

Abstract
Stressful life experiences are likely etiological factors in sporadic forms of Alzheimer's disease (AD). Many AD patients hypersecrete glucocorticoids (GCs), and their GC levels correlate with the rate of cognitive impairment and extent of neuronal atrophy. Severity of cognitive deficits in AD correlates strongly with levels of hyperphosphorylated forms of the cytoskeletal protein TAU, an essential mediator of the actions of amyloid β (Aβ), another molecule with a key pathogenic role in AD. Our objective was to investigate the sequential interrelationships between these various pathogenic elements, in particular with respect to the mechanisms through which stress might precipitate cognitive decline. We thus examined whether stress, through the mediation of GCs, influences TAU hyperphosphorylation, a critical and early event in the cascade of processes leading to AD pathology. Results from healthy, wild-type, middle-aged rats show that chronic stress and GC induce abnormal hyperphosphorylation of TAU in the hippocampus and prefrontal cortex (PFC), with contemporaneous impairments of hippocampus- and PFC-dependent behaviors. Exogenous GC potentiated the ability of centrally infused Aβ to induce hyperphosphorylation of TAU epitopes associated with AD and cytoplasmic accumulation of TAU, while previous exposure to stress aggravated the biochemical and behavioral effects of GC in Aβ-infused animals. Thus, lifetime stress/GC exposure may have a cumulative impact on the onset and progress of AD pathology, with TAU hyperphosphorylation serving to transduce the negative effects of stress and GC on cognition.
Introduction
The etiology of late-onset, sporadic (nonfamilial) forms of Alzheimer's disease (AD) is largely unknown, but there is growing consensus that lifetime events such as environmental stressors may increase the probability of risk for the disease (Wilson et al., 2003; Csernansky et al., 2006; Kang et al., 2007). This view is supported by reports of hypersecretion of stress hormones [glucocorticoids (GCs)] in AD patients (Hartmann et al., 1997; Weiner et al., 1997; Elgh et al., 2006), and indications that psychological distress may cause mild cognitive impairment (Wilson et al., 2003) and predispose affected individuals to AD (Wilson et al., 2007). Studies in humans and animals demonstrate a robust relationship among elevated GC secretion, cognitive impairment, and neuronal atrophy. The cognition-impairing actions of stress and high GC levels are largely ascribed to concomitant reductions in the volume of the hippocampus (Sousa et al., 2000; Landfield et al., 2007; Lupien et al., 2009), a brain area that displays some of the earliest neurodegenerative changes in AD. Stress and GC induce similar volumetric reductions in the prefrontal cortex (PFC) (Cerqueira et al., 2007; Schubert et al., 2008), which receives afferents from the hippocampus and is critical for the control of higher cognitive functions.
Amyloid β (Aβ) has a well established role in AD-associated neuropathology, although there is evidence that cognitive deficits are detectable in advance of Aβ deposition into senile plaques (Terry et al., 1991; Guillozet et al., 2003). Previous studies in mice carrying human transgenes implicated in familial AD reported that chronic stress and exogenous GC accelerate the production and deposition of Aβ and impair learning and memory (Green et al., 2006; Jeong et al., 2006); additionally, treatment of nontransgenic animals with either chronic stress or exogenous GC shifts the metabolism of amyloid precursor protein (APP) in favor of the amyloidogenic pathway (Catania et al., 2009). On the other hand, the cytoskeletal protein TAU appears to mediate the pathogenic actions of Aβ (Rapoport et al., 2002; Roberson et al., 2007) after its hyperphosphorylation by TAU kinases such as GSK3β and cdk5 (Takashima et al., 1998). Indeed, anomalous hyperphosphorylation of TAU is another pathogenic mechanism in AD: specifically, hyperphosphorylated TAU detaches from microtubules, oligomerizes, and accumulates in the somatodendritic compartment, resulting in neuronal dystrophy and degeneration and cognitive impairment (Grundke-Iqbal et al., 1986; Sengupta et al., 1998; Schneider et al., 1999).
The results of the present study in nontransgenic middle-aged rats show that stress and GC markedly compromise hippocampus-dependent reference memory and PFC-dependent behavioral flexibility and aggravate the behavioral effects of central infusions of Aβ1-40. These behavioral impairments occur concomitantly with increased levels of TAU kinases and hyperphosphorylated TAU, thus suggesting a new cellular mechanism through which stress and GC interfere with the neurostructural correlates of behavior.
Materials and Methods
Animal procedures.
Male Wistar rats, aged 14 months, were used in accordance with European Union Council Directive 86/609/EEC and local animal welfare regulations. Animals were housed four to five per cage under standard environmental conditions [temperature 22°C; relative humidity 70%; 12 h light/dark cycle (lights on at 6:00 A.M.); ad libitum access to food and water]. Subgroups (n = 6–7) were subjected to 1 month of chronic, unpredictable stress (Catania et al., 2009). Briefly, the stress paradigm involved random application of one of the following stressors, daily: hypertonic saline [9% NaCl, i.p., 1 ml/100 g of body weight (BW)], overcrowding for 1 h, placement in a confined environment (30 min), or placement on a vibrating/rocking platform (1 h). Animals were then subjected to a first behavioral testing (see below) before receiving intracerebroventricular infusions of either freshly solubilized Aβ1-40 or vehicle (see below) over 14 d. Subgroups of animals received subcutaneous GC injections [dexamethasone, 300 μg/ml/kg BW delivered in an oily suspension (depot) (1:10 Fortecortin, (Merck) in sesame oil (Sigma)] for 14 d. Animals were subjected to a second behavioral test at the end of the various treatments. Efficacy of the stress paradigm was verified at autopsy: basal serum corticosterone levels were 54.7 ± 4.8 and 384.6 ± 65.9 ng/ml, respectively, in control and stressed rats (p < 0.02); net body mass gain/loss was −12.1 ± 2.1 g in stressed rats and +6.1 ± 1.3 g in control animals (p ≤ 0.0001); thymus weights were 4.3 ± 0.4 mg/kg BW in stressed animals and 7.6 ± 0.9 mg/kg BW control animals (p < 0.01).
Intracerebral infusions.
Alzet miniosmotic pumps (delivering 0.5 μl/h over 14 d; model 2002, Charles River), filled and equilibrated with 4.2 nm Aβ1-40 (American Peptide) or vehicle (sterile distilled water), were placed subcutaneously under anesthetic. Alzet Brain Infusion sets were used to connect the outlet of the pumps to a cannula placed under stereotaxic control into a left lateral ventricle of the brain (stereotaxic coordinates: anteroposterior, −1.0 mm; dorsoventral, −2.5 mm; and mediolateral, +1.5 mm (right) with bregma as a reference, under pentobarbital (50 mg/kg) anesthesia. We infused Aβ1-40 rather than the more pathogenic Aβ1-42 species because the former aggregates less readily (Harper et al., 1997). None of the biometric parameters were significantly altered by the Aβ1-40 infusions.
Behavioral tests.
Spatial reference memory was tested using the Morris water maze over 4 consecutive days (4 trials per day). As previously described (Cerqueira et al., 2007), testing was conducted in a circular black tank (170 cm diameter) filled to a depth of 31 cm with opaque water (22°C) and placed in a dimly lit room with extrinsic clues. The tank was divided into virtual quadrants and had a black platform (12 cm diameter, at a height of 30 cm) placed in one of them. Data were collected using a video tracking system (Viewpoint). Assessment of reversal learning (four trial paradigm) was begun 1 d later using the above setup. For this, the escape platform was repositioned in the opposite (new) quadrant; and rats were tested in a four trial paradigm (1 trial per day), with distance and time spent swimming in each quadrant being recorded.
Immunocytochemistry.
Half of each brain was used for immunohistochemical analysis; hippocampi and PFCs from the contralateral side were processed for Western blotting. Brains excised at autopsy were snap frozen, post-fixed in 4% paraformaldehyde, immersion fixed for 48 h, paraffin embedded, and sectioned at 8 μm. Following antigen retrieval (microwaved in sodium citrate, 30 min), washing (TBS), and blocking endogenous peroxides with H2O2 (3%), slide-mounted sections were incubated in an appropriate nonimmune serum (1:10 in TBS, pH 7.6, 30 min). Sections were then incubated [room temperature (RT) overnight] with primary antibodies of interest [Tau-5 (1:200), CP-13, PHF1, PG5 (all at 1:50), and 12E8 (1:100)] before washing and incubation with biotinylated secondary antibody (BioGenex) (RT, 30 min.) After a further wash in TBS, slides were incubated (30 min, RT) with a horseradish peroxidase complex (BioGenex). Immunopositive structures were revealed with the chromogen diaminobenzidine (1 mg/ml in 0.01% H2O2); stain development was monitored using a microscope. Negative controls were performed by omitting the primary antibody (no staining was seen in any sections). Staining was scored by visual examination on a scale of 0.5–5 by an independent investigator who was blind to the treatments. The degree of antibody staining was scored by visual examination on a scale of 0.5–5 by two independent subjects who were blind to the treatments.
Western blotting.
Frozen hippocampi and PFC were homogenized in lysis buffer [100 mm Tris-HCl, 250 mm NaCl, 1 mm EDTA, 5 mm MgCl2, 1% NP-40, Complete Protease Inhibitor (Roche), and Phosphatase Inhibitor Cocktails I and II (Sigma)] using a Dounce glass homogenizer; extracts were cleared by centrifugation (14,000 × g), and their protein contents were estimated (Lowry assay) after reconstitution in Laemmli buffer (250 mm Tris-HCl, pH 6.8, containing 4% SDS, 10% glycerol, 2% b-mercaptoethanol, and 0.002% bromophenol blue). Thereafter, lysates were electrophoresed on 10% acrylamide gels, and transferred onto nitrocellulose membranes (Protran BA 85, Schleicher & Schuell). Membranes were blocked in Tris-buffered saline containing 5% nonfat milk powder and 0.2% Tween-20 before incubation with the following antibodies: Tau-5 (1:200, BD Biosciences), CP-13 (p-Ser202-Tau), CP-9 (p-Thr231-Tau), PHF1 (p-Ser396/404-Tau), or PG5 (p-Ser409-Tau), all kindly provided Dr. Peter Davies (Albert Einstein College of Medicine, New York, NY) and used at a dilution of 1:50, total and p-Thr202/204 ERK1/2 (1:2500, Cell Signaling Technology), total and p-Thr183/Tyr185 JNK (1:1000, Cell Signaling Technology), cdk5 (1:1000, Millipore), p35 (Santa Cruz Biotechnology), GSK3β (1:2000, Santa Cruz Biotechnology), p-Tyr216/279 GSK3β (1:2000, Biosource), CaMKII (1:1000, Cell Signaling Technology), and actin (1:2000, Millipore). Antigens were revealed by enhanced chemiluminescence (GE Healthcare) after incubation with appropriate horseradish peroxidase–IgG conjugates (GE Healthcare); blots were scanned and quantified using TINA 3.0 bioimaging software (Raytest) after ascertaining linearity. All values were normalized and expressed as percentages of control.
Detection of detergent-insoluble TAU.
Sarkosyl-insoluble fractions from tissue protein extracts were prepared as previously described (Kimura et al., 2010). Briefly, frozen hippocampi of P301L tau mice were homogenized in Tris-buffered saline (10 mm Tris, 150 mm NaCl, pH 7.4) containing protease inhibitors (1 mg/ml antipain, 5 mg/ml pepstatin, 5 mg/ml leupeptin, 2 mg/ml aprotinin, and 0.5 mm 4-[2-aminoethyl]benzenesulfonyl fluoride hydrochloride) and phosphatase inhibitors (1 mm NaF, 0.4 mm Na3VO4, and 0.5 mm okadaic acid). After centrifugation, the supernatant was retained and Sarkosyl-insoluble, paired helical filament-enriched fractions were prepared from TBS-insoluble pellets after rehomogenization in 5 volumes of sucrose buffer (0.8 m NaCl, 10% sucrose solution including protease and phosphatases inhibitors as mentioned above) and centrifugation (100,000 × g, 20 min). Sarkosyl (10%) was added to the supernatant (1:10 v/v), and, after incubation at 37°C (1 h) and centrifugation (150,000 × g, 1 h), the resulting pellet was considered as the Sarkosyl-insoluble fraction. TBS-soluble and Sarkosyl-insoluble materials were solubilized in Laemmli sample buffer and subjected to SDS-PAGE and Western blot analysis using rabbit polyclonal anti-TAU (phosphorylated and nonphosphorylated isoforms) as described previously (Kimura et al., 2010).
Statistical analysis.
With the exception of the reference memory test data (repeated-measures ANOVA), all data were evaluated by one-way ANOVA and appropriate post hoc pairwise tests using SPSS and SigmaStat (Systat) software packages; differences were considered to be significant if p < 0.05.
Results
Chronic stress and GC induce abnormal hyperphosphorylation of TAU
Abnormal hyperphosphorylation of tau is a critical event in the cascade leading to AD pathology (Grundke-Iqbal et al., 1986; Schneider et al., 1999; Kimura et al., 2007). Clinical reports of GC hypersecretion in AD patients and in cases of mild cognitive impairment, a condition with a high conversion rate to AD (Hartmann et al., 1997; Weiner et al., 1997; Elgh et al., 2006; Wilson et al., 2007), prompted us to examine how elevated GC secretion, induced by chronic unpredictable stress, influences the pattern of tau phosphorylation in the hippocampus and PFC, brain regions that are established targets of the deleterious morphological and behavioral actions of stress and GC (Sousa et al., 2000; Cerqueira et al., 2007; Schubert et al., 2008; Sotiropoulos et al., 2008a) and among the first to show signs of AD neuropathology (Lace et al., 2009).
Immunoblot analysis showed that chronic stress results in hyperphosphorylation of TAU at pSer202, pThr231, pSer396/404, and pSer409 in both brain regions of interest (p ≤ 0.05 in all cases) (Figs. 1, 2a,b); immunocytochemical confirmation of these results using the phosphorylation-dependent antibodies CP13 (pSer202-TAU) and 12E8 (pSer262/Ser356-TAU) are shown for the subiculum (Fig. 2c) and PFC (Fig. 2d). Importantly, administration of exogenous GC to nonstressed animals largely reproduced the effects of chronic stress (p < 0.05 in all cases) (Figs. 1, 2a,b). In addition to monitoring TAU phosphorylation profiles, we also examined several kinases implicated in TAU pathology associated with AD (Mazanetz et al., 2007). As shown in Figures 1 and 2, e and f, stress and/or GC activated pERK1/2, pGSK3β, p35 (the cdk5 activator), pJNK, and CaMKII in the hippocampus and PFC (p ≤ 0.05). Together, these results show that stress and GC promote a pattern of changes that closely resembles neuropathological findings in AD and in cellular and animal models of the disease (Green et al., 2006; Jeong et al., 2006 Sotiropoulos et al., 2008b).
Figure 1.
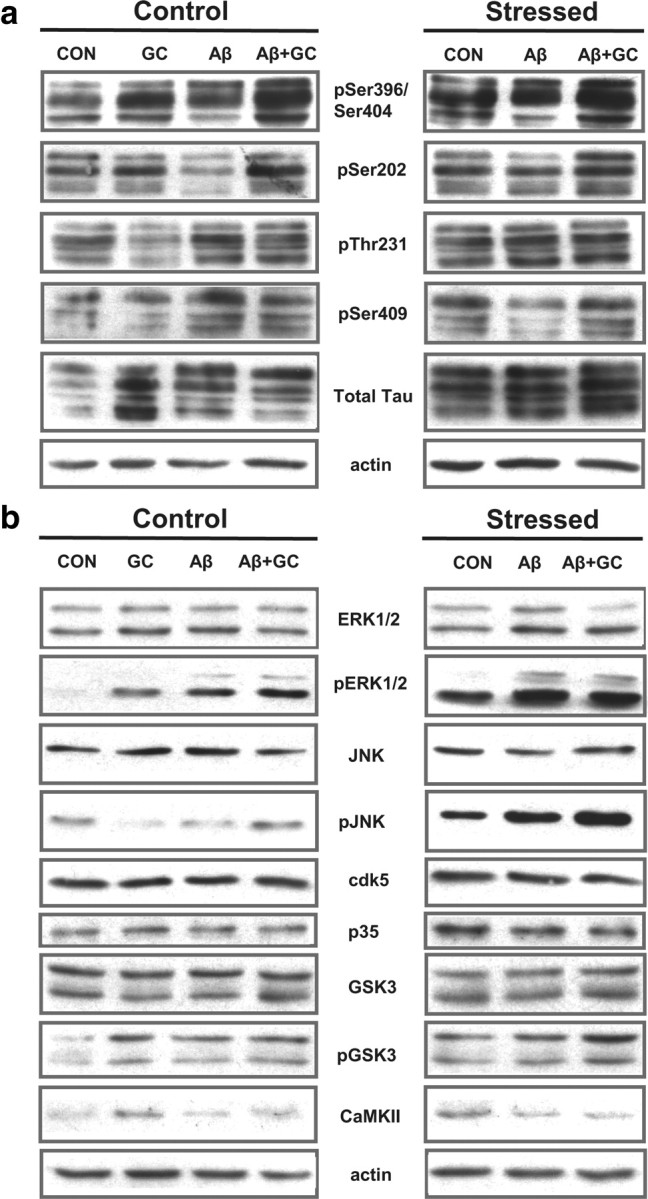
Chronic stress upregulates the expression of various phospho-TAU epitopes and TAU kinases. a, b, Depicted are representative immunoblots using a panel of phosphorylation-dependent TAU antibodies (a) and total and active isoforms of kinases (b).
Figure 2.

Stress and GCs induce TAU hyperphosphorylation in the hippocampus and PFC. TAU phosphorylation profiles in the hippocampus and PFC of chronically stressed or GC-treated rats were monitored by Western immunoblotting and immunocytochemistry. a, b, Western blot analysis of TAU phosphorylation at different epitopes in hippocampus and PFC. TAU phosphorylation levels were normalized against total TAU levels (recognized by TAU-5); control (CON) animals are shown as dotted lines (100%). c, d, Immunohistochemical staining of pSer202-TAU and pSer262/356-TAU, in the hippocampus (subiculum) and PFC, using antibodies CP-13 and 12E8 antibodies, respectively. Immunoreactive signals were confined to the cell bodies of pyramidal neurons. e, f, Both stress and GCs increased the expression of a number of kinases in the hippocampus and PFC of animals exposed to stress or GC, compared with controls. For all phosphorylated forms of kinases, data were normalized with respect to total levels of the respective kinase. g, Western blot analysis of sarkosyl-insoluble fractions from P301L-TAU animals showing that chronic stress increases the levels of sarcosyl-insoluble TAU. All numerical data shown represent mean ± SEM values (N = 7), depicted with respect to data obtained in control tissues. *Significant differences from CON values (p ≤ 0.05).
Since the expression of hyperphosphorylated TAU species correlates with the presence of insoluble TAU aggregates in AD brains (Wang et al., 1995; Alonso et al., 2001; Maeda et al., 2006), we next examined whether stress influences the aggregation of TAU. To that end, Sarkosyl-insoluble TAU was extracted from the brains of aggregation-prone P301L-Tau transgenic mice (Kimura et al., 2010) and analyzed by Western blotting. Expectedly (Kimura et al., 2010), brains from P301L-Tau mice showed detectable levels of detergent-insoluble TAU immunoreactivity; the latter was significantly increased in extracts from P301L-Tau mice that had been previously exposed to stress (Fig. 2g). These results indicate that stress aggravates TAU aggregation.
Potentiating effects of glucocorticoids
GCs were recently shown to stimulate the generation of Aβ in experimental animals (Green et al., 2006; Jeong et al., 2006; Catania et al., 2009) and neural cell cultures (Sotiropoulos et al., 2008b) and to potentiate the deleterious effects of Aβ on neuronal survival (Sotiropoulos et al., 2008b). Demonstrations that GCs trigger hyperphosphorylation of TAU in neuronal cultures (Sotiropoulos et al., 2008b) raised the question of whether this is also the case in vivo. We addressed this by monitoring the expression of pathogenically relevant phospho-TAU epitopes (Augustinack et al., 2002; Ewers et al., 2007; van der Vlies et al., 2009) in a nontransgenic rat model of early-stage AD (Stéphan and Phillips, 2005).
Chronic intracerebroventricular infusions of Aβ led to a significant increase in the expression of the majority of tested phosphorylated sites of tau protein in the hippocampus and PFC (p ≤ 0.05) (Fig. 3a,b), indicating tau hyperphosphorylation at different sites. These effects were potentiated by concomitant treatment with GCs, in particular with respect to pSer202-TAU in the hippocampus (Fig. 3a,c) and PFC (Fig. 3b), pSer396/404- and pSer409-TAU in the hippocampus (Fig. 3a), and pSer262/356 in the PFC (Fig. 3d). In addition, the combined treatment of GC and Aβ resulted in increased pSer396/404-TAU staining of the hippocampal mossy fiber network (GC+Aβ > Aβ alone) (Fig. 3e). Correspondingly, compared with Aβ alone, the combined Aβ and GC treatment regimen resulted in a greater upregulation of hippocampal levels of activated ERK1/2, pJNK, and pGSK3β (p ≤ 0.05 in all cases) (Fig. 3f).
Figure 3.

GCs potentiate Aβ-induced TAU hyperphosphorylation. a, b, Concomitant administration of Aβ and GCs resulted in increases in the levels of several phospho-TAU isoforms in the rat hippocampus (a) and PFC (b), as measured by Western blotting; these increases were greater than after treatment with Aβ alone (†p ≤ 0.05). Note the differential responses of the hippocampus and PFC in terms of number of phospho-TAU epitopes that were affected by the combined Aβ + GC treatment. c, d, Immunocytochemical analysis of pSer202-TAU (detected with antibody CP13; c) and pSer262/356-TAU (antibody 12E8; d) in hippocampus (subiculum) and PFC confirmed the above Western blot analysis data. e, Illustration of pSer396/404-TAU staining (using antibody PHF-1) in the mossy fiber system of control rats and rats treated with Aβ or Aβ + GCs. While CP-13 and 12E8 immunoreactivity was localized to neuronal perikarya, that of PHF-1 was predominantly found within mossy fibers. f, Significant increases in the expression of pERK1/2, pJNK and pGSK3β levels were observed in the hippocampus. Note that total levels of the respective TAU kinases were not altered by any of the treatments. Numerical data represent mean ± SEM values (N = 7), depicted after normalization to total Tau (a, b) or respective total kinases (f, g). *Significant differences from CON values (p ≤ 0.05); †significant difference (p ≤ 0.05) between corresponding pairs of values for each treatment group (Aβ vs Aβ + GC).
History of stress increases sensitivity to GC and Aβ
Individuals experience multiple lifetime stressors, usually with an amplification of the GC response to successive stressful episodes (Lupien et al., 2009). In an attempt to simulate this natural situation, middle-aged rats were subjected to chronic stress before receiving intracerebroventricular infusions of Aβ and/or peripheral GC injections.
Compared with animals that received Aβ alone, those receiving the combinatorial treatment of Aβ and GC showed an enhancement of phosphorylation of several TAU sites (pSer202, pSer396/404, pThr231, pSer409) in the PFC and hippocampus (Fig. 4a,b), with parallel increases in pERK1/2, pJNK, and pGSK3β (Fig. 4c,d) expression. In agreement with these findings, stressed animals that were subsequently treated with Aβ and GC displayed more intense immunostaining of PHF-1 (pSer396/404-TAU) in hippocampal neurons compared with those treated with Aβ alone (Fig. 4e). Furthermore, GC led to a greater accumulation of hyperphosphorylated TAU in the perikarya of hippocampal (compare Figs. 4e, 3e) and PFC (Fig. 4f) neurons of previously stressed animals (stress + GC + Aβ > GC+ Aβ); these findings are supported by the observation of graded increases in the level of total neuronal TAU detected with pan-TAU (TAU-5) antiserum (Table 1).
Figure 4.
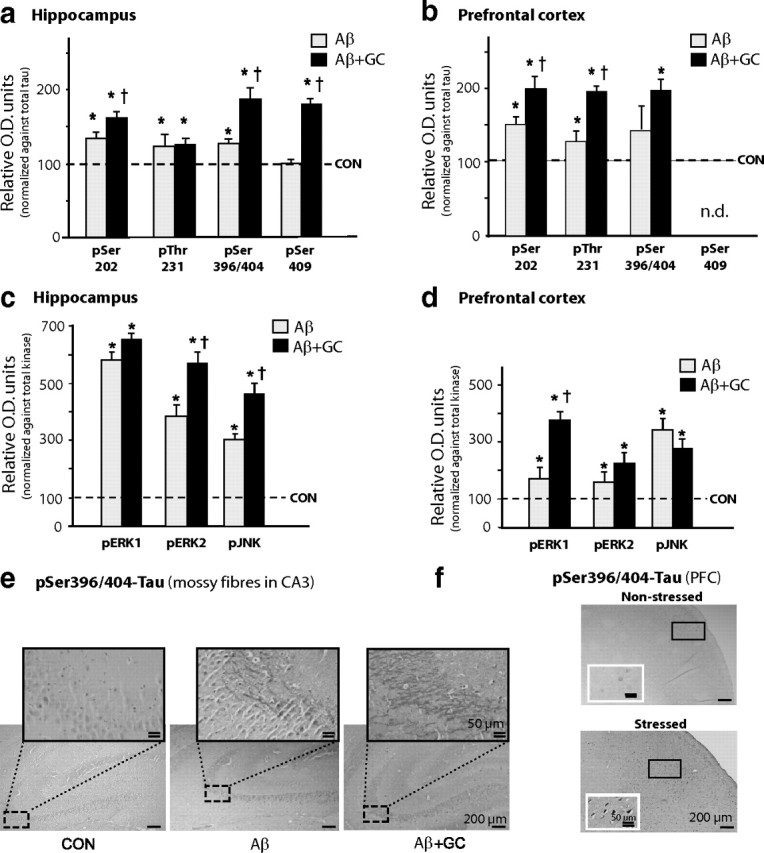
Previous stressful experience increases sensitivity to Aβ + GC. a–d, Animals that had experienced chronic unpredictable stress (1 month) responded to cotreatment with dexamethasone GCs, 300 μg/kg BW/d) and Aβ (0.3 nmol/d) with increases in TAU phosphorylation (a, b) and levels of TAU-related kinases (c, d); overall, stressed animals exhibited higher susceptibility to the combinatorial treatment than nonstressed animals. Immunoblotting revealed that, in contrast to the hippocampus, the PFC of previously stressed animals show a greater extent of TAU hyperphosphorylation (compare Fig. 2b) and expression of active forms of ERK1/2 and JNK (compare Figs. 4c,d, 3f,g) after treatment with GC + Aβ, indicating the cumulative nature and dissipation of the effects of stress and GCs from the hippocampus to the PFC. e, Immunohistochemical analysis demonstrated an enhancement of PHF-1 immunoreactivity in the mossy fiber network of the hippocampus of stressed rats receiving Aβ or Aβ + GC (Aβ + GC > Aβ > vehicle). f, Strong PHF-1 immunoreactivity is shown in the perikarya of PFC neurons of stressed + Aβ + GC-treated rats (micrograph of the PFC region from a nonstressed rat that had been treated with Aβ + GC is shown in panel f, for comparison). Insets in f are high-power magnifications of the rectangular areas marked in the respective low-magnification micrographs. All numerical data shown represent mean ± SEM values (N = 6–7), depicted with respect to data obtained in control tissues (CON, dotted line), set at 100%. *Significant differences from CON values (p ≤ 0.05); †Significant difference (p ≤ 0.05) between corresponding pairs of values for each treatment group (stress + Aβ vs stress + Aβ + GC).
Table 1.
Relative amounts of TAU staining in the hippocampus (CA3 subfield) and PFC of control and stressed animals treated with either vehicle, Aβ or Aβ and GC
| Hippocampal CA3 | PFC | |
|---|---|---|
| Control | ||
| Vehicle | ++ | + |
| Aβ | +++ | + |
| Aβ + GCs | ++++ | ++ |
| Stress | ||
| Vehicle | ++ | ++ |
| Aβ | +++ | +++ |
| Aβ + GCs | +++++ | +++++ |
Total TAU was detected using TAU-5, and scores were assigned by an observer who was blind to the treatments. +, Very low; ++, low; +++, moderate; ++++, high; +++++, very high.
Behavioral profiles
There is a strong correlation between the extent of TAU hyperphosphorylation and the severity of impairments of memory, speed of mental processing, and executive functions in humans (Augustinack et al., 2002; Ewers et al., 2007; Lace et al., 2009); in experimental animals, learning and memory are impaired when TAU is hyperphosphorylated (Gatz et al., 2006; Kimura et al., 2007).
Consistent with previous findings (Sousa et al., 2000; Cerqueira et al., 2007), tests of hippocampus-dependent spatial reference memory and PFC-dependent reversal learning showed that both stress and GC disturbed hippocampus-dependent reference memory (p < 0.05) (Fig. 5a,b) and PFC-dependent behavioral flexibility (p < 0.05) (Fig. 5c,d). Central infusions of Aβ significantly impaired reference memory (p < 0.001) (Fig. 5a), an effect aggravated by coadministration of GC. As is evident from Figure 5b, even more pronounced impairments of reference memory were observed when Aβ and GC were applied to animals that had been previously exposed to stress (stress + Aβ vs stress + Aβ + GC, p < 0.0001). Behavioral flexibility was impaired by centrally applied Aβ (p < 0.001) (Fig. 5c). Interestingly, superimposition of GC did not worsen this PFC-dependent behavioral parameter (Fig. 5c), matching the lack of changes in TAU phosphorylation in the PFC after the combined treatment (Fig. 2b). On the other hand, GC significantly exacerbated this PFC-dependent behavioral deficit in previously stressed animals (p < 0.05 (Fig. 4d). Together with the data on the hyperphosphorylation and accumulation of TAU (compare Figs. 3, 4) (Table 1), these findings highlight the cumulative and deleterious effects of stress/GCs.
Figure 5.

Stress exacerbates the cognition-impairing actions of Aβ and GC. a–d, Hippocampus-dependent spatial reference memory (a, b) and PFC-dependent working memory (reversal learning used to measure behavioral flexibility) (c, d) in nonstressed (a, c; N = 7) and stressed (b, d; N = 6) rats. Chronic, unpredictable stress (1 month) was imposed before subgroups of animals were treated with either vehicle, Aβ or Aβ+GC for 14 d. Aβ1-40 was chronically infused into the lateral ventricle at a dose of 0.3 nmol/d; dexamethasone (GC) was given as a daily depot injection at a dose of 300 μg/kg, s.c. In nonstressed animals, spatial reference memory was impaired after Aβ treatment (p < 0.001), and concomitant GC administration resulted in slight worsening of the deficit (a). While treatment with Aβ alone did not interfere with spatial memory in previously stressed rats, combined treatment with Aβ and GC markedly impaired spatial memory (p < 0.0001) (b). Compared with their nonstressed counterparts, stressed rats showed deficits in working memory (p < 0.05), as revealed by comparison of percentage distance swum in the new (W) and old (E) quadrant of the maze (c, d). Treatment of nonstressed rats with Aβ reduced performance in the reversal learning test (p < 0.05), an effect that was not changed in GC + Aβ-treated animals (c). In contrast, although reversal learning was not disrupted after administration of Aβ alone to previously stressed animals, the behavior was significantly impoverished in stressed animals that were subsequently given the combined Aβ + GC regimen (p < 0.05) (d). *Significant differences from CON values; †significant difference between stressed and nonstressed animals.
Discussion
Environmental factors account for 25–40% of the risk of developing AD (Gatz et al., 2006), a disease that develops progressively over decades. Stress, whose effects are mediated by GCs, is a plausible causal agent (Wilson et al., 2003; Csernansky et al., 2006; Kang et al., 2007). Clinical studies show a relationship between GC levels and severity of symptoms (Miller et al., 1998) and suggest that high GC levels accelerate disease progression (Csernansky et al., 2006).
The present study in rats delivers new insights into the link between stress and AD. Our results show that stress induces a biochemical anomaly found in AD, namely, hyperphosphorylation of the microtubule protein TAU; importantly, at least two of the phospho-TAU epitopes (pThr231 and pSer262) upregulated by stress are strongly implicated in the neuropathology of AD (Grundke-Iqbal et al., 1986; Sengupta et al., 1998; Kimura et al., 2007; Mazanetz and Fischer, 2007; Wang et al., 2007; Sotiropoulos et al., 2008b). Hyperphosphorylation of Thr231- and Ser262-TAU disrupts microtubule stability and leads to retrograde neuronal degeneration and synaptic loss; moreover, strong associations between increased levels of pThr231 and pSer262 and the appearance of neurofibrillary tangles have been reported previously (Sengupta et al., 1998; Schneider et al., 1999; Alonso Adel et al., 2006; Kimura et al., 2007; Mazanetz and Fischer, 2007). We here found that administration of exogenous GCs to middle-aged rats partly recapitulates the effects of stress on TAU hyperphosphorylation. Although specific TAU phosphoepitopes were differentially regulated by stress and GCs, the overall results obtained here and in earlier in vitro studies (Sotiropoulos et al., 2008b) clearly implicate GCs as a key mediator of the cellular response to stress. Nevertheless, contributions by other stress-related molecules such as corticotrophin-releasing hormone cannot be excluded (Rissman et al., 2007), and possible interactions between GC and catecholamines cannot be dismissed since stress stimulates the release of central norepinephrine (Nisenbaum et al., 1991). Understanding these interactions would be important given that TAU hyperphosphorylation and pathology is triggered by misprocessing of APP into Aβ and/or C99 (Takashima et al., 1998; Ghosal et al., 2009, Shipton et al., 2011) and that norepinephrine can reduce Aβ pathology (Heneka et al., 2010). Notwithstanding likely “ceiling effects” of the treatments (for the case of GC, see Green et al., 2006), physiological regulatory loops might also underpin our observations that the magnitude of effects of the combined treatments (e.g., stress and Aβ + GC) on TAU pathology are not merely additive.
The present finding that stress and GCs increase TAU hyperphosphorylation and accumulation in neuronal soma largely echoes and extends previous reports in AD transgenic models (Green et al., 2006; Jeong et al., 2006) and neuronal cell cultures (Sotiropoulos et al., 2008b). Specifically, our results show that stress/GCs induce extensive hyperphosphorylation of TAU at more epitopes (several considered critical in AD) than were described by Jeong et al. (2006); further, we show that stress aggravates the aggregation of Sarkosyl-insoluble TAU. Detergent-insoluble TAU aggregates eventually give rise to the neurofibrillary tangles characteristic of tauopathies, including AD (Wang et al., 1995; Kimura et al., 1996; Alonso et al., 2001); moreover, insoluble TAU and TAU oligomers are causally implicated in neurodegenerative processes (Berger et al., 2007) and are found during presymptomatic stages of AD (Maeda et al., 2006). The discrepancy between our results (stress/GC-induced TAU hyperphosphorylation) and those of Green et al. (2006) may be attributed to the fact that the latter authors used 3×Tg mice expressing the P301-mutated TAU; wild-type TAU and P301L-mutated TAU display distinct phosphorylation profiles and exert dissimilar effects on neuronal structure and function (Kimura et al., 2010) and may be differentially regulated by stress. Further, Green et al. (2006) studied whole-brain homogenates, whereas our analysis in wild-type animals focused on specific brain regions that are affected in AD and targeted by stress/GCs.
Dendritic and synaptic atrophy, as well as cognitive impairments, are well known consequences of chronic stress and the accompanying increases in GC secretion (Sousa et al., 2000; Cerqueira et al., 2007; Landfield et al., 2007; Schubert et al., 2008; Lupien et al., 2009), albeit through still poorly understood cellular mechanisms. Extending earlier reports on the deleterious effects of GCs on cytoskeletal proteins, including TAU (Stein-Behrens et al., 1994; Cereseto et al., 2006), the results of the present study demonstrate that stress and GCs induce TAU hyperphosphorylation at epitopes implicated in cytoskeletal pathology and synaptic loss in AD patients (e.g., pSer262) (Callahan et al., 2002; Lauckner et al., 2003) and correlated with hippocampal atrophy in AD patients (e.g., pThr231) (Hampel et al., 2005). Clinical studies report a strong correlation between the extent of TAU hyperphosphorylation (including that of the Thr231 and Ser262 residues) and severity of impairments of memory, speed of mental processing, and executive functions (Augustinack et al., 2002, Ewers et al., 2007; van der Vlies et al., 2009). In addition, TAU hyperphosphorylation is associated with synaptic loss and memory impairment in experimental animals (Kimura et al., 2007). Based on the above considerations, we suggest that TAU hyperphosphorylation might be part of the cellular mechanism through which stress and GCs cause dendritic and synaptic dysfunction. In this connection, it is relevant to mention that recently Hoover et al. (2010) showed that hyperphosphorylated TAU within dendritic spines induces synaptic abnormalities, while Ittner et al. (2010) demonstrated that TAU mediates the synaptotoxic actions of Aβ. Results of the present study, together with other reports that TAU is essential for the manifestation of the actions of Aβ (Busciglio et al., 1995; Rapoport et al., 2002; Roberson et al., 2007; Shipton et al., 2011) and our previous demonstrations that stress and GCs trigger APP misprocessing in animals (Catania et al., 2009) and neuronal cultures (Sotiropoulos et al., 2008b), provide a mechanistic sequence through which stress leads to the expression of AD-relevant biochemical and behavioral markers. Accordingly, Aβ and TAU are introduced as new players in the cascade of events responsible for stress-induced brain dysfunction.
There is growing consensus that stress renders individuals vulnerable to brain disorders by causing inappropriate remodeling of individual neurons and disconnection of critical neural circuits (Cerqueira et al., 2007; Sotiropoulos et al., 2008a). The results reported here show that animals with a history of stress are more sensitive to the deleterious actions of Aβ and GCs; moreover, previously stressed animals express higher levels of hyperphosphorylated TAU and display more overt signs of memory dysfunction. Thus, previous stressful experiences may leave a “trace of vulnerability” to the development of AD-like pathology, with vulnerability increasing with age (Sotiropoulos et al., 2008a). In addition, our findings that GCs and stress act cumulatively suggest that repeated adverse experiences over an individual's lifetime will increase the probability or severity of AD pathology (Fig. 6). Our demonstration that stress and GCs can induce TAU pathology in otherwise healthy, middle-aged nontransgenic animals reinforces the view that lifetime experience may be an etiological factor in sporadic forms of AD. In this respect, the growing body of evidence for epigenetic regulation of AD pathophysiology (for review, see Chouliaras et al., 2010) is worth noting. Last, the introduction of TAU into the cascade of events leading to stress-induced neuronal and behavioral dysfunction has implications that extend beyond AD itself: stress is considered to be a major trigger of major depression, a disease that places individuals at a significant risk for developing AD (Ownby et al., 2006; Sun et al., 2008).
Figure 6.

Working model of how stress acts cumulatively to induce TAU pathology and cognitive impairment. Repeated exposures to adverse life events (stress), accompanied by increased levels of GCs, induce TAU pathology in a cumulative manner. TAU hyperphosphorylation is probably stimulated secondarily to stress/GC-induced misprocessing of APP to amyloidogenic peptides such as amyloid β and C99 (Sotiropoulos et al., 2008a; Catania et al., 2009). Hyperphosphorylated TAU accumulates and aggregates in neuronal perikarya, leading to neuronal dystrophy and dysfunction. The first detrimental signs of stress are seen in the hippocampal and parahippocampal regions, gradually spreading to the PFC and other cortical regions; similar spatiotemporal patterns are observed in patients with Alzheimer's disease (Lace et al., 2009). Rat brain images were adapted from Swanson (1999).
Footnotes
C.C. and I.S. were supported by the Max Planck Society and European Union (EU) Marie Curie Training Fellowships at University College, London. The work was supported by the German-Portuguese Luso-Alemas Program and the EU CRESCENDO Consortium (Contract FP6-018652). Dieter Fischer and Jutta Waldherr provided excellent technical assistance; Isabel Matos blind scored behavioral and histochemical data; and Drs. Peter Davies and Peter Seubert generously supplied monoclonal antibodies.
The authors declare no competing financial interests.
References
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated TAU into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006;103:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific TAU phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–395. doi: 10.1093/jnen/61.5.384. [DOI] [PubMed] [Google Scholar]
- Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OF. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14:95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- Cereseto M, Reinés A, Ferrero A, Sifonios L, Rubio M, Wikinski S. Chronic treatment with high doses of corticosterone decreases cytoskeletal proteins in the rat hippocampus. Eur J Neurosci. 2006;24:3354–3364. doi: 10.1111/j.1460-9568.2006.05232.x. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouliaras L, Rutten BP, Kenis G, Peerbooms O, Visser PJ, Verhey F, van Os J, Steinbusch HW, van den Hove DL. Epigenetic regulation in the pathophysiology of Alzheimer's disease. Prog Neurobiol. 2010;90:498–510. doi: 10.1016/j.pneurobio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgh E, Lindqvist Astot A, Fagerlund M, Eriksson S, Olsson T, Näsman B. Cognitive dysfunction, hippocampal atrophy and glucocorticoid feedback in Alzheimer's disease. Biol Psychiatry. 2006;59:155–161. doi: 10.1016/j.biopsych.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Ewers M, Buerger K, Teipel SJ, Scheltens P, Schröder J, Zinkowski RP, Bouwman FH, Schönknecht P, Schoonenboom NS, Andreasen N, Wallin A, DeBernardis JF, Kerkman DJ, Heindl B, Blennow K, Hampel H. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology. 2007;69:2205–2212. doi: 10.1212/01.wnl.0000286944.22262.ff. [DOI] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106:18367–18372. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and TAU pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Hampel H, Bürger K, Pruessner JC, Zinkowski R, DeBernardis J, Kerkman D, Leinsinger G, Evans AC, Davies P, Möller HJ, Teipel SJ. Correlation of cerebrospinal fluid levels of tau protein phosphorylated at threonine 231 with rates of hippocampal atrophy in Alzheimer disease. Arch Neurol. 2005;62:770–773. doi: 10.1001/archneur.62.5.770. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer's and Parkinson's disease compared withnormal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997;18:285–289. doi: 10.1016/s0197-4580(97)80309-0. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KY, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. FASEB J. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Ono T, Takamatsu J, Yamamoto H, Ikegami K, Kondo A, Hasegawa M, Ihara Y, Miyamoto E, Miyakawa T. Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia. 1996;7:177–181. doi: 10.1159/000106875. [DOI] [PubMed] [Google Scholar]
- Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A. Hyperphosphorylated TAU in parahippocampal cortex impairs place learning in aged mice expressing wild-type human TAU. EMBO J. 2007;26:5143–5152. doi: 10.1038/sj.emboj.7601917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem. 2010;285:38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace G, Savva GM, Forster G, de Silva R, Brayne C, Matthews FE, Barclay JJ, Dakin L, Ince PG, Wharton SB. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain. 2009;132:1324–1334. doi: 10.1093/brain/awp059. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Blalock EM, Chen KC, Porter NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer's disease. Curr Alzheimer Res. 2007;4:205–212. doi: 10.2174/156720507780362083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner J, Frey P, Geula C. Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging. 2003;24:767–776. doi: 10.1016/s0197-4580(02)00228-2. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Miller TP, Taylor J, Rogerson S, Mauricio M, Kennedy Q, Schatzberg A, Tinklenberg J, Yesavage J. Cognitive and noncognitive symptoms in dementia patients: relationship to cortisol and dehydroepiandrosterone. Int Psychogeriatr. 1998;10:85–96. doi: 10.1017/s1041610298005171. [DOI] [PubMed] [Google Scholar]
- Nisenbaum LK, Zigmond MJ, Sved AF, Abercrombie ED. Prior exposure to chronic stress results in enhanced synthesis and release of hippocampal norepinephrine in response to a novel stressor. J Neurosci. 1991;11:1478–1484. doi: 10.1523/JNEUROSCI.11-05-01478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530–538. doi: 10.1001/archpsyc.63.5.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous TAU ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches TAU protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Schubert MI, Kalisch R, Sotiropoulos I, Catania C, Sousa N, Almeida OF, Auer DP. Effects of altered corticosteroid milieu on rat hippocampal neurochemistry and structure—an in vivo magnetic resonance spectroscopy and imaging study. J Psychiatr Res. 2008;42:902–912. doi: 10.1016/j.jpsychires.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos I, Cerqueira JJ, Catania C, Takashima A, Sousa N, Almeida OF. Stress and glucocorticoid footprints in the brain-The path from depression to Alzheimer's disease. Neurosci Biobehav Rev. 2008a;32:1161–1173. doi: 10.1016/j.neubiorev.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OF. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in neuronal cells expressing human TAU. J Neurochem. 2008b;107:385–397. doi: 10.1111/j.1471-4159.2008.05613.x. [DOI] [PubMed] [Google Scholar]
- Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994;14:5373–5380. doi: 10.1523/JNEUROSCI.14-09-05373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stéphan A, Phillips AG. A case for a non-transgenic animal model of Alzheimer's disease. Genes Brain Behav. 2005;4:157–172. doi: 10.1111/j.1601-183X.2004.00113.x. [DOI] [PubMed] [Google Scholar]
- Sun X, Steffens DC, Au R, Folstein M, Summergrad P, Yee J, Rosenberg I, Mwamburi DM, Qiu WQ. Amyloid-associated depression: a prodromal depression of Alzheimer disease? Arch Gen Psychiatry. 2008;65:542–550. doi: 10.1001/archpsyc.65.5.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. Brain maps: structure of the rat brain. Amsterdam: Elsevier Science; 1999. [Google Scholar]
- Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H. Activation of TAU protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of TAU in hippocampal neurons. Neurosci Res. 1998;31:317–323. doi: 10.1016/s0168-0102(98)00061-3. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- van der Vlies AE, Verwey NA, Bouwman FH, Blankenstein MA, Klein M, Scheltens P, van der Flier WM. CSF biomarkers in relationship to cognitive profiles in Alzheimer disease. Neurology. 2009;72:1056–1061. doi: 10.1212/01.wnl.0000345014.48839.71. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner MF, Vobach S, Olsson K, Svetlik D, Risser RC. Cortisol secretion and Alzheimer's disease progression. Biol Psychiatry. 1997;42:1030–1038. doi: 10.1016/s0006-3223(97)00165-0. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer's disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Schneider JA, Boyle PA, Arnold SE, Tang Y, Bennett DA. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68:2085–2092. doi: 10.1212/01.wnl.0000264930.97061.82. [DOI] [PubMed] [Google Scholar]