

Abstract
Microglia cells are the resident macrophages of the CNS, and their activation plays a critical role in inflammatory reactions associated with many brain disorders, including ischemia, Alzheimer's and Parkinson's diseases, and epilepsy. However, the changes of microglia functional properties in epilepsy have rarely been studied. Here, we used a model of status epilepticus (SE) induced by intraperitoneal kainate injections to characterize the properties of microglial cells in hippocampal slices from CX3CR1eGFP/+ mice. SE induced within 3 h an increased expression of inflammatory mediators in the hippocampus, followed by a modification of microglia morphology, a microglia proliferation, and a significant neurodegeneration in CA1. Changes in electrophysiological intrinsic membrane properties of hippocampal microglia were detected at 24–48 h after SE with, in particular, the appearance of new voltage-activated potassium currents. Consistent with the observation of an upregulation of purinergic receptor mRNAs in the hippocampus, we also provide pharmacological evidence that microglia membrane currents mediated by the activation of P2 receptors, including P2X7, P2Y6, and P2Y12, were increased 48 h after SE. As a functional consequence of this modification of purinergic signaling, motility of microglia processes toward a source of P2Y12 receptor agonist was twice as fast in the epileptic hippocampus. This study is the first functional description of microglia activation in an in vivo model of inflammation and provides evidence for the existence of a particular microglial activation state after a status epilepticus.
Keywords: glial cells, ATP, inflammation, epilepsy, cytokines, hippocampus
Introduction
Increasing evidence indicates that inflammation is a landmark for a number of acute and chronic brain disorders, including neuropathologies associated with seizures (Rock and Peterson, 2006; Choi and Koh, 2008). For instance, an increased expression of inflammatory markers has been observed in surgically resected temporal tissues from patients with temporal lobe epilepsy (Sheng et al., 1994; Crespel et al., 2002; Vezzani and Granata, 2005). A similar inflammatory reaction has also been reported in experimental models of temporal lobe epilepsy. In particular, an increase of proinflammatory cytokines and of markers of the innate immunity (IL-1β, IL-6, TNF-α, NF-κB system, COX-2, prostaglandins, and Toll-like receptors) has been detected in the rat hippocampus, starting within the first hour after the induction of a status epilepticus (SE) and lasting several days (Eriksson et al., 1999; De Simoni et al., 2000; Vezzani et al., 2008).
Microglia and astrocytes represent the major source of proinflammatory molecules, and they play a critical role in determining the spatial and temporal extent of inflammation as well as its impact on neuronal function and survival (Wyss-Coray and Mucke, 2002; Hanisch and Kettenmann, 2007; Vesce et al., 2007; Viviani et al., 2007). Accordingly, microglia and astrocytes become reactive after a status epilepticus. In the case of microglia, this activation has been well documented in terms of changes in morphology and expression of specific markers (Turrin and Rivest, 2004; Vezzani and Granata, 2005). However, activation of microglial cells is a complex process that includes changes in pharmacological and electrophysiological properties, migration, proliferation, and release of a variety of mediators (Färber and Kettenmann, 2005; Hanisch and Kettenmann, 2007). However, these functional properties have never been investigated in epileptic tissues. Furthermore, different stimuli and stimulatory contexts can trigger different activation states of microglial cells, and the concept of a continuum of activation between quiescent and fully activated microglia has recently emerged (Hanisch and Kettenmann, 2007). Thus, the specific state of microglia after a status epilepticus cannot be inferred from our knowledge of other models.
Among the key signals that govern microglia activation, several lines of evidence indicate that ATP released by glia and neurons and acting through ionotropic (P2X) and metabotropic (P2Y) purinergic receptors regulates numerous aspects of microglia activation (Inoue et al., 2007). In particular, specific functions of different purinergic receptors in microglia have been identified pointing at essential roles of P2Y6 in phagocytosis (Koizumi et al., 2007), P2Y12 in the polarization, migration, and process extension (Davalos et al., 2005; Haynes et al., 2006; Ohsawa et al., 2007), and P2X receptors in the secretion of proinflammatory molecules from microglial cells (Tsuda et al., 2003; Coull et al., 2005; Chessell et al., 2005; Skaper et al., 2006). Interestingly, activation of microglia can differentially regulate the expression of its purinergic receptors in different models, as reported for P2X4, P2X7, P2Y6, and P2Y12 (Tsuda et al., 2003; Franke et al., 2004; Haynes et al., 2006; Koizumi et al., 2007; Kobayashi et al., 2008).
The aim of the present work was to characterize the activation state of hippocampal microglia during the inflammatory reaction, which occurs in a noninvasive model of status epilepticus (i.e., intraperitoneal injection of kainate) in mice. We used CX3CR1eGFP/+ mice, in which microglia express the green fluorescent protein and retain normal functional properties (Cardona et al., 2006), allowing the comparison of cellular characteristics between nonactivated and activated microglia. Our data provide the first comprehensive functional characterization of activated microglia in the epileptic hippocampus.
Materials and Methods
Animals and seizure inductions.
All experiments followed European Union and institutional guidelines for the care and use of laboratory animals (council directive 86/609EEC). The heterozygous CX3CR1eGFP/+ mice used throughout this study were obtained by crossing CX3CR1eGFP/eGFP (Jung et al., 2000) with C57BL/6 (Janvier) wild-type mice. Intraperitoneal injection of kainate (18–22 mg/kg) in PBS was used to induce a status epilepticus in 30- to 40 d-old mice, and age-matched PBS-injected animals were used as control. Animals were observed and classified according to the Racine scale: (1) freezing behavior; (2) rigid posture with straight and rigid tail; (3) repetitive head bobbing, rearing into a sitting position with forepaws shaking; (4) rearing and falling, jumping, running with period of total stillness; (5) continuous level 4; and (6) loss of posture and generalized convulsion activity, usually preceding death. After kainate injection, mice showed progression through the different stages, usually entering in phase 1 in the first 15 min after the injection and reaching stage 3 in 30–45 min. Animals not showing the normal progression were reinjected with half dose. Only animals reaching at least stage 4 were considered for this study. The duration of crises varied from 2 to 4 h, and mortality was ∼20%.
Western blotting.
The hippocampi of three mice per condition were homogenized in a lysis buffer (10 mm HEPES buffer, pH 7.4), 0.5% Triton X-100, and protease inhibitor mixture (Roche Applied Science). Lysates were clarified by centrifugation, and protein concentration was determined using a protein assay kit (Biorad). Proteins were separated on reducing 12% SDS-PAGE gels and transferred on nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk/0.5% Tween 20 in Tris buffer saline (TBST) overnight at 4°C. The membrane was incubated overnight at 4°C with a rabbit anti-Iba1 (1:1000; Wako Chemicals) in TBST and with mouse anti-tubulin (1:2500; Sigma-Aldrich). After three washes in TBST, the membrane was then treated with HRP-conjugated secondary antibody for 1 h at room temperature and revealed with an ECL+ detection kit (GE Healthcare).
Histochemistry.
Mice were anesthetized with pentobarbital (50 mg/kg) or urethane (2 g/kg) and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were dissected out and postfixed for 2 h or overnight at 4°C, and 50-μm-thick slices were prepared with a VT 1000 Leica vibratome. Slices were washed three times with PBS and incubated for 2 h with PBS containing 2% bovine serum albumin (Sigma-Aldrich) and 0.1% Triton X-100 (Sigma-Aldrich). After three washes, slices were incubated overnight with primary antibodies for microglia markers Iba1 (rabbit polyclonal; 1:1000; Wako Chemicals), MAC2 [1:1000; rat monoclonal; kindly provided by M. Benhamou (Inserm U699, Paris, France), obtained from hybridomas (TIB-166) distributed by American Type Culture Collection-LGC Standards], P2Y12 [1:1000; kindly provided by David Julius (University of California, San Francisco, San Francisco, CA)]. Ki67 antibody (mice monoclonal; 1:20; BD Bioscience) was used to assess cell proliferation. Appropriate secondary antibodies were applied for 2 h at room temperature and included anti-rabbit/anti-rat IgG Alexa Fluor 568/555 (1:1000; Invitrogen) and anti-mouse IgG Alexa Fluor 555 (1:1000; Millipore Bioscience Research Reagents). After staining, brain slices were mounted in Vectashield H1400 (Vector Laboratories; distributed by Abcys) and examined with a confocal laser-scanning microscope (Zeiss 510).
To assess neuronal damage, slices were incubated with eosin–hematoxylin solution for 10 s followed by five washes with distilled water. For Fluoro-Jade B staining, slices were successively immersed at room temperature in a solution of 5% NaOH in 80% ethanol for 5 min, 2 min in 70% ethanol, 2 min in distilled water, 10 min in 0.06% potassium permanganate, and 2 min in distilled water. Slices were incubated for 20 min with 0.004% Fluoro-Jade B in a 0.1% acetic acid and rinsed by three washes with distilled water. Slices were mounted and images of the hippocampal rostral region were captured with a Leica DMRA2 fluorescence microscope equipped with a 20× objective. To quantify Fluoro-Jade B-positive cells, three mice per condition (PBS or 48 h after SE) were used. For each hippocampus, usually two fields (452 × 338 μm) were necessary to acquire the whole CA1 and CA3 regions, and five consecutive sections were analyzed. Fluoro-Jade B-positive neurons were manually counted in both areas. Results are reported as the number of positive neurons per field of view and expressed as mean ± SEM.
Quantification of the number of microglial cells.
GFP-positive cells were counted in confocal sections taken with 40× objective (size of the field of view, 230 × 230 μm). Similar laser intensity was used to acquire pictures of PBS- and kainate-injected animals. Cells were counted in 3–11 slices from each animal, and 3–7 animals were analyzed for each condition. The number of cells counted in different slices of each animal was quite homogeneous and followed a Gaussian distribution. For each condition, we calculated the weighted average and its relative SEM. ANOVA with post hoc Tukey test was applied to test significance between different groups.
RNA extraction and reverse transcription-PCR.
Hippocampi from PBS- or kainate-injected mice were collected, frozen in liquid nitrogen, and stored at −80°C until RNA extractions were performed. Total RNA were extracted using the Trizol reagent (Invitrogen), and then genomic DNA was digested using a RNase-free DNase set (Qiagen) according the to the manufacturer's protocols. Total RNA (2 μg) was reverse transcribed into cDNA with random hexamers in a 20 μl reaction by using a SuperScript III reverse transcriptase kit (Invitrogen). Quantitative real-time PCR was performed in duplicate using four to six independent RNA samples. The cDNA was quantified using the Quantitect SYBR Green PCR kit and the Quantitect Primer assay kit (Qiagen) according to the manufacturer's instructions. The PCR amplification was run in 10 μl on cDNA generated from 40 ng of total RNA and using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). For each reaction, we calculated the difference (ΔCt) between the cycle threshold (Ct) of the gene of interest and that of the housekeeping gene HPRT (hypoxanthine-guanine phosphoribosyltransferase). To quantify the fold changes in expression of each gene in kainate-injected animals compared with control, we first calculated the mean of ΔCt obtained in four PBS-injected animals (ΔCtmPBS). Then, for each kainate-injected animal, we calculated the ratio between 2−ΔCt and 2−ΔCtmPBS, i.e., 2−(ΔCt−ΔCtmPBS), which estimates the fold change in expression of a given gene relative to the control animals (Livak and Schmittgen, 2001). Results are presented as the mean ± SEM. Statistical significance compared with control was calculated using Student's t test with Welch's correction, because SDs are usually different, and was established at p < 0.05 and p < 0.01.
In vivo electrophysiological recordings.
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg) and placed into a stereotaxic frame. Skull surface was exposed and five electrodes were placed as follows: four extradural electrodes were inserted bilaterally in the parietal areas and one screwed at the level of the frontal area (ground electrode). Electrodes were plugged in a microconnector with acrylic cement. After surgery, mice were allowed to recover for 1 week before recording. Freely moving animals were placed into individual Plexiglas boxes in a Faraday cage and their microconnectors plugged to an electroencephalographic (EEG) preamplifier box (Reega mini8; Alvar). The electrical activity recorded by derivation between extradural electrodes was filtered (passband, 0.5–60 Hz) and acquired with a computer-based system (Micromed).
In vitro electrophysiological recordings.
Hippocampal slices were obtained from 30- to 40-d-old mice. Animals were killed by cervical dislocation, and the brain was then quickly removed and placed in ice-cold artificial CSF (aCSF) bubbled with carbogen (95% CO2/5% O2) and in which NaCl was replaced with sucrose (in mm: 210 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 25 glucose, 1 CaCl2, and 7 MgSO4, pH 7.4, osmolarity ∼310 mOsm). Transverse 350-μm-thick slices were cut using a vibratome, transferred to a heated (34°C) holding chamber containing oxygenated (95% CO2/5% O2) standard aCSF (in mm: 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 2 CaCl2, and 1 MgCl) for 1 h, and then subsequently maintained at room temperature.
Individual slices were transferred to a recording chamber on the stage of an Olympus microscope (BX50WI) with a 40× water-immersion objective, equipped with cell-R imaging station including MT20 illumination system (Olympus) and a CCD camera (Hamamatsu ORCA2-AG). Slices were constantly perfused at room temperature (21–24°C) with oxygenated aCSF (3 ml/min). Unless otherwise stated, all drugs were applied through the general perfusion. Visually identified GFP-expressing microglial cells were patched in whole-cell configuration in the stratum radiatum of the CA1 region of hippocampus. Micropipettes (5–7 MΩ) were filled with a solution containing the following (in mm): 132 K-gluconate, 11 HEPES, 0.1 EGTA, and 4 MgCl (pH 7.35 adjusted with KOH, osmolarity ∼300 mOsm), or 125 Cs-gluconate, 10 HEPES, 0.2 EGTA, and 1 MgCl (pH 7.35 adjusted with CsOH, osmolarity ∼300 mOsm). Voltage-clamp recordings were performed using an Axopatch 200B (Molecular Devices). Currents were low-pass filtered at 1–5 kHz, collected using PClamp 9 (Molecular Devices) at a frequency 2–10 kHz and analyzed off-line. An electrophysiological characterization in voltage clamp was made at the beginning of the recording. Hyperpolarizing and depolarizing steps (from −140 to +40 mV for 50 ms) were used to determine I/V relationship of each recorded cell. Membrane input resistance and capacitance of the cells were determined from the current responses to voltage pulses ranging from −20 to +20 mV from a holding potential of −60 mV.
The responses induced by the activation of purinergic receptors were studied by bath applying ATP or selective agonists. In preliminary experiments, we observed that the responses to a second application, even performed after a delay of 15–20 min, could be different from the first one. This was particularly obvious for responses mediated by the activation of P2Y receptors studied with whole-cell recordings, during which a washout of the evoked potassium currents occurred after ∼10 min of dialysis. Changes in amplitude or kinetics were sometimes also observed for P2X receptor-mediated currents. Bath applications of agonists were therefore tested only once per slice. Consequently, all comparisons were done by testing differences between mean responses obtained from one microglial cell per slice, three to four slices per animal, and three to six animals per experimental group. The I/V curves of the responses induced by the activation of purinergic receptors were determined by subtracting the current responses to the same voltage step protocol applied in control conditions from those obtained in the presence of the agonists.
Data were analyzed off-line using Clampfit (Molecular Devices). Data values are presented as mean ± SEM. Statistical significance was tested with the program GraphPad Instat. First we tested whether data were sampled from populations that follow Gaussian distributions using the method of Kolmogorov and Smirnov. Then according to the result, nonparametric test (Mann–Whitney) or parametric test that assumes the SD can be different (t test with Welch's correction) was used to compare results between the two groups of data. Statistical significance was established at p < 0.05 and p < 0.01 (GraphPad Instat 3.06).
Imaging experiments.
Slice were placed in the recording chamber at 34°C, and after 5 min (taken as t 0), a pipette containing 2-(methylthio)adenosine-5′-(trihydrogen diphosphate) (2-MeSADP) (100 μm) was placed in the stratum radiatum in the center of the field of view. Movement of microglia processes was followed by taking a fluorescent image every minute through a 40× water-immersion objective (Olympus; numerical aperture, 0.8). To quantify the elongation of the GFP-positive processes toward the pipette, we measured the fluorescence in two concentric circles, one close to the pipette (Fcint; blue circle in Fig. 8) and a second covering almost the whole field of view (Fcext; light blue circle in Fig. 8). We then calculated the ratio of the fluorescence in the two circles at different time points (t) and subtracted the ratio at t 0: Fcint(t)/Fcext(t) − Fcint(t 0)/Fcext(t 0). The obtained values were then normalized to the maximum.
Figure 8.

Higher motility of microglia processes after a status epilepticus. A, Examples of fluorescence images at different time points after the insertion (at t = 0; first column) in the slice of a pipette containing 2-MeSADP (100 μm) in control (top row) and 48 h after the induction of a status epilepticus (bottom). The pictures at t = 15 min (second column) show the formation of a ring of fluorescence corresponding to microglia processes elongated from cells at the periphery of the field of view toward the pipette tip in the center. At t = 25 min (right column), the processes of activated microglia have reached the pipette (bottom picture), whereas those of microglia from control mice have not yet completed their extension (top picture). Scale bar, 50 μm. B, Normalized (Norm.) ratio of the internal over the external circle fluorescence (top, left; see Materials and Methods) in experiments on control (black; n = 5 slices) and epileptic (red; n = 6 slices) mice 48 h after status epilepticus. This ratio increases when the fibers approach the pipette and reaches its maximum when all the processes are within the central circle. C, Temporal evolution of the total fluorescence measured in concentric rings (orange, second; red, third; blue, fourth; green, fifth; order starting from the external light blue ring). Left and right panels correspond to the example in control (top in A) and 48 h after status epilepticus induction (bottom in A), respectively. The distance between peaks provides a measure of the velocity of the processes. Inset, Histogram showing averages of the velocity measured in slices of control (black; n = 7) and epileptic (red; n = 4) mice. **p < 0.01.
To estimate the velocity at which the fluorescent processes elongated toward the pipette, a series of concentric circles were drawn as region of interest, and the fluorescence was measured in concentric rings (Fr). Because the microglia processes approach the pipette with approximately a circular symmetry, it was possible to observe for each ring a maximum in the temporal course of the fluorescence. Fr was normalized to the fluorescence of the external circle, and the following ratio was calculated for each ring: Fr(t)/Fcext(t) − Fr(t 0)/Fcext(t 0), which was then normalized to its maximum.
To define the velocity, the distance between two coronal sections was divided by the time difference between the corresponding two peaks of fluorescence. We considered only the experiments in which we could clearly identify at least three peaks of fluorescence in three coronal sections, thus two velocities, and the mean was taken as the representative velocity of each experiment. Data are given as mean ± SEM, and a Student's t test with Welch's correction was used to compare results between control and kainate-injected mice. Statistical significance was established at p < 0.05 and p < 0.01.
Reagents.
Tetrodotoxin (TTX), d-(−)-2-amino-5-phosphonopentanoic acid (APV), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), gabazine, kainate, pyridoxalphosphate-6-azophenyl-2′,5′-disulfonic acid (iso-PPADS), 4-aminopyridine, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), and 8-cyclopentyl-1–3-dipropylxanthine (DPCPX) were purchased from Tocris Bioscience or Ascent Scientific; 2-MeSADP, Brilliant Blue G (BBG), ATP, UDP, and ivermectin were purchased from Sigma-Aldrich.
In experiments involving BBG, slices were incubated in a solution containing BBG (3 μm) for 30 min at 37°C and then kept at room temperature until the experiment.
Results
Characterization of kainate-induced status epilepticus model in C57BL/6 CX3CR1eGFP/+ mice
To study microglia activation during the early phases of epilepsy, we used intraperitoneal injections of kainate as a noninvasive model of SE. In rats, systemic injection of kainate has been considered a valuable model of temporal lobe epilepsy (Ben-Ari and Cossart, 2000). However, models of experimental epilepsy are less well characterized in mice, and the susceptibility to seizures varies dramatically across mouse strains (Schauwecker, 2002). Therefore, we first characterized the inflammatory reaction and the extent of neuronal damage in the hippocampus of the CX3CR1eGFP/+ mice after a status epilepticus.
Eighty percent of the kainate-injected mice survived, of which 70% developed a status epilepticus corresponding to stages 4 or 5 on the Racine scale (see Materials and Methods) and were used for subsequent analysis. Quantitative PCR experiments performed on hippocampi from four control mice and four to six kainate-injected mice revealed a 10- to 22-fold increase in the expression of the mRNAs coding for the proinflammatory molecules IL-1β, IL-6, TNF-α, and COX-2 (Fig. 1 A) 3 h after SE. At 24 h after SE, the expression of TNF-α was further increased (80. 7 ± 9.2-fold) compared with 3 h (19.7 ± 5-fold), whereas that of COX-2 was decreased (5.7 ± 1.6 vs 22.6 ± 1.9 at 3 h), but remained higher than in control animals. At 48 h after SE, the increase in expression of the four genes was more variable and remained significant only for TNF-α and COX-2. These results confirmed the occurrence of an inflammatory reaction during the 48 h that follow an SE that is comparable with that observed in other experimental models of epilepsy (Vezzani and Granata, 2005).
Figure 1.
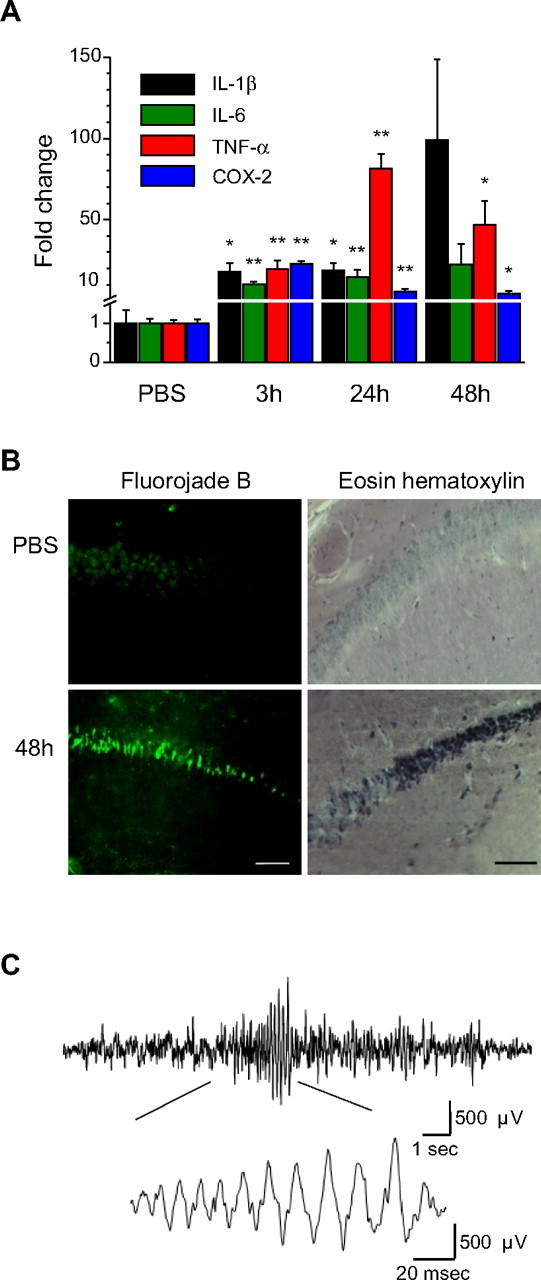
Status epilepticus induced by intraperitoneal kainate injection triggers inflammation, neuronal death in the hippocampus, and long-term changes in EEG activity in CXCR1eGFP/+ mice. A, Changes in expression of proinflammatory markers in the hippocampus analyzed by qPCR at different time points after induction of the status epilepticus. Tenfold to twentyfold increases in mRNA expression for IL-1β, IL-6, TNF-α, and COX-2 were observed at 3 h (n = 4 mice) compared with control (PBS; n = 4). Significant increased expression persisted at 24 h (n = 6) for all mRNA species and became more variable at 48 h (n = 6). B, Fluoro-Jade B (left) and eosin–hematoxylin (right) staining in the CA1 area of the ventral hippocampus in PBS- (top) and kainate-injected mice (bottom) 48 h after the status epilepticus. Note the strong staining of the pyramidal cell layer with both techniques in kainate-injected mice. Scale bars, 200 μm. C, Representative cortical EEG-recording from a mouse 2 months after induction of status epilepticus. The representative trace exhibits spontaneous collective activity in the form of a burst of spike discharges. The burst is shown at higher temporal resolution in the inset, where individual spike discharges can be identified. *p < 0.05; **p < 0.01.
We then examined neuronal damage in the hippocampus of CX3CR1eGFP/+ mice at 24 and 48 h after SE with eosin–hematoxylin staining. After SE, there was a time-dependent increase of the number of black and large neuronal morphologies observed in the cell layers of the hippocampus. Neuronal degeneration was first observed in CA3 24 h after SE (data not shown) and extended to the CA1 area 48 h after SE (Fig. 1 B). Similar observations were made using the specific degeneration marker Fluoro-Jade B (Schmued and Hopkins, 2000). Whereas no staining was observed in control mice, many neurons were labeled at 24 h (data not shown) and 48 h after the induction of SE. The number of cells per field of view (442 × 338 μm) counted 48 h after SE was 31.5 ± 2.7 and 18 ± 2.42 in CA1 and CA3 regions, respectively. These observations are in good agreement with a previous study reporting neuronal damage in the hippocampus of C57BL/6 mice after kainate-induced seizures (Benkovic et al., 2004), although neuronal degeneration in these mice is clearly less massive than in other mouse strains as previously shown (McKhann et al., 2003). Furthermore, in vivo EEG recordings performed 48 h after SE (n = 4 animals) but also 50 d after SE (n = 2 animals) revealed the occurrence of bilateral synchronized bursting activities (Fig. 1 C), which were not observed in control mice (n = 4 animals).
Altogether, these data indicate that kainate-induced seizures in CX3CR1eGFP/+ mice lead to an inflammatory reaction associated with neuronal death in the hippocampus and trigger long-term changes of electrical brain activity. Because neuronal damage appeared more pronounced in CA1, we focused our investigation of microglia activation on this region of the hippocampus.
Activation of microglia after kainate-induced SE
As previously reported for the CX3CR1eGFP/+ mouse line (Cardona et al., 2006), GFP colocalized with the specific marker of microglia Iba1 (Fig. 2 A), but not with the neuronal marker NeuN, the astrocytic marker GFAP, nor NG2, a marker of oligodendrocyte precursors (data not shown). We then investigated changes in microglia morphology and immunoreactivity in the hippocampus 3, 24, and 48 h after the induction of the SE. A higher Iba1 immunoreactivity was first observed by immunofluorescence in the hippocampus of treated animals 24 h after the kainate injection (Fig. 2 A). At this stage, microglia had larger somata and thicker primary processes compared with control animals (Fig. 2 A). We never observed, however, GFP-positive cells with amoeboid macrophage-like morphology. At 48 h after SE, similar changes of morphology and Iba1 immunoreactivity were observed, and the increased expression of Iba1 was confirmed by a Western blot analysis performed on hippocampal extracts (Fig. 2 B) (three control and three epileptic mice).
Figure 2.

Increase in size and number of microglia in the hippocampus after status epilepticus. A, Confocal images (stack of 21 z-sections, 8.28 μm total thickness) of Iba1 immunostaining and GFP fluorescence in the CA1 stratum radium of CX3CR1eGFP/+ mice in control conditions (top) and 24 h after induction of the status epilepticus (bottom). The two markers are colocalized, confirming that GFP-expressing cells are microglia. Note the stronger Iba1 immunoreactivity, the larger somata, and the thicker proximal processes of activated microglia. Scale bar, 15 μm. B, Western blot analysis of Iba1 expression in the hippocampus of control and kainate-injected animals after 24 and 48 h. Samples were obtained from hippocampi of three animals for each condition. C, Quantification of microglial cell number in fields of view of 230 × 230 μm in the CA1 area of control (n = 4) and treated mice 24 (n = 3) and 48 h (n = 7) after the induction of the status epilepticus. The number of microglial cells was significantly larger already at 24 h and had doubled at 48 h. **p < 0.01.
This increased expression of Iba1 proteins in the hippocampus of epileptic mice could be in part explained by a change in the number of microglia. We thus evaluated whether there was a proliferation of microglial cells in kainate-injected mice. At 24 and 48 h after the induction of SE, there was a significant increase (ANOVA test; p < 0.001) in the number of microglial cells in CA1 (Fig. 2 C). The number of cells per field of view (230 × 230 μm) rose from 16.96 ± 0.54 in control to 25.31 ± 0.61 after 24 h and to 31.80 ± 0.54 after 48 h. Post hoc Tukey test revealed significant difference (p < 0.001) between all groups. This proliferation of microglia was similar in the dorsal and ventral hippocampus (data not shown). Consistent with the increased number of microglia, immunostainings for the proliferation marker Ki67 and for MAC2, a specific marker of activated microglia in proliferation (Lalancette-Hébert et al., 2007), were observed in microglial cells in the CA1 area 24 h after the kainate injection and became more evident at 48 h (Fig. 3).
Figure 3.

Microglia proliferates after status epilepticus. Confocal images of GFP fluorescence and of immunostaining of the proliferation markers Ki67 (left series of panels; scale bar, 25 μm) and MAC2 (right series of panels; scale bar, 40 μm). Representative pictures (stack of 5 z-sections, 4.5 μm total thickness) of the CA1 hippocampal region obtained from PBS-injected mice and from mice killed 3, 24, and 48 h after the induction of status epilepticus are shown. Immunostaining for both markers, highly colocalized with GFP, is first observed at 24 h and increases at 48 h.
Thus, status epilepticus evoked by systemic injection of kainate in CX3CR1eGFP/+ mice triggers an activation of hippocampal microglia characterized by a change in morphology and a cell proliferation.
Intrinsic membrane properties of activated and nonactivated microglia
There have been a limited number of studies on the electrophysiological properties of microglia in situ, but work in cultures showed that changes of intrinsic membrane properties occurs during microglia activation (Färber and Kettenmann, 2005). We thus used whole-cell recordings in acute hippocampal slices of control and epileptic CX3CR1eGFP/+ mice to characterize electrophysiological properties of microglial cells identified by the expression of GFP. Because the slicing procedure might result in activating microglia, in particular near the surface of the slice, recordings were obtained from cells usually at a depth below 30 μm from the slice surface. In addition, experiments were completed within the first 4 h after slicing.
Resting microglial cells in slices of control mice had a high membrane input resistance of 3.6 ± 0.2 GΩ and a membrane capacitance of 28.1 ± 1.0 pF (n = 37) (Fig. 4 A). Application of hyperpolarizing and depolarizing voltage steps from a holding potential of −60 mV did not induce large voltage-dependent membrane currents (Fig. 4 B), and the membrane I/V relationship was almost linear (Fig. 4 C). Microglial cells recorded 3 h after SE had similar intrinsic membrane properties (n = 7) (Fig. 4 B,C), whereas at 24 h after SE, microglia had a larger membrane capacitance (32.9 ± 1.8 pF; p = 0.018; n = 15) compared with control mice but a similar input resistance (3.0 ± 0.3 GΩ; p = 0.17). In addition, voltage-activated currents were evoked when applying hyperpolarizing and depolarizing pulses (Fig. 4 B), leading to rectifications in the I/V curve (Fig. 4 C). Forty-eight hours after SE, microglia cells had larger membrane capacitance (35.6 ± 1.6; p < 0.001; n = 42) and lower input resistance (2.2 ± 0.1 GΩ; p < 0.001) compared with control mice. Moreover, inward currents evoked by hyperpolarizing pulses were smaller, whereas outward currents evoked by depolarizing pulses were larger, than at 24 h after SE (Fig. 4 C). As illustrated in Figure 4 C, the difference in amplitude described for voltage-activated currents remained significant when normalizing the current values to the cell capacitance to take into account the difference in cell size between nonactivated and activated microglia. Both inward and outward currents were absent when microglia was recorded with an intracellular solution containing the nonselective potassium channel blocker cesium, whereas outward currents were abolished by 4-aminopyridine (1 mm; n = 4; data not shown), a blocker of outward-rectifying potassium channels. These results indicate that intrinsic membrane properties of microglia gradually evolve during the first 2 d that follow an SE.
Figure 4.
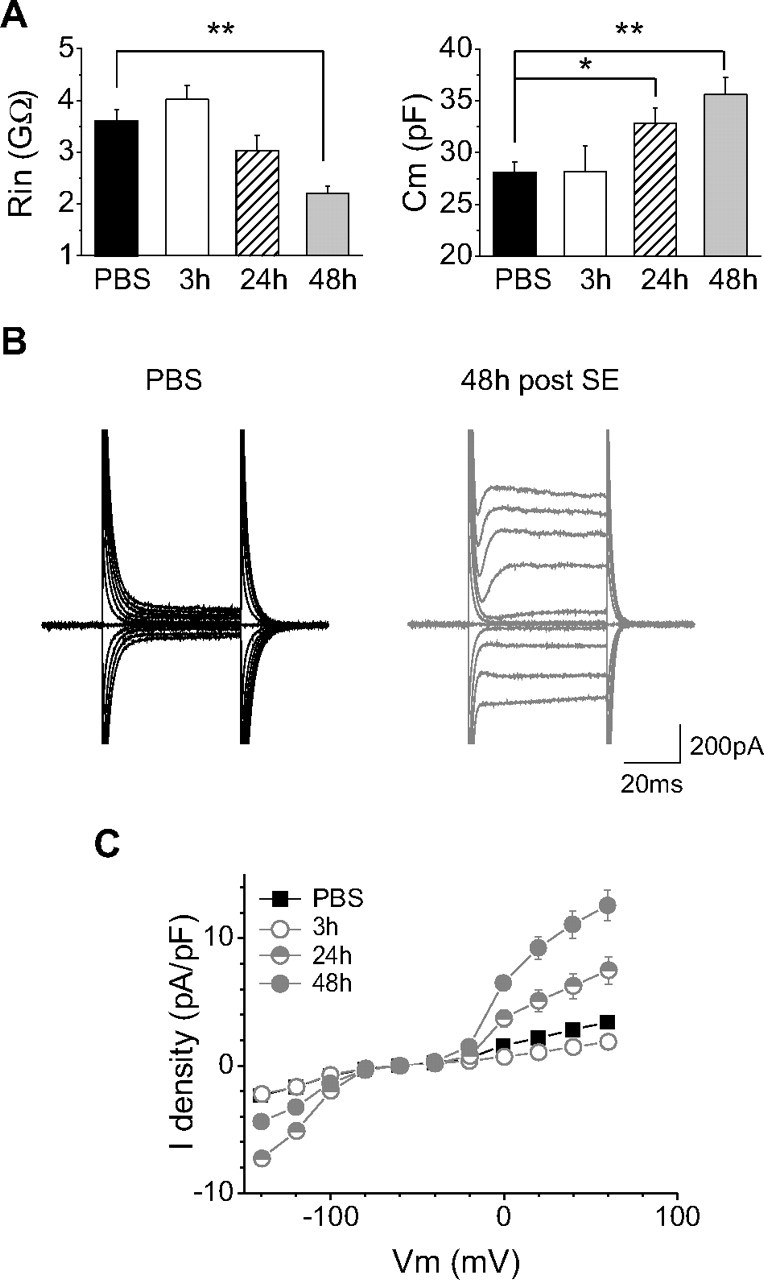
Time-dependent changes in intrinsic electrophysiological properties of microglia after status epilepticus. A, Temporal course of input resistance (left) and membrane capacitance (right) changes of CA1 hippocampal microglial cells after status epilepticus. *p < 0.05; **p < 0.01. B, Examples of current responses induced by voltage steps of 20 mV increment from −140 to +60 mV (holding potential, −60 mV) in microglia cells of control (left, black traces) and kainate-injected animals 48 h after status epilepticus (right, gray traces). C, I/V curves obtained from microglial cells recorded from control mice (black squares; n = 35) and from mice 3 (open circles; n = 7), 24 (half-filled circles; n = 15), and 48 h (filled circles; n = 39) after status epilepticus. Current densities were considered to take into account changes in membrane capacitance in activated microglia. Note the appearance of inward-rectifying currents at hyperpolarized potentials and of outward currents at depolarized potentials 24 h after the status epilepticus. Note also that the inward-rectifying currents decrease, whereas the outward currents increase, at 48 h. There was no significant difference between the I/V curves of control and 3 h conditions.
Increased expression of purinergic receptor mRNAs in the hippocampus after status epilepticus
Activation of purinergic receptors plays an essential role in different processes of microglia activation (Sperlágh et al., 2006; Färber et al., 2008). We therefore tested whether their expression was changed after SE. Quantitative PCR was performed on whole hippocampi from control animals and 3, 24, and 48 h after the induction of status epilepticus to compare the mRNA expression of ionotropic P2X1, P2X4, and P2X7 and of metabotropic P2Y6, P2Y12, and P2Y13 purinergic receptors. This specific set of receptors has been selected on the basis of previous studies that have established their expression in microglia or macrophages (Inoue et al., 2007). Already 3 h after SE there was a small but significant increase (1.60 ± 0.05-fold) in the expression of P2Y6 mRNAs, whereas that of P2Y13 mRNAs decreased and that of P2X1, P2X7, and P2Y12 mRNAs did not change significantly (Fig. 5). The pattern of expression of these receptors 24 h after status epilepticus was characterized by an important increase of P2Y6 mRNA expression (9.2 ± 1.5-fold). Forty-eight hours after SE, the expression of all four receptors was above control values with a 7.5 ± 1.2-fold increase for P2Y6, a 3.5 ± 0.2-fold increase for P2Y12, a 3.4 ± 0.2-fold increase for P2X1, a 2.0 ± 0.2-fold increase for P2Y13, a 2.0 ± 0.2-fold increase for P2X4, and a 1.9 ± 0.2-fold increase for P2X7 receptor mRNAs.
Figure 5.

Status epilepticus induces an increase of purinergic receptor mRNA expression in the hippocampus. Quantitative PCR of purinergic receptors in whole hippocampi from control animals (n = 4) and from treated animals at 3 (n = 4), 24 (n = 6), and 48 h (n = 6) after the status epilepticus. Note that the expression of P2Y6 was already increased at 3 h, whereas that of other receptors was increased at 24 or 48 h. *p < 0.05; **p < 0.01.
These results suggest that all purinergic receptors known to be expressed in microglia are upregulated in the hippocampus 48 h after SE. In the case of P2Y12 receptors, our results contrast markedly with the downregulation of this receptor in some other models of microglia activation (Möller et al., 2000; Haynes et al., 2006). Because the quantitative PCR (qPCR) analysis was performed on total RNA from whole hippocampi and not on isolated microglia, we verified by immunocytochemistry that the expression of this receptor was indeed restricted to microglia and was not downregulated in the hippocampus 48 h after SE (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Enhanced purinergic responses of activated microglia after status epilepticus
We then tested whether the enhanced expression of purinergic receptor mRNAs in the whole hippocampus after SE correlated with changes in responses of microglia to purinergic receptor activation. A preliminary series of experiments in control animals showed that hippocampal microglia cells respond to bath-applied ATP (1 mm for 2 min) with a complex response composed of inward and outward currents probably mediated by P2X and P2Y receptors, respectively. These responses were not altered by blocking synaptic transmission (n = 5) with TTX (1 μm), NBQX (10 μm), APV (50 μm), and gabazine (5 μm), whereas they were greatly reduced by the large-spectrum antagonist of purinergic receptors PPADS (100 μm; n = 3; data not shown). New series of experiments were therefore designed to analyze the effect of status epilepticus on isolated P2X and P2Y receptor-mediated components of these responses.
Responses mediated by the activation of P2X receptors were isolated by recording microglia with an intracellular solution containing cesium to block potassium currents induced by P2Y receptor activation (Fig. 6 A). Inward currents evoked by ATP (1 mm for 2 min) at a holding potential of −40 mV had an average amplitude of 4.8 ± 0.6 pA (n = 12) in microglia of control mice. These currents had an I/V curve that presented a slight inward rectification with a reversal potential near 0 mV, as expected for nonselective cation-permeable channels (Fig. 6 A). The amplitude and the I/V curve of these responses (n = 4) were not altered in the presence of a mixture aimed at reducing indirect effects through neuronal activation and including TTX (1 μm), NBQX (10 μm), APV (50 μm), and gabazine (5 μm) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Removing extracellular divalent ions greatly increased the amplitude of the ATP-induced currents (49.3 ± 7.6 pA at −40 mV; n = 6) without changing the form of the I/V curve (Fig. 6 B,C). The antagonist of P2X7 receptors BBG (3 μm) blocked a large part of the ATP currents (residual current at −40 mV: 1.6 ± 0.3; n = 3), and the further addition of the antagonist of ionotropic purinergic receptors iso-PPADS (50 μm) did not antagonize more the responses (Fig. 6 C). The sensitivity of the responses to extracellular divalent cations and to BBG suggests that P2X7 receptors were responsible for most of the response induced by ATP in nonactivated microglia.
Figure 6.

Larger P2X receptor-mediated responses in activated microglia. A, B, Examples of currents induced by bath application of ATP (black bars; 1 mm, 2 min) in microglia from control animals (left; black traces) and from mice 48 h after a status epilepticus (right; gray traces) in normal (A) and Mg2+-, Ca2+-free (B) medium. Cells were patched with a Cs-gluconate-based internal solution and held at −40 mV. The vertical fast deflections correspond to currents generated by voltage step series applied to obtain the I/V curves of the ATP responses shown on the right. Graphs on the right represent the average of ATP-induced current density/voltage relation in control (black squares; n = 12 for control medium, and n = 6 for Ca2+-, Mg2+-free medium) and 48 h after status epilepticus (gray circles; n = 13 for control medium, and n = 9 for Ca2+-, Mg2+-free medium) obtained by subtracting the I/V curve obtained before from that obtained during ATP application. C, Pharmacology of the P2X receptor-mediated responses induced by ATP (1 mm) application at −40 mV in control (black bars) and 48 h after status epilepticus induction (gray bars). Responses were enhanced in Mg2+-, Ca2+-free medium and strongly reduced by BBG (3 μm) in control as in activated microglia. However, a residual current, which was blocked by further addition of iso-PPADS (50 μm), was present in activated microglia. *p < 0.05; **p < 0.01.
The responses resulting from the activation of ionotropic purinergic receptors in activated microglia were then analyzed 48 h after SE, when the enhanced expression of the corresponding mRNAs was most pronounced (see above and Fig. 5). The amplitude of the currents induced in standard extracellular medium by ATP was larger in activated microglia than in control mice (13.2 ± 1.9 pA; n = 14; p < 0.01). As illustrated in Figure 6, A and C, this difference remained significant when normalizing the current values to the cell capacitance to take into account the difference in cell size between nonactivated and activated microglia. As shown in Figure 6 C, the responses were greatly reduced by BBG (3 μm), but in contrast to the responses of nonactivated microglia, the residual current (3.9 ± 0.4 pA at −40 mV; n = 5) was further decreased by adding iso-PPADS (residual current: 1.7 ± 0.4 pA; n = 4). As in nonactivated microglia, removing divalent cations from the extracellular solution enhanced ATP responses of microglia from epileptic mice, and these responses were larger (156.9 ± 23.0 pA; n = 9) (Fig. 6 B,C) compared with control animals (p < 0.01). BBG (3 μm) decreased ATP responses in Mg-, Ca-free solutions, and the residual current (28.8 ± 6.8 pA; n = 4) was further reduced by adding iso-PPADS (5.8 ± 1.0 pA; n = 3). These results indicate a higher expression of functional P2X7 receptors in activated microglia and suggest the presence of another type of P2X receptor responsible of the BBG-resistant component of the response. It is worth noting that ATP responses recorded in our experimental conditions were not sensitive to the positive modulator of P2X4 receptors, ivermectin (5 μm; data not shown), suggesting that these receptors did not mediated the BBG-resistant component observed in activated microglia.
We then investigated the responses to P2Y receptor activation. Microglia cells were voltage clamped at −20 mV with a K-gluconate-based internal solution, and the responses to bath-applied selective agonists of P2Y receptors were tested. The P2Y6 agonist UDP (1 mm, 1 min) induced an outward current in all microglia recorded in hippocampal slices of both control and epileptic animals (Fig. 7 A). However, the amplitude current induced in activated microglia was approximately six times larger in epileptic animals (7.1 ± 1.1 pA, n = 9, and 44.3 ± 11 pA, n = 8, in control and kainate-injected animals, respectively; p = 0.01). As for P2X receptor-mediated responses, this difference was not attributable to the larger size of activated microglial cells, because it remained significant when the responses were expressed in current densities (Fig. 7).
Figure 7.

Larger P2Y6 and P2Y12 receptor-mediated responses in activated microglia. A, Examples of currents induced by bath application of the P2Y6 receptor agonist UDP (black bars; 1 mm, 1 min) in microglia from a control animal (left, black trace) and from a mouse 48 h after the status epilepticus (right, gray trace). Cells were patched with K-gluconate-based solution, and responses were tested at −20 mV. B, I/V curve of the UDP-induced responses in control (black symbols; n = 9) and epileptic (gray symbols; n = 8) mice. C, Same as in B for the P2Y12 receptor-mediated responses induced by the selective agonist 2-MeSADP application (100 μm, 1 min). Numbers of microglia are 11 in control (black bar/symbols) and 7 in treated mice (gray bar/symbols).
Differences between control and epileptic mice were also observed with an agonist of both P2Y12 and P2Y13 receptors, 2-MeSADP (100 μm, 1 min): the currents induced in activated microglia (46.8 ± 13.8 pA; n = 7) were significantly larger than those recorded in slices of control mice (6.5 ± 1.0; n = 11; p < 0.05). The amplitude of the currents induced by both agonists decreased with hyperpolarization and reached their zero value close to −90 mV, as expected for potassium currents (Fig. 7 A,B, I/V curves). In addition, the amplitude and the I/V of responses to UDP (n = 4) and to 2-MeSADP (n = 4) were not altered in the presence of TTX (1 μm), NBQX (10 μm), APV (50 μm), gabazine (5 μm), LY341495 (100 μm), and DPCPX (1 μm) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Thus, together with the results of quantitative PCR, these observations indicate the occurrence of an increase in expression of functional P2Y receptors in activated microglia 2 d after a status epilepticus.
Activated microglial cells have higher motility in the hippocampus of epileptic mice
Microglia can readily send their processes toward an ATP source (Davalos et al., 2005; Haynes et al., 2006) that, in pathological conditions, would be generated by damaged cells or a prolonged hyperactivity. This phenomenon is partially mediated by P2Y12 receptors (Haynes et al., 2006). We therefore compared the elongation velocity of the processes of GFP-expressing microglia in acute hippocampal slices of control and epileptic mice. In time-lapse video-microscopy experiments, we observed that processes of microglia elongated toward a pipette containing ATP (1 mm, data not shown) or 2-MeSADP (100 μm) in slices from both epileptic and control mice (see supplemental Movies 1, 2, available at www.jneurosci.org as supplemental material). No directional movement was observed when the pipette contained extracellular solution without purinergic receptor agonist (data not shown). The velocity with which the processes reached the pipette was significantly faster in slices of epileptic animals compared with control (Fig. 8 A,B). To quantify these differences, we measured the temporal course of the ratio between the fluorescence in a circle around the pipette and that in a circle that covers almost the entire field of view. This ratio increased as the processes approach the pipette. The mean time to reach the maximum of this ratio was 54.7 ± 2.1 min in control animals (n = 13) and 28.4 ± 2.8 min in epileptic mice (n = 14; p < 0.01).
There is a higher density of microglia in epileptic animals (Fig. 2 C). Therefore, this difference could be attributable to the fact that more microglial cells are closer to the pipette containing purinergic agonist. We thus estimated the velocity by measuring the time differences at which peaks of fluorescence appeared in concentric rings along the elongation path toward the pipette (Fig. 8 A,C) (see Materials and Methods for details). The estimated velocity in slices of control mice was 3.32 ± 0.24 μm/min (n = 10), whereas that in kainate-injected mice was 5.85 ± 0.6 μm/min (n = 10; p < 0.01). To evaluate a possible contribution of neuronal activity, experiments with 2-MeSADP were repeated in the presence of TTX (1 μm), NBQX (10 μm), APV (50 μm), and gabazine (5 μm). The velocity with which microglia processes elongated toward the pipette containing the agonist was 3.19 ± 0.12 μm/min (n = 7) in control animals and increased to 4.97 ± 0.49 μm/min (n = 4) in epileptic mice (p < 0.01). These values were not significantly different from those measured in normal medium (p > 0.4). These observations indicate that increased velocity of microglia processes observed after SE was a consequence of changes in P2Y receptor signaling in microglial cells.
Discussion
In addition to the observed changes in morphology and in Iba1 expression, our study provides the first description of microglia functional changes during the inflammatory reaction that follows a status epilepticus. We show that this activation is characterized by a proliferation, a modification of intrinsic membrane properties, an increase of purinergic receptor-mediated responses, and an enhanced process motility of microglia.
The model of status epilepticus in CX3CR1eGFP/+ C57BL/6 mice
There is an important variability in the susceptibility of different mouse strains, in response to systemic injections of kainate, to develop seizures and to display the characteristic pattern of hippocampal neurodegeneration observed in rats (Pisa et al., 1980; Sperk et al., 1983; Ben-Ari, 1985), and the mouse strain C57BL/6 has been considered as one of the most resistant (Schauwecker and Steward, 1997; Schauwecker, 2002; McKhann et al., 2003; Mazarati et al., 2004; Shikhanov et al., 2005). However, recent evidence indicates that seizures and some degree of hippocampal neurodegeneration also occur in these mice (Benkovic et al., 2004, 2006; McLin and Steward, 2006). In agreement with these latter observations, we could trigger stage 4–5 seizures in CX3CR1eGFP/+ C57BL/6 mice, and Fluoro-Jade B labeled a significant number of CA1 neurons after kainate-induced SE in these mice. This susceptibly was probably not a consequence of the use of mice heterozygotes for CX3CR1, because similar increases in the expression of inflammatory mediators were also observed in wild-type C57BL/6 mice (data not shown). A previous study on the role of CX3CR1 receptors showed that the functions of microglia in various experimental models of brain disorders are impaired in CX3CR1eGFP/eGFP but not in CX3CR1eGFP/+ mice (Cardona et al., 2006). Together with the rapid and prolonged increase expression of proinflammatory molecules and the activation of microglia, our results thus validate the use of intraperitoneal injection of kainate in these mice as a model of status epilepticus. Whether this status epilepticus leads to the development of chronic epilepsy remains to be established, but our data suggest that kainate-treated mice have abnormal hippocampal hyperexcitability up to 2 months after the induction of SE.
Intrinsic membrane properties of microglia
Until recently, the difficulty to identify microglia has precluded the analysis of their functional state in situ, and most of our current knowledge on electrophysiological properties of microglia and their changes during activation relies on studies performed in culture systems. There are a few exceptions, however, and similarly to what we observed in hippocampal slices of control mice, other studies reported that nonactivated microglia in acute brain slices has almost linear I/V curves with virtually no voltage-activated channels (Boucsein et al., 2000; Wu et al., 2007). However, cultured microglia expresses a prominent inward-rectifying current at hyperpolarizing potentials in the absence of any specific activation stimulus (Färber and Kettenmann, 2005). Boucsein et al. (2000) also observed in slices of the facial nucleus after lesion of the facial nerve that in activated microglia an inward-rectifying current is first expressed, followed by the expression of additional outward currents. Our present study provides further support for this sequential activation of different voltage-activated channels during the development of inflammation in situ, with the inward-rectifying current being maximum first (i.e., at 24 h after SE) and a delayed expression of the outward current. Furthermore, we observed a decrease in amplitude of the inward-rectifying current at 48 h after SE, when the outward current reaches its maximum amplitude. This pattern of activation resembles that observed in cultured microglia, in which application of the proinflammatory toxin lipopolysaccharide (LPS) induces the expression of outward currents together with the decrease in amplitude of the already present inward-rectifying current (Boucsein et al., 2000). Thus, changes in intrinsic membrane properties during the activation of microglia follow the same sequence of events in the three experimental conditions in which they have been studied (i.e., in cultures, in vivo after axotomy, and after status epilepticus). Therefore, in contrast with some other aspects of microglia activation [for review, see below and Hanisch and Kettenmann (2007)], upregulation of potassium channels does not seem to be differentially regulated according to the context of the activation. It is worth noting, however, that the upregulation of different potassium channels may be a prerequisite for triggering different subsequent aspects of microglia activation (Fordyce et al., 2005; Pannasch et al., 2006; Franciosi et al., 2006; Kaushal et al., 2007).
Enhanced purinergic responses in activated microglia
Microglial cells express both metabotropic (P2Y) and ionotropic (P2X) purinergic receptors. Accordingly and in agreement with previous studies (Boucsein et al., 2003; Wu et al., 2007), we found that ATP application evoked complex responses composed of inward and outward currents. By investigating separately P2X and P2Y receptor-mediated responses, we found that both ionotropic and metabotropic components of purinergic responses were enhanced in activated microglia after SE. This enhancement of purinergic responses probably involves an increased expression of the corresponding receptor, because P2X and P2Y mRNAs were upregulated in the epileptic hippocampus. Our study provides electrophysiological evidence that activation of microglia in situ involves an increase of all types of purinergic responses.
Pharmacological investigations point toward a major role of P2X7 receptors in the ATP response mediated by ionotropic receptors, which were increased in divalent cation-free solution and strongly reduced by BBG. However, an iso-PPADS-sensitive but BBG-resistant component was found in activated microglia, suggesting that the enhanced purinergic response would be partially mediated also by another type of P2X receptors, among which P2X1 is a possible candidate. However, the rapid desensitization and the poor selectivity of pharmacological tools available for ionotropic purinergic receptors makes their study difficult in slices and imposes caution in the interpretation of results about the role of specific receptor subtype. In models of LPS-induced inflammation, P2X7 receptors support the release of IL-1β by microglia (Ferrari et al., 1997; Rampe et al., 2004; Choi et al., 2007; Mingam et al., 2008), but they probably also regulate indirectly the expression or release of other inflammatory factors such as cyclooxygenase-2, IL-6, IL-12, and TNF-α (Rampe et al., 2004; Choi et al., 2007). P2X7 receptors, probably in association with Pannexin-1 (Pelegrin and Surprenant, 2006), could therefore play a central role in the secretion of inflammatory mediators that occurs during the 48 h that follow SE, because these receptors could also control the microglia proliferation (Bianco et al., 2006). Finally, the observation that P2X7 (and other P2 receptors; see below) expression is significantly enhanced after the induction of the SE, while the cytokine expression is also enhanced, suggests the occurrence of a positive feedback mechanism exacerbating the activation process.
The most dramatic difference we observed between nonactivated and activated microglia is the upregulation of P2Y6 receptors. The mRNA expression had already increased 3 h after SE and reached a plateau between 24 and 48 h, when electrophysiological responses to UDP are six times larger than in nonactivated microglia. This increased expression of P2Y6 receptors has been observed in other models of microglia activation in vitro but also in vivo (Bianco et al., 2005; Koizumi et al., 2007). Accordingly, this receptor sustains a key function of microglia, because its activation is essential in triggering phagocytosis (Koizumi et al., 2007). Thus, enhanced UDP-mediated currents observed in the present study would mediate the enhanced phagocytic action observed in microglia of epileptic animals (Koizumi et al., 2007).
Nonactivated microglia constantly move their processes to probe their environment (Davalos et al., 2005; Nimmerjahn et al., 2005). After a local lesion, microglia cells send their process toward the lesion, and ATP has been proposed to be one of the “alarm signals,” because a source of exogenous ATP (or ATP analog) mimics this phenomenon in vivo (Davalos et al., 2005; Haynes et al., 2006) as well as in slices (Wu et al., 2007). P2Y12 receptors mediate this chemotaxis (Haynes et al., 2006), and the potassium currents induced by the activation of P2Y12 receptor seem also to be essential (Wu et al., 2007). Our results show that activated microglial cells respond to a P2Y12–13 receptor agonist (2-MeSADP) with larger outward potassium currents and, as a functional consequence, the velocity with which their processes reach a source of 2-MeSADP is twice as fast as for nonactivated microglia. Despite the lack of evidence for the involvement of P2Y13 receptors in microglia chemotaxis and the fact that upregulation of P2Y12 receptor mRNAs is twice as large as that of P2Y13 receptors in the epileptic hippocampus, we cannot totally rule out the possibility that P2Y13 receptors contribute to the increase in velocity of microglia processes after SE. It worth noting, however, that P2Y13 transcripts but not receptor proteins were detected in brain microglia (Möller et al., 2000; Haynes et al., 2006). Thus, increase in both P2Y6 and P2Y12 receptor-mediated responses confers to activated microglia the ability to intervene faster onto damaged cells, because they would have larger responses to both the “eat-me” and “find me” signals.
A specific activation state of microglia after status epilepticus
The analysis of purinergic responses reveals striking differences between models of microglia activation. In contrast to the upregulation of P2X7 receptors that we observed, a downregulation of P2X7 receptors has been reported in cultured microglia stimulated with LPS (Bianco et al., 2005), suggesting that regulation of P2X7 receptors is dependent on the stimulus or on the context in which activation is induced. Along the same line, we observed that P2Y12 mRNAs are upregulated and P2Y12 receptor-mediated responses are increased in activated microglia of the epileptic hippocampus. This is in agreement with the increased expression of P2Y12 receptor observed in spinal cord microglia in a model of neuropathic pain (Kobayashi et al., 2008) but contrasts markedly with the downregulation of this receptor in models of neuronal injury in vitro and of LPS injection in vivo and in vitro (Möller et al., 2000; Haynes et al., 2006). In these latter models, the downregulation of P2Y12 receptors correlates with the progression of microglia morphology toward an amoeboid state (Haynes et al., 2006). Interestingly, microglial cells in the hippocampus after SE, which clearly maintain their expression of P2Y12 receptors (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material), undergo morphological changes but never progress toward a full amoeboid morphology. Thus, the absence of amoeboid microglia together with the upregulation of P2X7 and P2Y12 receptors indicate that microglia in the epileptic hippocampus is in a specific activation state that differs from that occurring during stronger inflammatory reactions triggered by severe neuronal injury or by LPS. This state, characterized by a maintained ability to release cytokines (P2X7) and by an enhanced chemotaxis (P2Y12), might be correlated with the persistent hyperexcitability of the hippocampal neuronal networks, which outlasts the duration of the status epilepticus per se. Whether microglia acquire a similar functional state in other brain disorders associated with chronic and probably milder inflammation remains to be studied.
Footnotes
This work was funded by grants from Inserm (Avenir program), Agence Nationale de la Recherche (ANR-05-NEUR-037-01/02), Fédération Française de la Recherche pour l'Epilepsie, and Fondation Bettencourt-Schueller. We thank Mireille Lerner-Natoli and Fédérica Bertaso for helping with the in vivo recordings and for encouraging discussions. We thank David Julius for the gift of the P2Y12 antibody, Alexis Menteyne for some in vitro tests, and Serge Charpak for critical comments on this manuscript.
References
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Benkovic SA, O'Callaghan JP, Miller DB. Sensitive indicators of injury reveal hippocampal damage in C57BL/6J mice treated with kainic acid in the absence of tonic-clonic seizures. Brain Res. 2004;1024:59–76. doi: 10.1016/j.brainres.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Benkovic SA, O'Callaghan JP, Miller DB. Regional neuropathology following kainic acid intoxication in adult and aged C57BL/6J mice. Brain Res. 2006;1070:215–231. doi: 10.1016/j.brainres.2005.11.065. [DOI] [PubMed] [Google Scholar]
- Bianco F, Fumagalli M, Pravettoni E, D'Ambrosi N, Volonte C, Matteoli M, Abbracchio MP, Verderio C. Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Brain Res Rev. 2005;48:144–156. doi: 10.1016/j.brainresrev.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Bianco F, Ceruti S, Colombo A, Fumagalli M, Ferrari D, Pizzirani C, Matteoli M, Di Virgilio F, Abbracchio MP, Verderio C. A role for P2X7 in microglial proliferation. J Neurochem. 2006;99:745–758. doi: 10.1111/j.1471-4159.2006.04101.x. [DOI] [PubMed] [Google Scholar]
- Boucsein C, Kettenmann H, Nolte C. Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci. 2000;12:2049–2058. doi: 10.1046/j.1460-9568.2000.00100.x. [DOI] [PubMed] [Google Scholar]
- Boucsein C, Zacharias R, Färber K, Pavlovic S, Hanisch UK, Kettenmann H. Purinergic receptors on microglial cells: functional expression in acute brain slices and modulation of microglial activation in vitro. Eur J Neurosci. 2003;17:2267–2276. doi: 10.1046/j.1460-9568.2003.02663.x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–4968. doi: 10.1523/JNEUROSCI.5417-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Koh S. Role of brain inflammation in epileptogenesis. Yonsei Med J. 2008;49:1–18. doi: 10.3349/ymj.2008.49.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Crespel A, Coubes P, Rousset MC, Brana C, Rougier A, Rondouin G, Bockaert J, Baldy-Moulinier M, Lerner-Natoli M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002;952:159–169. doi: 10.1016/s0006-8993(02)03050-0. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, De Luigi A, Garattini S, Vezzani A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci. 2000;12:2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Van Dam AM, Lucassen PJ, Bol JG, Winblad B, Schultzberg M. Immunohistochemical localization of interleukin-1beta, interleukin-1 receptor antagonist and interleukin-1beta converting enzyme/caspase-1 in the rat brain after peripheral administration of kainic acid. Neuroscience. 1999;93:915–930. doi: 10.1016/s0306-4522(99)00178-5. [DOI] [PubMed] [Google Scholar]
- Färber K, Kettenmann H. Physiology of microglial cells. Brain Res Brain Res Rev. 2005;48:133–143. doi: 10.1016/j.brainresrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Färber K, Markworth S, Pannasch U, Nolte C, Prinz V, Kronenberg G, Gertz K, Endres M, Bechmann I, Enjyoji K, Robson SC, Kettenmann H. The ectonucleotidase cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia. 2008;56:331–341. doi: 10.1002/glia.20606. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997;159:1451–1458. [PubMed] [Google Scholar]
- Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciosi S, Ryu JK, Choi HB, Radov L, Kim SU, McLarnon JG. Broad-spectrum effects of 4-aminopyridine to modulate amyloid β1–42-induced cell signaling and functional responses in human microglia. J Neurosci. 2006;26:11652–11664. doi: 10.1523/JNEUROSCI.2490-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke H, Günther A, Grosche J, Schmidt R, Rossner S, Reinhardt R, Faber-Zuschratter H, Schneider D, Illes P. P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Exp Neurol. 2004;63:686–699. doi: 10.1093/jnen/63.7.686. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Inoue K, Koizumi S, Tsuda M. The role of nucleotides in the neuron–glia communication responsible for the brain functions. J Neurochem. 2007;102:1447–1458. doi: 10.1111/j.1471-4159.2007.04824.x. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Lu X, Shinmei S, Badie-Mahdavi H, Bartfai T. Patterns of seizures, hippocampal injury and neurogenesis in three models of status epilepticus in galanin receptor type 1 (GalR1) knockout mice. Neuroscience. 2004;128:431–441. doi: 10.1016/j.neuroscience.2004.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- McLin JP, Steward O. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur J Neurosci. 2006;24:2191–2202. doi: 10.1111/j.1460-9568.2006.05111.x. [DOI] [PubMed] [Google Scholar]
- Mingam R, De Smedt V, Amédée T, Bluthé RM, Kelley KW, Dantzer R, Layé S. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1 beta in the murine brain. Brain Behav Immun. 2008;22:234–244. doi: 10.1016/j.bbi.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller T, Kann O, Verkhratsky A, Kettenmann H. Activation of mouse microglial cells affects P2 receptor signaling. Brain Res. 2000;853:49–59. doi: 10.1016/s0006-8993(99)02244-1. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Färber K, Nolte C, Blonski M, Yan Chiu S, Messing A, Kettenmann H. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol Cell Neurosci. 2006;33:401–411. doi: 10.1016/j.mcn.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisa M, Sanberg PR, Corcoran ME, Fibiger HC. Spontaneously recurrent seizures after intracerebral injections of kainic acid in rat: a possible model of human temporal lobe epilepsy. Brain Res. 1980;200:481–487. doi: 10.1016/0006-8993(80)90938-5. [DOI] [PubMed] [Google Scholar]
- Rampe D, Wang L, Ringheim GE. P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol. 2004;147:56–61. doi: 10.1016/j.jneuroim.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. J Neuroimmune Pharmacol. 2006;1:117–126. doi: 10.1007/s11481-006-9012-8. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res. 2002;135:139–148. doi: 10.1016/s0079-6123(02)35014-3. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Boop FA, Mrak RE, Griffin WS. Increased neuronal beta-amyloid precursor protein expression in human temporal lobe epilepsy: association with interleukin-1 alpha immunoreactivity. J Neurochem. 1994;63:1872–1879. doi: 10.1046/j.1471-4159.1994.63051872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikhanov NP, Ivanov NM, Khovryakov AV, Kaspersen K, McCann GM, Kruglyakov PP, Sosunov AA. Studies of damage to hippocampal neurons in inbred mouse lines in models of epilepsy using kainic acid and pilocarpine. Neurosci Behav Physiol. 2005;35:623–628. doi: 10.1007/s11055-005-0102-5. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Culbert AA, Evans NA, Chessell I, Davis JB, Richardson JC. P2X(7) receptors on microglial cells mediate injury to cortical neurons in vitro. Glia. 2006;54:234–242. doi: 10.1002/glia.20379. [DOI] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O. Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- Sperlágh B, Vizi ES, Wirkner K, Illes P. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–346. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16:321–334. doi: 10.1016/j.nbd.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol. 2007;82:57–71. doi: 10.1016/S0074-7742(07)82003-4. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Ravizza T, Balosso S, Aronica E. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(Suppl 2):24–32. doi: 10.1111/j.1528-1167.2008.01490.x. [DOI] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. Int Rev Neurobiol. 2007;82:247–263. doi: 10.1016/S0074-7742(07)82013-7. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Vadakkan KI, Zhuo M. ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia. 2007;55:810–821. doi: 10.1002/glia.20500. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]