 mm K+ to demonstrate exocytotic release and washout of the endocytosed FM dye. After fixation, and immunostaining with either VGLUT1 or VGAT to distinguish dye uptake at glutamatergic or GABAergic synapses, the neurons were viewed by laser confocal microscopy. Representative confocal fluorescent images are shown in A–H, with A, C, E, and G on the left depicting merged images of FM1-43 dye fluorescence and transporter immunostaining taken at 63× (optical slice, 0.81 μm; scale bar, 50 μm). The panels on the right (B1–B3, D1–D3, F1–F3, H1–H3) are series of separated color and merged images of the areas outlined in the low-power images (white box) that were digitally magnified 7× (Adobe Photoshop; scale bar, 7 μm). Insets in the far right merged panels are further digitally enlarged images of the puncta indicated by white arrowheads. Note that FM1-43 dye (green in all panels) is taken up readily by wild-type neurons with equal and consistent colocalization in both glutamatergic (red, B1–B3) and GABAergic (red, F1–F3); whereas in SNAP-25-deficient terminals, there is no detectable FM1-43 dye uptake regardless of their neurotransmitter phenotype (VGLUT, red, D1–D3; VGAT, red H1–H3), consistent with of the lack of stimulus-evoked vesicular recycling in these mutant neurons. I shows the quantitative data of FM1-43 dye intensity over immunoreactive puncta obtained for each transporter after high K+ stimulation. Note that levels of FM1-43 dye fluorescence in loaded wild-type terminals were highly significant (p < 0.001) compared with both destained control synapses and to knock-out mutant synapses, which did not differ from background fluorescence. J compares the relative amount of FM1-43 fluorescence between wild-type and control neurons in a similar series of experiments using hyperosmotic sucrose to promote exocytosis. As in I, although wild-type neurons showed robust vesicular recycling, no detectable fluorescence was observed over VGLUT- or VGAT-immunoreactive puncta of Snap25−/− mutant neurons, indicating that there was no loading of a readily releasable pool of synaptic vesicles at either glutamatergic or GABAergic terminals in these neurons. Error bars indicate SEM.
SNAP-25 is localized to the presynaptic terminals of cultured fetal GABAergic neurons
To confirm that SNAP-25 is localized in the presynaptic terminal of fetal GABAergic neurons, we performed immunocytochemical analysis of dispersed neuronal cultures to compare the expression of SNAP-25 in GABAergic and glutamatergic synapses. As described in Materials and Methods, the monocolonal antibody SMI81 that recognizes an epitope within the N-terminal 31 residues of SNAP-25 (Washbourne et al., 2002) is highly specific and shows no detectable staining in SNAP-25 knock-out neurons (Fig. 1). Transmitter-specific presynaptic terminals and synaptic contacts were identified using antibodies to the GABA synthetic enzyme GAD65/67 and to the GABA and glutamate vesicular transport proteins as described above. To visualize the presynaptic localization of these transmitter phenotype specific markers with SNAP-25, we used hippocampal neurons prepared from E17.5 mouse fetuses and grown as mixed neuronal and glial cell cultures for 9–21 d. These cultures develop extensive processes and sufficiently fine networks to resolve well defined immunoreactive punctate staining of GABAergic and glutamatergic synapses.
GABAergic neurons were first identified by dual immunocytochemical staining with antibodies to the GABA-specific GAD65/67 and SNAP-25. GABAergic neurons express GAD in two different isoforms, encoded by separate genes. These two isoforms are expressed in varying proportions, which prompted the use of a rabbit polyclonal antibody raised against a rat fusion protein that recognizes common determinants between the two GAD proteins (Solimena et al., 1993). As shown in Figure 5A–B3, confocal images of fluorescent immunostaining with the SNAP-25 monoclonal antibody SMI81 was distributed throughout neuronal processes, but also showed punctate staining consistent with the localization of SNAP-25 in presynaptic terminals. In these cultures, immunostaining for GAD65/67 overlapped extensively with some of these SNAP-25-positive processes. Specifically, colocalization between the punctate staining for GAD65/67 and SNAP-25 was revealed, suggesting their expression within the same synapse (Fig. 5A–B3, arrows and inset in merged image).
Figure 5.
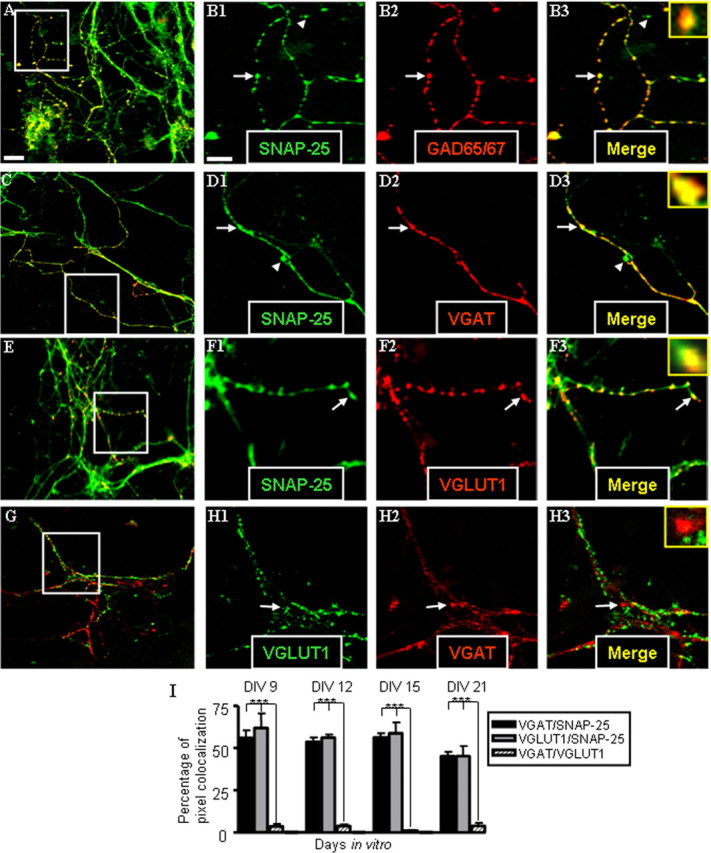
Immunostaining of cultured hippocampal neurons reveals SNAP-25 colocalization with GAD65/67, VGAT, and VGLUT1 in presynaptic terminals. Hippocampal neurons prepared from E17.5 mouse fetuses were cultured, probed with antibodies to the indicated proteins, and viewed by confocal microscopy as described in Materials and Methods. A–H are representative confocal fluorescent images of stained neurons at DIV 15. A, C, E, and G are merged images of dual staining taken at 63× (optical slice, 0.81 μm; scale bar, 50 μm). B1–B3, D1–D3, F1–F3, and H1–H3 are series of separated color and merged images at higher digital magnification of the areas outlined in the low-power images (scale bar, 20 μm). Insets in the far right merged panels are digitally enlarged images of the puncta indicated by white arrows. Note that dual immunostaining for SNAP-25 (green), and GAD65/67 (red; A, B1–B3), VGAT (red; C, D1–D3), and VGLUT1 (red; E, F1–F3) show marked colocalization (yellow in merged images) of punctate fluorescence for transmitter-specific proteins with SNAP-25, consistent with expression of the SNARE protein in GABAergic and glutamatergic terminals (and see I). Arrowheads indicate punctate staining of SNAP-25 that is not colocalized with transmitter-specific antibodies indicating accumulation of SNAP-25 outside terminals of the indicated transmitter phenotype. G, H, Dual staining for transporters VGLUT1 (green) and VGAT (red) show little or no colocalization. I summarizes quantitative data of colocalized punctate staining from dual-stained cultures at DIV 9, 12, 15, and 21 obtained using MetaMorph software (∗∗∗p < 0.001). Error bars indicate SEM.
To confirm that the observed punctate staining pattern of SNAP-25 represented presynaptic terminals in GABAergic neurons, we further stained neuronal cultures with antibodies to SNAP-25 and to the vesicular transporters VGAT and VGLUT1, as described above, to distinguish GABAergic and glutamatergic presynaptic terminals, respectively. As with GAD65/67, immunostaining for the two transporters, VGAT and VGLUT1, resulted in a punctate pattern, consistent with synaptic localization of these vesicular proteins (Fig. 5C–F3). In comparison, SNAP-25 staining in many fibers appeared to be less punctate and more continuous with neurites, which is likely to represent plasma membrane association of this target membrane-associated SNARE (t-SNARE) in fasciculating axons as well as its accumulation in presynaptic terminals (Garcia et al., 1995). Importantly, however, the immunoreactive punctate staining for either transporter coincided with focal immunoreactivity for SNAP-25 (Fig. 5C–F3, arrows and digitally magnified inset), indicating that SNAP-25 is expressed in both terminals with GABA- and glutamate-containing vesicles. As shown in Figure 5I, there was no difference in the extent of colocalization between these two transporters with SNAP-25, suggesting that the SNARE protein expression occurs comparably in GABAergic and glutamatergic terminals. Interestingly, the level of colocalization for VGAT and SNAP-25 immunoreactivity remained remarkably constant throughout 21 d of culture, indicating a persistent expression of this SNARE protein by GABAergic neurons.
Although these results indicated that SNAP-25 expression occurs in both glutamatergic and GABAergic fetal hippocampal presynaptic terminals, a recent study has shown that, in neonatal brainstem, GABAergic/glycinergic synapses transiently express glutamate transporters and are capable of eliciting glutamatergic transmission (Gillespie et al., 2005). Although perhaps specific to certain developmental transitions in neuronal circuitry, such a combined neurotransmitter phenotype in hippocampal cells might complicate any assignment of neurotransmitter phenotype to SNAP-25-expressing neurons. Therefore, to confirm that cultured hippocampal neurons did not simultaneously express both VGLUT1 and VGAT within the same terminal, we immunostained with antibodies to both transporters. In contrast to the colocalization seen with either of the two transporters and SNAP-25, the punctate pattern obtained for VGAT and VGLUT1 dual staining revealed little or no colocalization of the transporters themselves, even within fasciculated bundles of both GABAergic and glutamatergic fibers (Fig. 5G,H; quantitated in I). This suggests that GABAergic and glutamatergic synapses are distinct in these cultured hippocampal neurons, and that few if any terminals contain substantial amounts of both GABAergic and glutamatergic vesicles. Quantitating the pixel overlap of the images confirmed that there was minimal (<5%) colocalization of the two vesicular transporters. This nonoverlapping pattern was found even at the earliest time point analyzed (DIV 9). Overall, the colocalization of the transporters with SNAP-25, but not between each other, indicate that these two distinct neurotransmitter phenotypes are expressed in cultured hippocampal neurons and that SNAP-25 is present in the presynaptic terminals of both developing GABAergic and glutamatergic neurons.
Mature GABAergic neurons express SNAP-25
The previous results demonstrated that SNAP-25 expression is required for synaptic transmission in fetal GABAergic neurons. We performed additional evaluation of SNAP-25 expression by mature GABAergic neurons by using immunocytochemistry and FISH to examine protein and mRNA expression in adult mouse brain. We examined SNAP-25 accumulation in synaptic terminals of GABAergic and glutamatergic neurons in adult mice, by performing fluorescent immunohistochemistry with the same SNAP-25, VGAT, and VGLUT1 antibody combinations used above for colocalization studies in cultured neurons. In coronal sections of hippocampus, we observed marked differences in the distribution of glutamatergic and GABAergic synapses, with VGLUT1 immunoreactivity found primarily in the stratum oriens and stratum radiatum layers and VGAT staining mainly localized to the stratum pyramidale layer. Despite the overall anatomical segregation of these terminals, VGAT- and VGLUT1-positive staining was interspersed at the borders of these hippocampal layers, consistent with the intermingling of excitatory and inhibitory synapses. Quantification of the pixel overlap of the punctate immunostaining for the transporters with SNAP-25 detected with the mAb SMI81 showed comparable colocalization between SNAP-25 and VGLUT1 (53%) or for SNAP-25 and VGAT (47%) (Fig. 6A–D,G). In contrast, little or no pixel colocalization was found after costaining for VGLUT1 and VGAT (Fig. 6E–G), consistent with separate and distinct GABAergic and glutamatergic synaptic terminals. To further assess the colocalization of SNAP-25 with punctate staining that could represent glutamatergic or GABAergic synaptic boutons, we selected a minimal size for pixel clusters of the immunostainning for the vesicular transporters before evaluating their colocalization with SNAP-25. This indicated that ∼95% of clustered pixels representing either VGAT- or VGLUT1-positive terminals overlapped with SNAP-25 reactivity (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 6.
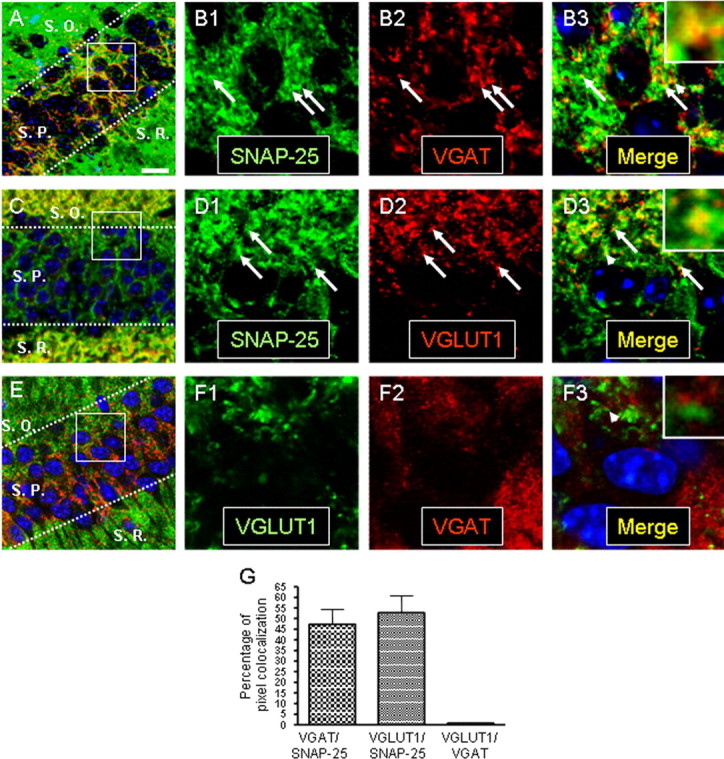
SNAP-25 immunoreactivity colocalizes with VGAT in the CA1 pyramidal layer of the adult hippocampus. Thirty micrometer coronal sections of adult mice (>60 d of age) were coimmunostained with antibodies to SNAP-25 and either vesicular transporters VGAT or VGLUT1, and as a control for specificity coimmunostained with anti-VGLUT1 and VGAT antibodies. After nuclear counterstaining with ToPro3 (blue), the sections were imaged by laser confocal microscopy as described in Materials and Methods. A, C, and E on the right are images taken using a 63× objective (optical slice, 0.81 μm), and the series of panels on the left (B1–B3, D1–D3, F1–F3) show separate channel and merged images of the boxed areas with overlapping red and green pixels depicted as yellow after digital magnification to 420×. A, B, SNAP-25 (green) colocalizes with VGAT (red) within the stratum pyramidale (S.P.) layer of the CA1 region. C, D, VGLUT1 immunoreactivity (red) localized to the stratum oriens (S.O.) and stratum radiatum (S. R.), also colocalizes with SNAP-25 (green). E, F, Nonoverlapping immunoreactivity for VGLUT1 (green) and VGAT (red) seen throughout the hippocampus. Note that, although the patterns of immunostaining for the two transporters are predominantly located in different layers of the hippocampus (E), in regions in which VGAT- and VGLUT1-immunoreactive puncta are interspersed, such as the border of the stratum oriens and pyramidale (boxed area in E; magnified in F), there is virtually no colocalization of these transporters. G, VGLUT1 and VGAT colocalize to a similar extent with SNAP-25. The proportion of pixel overlap in representative immunofluorescent images for transporters VGLUT1 and VGAT with SNAP-25 (VGLUT1/SNAP-25 and VGAT/SNAP-25, respectively) and between the two transporters (VGLUT1 and VGAT) was quantified using MetaMorph software. The histogram represents average values (error bars indicate SEM) obtained from 12 images taken from three animals (∗∗∗p < 0.001). Scale bar: (in A) A, C, E, 20 μm; B, D, F, 3.0 μm.
As shown in Figure 7, A and B, immunofluorescent staining revealed a rich abundance of GABAergic synapses within the ventral posteriolateral (VPL) nucleus of the thalamus, which likely represents afferent projections from the reticular nucleus. Within this region, SNAP-25 immunoreactivity was also widespread, and its colocalization with VGAT was consistent and robust throughout. In addition, punctate staining for VGLUT1 found in the VPL also overlapped with SNAP-25 immunoreactivity, consistent with colocalization of SNAP-25 with these interspersed glutamatergic terminals (Fig. 7C,D). Overlapping punctate staining reflecting colocalized expression of VGAT/SNAP-25 was also observed throughout the cortex and caudate–putamen (data not shown). As seen in immunostaining of fetal neuronal cultures, the punctate colocalized staining of VGAT and VGLUT1 with SNAP-25, coupled with the nonoverlapping pattern of the two transporters, indicated that SNAP-25 is translocated to presynaptic terminals of both glutamatergic and GABAergic neurons in the adult CNS.
Figure 7.

Colocalization of SNAP-25 and VGAT immunoreactivity occurs in the thalamus. Confocal images of immunofluorescent staining for SNAP-25 (green), VGAT (red), and VGLUT1 (red) in 30 μm sections of >P60 wild-type mice were obtained as described in Materials and Methods. A, B1–B4, SNAP-25 and VGAT immunostaining within the VPL of the thalamus. The separate color channel and merged images illustrate the extensively overlapping punctate pattern of robust immunoreactivity observed for VGAT and SNAP-25 (arrows) in this region; consistent cocompartmentalization of SNAP-25 and the GABA transporter within presynaptic terminals of GABAergic neurons. The arrowhead indicates an example of colocalized punctate stained structure after 5× digital enlargement in far upper right corner. C, D1–D4, In contrast, VGLUT1 staining was scarce within the VPL, although small regions with prominent immunoreactivity were evident. Immunofluorescent staining for the glutamate transporter within these patches also appeared punctate and overlapped with SNAP-25 immunostaining, again reflecting the expression of SNAP-25 in glutamatergic presynaptic terminals. A, C, Magnification, 20×; scale bar, 50 μm. B, D, Magnification, 63×; scale bar, 20 μm.
Because comparable levels of pixel overlap were found in both glutamatergic and GABAergic synapses, we used FISH analysis to examine the colocalized expression of SNAP-25 and GAD65/67 mRNAs. FISH analysis of multiple, differently labeled probes provides a complementary method with increased resolution and clarity, because it allows for cell-by-cell comparison and avoids the ambiguity found when probing for protein expression in overlapping and interspersed synaptic connections. Therefore, we used a double-label FITC and Cy3 amplification (Guzowski et al., 1999; Vazdarjanova et al., 2002; Vazdarjanova and Guzowski, 2004) of fluorescein-labeled GAD65/67 and digoxigenin-labeled SNAP-25 antisense riboprobes to identify cells coexpressing both mRNAs in four separate anatomical regions. For these experiments, a mixture of both GAD65 and GAD67 fluorescein-labeled riboprobes were transcribed separately, and equal amounts of the two probes were pooled before hybridization. As shown in Figure 8A, GAD65/67 was readily detected in neurons dispersed throughout layers I–V of cortex, consistent with the distribution of GABAergic interneurons and the prominent hybridization for SNAP-25 mRNA within these neurons. In hippocampus, a similar colocalization of double-labeled GAD65/67 and SNAP-25-positive neurons was found with GABAergic neurons throughout the stratum oriens, stratum pyramidale, and stratum radiatum of the CA1–CA3 regions (Fig. 8B). As shown in Figure 8C, a striking pattern was also observed in thalamus where GAD65/67-positive neurons that are distinctly partitioned within the thalamic reticular nucleus also exhibited robust expression of SNAP-25 mRNA (Fig. 8C). In contrast, throughout neighboring regions, such as the VPL nucleus and the internal capsule, SNAP-25 mRNA was clearly detected in the absence of GAD65/67 hybridization, consistent with the production of this t-SNARE in glutamatergic neurons. Similarly, neurons within the caudate–putamen hybridized with both SNAP-25 and GAD65/67 probes (Fig. 8D). Quantitative analysis of the FISH images revealed that virtually all GAD65/67-positive neurons were also SNAP-25 positive throughout all four brain regions (Fig. 8E).
Figure 8.

Adult GABAergic neurons express both SNAP-25 and GAD65/67 mRNA in several brain regions. FISH of SNAP-25 and GAD65/67 cRNA probes, followed by nuclear counterstaining with DAPI (blue), was performed on 30 μm coronal brain sections of >P60 wild-type mice as described in Materials and Methods. Sections were visualized using a wide-field fluorescent microscope and imaged with MetaMorph software. A–D, Fluorescent images of hybridization for SNAP-25 (red) and GAD65/67 (green), and ToPro3 staining in selected brain regions in separate color channels, and a color merged micrograph (far right). A, Primary motor cortex (layers I–V). B, Hippocampal CA1 region (SO, stratums oriens; SP, stratums pyramidale; SR, stratums radiatum, and SL, lacunosum molecular). C, Thalamus (RT, reticular nucleus; ic, internal capsule). D, Caudate–putamen. GAD65/67-positive nuclei throughout these selected anatomical regions display robust expression and colocalization with SNAP-25 mRNA (white arrows, digitally enlarged in white box in upper far right corner of the merged image). Images were taken with a 20× objective. Scale bar: A–D, 50 μm. E, Quantitation of colocalized GAD65/67 and SNAP-25 hybridization with 20× magnification. The fraction of GAD65/67-positive cells with overlapping SNAP-25 hybridization, determined using ImageJ software, demonstrates that nearly all cells expressing GAD65/67 mRNA coexpressed SNAP-25 mRNA in each brain region assayed. Error bars indicate SEM. Statistical analysis was performed by one-way ANOVA with Bonferroni’s post hoc comparisons.
Discussion
SNAP-25 is a critical component of the neural SNARE complex that contributes to the calcium-triggered exocytosis of neurotransmitter by facilitating membrane fusion between synaptic vesicles and the presynaptic plasma membrane. Numerous studies have implicated the involvement of SNAP-25 and neural SNARE complex in the release of a wide range of neurotransmitters. Here, we provide electrophysiological evidence from SNAP-25-deficient mutant and wild-type mice that extends the repertoire of this t-SNARE to a role in GABAergic transmission. Moreover, we observed SNAP-25 localization in the presynaptic terminals of GABAergic neurons throughout maturation, inferring that GABAergic function is dependent on SNARE-mediated neurotransmission throughout development.
Interestingly, in contrast to the evidence presented here, it has been reported recently that maturation of GABAergic neurons may be accompanied by a decrease in detectable SNAP-25 expression and an increase in resistance of GABA transmission and vesicular cycling to BoNT/A and BoNT/E. These observations have been interpreted to indicate that SNAP-25 does not participate in the SNARE complexes responsible for GABAergic transmission (Verderio et al., 2004; Frassoni et al., 2005). The activity of Clostridium neurotoxins in blocking transmitter release, however, varies greatly depending on the serotype of toxin, as well as the susceptibility of different neurons to the surface binding and activation of the holotoxin (Purkiss et al., 2001) (for review, see Montecucco et al., 2004). The spatial orientation, grouping, and organization of specialized gangliosides within a lipid raft have been shown to alter the plasma membrane binding affinity of botulinum neurotoxins (Yowler et al., 2002). Differential vulnerability to toxins, therefore, can depend on the regulated expression of gangliosides that act as neuron-specific, toxin coreceptors (Vyas et al., 2001). For example, although BoNT/A effectively binds to and blocks transmitter release at peripheral cholinergic synapses (Black and Dolly, 1987), there is considerable variability in the binding and uptake of neurotoxins into neurons across different anatomical regions in the adult rat brain (Williams et al., 1983; Black and Dolly, 1987). Thus, the inability to detect SNAP-25-mediated GABA release, when assayed by neurotoxin sensitivity, could be attributable to changes in cell surface receptors during the maturation of inhibitory neurons, which result in decreased binding and increased resistance to BoNT/A and BoNT/E intoxication. Use of Snap25-null mutants in which the expression of this SNARE protein is uniformly ablated in all neurons, therefore, circumvents possible variability in toxin efficacy. Nevertheless, because the mutants do not survive birth, functional studies of integrated neuronal connectivity are limited to the developing fetal brain.
As did Frassoni et al. (2005), we observed SNAP-25 immunoreactivity in cultured GABAergic neurons after 9–10 d in culture. However, in contrast to their observation of decreasing coexpression in GAD-positive neurons over several days in culture, we found no change in the extent of colocalization of SNAP-25 with immunoreactive GAD65/67 and VGAT (or of comparable levels of VGLUT1 for glutamatergic neurons) in hippocampal neurons cultured for up to 21 d. Similarly, we show extensive coexpression of SNAP-25 with GAD65/67 and VGAT, which define GABAergic neurons, within several anatomically distinct regions of the adult mouse brain. Based on these results, we propose that GABAergic neurons maintain SNAP-25 expression for stimulus-driven GABA release throughout synaptic maturation.
Although all inhibitory neurons that we examined expressed SNAP-25 mRNA, there were differences in the level of hybridization distinguished by fluorescent intensity among different subpopulations of GABAergic neurons, consistent with the differential expression of this t-SNARE across brain regions (Oyler et al., 1989). Interestingly, regional differences have also been detected in the expression of SNAP-25 isoforms, generated through alternative splicing (Bark et al., 1995; Boschert et al., 1996). This indicates that the expression of SNAP-25 may be coupled with distinct physiological properties of neurotransmission in different neurons. The two SNAP-25 isoforms have been shown to significantly affect the recruitment of catecholaminergic vesicles into the readily releasable pool of chromaffin cells (Sørensen et al., 2003). Additional studies will examine the possibility that regulated expression of SNAP-25 and its isoforms may confer differences that suit the differing requirements of transmission by various GABAergic interneurons and projection neurons.
The absence of stimulus-evoked GABA postsynaptic responses in SNAP-25-deficient fetal brain indicates the role of SNAP-25-containing SNARE complexes mediating AP-dependent GABAergic synaptic transmission. This is further supported by FM1-43 dye uptake experiments that demonstrated the lack of evoked vesicular recycling at both glutamatergic and GABAergic terminals in SNAP-25 knock-out mutant neuronal cultures. Moreover, recent ongoing studies have shown that the expression of SNAP-25 transgenes is able to rescue and restore both evoked GABAergic and glutamatergic transmission to Snap25-null mutant neurons in culture (I. D. Martinez and J. B. Sørensen, personal communication). The observation of spontaneous GABAergic mPSC activity is, however, also consistent with the AP-independent release of other transmitters previously seen in this mutant (Washbourne et al., 2002). The amplitude and frequency of these spontaneous events, however, appears to vary considerably between transmitter systems. For example, although both the frequency and amplitude of GABAergic mPSCs recorded in mutant cortex were significantly reduced when compared with controls, cholinergic miniature endplate potentials at mutant neuromuscular junctions were found to have increased frequency and amplitude (Washbourne et al., 2002). This contrasts with the comparable glutamatergic mEPSCs recorded from cortex of mutant and control fetal brain, and the similar level of the slow, non-Ca2+-triggered component of catecholamine secretion of mutant chromaffin cells that is thought to represent the slow release from an unprimed pool of secretory granules (Sørensen et al., 2003). Interestingly, mEPSCs recorded from cultured hippocampal neurons occur at significantly lower frequency, but maintain the same amplitude as control mEPSCs This does suggest, however, that although quantal release of transmitter does occur in the absence of SNAP-25, presumably promoted by other SNARE complexes, changes in presynaptic and postsynaptic properties of these spontaneous events can be influenced by the loss of evoked transmission.
The reason for the decreased frequency of mPSCs in both mutant GABAergic and mutant glutamatergic neurons is not clear. It has been shown that in fetal Snap25-null mutants, calbindin- and calretinin-positive populations of GABAergic neurons are generated and migrate appropriately within the cortex (Molnar et al., 2002). However, the possibility remains that mutant neurons may exhibit alterations in activity-dependent synaptic refinement, resulting in less efficient formation of functional synapses, thereby causing a decreased mPSC frequency.
The decreased amplitude of GABAergic mPSCs in SNAP-25-deficient brain slices was not accompanied by a corresponding decrease in either VGAT expression or a decrease in the overall GABAA receptor responsivity. In fact, application of exogenous GABA elicited a markedly higher level of receptor activation in mutant slices compared with controls, suggesting upregulation of GABAA receptors in response to the lack of evoked GABAergic transmission. The reduced amplitude recorded from these spontaneous events nevertheless may reflect deficits in filling, trafficking, and/or targeting of GABA-containing synaptic vesicles. For example, the amplitude of mIPSCs can be dependent on the efficient uptake and conversion of extracellular glutamate to GABA (Mathews and Diamond, 2003). The abolition of evoked glutamatergic transmission in Snap25-null mutants could lead to decreased availability of glutamate within the synaptic cleft, thus diminishing the uptake of this amino acid by GABAergic neurons. Impaired glutamatergic release may therefore depress GABA synthesis and contribute to a decreased quantal content of GABAergic vesicles. In addition, reuptake of GABA appears to be regulated by the interaction of syntaxin 1a with the GABA transporter 1 (GAT1) (Wang et al., 2003). In the absence of SNAP-25, syntaxin 1a that is no longer recruited into SNARE complexes may be available to interact and downregulate GAT1, thereby leading to decreased GABA reuptake and repackaging into GABAergic vesicles. Decreased levels of phospho-VGAT, in addition to the decreased mPSC frequency discussed above, may further reflect alterations in the trafficking and turnover of a pool of GABA-containing vesicles that are compromised in SNAP-25-deficient terminals.
Identifying proteins that mediate the release of different neurotransmitters provides a means to differentiate between those proteins that are central to the basic mechanisms of neuroexocytosis from those that are modulatory and shape the physiological characteristics of transmitter-specific synapses. Demonstration of the expression of SNAP-25 in GABA-containing terminals and the dependence on this SNARE protein for GABAergic transmission therefore support an essential role for SNAP-25 in a universal neural SNARE complex necessary for calcium-triggered release within diverse neurotransmitter systems. However, although SNAP-25 is a key element for evoked transmitter release, the inclusion of its developmentally regulated isoforms into neural SNARE complexes also affects the release properties of neurosecretion (Sørensen et al., 2003; Nagy et al., 2005) and may participate in short-term plasticity and maturation of excitatory synaptic transmission (Bark et al., 2004). This suggests that SNAP-25 can serve a pivotal role, both as a fundamental constituent and as a modulatory component of the presynaptic membrane fusion machinery underlying neurotransmission.
Footnotes
M. Mameli’s present address: Department of Basic Neuroscience, University of Geneva, 1 Michel Servet, 1211 Geneva, Switzerland.
T. Miyashita and J. F. Guzowski’s present address: Department of Neurobiology and Behavior and Center for the Neurobiology of Learning and Memory, University of California, Irvine, CA 92697-3800.
This work was supported by National Institutes of Health Grants MH48989 (M.C.W.), MH60123 (J.F.G.), and AA12684 (C.F.V.), and in part by the Dedicated Health Research Funds of the University of New Mexico (UNM) School of Medicine (M.C.W.). Images in this paper were generated in the UNM Cancer Center Fluorescence Microscopy Facility, which received support from National Center for Research Resources (NCRR) Grant 1 S10 RR14668, National Science Foundation Grant MCB9982161, NCRR Grant P20 RR11830, National Cancer Institute Grant R24 CA88339, NCRR Grant S10 RR19287, NCRR Grant S10 RR016918, the UNM Health Sciences Center, and the UNM Cancer Center. We thank Donald Partridge and Michela Matteoli for stimulating discussions, Michele Solimena for the GAD65/67 polyclonal antisera, and Donald Patridge and Chessa Scullin for their careful reading and comments on this manuscript. We also thank Julie Torres and Erika Langsfeld for technical assistance in genotyping, and Ed Padilla at the UNM Health Sciences Center Animal Resource Facility for maintaining the mouse colony. We also thank the reviewers for their suggestions, which have helped to strengthen this manuscript.
References
- Augustin I, Rosenmund C, Sudhof TC, Brose N (1999). Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400:457–461. [DOI] [PubMed] [Google Scholar]
- Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC (2004). Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci 24:8796–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, Wilson MC (1994). Regulated vesicular fusion in neurons: snapping together the details. Proc Natl Acad Sci USA 91:4621–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC (1995). Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA 92:1510–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedet C, Isambert MF, Henry JP, Gasnier B (2000). Constitutive phosphorylation of the vesicular inhibitory amino acid transporter in rat central nervous system. J Neurochem 75:1654–1663. [DOI] [PubMed] [Google Scholar]
- Black J, Dolly J (1987). Selective location of acceptors for botulinum neurotoxin A in the central and peripheral nervous systems. Neuroscience 23:767–779. [DOI] [PubMed] [Google Scholar]
- Bock JB, Matern HT, Peden AA, Scheller RH (2001). A genomic perspective on membrane compartment organization. Nature 409:839–841. [DOI] [PubMed] [Google Scholar]
- Boschert U, O’Shaughnessy C, Dickinson R, Tessari M, Bendotti C, Catsicas S, Pich EM (1996). Developmental and plasticity-related differential expression of two SNAP-25 isoforms in the rat brain. J Comp Neurol 367:177–193. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Lieber JL, Angleson JK, Betz WJ (2004). Using FM1-43 to study neuropeptide granule dynamics and exocytosis. Methods 33:287–294. [DOI] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF (2004). Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci 24:3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998). Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA 95:15781–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassoni C, Inverardi F, Coco S, Ortino B, Grumelli C, Pozzi D, Verderio C, Matteoli M (2005). Analysis of SNAP-25 immunoreactivity in hippocampal inhibitory neurons during development in culture and in situ. Neuroscience 131:813–823. [DOI] [PubMed] [Google Scholar]
- Garcia EP, McPherson PS, Chilcote TJ, Takei K, De Camilli P (1995). rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol 129:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie DC, Kim G, Kandler K (2005). Inhibitory synapses in the developing auditory system are glutamatergic. Nat Neurosci 8:332–338. [DOI] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ (2004). Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci 24:11368–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, Washbourne P, Wilson MC, Burgoyne RD (2002). Molecular analysis of SNAP-25 function in exocytosis. Ann NY Acad Sci 971:210–221. [DOI] [PubMed] [Google Scholar]
- Gras C, Vinatier J, Amilhon B, Guerci A, Christov C, Ravassard P, Giros B, El Mestikawy S (2005). Developmentally regulated expression of VGLUT3 during early post-natal life. Neuropharmacology 49:901–911. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, McNaughton BL, Barnes CA, Worley PF (1999). Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci 2:1120–1124. [DOI] [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S (2001). The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. In: J Neurosci 21: pp. 1–6. RC181 :. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W (2005). SNAREs and traffic. Biochim Biophys Acta 1744:493–517. [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P (1983). Synapsin I (protein I), a nerve terminal-specific phosphoprotein III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC (2003). Membrane fusion. Cell 112:519–533. [DOI] [PubMed] [Google Scholar]
- Keller JE, Cai F, Neale EA (2004). Uptake of botulinum neurotoxin into cultured neurons. Biochemistry 43:526–532. [DOI] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouille P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, Galli T (2001). A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci 21:3830–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews GC, Diamond JS (2003). Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci 23:2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, Mahal LK, Sollner TH, Rothman JE (2002). Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J Cell Biol 158:929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar Z, Lopez-Bendito G, Small J, Partridge LD, Blakemore C, Wilson MC (2002). Normal development of embryonic thalamocortical connectivity in the absence of evoked synaptic activity. J Neurosci 22:10313–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco C, Rossetto O, Schiavo G (2004). Presynaptic receptor arrays for clostridial neurotoxins. Trends Microbiol 12:442–446. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G, Pantano S (2005). SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem Sci 30:367–372. [DOI] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sørensen JB (2005). Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell 16:5675–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Hioki H, Fujiyama F, Kaneko T (2005). Postnatal changes of vesicular glutamate transporter (VGluT)1 and VGluT2 immunoreactivities and their colocalization in the mouse forebrain. J Comp Neurol 492:263–288. [DOI] [PubMed] [Google Scholar]
- Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC (1989). The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J Cell Biol 109:3039–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkiss JR, Friis LM, Doward S, Quinn CP (2001). Clostridium botulinum neurotoxins act with a wide range of potencies on SH-SY5Y human neuroblastoma cells. Neurotoxicology 22:447–453. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Lang J, Montecucco C, Weller U, Regazzi R, Catsicas S, Wollheim CB, Halban PA (1995). SNAP-25 is expressed in islets of Langerhans and is involved in insulin release. J Cell Biol 128:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Mozhayeva MG, Liu X, Kavalali ET (2002). Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J Neurosci 22:1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC (2002). RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415:321–326. [DOI] [PubMed] [Google Scholar]
- Solimena M, Aggujaro D, Muntzel C, Dirkx R, Butler M, De Camilli P, Hayday A (1993). Association of GAD-65, but not of GAD-67, with the Golgi complex of transfected Chinese hamster ovary cells mediated by the N-terminal region. Proc Natl Acad Sci USA 90:3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003). Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114:75–86. [DOI] [PubMed] [Google Scholar]
- Sudhof TC (2004). The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 395:347–353. [DOI] [PubMed] [Google Scholar]
- Vazdarjanova A, Guzowski JF (2004). Differences in hippocampal neuronal population responses to modifications of an environmental context: evidence for distinct, yet complementary, functions of CA3 and CA1 ensembles. J Neurosci 24:6489–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazdarjanova A, McNaughton BL, Barnes CA, Worley PF, Guzowski JF (2002). Experience-dependent coincident expression of the effector immediate-early genes arc and Homer 1a in hippocampal and neocortical neuronal networks. J Neurosci 22:10067–10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Pozzi D, Pravettoni E, Inverardi F, Schenk U, Coco S, Proux-Gillardeaux V, Galli T, Rossetto O, Frassoni C, Matteoli M (2004). SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 41:599–610. [DOI] [PubMed] [Google Scholar]
- Vyas KA, Patel HV, Vyas AA, Schnaar RL (2001). Segregation of gangliosides GM1 and GD3 on cell membranes, isolated membrane rafts, and defined supported lipid monolayers. Biol Chem 382:241–250. [DOI] [PubMed] [Google Scholar]
- Wang D, Deken SL, Whitworth TL, Quick MW (2003). Syntaxin 1A inhibits GABA flux, efflux, and exchange mediated by the rat brain GABA transporter GAT1. Mol Pharmacol 64:905–913. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002). Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. [DOI] [PubMed] [Google Scholar]
- Williams RS, Tse CK, Dolly JO, Hambleton P, Melling J (1983). Radioiodination of botulinum neurotoxin type A with retention of biological activity and its binding to brain synaptosomes. Eur J Biochem 131:437–445. [DOI] [PubMed] [Google Scholar]
- Yowler BC, Kensinger RD, Schengrund CL (2002). Botulinum neurotoxin A activity is dependent upon the presence of specific gangliosides in neuroblastoma cells expressing synaptotagmin I. J Biol Chem 277:32815–32819. [DOI] [PubMed] [Google Scholar]