

Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder primarily affecting regions of the brain responsible for higher cognitive functions. Immunization against β-amyloid (Aβ) in animal models of AD has been shown to be effective on the molecular level but also on the behavioral level. Recently, we reported naturally occurring autoantibodies against Aβ (NAbs–Aβ) being reduced in Alzheimer's disease patients. Here, we further investigated their physiological role: in epitope mapping studies, NAbs–Aβ recognized the mid-/C-terminal end of Aβ and preferentially bound to oligomers but failed to bind to monomers/fibrils. NAbs–Aβ were able to interfere with Aβ peptide toxicity, but NAbs–Aβ did not readily clear senile plaques although early fleecy-like plaques were reduced. Administration of NAbs–Aβ in transgenic mice improved the object location memory significantly, almost reaching performance levels of wild-type control mice. These findings suggest a novel physiological mechanism involving NAbs–Aβ to dispose of proteins or peptides that are prone to forming toxic aggregates.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder primarily affecting regions of the brain responsible for higher cognitive functions, such as learning and memory. Plaque deposition and neurofibrillary tangles are the histopathological hallmarks of AD. β-Amyloid (Aβ) peptides become deposited in those plaques, and hence their clearance has been discussed as a major therapeutic goal.
Emerging experimental evidence detected the early acidic endosomes as the principal generation site for Aβ peptides (Kaether et al., 2006; Rajendran et al., 2006), and dimers, trimers, and multimeric aggregates have been shown recently in vitro and in transgenic (Tg) mice models to be crucial toxic species (Cleary et al., 2005; Klyubin et al., 2005; Lesné et al., 2006; Townsend et al., 2006; Glabe, 2008; Shankar et al., 2008; Tomic et al., 2009). It was further suggested that small Aβ oligomers may form intracellularly before being released into the extracellular medium, in which they may interfere with synaptic activity or act as seeds to accelerate fibril formation (Selkoe, 2004; Khandogin and Brooks, 2007). Thus, preventing or reversing the formation of aggregated amyloid would appear to be a promising strategy for AD treatment.
Several therapeutic approaches are currently under consideration, including active/passive immunization against Aβ as pioneered by Schenk et al. (1999). In transgenic amyloid precursor protein (APP)-expressing mice, immunization against Aβ peptides has been shown to be effective on both molecular and behavioral levels. Active immunization in transgenic mice reduced fibril formation, promoted clearance of Aβ plaques, and also interfered with tau phosphorylation (Schenk et al., 1999; Morgan et al., 2000). Moreover, passive immunization was also effective with antibodies that recognized the N-terminal and the mid-terminal domains of Aβ peptides (DeMattos et al., 2001). Based on these data, several clinical trials have been started (Mangialasche et al., 2010). In patients treated with antibodies directed against the N terminus of Aβ, a considerable decrease in plaque load has been reported, but clearance of already formed plaques might not be sufficient to improve cognitive function in AD patients (Holmes et al., 2008).
Recently, we and others identified naturally occurring autoantibodies against Aβ (NAbs–Aβ) being reduced in patients with AD (Du et al., 2001; Weksler et al., 2002). Naturally occurring autoantibodies make up to two-thirds of the human antibody pool and are known to have many functions; however, the underlying mechanisms are far from being completely understood (Shoenfeld et al., 2007). NAbs–Aβ have been characterized in different experimental settings to inhibit the propensity of Aβ to fibrillize, thereby blocking its toxicity and to affect the clearance of Aβ (Dodel et al., 2004; Taguchi et al., 2008; Bacher et al., 2009; Relkin et al., 2009). However, how NAbs–Aβ interact with Aβ and promote their clearance remains to be elucidated. Here, we show for the first time that NAbs–Aβ interfered with, and preferentially bound to early oligomerization products of Aβ peptide. Moreover, in a mouse model of AD, plaque formation was reduced after passive immunization with NAbs–Aβ and the subsequent clearance of Aβ led to a rapid improvement of mice behavior.
Based on the concept of NAbs–Aβ, commercially available human Ig preparations (IVIg) have been used in small pilot trials for the treatment of patients with AD (Dodel et al., 2010), which showed promising effects on cognition, thus leading to a phase III trial in the United States (Relkin et al., 2009).
Materials and Methods
Isolation of NAbs–Aβ.
We used purified human intravenous IgG (Octagam 5%) for the isolation of NAbs–Aβ, which was kindly provided by Octapharma AG. Octagam 5% liquid is a solvent/detergent-treated, sterile preparation of highly purified IgG derived from large pools of human plasma (10,000–20,000 of donations). The product is prepared by using cold Cohn–Oncley ethanol fractionation process, followed by ultrafiltration and chromatography and contains ∼50 mg of protein per milliliter (5%). Ninety-six percent of protein represents human normal IgG (IgA < 0.2 mg; IgM < 0.1 mg). It contains no more than 3% aggregates and fragments, respectively, and no more than 90% monomers and dimers with an Fc portion maintained intact. IgG subclasses are fully represented (IgG 1, 65%; IgG 2, 30%; IgG 3, 3%; IgG 4, 2%). For the following isolation steps, this preparation was mixed with an equal volume of PBS and loaded directly onto an affinity column.
Because the Aβ1-40 sequence contains internal lysine residues that might lead to side reactions in immobilization procedures using amino groups, a specific affinity column was prepared using a cysteine residue attached to the Aβ N terminus to ensure homogeneous orientation of peptide molecules on the column support by immobilization through cysteinyl-S-thioether linkage. The azlactone-activated support contains an iodoacetyl group (Ultralink; Perbio) at the end of a hexadecyl-spacer group, which reacts with the cysteinyl-sulfhydryl group to yield a stable thioether linkage to reduce steric hindrance and provide maximum binding capacity of the antibodies. For covalent attachment of the Cys–Aβ1-40, 3.7 mg of peptide was dissolved in 50 mm Tris and 5 mm EDTA-Na coupling buffer, pH 8.5, to a final concentration of 0.37 mg/ml. The solution was added to 1 ml of drained Ultralink-Iodoacetyl gel, and the coupling reaction was performed for 1 h at room temperature under gentle mixing, followed by 30 min reaction time without mixing. An aliquot of 0.5 ml of the Cys–Aβ1-40 coupled support was packed into a column (2.5 ml; MoBiTec) allowing the solution to drain. The column was washed with 3 ml of coupling buffer, and nonspecific binding sites on the gel were blocked two times for 45 min with 1 ml of 50 mm l-cysteine-HCl in coupling buffer. Subsequently, the column was washed with 5 ml of 1 m NaCl, 5 ml of 0.l m Na-phosphate, and 0.15 m NaCl, pH 7.2, and stored at 4°C. The gel support (0.5 ml) was transferred into a 15 ml Falcon vial using 5 ml of PBS and mixed with 5 ml of IVIg. After gentle shaking overnight at 4°C, this suspension was transferred to the column using the effluent to completely rinse the matrix back into the column. The column was washed eight times with 10 ml of PBS, followed by two wash cycles with 10 ml of ultrapure water.
The bound antibodies were eluted from the column with 10 × 0.5 ml of 0.1 m glycine buffer, pH 2.8. Each fraction was collected in a microreaction tube containing 35 μl of 1 m Tris-HCl, pH 9. The flow-through (FT) of this isolation procedure, which contains IgG depleted of NAbs–Aβ, was also collected and was used as a control in the respective experimental settings.
To maintain the integrity of the antibodies, a neutral pH was adjusted immediately after elution by adding the appropriate amount of Tris-HCI or glycine buffer. To regenerate the column for additional use, the column was washed once with 10 ml of 10 mm Na-phosphate buffer, pH 6.8, followed by two wash cycles with 10 ml of PBS containing 1 m NaCl and finally two wash cycles with 10 ml of PBS.
Antibody concentrations in the elution fractions were determined by the Micro BCA Protein Assay kit method (Pierce; Perbio). The stock solution of 2 mg/ml bovine albumin supplied within the Micro BCATM kit was used to prepare fresh standard dilutions within the range 40–0.5 μg/ml. The antibodies were eluted between fractions 1 and 6, with the highest concentrations in fractions 1 and 3. For quantification of each set of 10 elutions, fresh albumin standard dilutions were prepared. Results were read at 562 nm with an ELISA reader.
Cell culture and toxicity assay.
SH-SY5Y human neuroblastoma cells were grown in RPMI 1640 medium supplemented with 10% FCS, 10 mm HEPES, 4 mm glutamine, penicillin (200 U/ml), and streptomycin (200 μg/ml) at 37°C in a 5% CO2 atmosphere. Cells were plated and kept overnight at a density of 30,000 cells per well in a 96-well microtiter plate. After removal of medium, cells were washed with PBS, and toxic Aβ oligomers (2 μm final concentration) (Kayed et al., 2003) were added in 100 μl of fresh medium with either 7.5 or 15 μm NAbs–Aβ or without NAbs–Aβ. Each data point was determined in hexaplicate. MTT toxicity assay with SH-SY5Y cells was performed according to the protocol of the manufacturer.
Preparation of Aβ.
The lyophilized synthetic Aβ1-40 (PSL GmbH) was resuspended in 100% 1,1,1,3,3,3 hexafluoro-2-propanol (HFIP) at 1 mg/ml and incubated for 1 h at room temperature. After evaporation of HFIP, the peptide was resuspended according to the species of interest. For preparation of the monomer, the synthetic Aβ1-40 peptide was suspended in ultrapure or double-distilled water at 1 mg/ml just before the experiment. Aβ dimers were prepared using Aβ1-40 peptide with N-terminal cysteine (Cys–Aβ1-40) in 1× PBS at 1 mg/ml concentration and centrifuged for 30 min at 12,000 rpm. The protein content of the supernatant was quantified using Nanodrop and sonicated for 15 min at 65°C. The dimer was generated by incubation at 37°C for 3 h with shaking (900 rpm). The trimer was generated by a double mutation of Aβ1-40 peptide and resuspended in sodium carbonate buffer (1 mg/ml), pH 9.6, after the HFIP treatment, which instantly formed the SDS-stable trimer. Low-binding minicentrifuge tubes were used (Eppendorf) throughout the experiments.
Characterization of Aβ epitopes recognized by NAbs–Aβ.
Ninety-six-well plates were coated with Aβ1-40 and other peptides as outlined in the respective figure [5 μg/ml; freshly resuspended in coating buffer (1.7 mm NaH2PO4, 98 mm Na2HPO4, pH 7.4, and 0.05% sodium azide)], and incubated at 37°C for 2 h. The plates were then incubated with blocking buffer (Pierce) at 37°C for 2 h.
HRP-labeled heavy and light chain-specific goat anti-human antibodies were used as detection antibodies at 1:3000 dilution and incubated for 1 h at 37°C (Calbiochem). The reaction with a chromogenic substrate, 3,3′,5,5′-tetramethylbenzidine (Calbiochem), was terminated with 2N H2SO4, and plates were read at 450 nm using a plate reader (Thermo Fisher Scientific).
Size exclusion chromatography.
A Superdex 75 10/30 HR column attached to an AKTA FPLC system (GE Healthcare) was used for all experiments and calibrated using unbranched dextran standards of the following molecular masses: 123,600; 66,700; 43,800; 21,400; 9890; and 4440 Da (Pharmacosmos). β-Amyloid (1 mg/ml) was injected onto the Superdex 75 column and eluted with 50 mm ammonium acetate, pH 8.5, at a flow rate of 1 ml/min. One milliliter fractions were lyophilized, resuspended in 2× SDS-tricine sample buffer (20 μl), boiled for 10 min, and then subjected to electrophoresis on 10–20% tricine gels. Proteins were transferred onto 0.2 μm nitrocellulose membranes and used for Western blotting.
Surface plasmon resonance experiments.
Immobilization of antibodies and antibody/Aβ interaction analyses were performed on a Biacore 2000 optical biosensor system. Each antibody was immobilized via amino coupling (Biacore) up to 8500–10,000 response units on CM5 sensor chips (Biacore). Then, 60 μl of analyte (Aβ oligomers) was injected on sensor chips surfaces at a flow rate of 15 μl/min and in various concentrations (0.1–100 μg/ml). PBS and a carbonate-bicarbonate buffer were used as running buffers as specified. Aβ samples were kept at 4°C before each injection over the sensor chip at 25°C. Surface plasmon resonance (SPR) chip regeneration was performed using 6.25–50 mm NaOH. Binding analysis was performed using BIA evaluation 3.2RC1 software (Biacore).
Animals and drug treatment.
We investigated female CRND8 transgenic mice carrying a double-mutated form of the human amyloid precursor protein 695 (APP695; the Swedish and Indiana mutations) under the control of the Syrian hamster prion promoter, on a hybrid C57BL/6–C3H/ HeJ background (courtesy of D. Westaway, University of Toronto, Toronto, Canada) (Chishti et al., 2001). Compared with other murine models of AD (Röskam et al., 2010), TgCRND8 mice exhibit Aβ plaques at a very young age (at ∼3 months), accompanied by cognitive deficits (Lovasic et al., 2005; Bacher et al., 2008). At 3 months (“young”; n = 15) or 12 months (“old”; n = 15) of age, mice were injected intraperitoneally once a week for the duration of 4 weeks with either a low (80 μg) or high (200 μg) dose of NAbs–Aβ or with 200 μm FT (n = 5). In all experiments, the column FT was used as negative control (see above). In addition, female APP23 transgenic mice, which express the human APP751 isoform with the Swedish double mutation (K670N–M671L) under control of a murine Thy1.2 expression cassette (courtesy of Dr. Matthias Staufenbiel, Novartis Institutes for Biomedical Research, Basel, Switzerland) were used. The animals were hemizygous or littermate controls and had been backcrossed with C57BL/6J mice for at least eight generations (Sturchler-Pierrat et al., 1997). Mice were housed in groups of three to four animals in standard cages (37 cm length × 21 cm width × 15 cm height) together with wild-type littermates with food and water available ad libitum. All experimental procedures were in accordance with the guidelines of the local animal care commission.
Brain tissue preparation.
Mice were decapitated 5 d after the last injection. Brains were removed, and one hemisphere was immediately snap-frozen in liquid nitrogen and kept at −80°C until use. The other hemisphere was fixed in 4% buffered formaldehyde for 24 h, followed by dehydration and paraffin embedding.
Immunohistochemistry.
For Aβ staining, six pairs of 2 μm sagittal brain sections of each transgenic animal were pretreated with formic acid and incubated in a TechMate instrument (Dako) with 6F/3D (anti-Aβ monoclonal antibody directed against residues 8–17; 1:100; Dako). The second step used the Dako StreptABC complex horseradish peroxidase-conjugated anti-mouse/rabbit antibody kit, and staining was developed with 3,3-diaminobenzidine. Counterstaining was performed with hematoxylin. The pairs of sections (10 μm distances) were situated 100, 200, 300, 400, 500, and 600 μm laterally from the midsagittal fissure. All slides were stained in five consecutive procedures, ensuring that the brains of all three experimental groups were equally distributed.
Morphometry of the Aβ plaques.
To quantify the Aβ plaque burden, neocortices and hippocampi of all stained sections were digitized (Olympus BX50, ColorView II, charge-coupled device camera; Olympus) under constant light and filter settings. Color images were converted to grayscale by extracting blue to gray values to obtain the best contrast between positive immunoreactivity and the background. A constant threshold was chosen for all images to detect immunoreactive staining (analySIS 5; Soft Imaging System). The total number and size of plaques were determined. Absolute values of plaque burden were related to the investigated area.
Behavioral experiments.
Fourteen- to 16-month-old Tg2576 mice expressing the Swedish mutation (K670N/M671L) of APP695 under the control of the hamster prion promoter, on a hybrid C57BL/6–SJL background (Hsiao et al., 1996), were intraperitoneally injected with either 400 μg of NAbs–Aβ dissolved in 200 μl of PBS (n = 21) or 200 μl of PBS only (n = 20). An additional group of age-matched wild-type mice were treated with PBS (n = 20).
To investigate the treatment efficacy of NAbs–Aβ on spatial memory, we performed the object location test (Hale and Good, 2005). In brief, animals are introduced to two identical objects in an experimental apparatus and, after a delay, are exposed again to the same two objects, one of which has been displaced to a new location. Animals that remember the previous exposure spontaneously spend more time exploring the object in the new position. On the day of testing, mice were submitted to two 10 min trials with a 30 min intertrial delay. In trial 1, mice were allowed to explore two similar objects located on one side of the cage for 10 min. In trial 2, one of these objects was moved to a different location, and the mice were allowed to explore the object for another 10 min. For analysis, a discrimination ratio (DR) (time spent on the object in the new location versus the total time spent exploring both objects in the test trial) was calculated for each mouse. Object location test was reflected by more time spent interacting with the object in the new location than with the object in the familiar position and a DR above 0.5. Analysis of DRs is less subject to the influence of individual variability in contact times with objects and provides a measure of the extent of the discrimination between the novel and sample objects.
Murine anti-human antibody response.
To investigate the murine anti-human antibody (MAHA) response, we treated 10 wild-type mice (C57 BL/6 genetic background of TgCRND8 mice used in drug treatment experiments) with 200 μg of NAbs–Aβ once a week for a period of 28 d (Imboden et al., 2001). Blood was taken before and after the treatment and investigated by MAHA ELISA. Briefly, plates were coated overnight at 4°C with 5 μg/ml human IgG1, diluted in sodium bicarbonate buffer, pH 9.6. After washing four times with 1× PBS, 0.05% Tween 20, pH 7.4, plates were blocked for 3 h with SuperBlock (Thermo Fisher Scientific). After washing four times with 1× PBS, 0.05% Tween 20, pH 7.4, the serum samples were diluted 1:100 and added to the wells for overnight incubation at 4°C. The wells were then washed four times with 1× PBS and 0.05% Tween 20, pH 7.4, and then 0.1 μg/ml HRP-conjugated goat anti-mouse IgG was added and plates were incubated for 1 h at room temperature. Plates were then washed four times with 1× PBS and 0.05% Tween 20, pH 7.4. A total of 100 μl of tetramethylbenzidine (Calbiochem) substrate was added to each well. The plates were incubated for 10 min, and then the reaction was stopped by the addition of 25 μl/well of 2N H2SO4. The plates were read at 450 nm with a 570 nm reference filter. The standard curve reagent used for this assay was obtained by performing these measurements on serial dilutions of mouse anti-human IgG1 (BD Biosciences Pharmingen) in concentrations between 1 and 1000 ng/ml in twofold increments.
Statistical analysis.
Data were analyzed using the Student's t test or ANOVA as appropriate. All tests were applied (two tailed) using the software package SPSS (version 12.0.1). Differences were considered significant at p < 0.05.
Results
To assess whether purified NAbs–Aβ were protective against Aβ-induced cell death, we used Aβ preparations consisting of oligomerized products (Kayed et al., 2003). NAbs–Aβ were able to block the toxic effects of Aβ oligomers in SH-SY5Y cells in a concentration-dependent manner (Fig. 1). Cell death was reduced by >80% in the presence of NAbs–Aβ. Ig preparations lacking NAbs–Aβ (flow-through of IVIg after isolation of NAbs–Aβ) administered at concentrations up to 15 μm were not able to block the cytotoxic effects in this assay (Fig. 1).
Figure 1.

NAbs–Aβ were able to block the neurotoxic effect of oligomeric Aβ1-40 in neuroblastoma SH-SY5Y cells. SH-SY5Y cells (30,000 cells per well) were incubated alone or in the presence of oligomerized Aβ1-40 (prepared as described in Material and Methods). The viability of the cells was determined by the MTT assay. There was a concentration-dependent inhibition of Aβ-induced cell death in the presence of NAbs–Aβ. At a concentration of 15 μm NAbs–Aβ, an almost complete inhibition of Aβ-induced cell death was observed. Using the FT in a concentration up to 15 μm did not block Aβ-induced cytotoxicity. Values are expressed as percentage of control cultures for each experiment, and the data represent the mean ± SE of triplicate determinations from a representative experiment repeated at least three times with similar results.
Therapeutic efficacy of immunization has been associated with the clearance of plaques in the brain of transgenic mice (Schenk et al., 1999). Therefore, we investigated the ability of NAbs–Aβ to resolve plaque burden in APP TgCRND8 transgenic mice (Chishti et al., 2001). We examined the effect of a low (80 μg) and high (200 μg) dose of purified NAbs–Aβ on Aβ plaque deposition by immunohistochemical staining and image analysis in young (4 months) and old (13 months) TgCRND8 mice. To avoid the MAHA response, treatment with the foreign antibody was limited to 4 weeks to prevent an adverse MAHA immune response in the animal model. To investigate the MAHA response, we injected 10 mice with 200 μg of NAbs–Aβ once a week for 28 d. As expected, all animals developed Ig against human IgG (baseline, 0.001 mg/ml; at 28 d, 0.017 mg/ml). However, the titers were low within the group of 10 animals, seven animals had a titer of <0.01 mg/ml, and only one animal (0.06 mg/ml) had a titer above 0.05 mg/ml. None of the animals died or exhibited clinical or behavioral changes. None of the inspected organs showed changes as investigated by pathological and histopathological examinations.
The cerebral plaque load in senescent animals remained unaffected after 4 weeks of treatment with either low or high doses of NAbs–Aβ when compared with age-matched sham-treated animals (Fig. 2A–D). In contrast, both low- and high dose-treated young transgenic mice had a significant reduction of plaque number (high dose, by 24%, p = 0.045; low dose, by 34%, p = 0.026) and plaque area (high dose, by 47%, p = 0.029; low dose, by 47%, p = 0.028) (Fig. 2E–H). The differences between the high and the low doses in young animals were not significant.
Figure 2.

Effect of NAbs–Aβ on plaque deposition and plaque load in transgenic APP TgCRND8 mice. A–D, Effect of NAbs–Aβ on plaque number (C) and plaque area (D) in old (age, 13 months) transgenic APP TgCRND8 mice. Transgenic mice were injected intraperitoneally once a week for the duration of 4 weeks with either FT after NAbs–Aβ purification as control (control, 200 μg; A; n = 10) or with a low (80 μg; C, D; n = 7) and high (200 μg; B–D; n = 8) dose of NAbs–Aβ, respectively. E–H, Effect of NAbs–Aβ on plaque number (G) and plaque area (H) in young transgenic mice (age, 4 months). Transgenic mice were injected intraperitoneally once a week for the duration of 4 weeks with either FT (flow-through depleted of NAbs–Aβ; 200 μg; control; E; n = 7) or a low (80 μg; G, H; n = 7) and high (200 μg; F–H; n = 8) dose of NAbs–Aβ, respectively. In old TgCRND8 mice, NAbs–Aβ did not affect the plaque load when compared with vehicle (FHT)-treated animals (B–D), whereas in young mice plaque number and plaque area was significantly suppressed after 4 weeks of treatment (F–H). The plaque density was significantly reduced after 4 weeks of both high (by 24%) and low (by 34%) dose treatment with NAbs–Aβ (p = 0.045 and 0.026, respectively; the difference between the low and the high dose in the respective figures was not significant). Furthermore, a reduction of plaque area by 47% was detected after high (p = 0.029) and low (p = 0.028) dose applications of NAbs–Aβ (H). Animals were killed 5 d after the last injection. Sagittal sections from FT- and NAbs–Aβ-treated young and old mice were stained for Aβ using anti-human Aβ monoclonal antibody 6F/3D. The average number (C, G) and area (D, H) of Aβ-immunoreactive deposits were quantified semiautomatically in the cortex and hippocampus (12 sections per brain were analyzed). Results were analyzed using a two-tailed Student's t test. Error bars represent the SD. I, J, Concentration of Aβ in CSF and plasma of APP transgenic mice after treatment with NAbs–Aβ. The concentration of Aβ in CSF (I) and plasma (J) was determined after the last treatment with NAbs–Aβ. There was an increase of the Aβ concentration in the plasma and a decrease of the Aβ concentration in CSF.
A disturbance in the equilibrium of the central and the peripheral pool of Aβ was assumed to take place after immunization against Aβ; a phenomenon termed “peripheral sink hypothesis” (Dodart et al., 2002; Dodel et al., 2002). Because the NAbs–Aβ had no plaque-dissolving benefit in the old animals, we further investigated their effect on the concentration of Aβ in CSF and plasma of immunized animals. We detected a significant reduction of CSF Aβ in the animal group treated with NAbs–Aβ (Fig. 2). In plasma, an increase of Aβ was detectable, indicating an efflux of Aβ from the central pool into the periphery (Fig. 2J). In contrast, CSF and plasma levels of Aβ did not change considerably when treated with FT for the same period.
Interestingly, NAbs–Aβ failed to detect plaques in brain samples of transgenic APP-expressing mice (Fig. 3A,B), AD patients (Fig. 3C), and patients with cerebral amyloid angiopathy (Fig. 3D). In contrast, antibodies such as 6F/3D detected plaques in both animal and human brain samples. Similar results were found when NAbs–Aβ was used to detect Aβ in the brain vasculature of patients with cerebral amyloid angiopathy (Fig. 3D).
Figure 3.

Immunohistochemistry to investigate the binding properties of the N-terminal monoclonal antibodies and NAbs–Aβ to amyloid deposits in transgenic APP23 mice and in brain samples of AD and human cerebral amyloid angiopathy. APP23 mice were used in this set of experiments because of their higher deposition of Aβ in vasculature. Similar results were detected with respect to amyloid deposition in TgCRND8 mice. A, Immunostaining of a brain sample from an APP23 mouse with NAbs–Aβ: no specific staining of the plaque (P) in the cortex is detectable. The inset represents the identical area with a positive staining using the 6F/3D monoclonal antibody. B, Immunostaining of a brain sample from an APP23 mouse with NAbs–Aβ: no specific staining of amyloid deposits in the vessel (V+) is detectable (the inset represents the identical area with positive staining using the 6F/3D monoclonal antibody). C, Immunostaining of a brain sample from an AD patient with NAbs–Aβ: no specific staining of a plaque (P) is detectable. The inset represents the identical area with positive staining using the 6F3D monoclonal antibody. D, Immunostaining of a brain sample from a cerebral amyloid angiopathy patient with NAbs–Aβ: no specific staining of amyloid deposits in the small vessels (V+) is visible, but there are some unspecific chromogen reactions with the Nissl substance. The inset represents the identical area with positive staining using the 6F3D monoclonal antibody.
Antibodies raised against the Aβ epitopes 4–10 have been reported to bind to plaques in the brains of transgenic animals (McLaurin et al., 2002). Therefore, we determined the linear sequences and the conformational epitopes that were recognized by NAbs–Aβ using a large set of synthetic peptides derived from the Aβ sequence (Fig. 4A). The NAbs–Aβ bound to the full-length Aβ but not to the albumin control. The epitope region recognized by NAbs–Aβ encompassed the residues 28–40(42) of the Aβ sequence (Fig. 4A), whereas N-terminal fragments were not recognized by NAbs–Aβ. The peptides with an altered amino acid as well as the scrambled peptide did not show a binding signal. The relevant amino acids within the epitope were confirmed by an alanine scan of the C-terminal region of Aβ.
Figure 4.
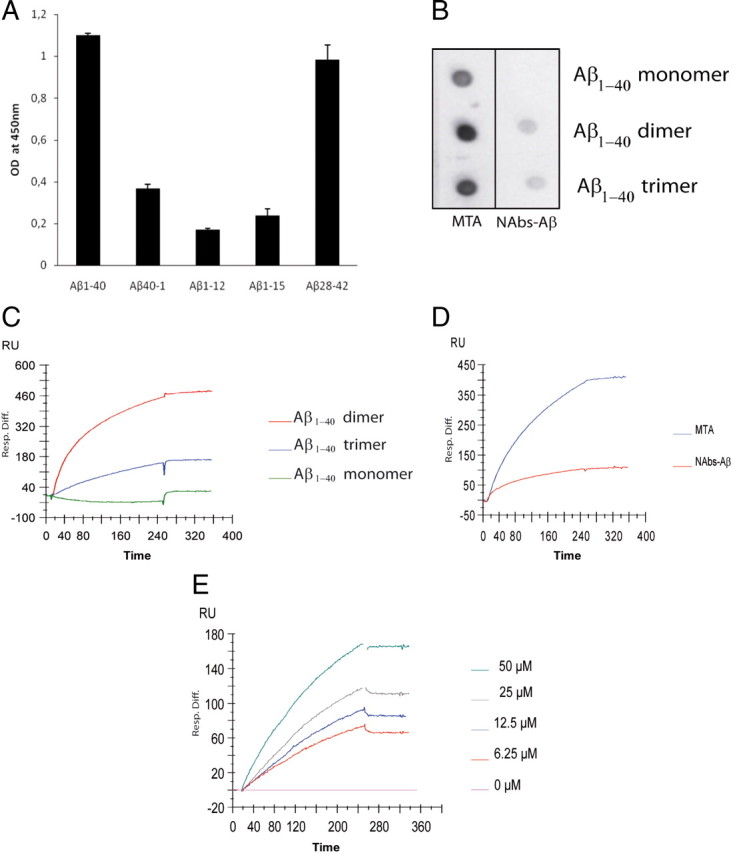
A, Mapping of the Aβ epitope recognized by NAbs–Aβ using ELISA. Various peptides representing different parts of the Aβ sequence were tested for the avidity of NAbs–Aβ to bind in ELISA. The binding experiments showed an increased binding of the NAbs–Aβ toward the C-terminal region. We identified sequence AA28–40(42) as the major binding site. N-terminal regions of the Aβ peptide showed only low binding with the Aβ reversed sequence (Aβ40-1). The following peptides (with and without exchanged amino acids) were also investigated: Aβ1-40; Aβ1-42; Aβ1-12; Aβ1-15; Aβ10-28; Aβ20-38; Aβ20-31; Aβ25-40; Aβ26-42; Aβ28-42; Aβ30-42; Aβ1-40;G25A; Aβ1-40;S26A; Aβ1-40;G29I;G33I; Aβ1-42;G29I;G33I; Aβ1-40;D23K;K28D; Aβ1-40;G29I;G33I;A42I; Aβ1-40;M35A;V40A; Aβ1-40;I32A;M35A; Aβ1-40;I32A;V40A; Aβ1-40;I32A; Aβ28-42;A30G; Aβ28-42;G33A; Aβ28-42;M35A; Aβ28-42;V39C; Aβ28-42;I32A; Aβ28-42;I32A;M35A;V40A. B, Dot blots of various peptide samples of SEC fractions of Aβ were applied on nitrocellulose membrane (0.25 μg/3 μl spot). The mid-terminal antibody (MTA) directed against residues AA16–24 readily recognized monomeric and oligomeric forms of Aβ, whereas NAbs–Aβ only interacted with dimeric and trimeric forms but not monomeric forms. C–E, Immobilization and interaction analyses of monoclonal antibodies and NAbs–Aβ to Aβ1-40 and its different oligomeric forms were performed on a Biacore 2000 optical biosensor system. C, NAbs–Aβ preferentially bound to dimers, and also to trimers, but not to the monomeric form of Aβ. D, The on-rate of the monoclonal antibody MTA to Aβ was higher compared with the on-rate of NAbs–Aβ. E, There was a dose-dependent on-rate of the binding of NAbs–Aβ to Aβ1–40. OD, Optical density; RU, response units.
Using size exclusion chromatography to separate monomeric, dimeric, and trimeric forms of Aβ as described previously (Walsh et al., 2005), we found that NAbs–Aβ preferentially recognized dimers and trimers and, to a lesser extent, monomeric forms of Aβ (Fig. 4B). In contrast, a monoclonal antibody recognizing the mid-terminal part of the Aβ sequence detected all three investigated Aβ oligomers. The affinity of NAbs–Aβ to the different Aβ assemblies using SPR are shown in Figure 4C–E.
Furthermore, in behavioral experiments in the Tg2576 transgenic mice, the object location memory improved significantly after the administration of NAbs–Aβ and almost reached performance levels of wild-type control mice. Mice treated with PBS regularly performed at chance levels (discrimination ratio: 0.5 = 50%), whereas NAbs–Aβ-treated animals and age-matched wild-type mice performed above chance (p < 0.001, ANOVA) (Fig. 5).
Figure 5.

Neuropsychological testing in Tg2576 mice. Mean discrimination ratios for 14- to 16-month-old Tg2576 mice treated with either NAbs–Aβ or PBS. NAbs–Aβ-immunized Tg2576 mice but not PBS-treated Tg2576 mice showed a preference for exploring objects moved to a new location, indicated by a discrimination ratio above 0.5 (NAbs–Aβ group, n = 21; PBS group, n = 20; wild-type group, n = 20; ***p < 0.001). Error bars show SEM. WT, Wild type.
Discussion
To the best of our knowledge, this is the first report to link naturally occurring autoantibodies to a physiological effect in the metabolism of peptides that are toxic to humans. We found that NAbs–Aβ were readily detectable in healthy individuals, interacted with Aβ, and promoted its degradation. In epitope mapping, NAbs–Aβ detected the mid-/C-terminal epitope of Aβ, starting at the amino acid AA28. Interestingly, they were not able to bind and clear plaques, but they were able to improve behavioral changes in a transgenic mice model. Therefore, the question is, how may NAbs–Aβ work if they can only recognize the mid-/C-end-terminal part of Aβ? According to recent solid-nuclear magnetic resonance spectroscopy studies of Aβ oligomerization, the Aβ residues 12–24 and 30–40 adopt β-strand conformations and form parallel [or anti-parallel, according to a proposed model (Petkova et al., 2005)] β-sheets through intermolecular hydrogen bonding (Petkova et al., 2002). This process is followed by oligomerization of the Aβ peptides, as shown in Figure 6. After fibrillation, the C-terminal part of the Aβ peptide is masked. However, if an antibody is intended to block oligomerization, the C-terminal part of the Aβ peptide must be recognized for the antibody to bind to it. If an antibody recognizes the N-terminal part of the Aβ peptide, oligomerization and fibrillation of the peptide may still continue. Therefore, to block these processes that result in plaque formation, an antibody appears to be useful that interferes with the C-terminal part of the Aβ peptide.
Figure 6.

A, Currently known epitopes of the Aβ sequence. The NAbs–Aβ recognized a distinct C-terminal epitope compared with antibodies generated against the N terminus by active immunization in transgenic mice and promoted clearance of amyloid plaques (McLaurin et al., 2002). The T-cell activation sites and the B-cell epitope have been identified at AA14–34 and AA4–10, respectively (Monsonego et al., 2001). B, Assembly of Aβ as proposed by Petkova et al. (2002, 2005). Central Aβ1--40 molecules from the energy-minimized, five-chain system, viewed down the long axis of the fibril. Residues are color coded according to their side chains as hydrophobic (green), polar (magenta), positive (blue), or negative (red). C, Possible mode of lateral association for generating fibrils with greater mass-per-length and greater cross-sectional dimensions. Assembly of oligomers started at mid-terminal and C-terminal parts of the Aβ peptide. Therefore, active immunization with Aβ led to the generation of N-terminal antibodies because only the N-terminal part was available for T-cell interaction. Accordingly, only N-terminal antibodies were effective in removing plaques because only N-terminal parts were easily accessible to antibodies. In contrast, NAbs–Aβ recognized the dimeric form of Aβ and interfered with further assembly of monomers to oligomers. B and C are reproduced with permission from Proceedings of the National Academy of Sciences of the United States of America.
In the active immunization studies, fibrillated Aβ peptide was used as antigen. It is likely that N-terminal epitopes of the Aβ peptide were predominantly exposed and were available as binding sites. Accordingly, active immunization generated primarily antibodies that recognized the N-terminal epitope (AA4–10) of the Aβ sequence (McLaurin et al., 2002). Furthermore, Aβ peptide is deposited in a fibrillar form in the plaques, and the N-terminal part of the Aβ peptide is predominantly available at the “outside” of the plaque as an “epitope” for N-terminal active antibodies.
In contrast, NAbs–Aβ—as mainly directed to the mid-/C-terminal epitope of Aβ—were in fact not capable of binding to the N terminus of Aβ. This finding could also explain why our NAbs–Aβ did not clear plaques.
Furthermore, we found that our NAbs–Aβ preferentially bound to dimers and trimers and interfered with oligomerization (Britschgi et al., 2009). This may be the mechanisms why treatment with NAbs–Aβ in young animals led to a reduction in plaque number and size, suggesting that they acted by preventing plaque formation.
Interestingly, NAbs–Aβ failed to interact with other aggregating/β-sheet promoting proteins, such as α-synuclein and prion proteins (data not shown) (Du et al., 2003). Because naturally occurring antibodies do not bind to distinct protein sequences but rather recognize specific patterns of conservative molecules, we hypothesized that NAbs–Aβ may recognize a common conformational epitope among those proteins; our current data, however, suggest that NAbs–Aβ display a rather narrow epitopal recognition pattern.
Recently, Shankar et al. (2008) pinpointed small assemblies of Aβ as “disease causation of AD” (Shankar et al., 2008). This interaction with small oligomers is pivotal if NAbs–Aβ play a role in the metabolism of Aβ peptides. However, targeting dissolved plaques is not the primary goal because these plaques are inert reservoirs (“garbage dump”) and they help to trap toxic oligomers in the brain. Recent evidence from preclinical experimental studies as well as data from clinical trials support this notion (Holmes et al., 2008).
Although our data showed an efflux of central Aβ into the periphery that favored the efflux hypothesis of Aβ clearance (peripheral sink) (DeMattos et al., 2002), it was noticeable that a small amount of NAbs–Aβ was capable of crossing the blood–brain barrier and penetrating into the brain, as was shown using radiolabeled NAbs–Aβ (Bacher et al., 2009). In previous studies, this small amount of autoantibodies available in the brain was suggested to be sufficient to induce uptake by opsonization in the brain (Schenk et al., 1999; Bard et al., 2000; Hock et al., 2002). Therefore, we cannot rule out that additional mechanisms, including a central degradation of Aβ oligomers in the presence of NAbs–Aβ by microglial cells, may occur in the brain.
In summary, we suggest that NAbs–Aβ, as part of the autoantibody pool, have a physiological role and convey a protective effect by inhibiting oligomerization of Aβ peptides and consequently degrading Aβ. We further postulate that this protective effect is primarily mediated through interactions with the mid-/C-terminal parts of the Aβ peptide and may involve oligomerized products of Aβ. Furthermore, the increase of NAbs–Aβ in the serum probably disrupts the equilibrium between central and peripheral Aβ pools, leading to a shift from the CNS to the periphery.
Finally, supportive evidence for the pivotal role that NAbs–Aβ may play in the protection from AD comes from clinical studies using IVIg preparations that contain NAbs–Aβ. IVIg has shown a potentially positive effect on behavioral symptoms in AD patients (Dodel et al., 2004; Relkin et al., 2009). We are fully aware that these pilot studies only included a small number of patients. Nevertheless, these preliminary clinical results, along with our experimental results reported here, warrant a large clinical trial to investigate the effects of IVIg and human NAbs–Aβ in patients with AD. A respective phase-III trial is currently underway in the United States of America and results will be most likely available by 2013.
Footnotes
This study was supported by an unrestricted research grant of the University Medical Center Giessen and Marburg (UKGM) (PN: 155/06-07MR) and in part by an unrestricted grant of the Deutsche Forschungsgemeinschaft Grant DO784/2-1. We also received an unrestricted grant from Prof. Dr. Reinfried Pohl, Marburg, Germany. We thank Caroline Gäckler for the excellent technical assistance and Tanja Rausch for the histopathological work.
R.D. and M.B. hold patents on immunization with autoantibodies in neurodegenerative disorders.
References
- Bacher M, Dodel R, Aljabari B, Keyvani K, Marambaud P, Kayed R, Glabe C, Goertz N, Hoppmann A, Sachser N, Klotsche J, Schnell S, Lewejohann L, Al-Abed Y. CNI-1493 inhibits Abeta production, plaque formation, and cognitive deterioration in an animal model of Alzheimer's disease. J Exp Med. 2008;205:1593–1599. doi: 10.1084/jem.20060467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher M, Depboylu C, Du Y, Noelker C, Oertel WH, Behr T, Henriksen G, Behe M, Dodel R. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer's disease. Neurosci Lett. 2009;449:240–245. doi: 10.1016/j.neulet.2008.08.083. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Britschgi M, Olin CE, Johns HT, Takeda-Uchimura Y, LeMieux MC, Rufibach K, Rajadas J, Zhang H, Tomooka B, Robinson WH, Clark CM, Fagan AM, Galasko DR, Holtzman DM, Jutel M, Kaye JA, Lemere CA, Leszek J, Li G, Peskind ER, Quinn JF, Yesavage JA, Ghiso JA, Wyss-Coray T. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:12145–12150. doi: 10.1073/pnas.0904866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Dodel R, Hampel H, Depboylu C, Lin S, Gao F, Schock S, Jäckel S, Wei X, Buerger K, Höft C, Hemmer B, Möller HJ, Farlow M, Oertel WH, Sommer N, Du Y. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann Neurol. 2002;52:253–256. doi: 10.1002/ana.10253. [DOI] [PubMed] [Google Scholar]
- Dodel R, Neff F, Noelker C, Pul R, Du Y, Bacher M, Oertel W. Intravenous immunoglobulins as a treatment for Alzheimer's disease: rationale and current evidence. Drugs. 2010;70:513–528. doi: 10.2165/11533070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Depboylu C, Hampel H, Frölich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B, Bales K, Gao F, Moeller HJ, Oertel W, Farlow M, Paul S. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57:801–805. doi: 10.1212/wnl.57.5.801. [DOI] [PubMed] [Google Scholar]
- Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale G, Good M. Impaired visuospatial recognition memory but normal object novelty detection and relative familiarity judgments in adult mice expressing the APPswe Alzheimer's disease mutation. Behav Neurosci. 2005;119:884–891. doi: 10.1037/0735-7044.119.4.884. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Imboden M, Murphy KR, Rakhmilevich AL, Neal ZC, Xiang R, Reisfeld RA, Gillies SD, Sondel PM. The level of MHC class I expression on murine adenocarcinoma can change the antitumor effector mechanism of immunocytokine therapy. Cancer Res. 2001;61:1500–1507. [PubMed] [Google Scholar]
- Jia CY, Nie J, Wu C, Li C, Li SS. Novel Src homology 3 domain-binding motifs identified from proteomic screen of a Pro-rich region. Mol Cell Proteomics. 2005;4:1155–1166. doi: 10.1074/mcp.M500108-MCP200. [DOI] [PubMed] [Google Scholar]
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006;7:408–415. doi: 10.1111/j.1600-0854.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Khandogin J, Brooks CL., 3rd Linking folding with aggregation in Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci U S A. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lovasic L, Bauschke H, Janus C. Working memory impairment in a transgenic amyloid precursor protein TgCRND8 mouse model of Alzheimer's disease. Genes Brain Behav. 2005;4:197–208. doi: 10.1111/j.1601-183X.2004.00104.x. [DOI] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, St George-Hyslop P. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsiveness to amyloid beta-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:10273–10278. doi: 10.1073/pnas.191118298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Röskam S, Neff F, Schwarting R, Bacher M, Dodel R. APP transgenic mice: the effect of active and passive immunotherapy in cognitive tasks. Neurosci Biobehav Rev. 2010;34:487–499. doi: 10.1016/j.neubiorev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoenfeld Y, Gershwin ME, Meroni PL. Autoantibodies. Burlington: Elsevier Science and Technology; 2007. [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi H, Planque S, Nishiyama Y, Symersky J, Boivin S, Szabo P, Friedland RP, Ramsland PA, Edmundson AB, Weksler ME, Paul S. Autoantibody-catalyzed hydrolysis of amyloid beta peptide. J Biol Chem. 2008;283:4714–4722. doi: 10.1074/jbc.M707983200. [DOI] [PubMed] [Google Scholar]
- Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002;37:943–948. doi: 10.1016/s0531-5565(02)00029-3. [DOI] [PubMed] [Google Scholar]