

A novel Drosophila-based approach resulted in a significant response by a patient with KRAS-mutant colorectal cancer.
Abstract
Colorectal cancer remains a leading source of cancer mortality worldwide. Initial response is often followed by emergent resistance that is poorly responsive to targeted therapies, reflecting currently undruggable cancer drivers such as KRAS and overall genomic complexity. Here, we report a novel approach to developing a personalized therapy for a patient with treatment-resistant metastatic KRAS-mutant colorectal cancer. An extensive genomic analysis of the tumor’s genomic landscape identified nine key drivers. A transgenic model that altered orthologs of these nine genes in the Drosophila hindgut was developed; a robotics-based screen using this platform identified trametinib plus zoledronate as a candidate treatment combination. Treating the patient led to a significant response: Target and nontarget lesions displayed a strong partial response and remained stable for 11 months. By addressing a disease’s genomic complexity, this personalized approach may provide an alternative treatment option for recalcitrant disease such as KRAS-mutant colorectal cancer.
INTRODUCTION
Colorectal cancer (CRC) remains the second leading cause of cancer mortality in the United States. Current standard of care includes surgery and 5-fluorouracil (5-FU)–based chemotherapy combinations such as FOLFIRI (5-FU/leucovorin/irinotecan) and FOLFOX (5-FU/leucovorin/oxaliplatin); recalcitrant or recurrent disease is then treated with one of several targeted therapies. Despite an increasing number of therapeutic options for patients with CRC, those diagnosed with metastatic disease (mCRC) have a 5-year survival rate of 11%. Furthermore, toxicities from targeted therapies are substantial: For example, many approved therapies inhibit FLT1, which is closely associated with kidney toxicity and hypertension (1–3).
Tumors with oncogenic RAS isoforms (“RAS-mutant” tumors) represent a particular challenge. An estimated 30 to 50% of CRC patient tumors include an oncogenic KRAS mutation; an additional ~6% of colorectal tumors contain mutations in NRAS or HRAS (4, 5). Several studies, although not all, have associated RAS-mutant tumors with more aggressive metastatic disease and reduced survival (6–10). RAS-mutant CRC affects more than 60,000 patients annually, leading to more than 20,000 cancer deaths in the United States alone (11, 12). More broadly, despite recent advances (12–14), therapeutic options for targeting RAS-dependent cancers remain limited (15–17).
U.S. Food and Drug Administration (FDA)–approved therapies that target the RAS pathway have shown limited efficacy in patients with KRAS-mutant mCRC. For example, the FDA-approved kinase inhibitor regorafenib (Stivarga) provides limited mCRC patient survival benefit (1.4 to 2.5 months), with substantial and highly penetrant adverse events (3). Other FDA-approved RAS pathway inhibitors such as trametinib (Mekinist) and immune checkpoint inhibitors have failed in CRC clinical trials for microsatellite stable disease (18, 19), leading to new interest in combinations of targeted therapies (20, 21). Patients with KRAS-mutant mCRC, typically presenting with right-sided tumors that are more aggressive on recurrence, are resistant to or even harmed by therapies targeting epidermal growth factor receptor (EGFR), and testing for RAS mutations are standard exclusionary criteria (22–26). Overall, patients with KRAS-mutant mCRC that develop recurrent disease have few good therapeutic options.
Our previous work demonstrated that complex RAS-mutant Drosophila CRC models exhibit broad aspects of transformation, including hyperproliferation, multilayering, altered apoptosis and senescence, and dissemination of transformed cells to distant sites (27). Here, we present the results of treating a patient with KRAS-mutant colorectal adenocarcinoma using Drosophila as a personalized cancer drug discovery platform (Fig. 1A). Before our treatment, the patient had shown initial partial response to chemotherapy and then tumor progression. We developed a “personalized Drosophila model” that altered orthologs of nine genes identified in the patient’s tumor. Robotics-based high-throughput screening was then used to identify a novel combination, trametinib plus zoledronate, that improved survival of the personalized Drosophila model. Treating the patient with trametinib plus zoledronate led to a partial response: Target lesions were reduced by 45% and remained stable for several months; nontarget lesions showed a similar response. Eventually, new lesions emerged that were nonresponsive to trametinib/zoledronate therapy. Given the typically poor third-line response observed in patients with KRAS-mutant mCRC, this work suggests that personalized screening using a model organism platform merits further investigation. Our results also demonstrate that drug combinations can provide a useful therapeutic alternative to single-agent targeted therapies in KRAS-mutant mCRC.
Fig. 1. Overview and construction of Drosophila patient model.
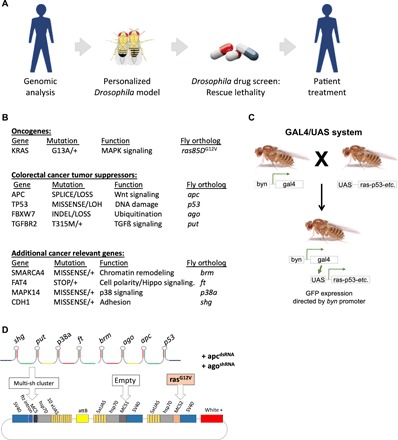
(A) An outline of our approach. First, a comprehensive genomic analysis of the patient’s tumor and normal DNA [copy number, whole-exome sequencing (WES), and targeted HotSpot panel] was performed. Then, a personalized Drosophila model that captures a portion of the patient’s tumor’s genomic complexity was generated by targeting each Drosophila ortholog specifically in the Drosophila hindgut. After the model was validated, a high-throughput “rescue from lethality” drug screen was performed on FDA-approved drugs as single agents and in combination. Findings were then presented to a multidisciplinary tumor board. A personalized treatment plan based on the multidisciplinary tumor board’s recommendation was prepared and institutional review board–approved, followed by patient treatment. (B) Patient’s genomic landscape: Genes altered in the patient’s tumor, their functions, and Drosophila orthologs are indicated. LOH, copy number neutral loss of heterozygosity. MAPK, mitogen-activated protein kinase. (C) GAL4/UAS system used for targeted genetic manipulations in Drosophila. Transgenes targeting nine genes (ras85DG12V, etc.) were cloned downstream of a GAL4-responsive UAS promoter and transgenic flies generated. Transgene expression was then induced in a tissue-specific manner by crossing transgenic flies to byn-gal4 for hindgut epithelium and to tubulin-gal4 for ubiquitous expression. GFP, green fluorescent protein. (D) Personalized construct generated for the patient, targeting nine genes. This construct expressed a GAL4-inducible (i) UAS-ras85DG12V transgene and (ii) synthetic eight-hairpin cluster targeting the Drosophila orthologs of the eight tumor suppressor genes. After transgenic flies were generated, transgenic constructs UAS-agoRNAi and UAS-apcRNAi were genetically introduced by standard genetic crosses to increase overall ago and apc knockdown.
RESULTS
Clinical synopsis and treatment history
A 53-year-old man without previous comorbidities was found to have a large partially obstructing mass of the distal sigmoid colon. A biopsy confirmed the diagnosis of colorectal adenocarcinoma. Intraoperatively, he was noted to have synchronous liver metastases. A laparoscopic lower anterior resection was performed with creation of a sigmoid end colostomy. Surgical pathology identified a moderately differentiated pT3N2a adenocarcinoma of the rectosigmoid colon with proficient DNA mismatch repair protein expression, lymphovascular and perineural invasion, and negative margins. A targeted next-generation sequencing panel identified a KRAS(G13A) mutation; BRAF, NRAS, and PIK3CA were wild type.
Six weeks after surgery, the patient initiated systemic therapy with FOLFOX and bevacizumab. Serum carcinoembryonic antigen (CEA), which was 9.6 ng/ml on the day of surgery, decreased to 7.1 ng/ml at the start of chemotherapy. After 6 months of therapy, his CEA normalized, and a repeat computed tomography (CT) of the chest, abdomen, and pelvis showed a partial response by the liver metastases. He underwent a segment 8 hepatectomy, en bloc diaphragm resection, and colostomy reversal, followed by 3 months of postoperative FOLFOX.
On a repeat CT 1 month later, multiple new lung nodules and left superior mediastinal adenopathy were identified. Serum CEA was normal at 1.8 ng/ml. The patient resumed chemotherapy with FOLFIRI and bevacizumab for an additional 6 months. Serial imaging performed during the chemotherapy regimen initially showed a slight decrease in size of the pulmonary nodules. Subsequent imaging 3 months later showed a mixed response: slight interval progression of some pulmonary nodules and stability in others. There was also an increase in scant subcentimeter retroperitoneal lymphadenopathy and a more prominent left supraclavicular lymph node. A subsequent CT 2 months later revealed progression of lung metastases plus new left axillary, subpectoral, and mediastinal adenopathy. Previously noted retroperitoneal and pelvic adenopathy had increased. Serum CEA was 2.3 ng/ml.
Anticipating possible emergence of resistant disease, we initiated an experimental personalized treatment platform (Fig. 1A) while the patient received chemotherapy. Given the limited expected efficacy of available third-line options should FOLFIRI/bevacizumab fail (28–32), the patient elected to enroll in the experimental study 2 months after the colostomy.
Genomic analysis and mutant selection
As a first step toward developing a personalized Drosophila model, we carried out a comprehensive analysis of the patient’s tumor genomic landscape (Fig. 1A). To this end, we extracted DNA from the primary tumor specimen and patient’s blood (patient-specific normal control) and performed whole-exome sequencing (WES), targeted HotSpot panel, and copy number analysis (CNA) assays. The patient’s tumor exhibited a large number of variants: 132 somatic and 965 rare germline variants.
To build a patient-specific Drosophila model, we focused our analysis on mutations in recurrently mutated cancer driver genes and genes that regulate cancer-relevant signaling pathways and cellular processes. In addition to confirming the oncogenic KRAS(G13A) mutation, WES analysis of the patient’s tumor showed biallelic loss of the well-established CRC drivers APC, TP53, and FBXW7 and a germline heterozygous missense mutation in TGFBR2 (Fig. 1B). We also identified heterozygous somatic mutations in SMARCA4, FAT4, and MAPK14 and a heterozygous germline mutation in CDH1 (Fig. 1B). While these genes are not frequently mutated in tumors, they regulate important cancer-relevant biological processes including chromatin remodeling, cell polarity, and adhesion.
CNA identified a large number of alterations that included hundreds of genes. Using immunohistochemistry to assess gene expression levels, we focused our analysis on copy number alterations recurrently observed in colon tumors (33). The patient’s tumor included a copy gain event in a region that encompassed receptor tyrosine kinases FLT1 and FLT3. However, immunohistochemistry analysis of the tumor specimen did not reveal an increase in the levels of either protein, and they were not included in the Drosophila model.
Model building and validation
To build a Drosophila model that reflected the patient’s specific genomic complexity, we altered Drosophila orthologs of the nine genes identified in our genomic analysis (Fig. 1B) in the fly’s hindgut using the GAL4/UAS expression system (Fig. 1C) (34). Specifically, we cloned transgenes downstream of UAS, a yeast-derived promoter that is responsive specifically to the yeast GAL4 transcription factor. To target transgenes to the hindgut, we then crossed together (i) transgenic flies containing a stable genomic insertion of UAS-transgenes with (ii) flies directing GAL4 expression in the hindgut (byn-GAL4; Fig. 1C). We included a UAS-GFP reporter to visualize transformed tissue.
We modified a previously developed transformation vector (35) to contain three UAS cloning cassettes (Fig. 1D). Oncogenic Drosophila ras85D(G12V) was placed under the control of one UAS promoter. To simultaneously reduce activity in eight tumor suppressors, we generated synthetic clusters of sequences encoding short hairpin RNAs (shRNAs) targeting each gene; we modeled the sequences on endogenous microRNA clusters found in Drosophila and human genomes (see Materials and Methods and table S1). For genes biallelically inactivated in the patient—APC, TP53, and FBXW7—we selected hairpins predicted to provide strong knockdown; for the remaining genes with heterozygous variants, hairpins predicted to provide moderate knockdown were used. We assembled hairpin sequences into a single oligonucleotide and placed them under the control of a separate UAS promoter (see Materials and Methods and Fig. 1D). Two stable transgenic Drosophila lines were generated to assess different hairpin predictions: 006.1 and 006.2 each with ras85D(G12V) but a different set of shRNA-based hairpin oligonucleotides targeting the same eight genes. After transgenic lines were established, we also introduced additional RNA interference (RNAi) constructs for apc and ago by standard genetic crosses to ensure strong knockdown. While both models showed effective knockdown of most target genes, model 006.1 showed a more favorable knockdown profile (fig. S1). We also used hindgut lysates to analyze knockdown of Shg and P53 proteins using commercially available antibodies; these results were consistent with our quantitative polymerase chain reaction (qPCR) data (fig. S2, A and B). Expression of the transgenes in the larval hindgut with the byn-GAL4 driver led to substantial expansion of the anterior portion of the hindgut (Fig. 2, A and B), reflecting aspects of transformation, as we previously published (27). We therefore selected model 006.1 for drug screening.
Fig. 2. Validating and screening Drosophila patient model.
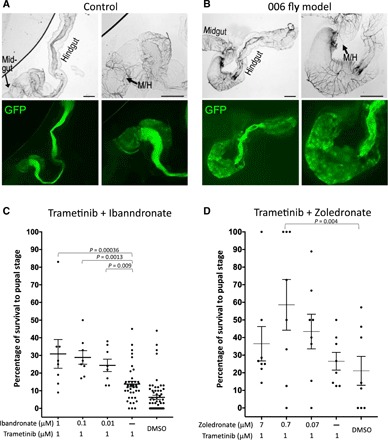
(A) Expressing byn > GFP in control animals highlighted the hindgut in bright-field (top panels) and expression of the byn-GAL4 driver specific to the hindgut (bottom panels). Microscope magnifications (5× and 10×) are shown. (B) Expressing the 006.1 transgene set in the hindgut led to strong expansion of the anterior hindgut. The midgut/hindgut (M/H) boundaries are indicated; the dark regions in the 006 bright-field images likely reflect cell death. Image contrast was enhanced equally using Preview software for clarity. Scale bars, 100 μm. (C and D) Trametinib in combination with ibandronate or zoledronate rescued the lethality observed by the patient’s personalized Drosophila model. Concentrations indicate final food concentrations. Each data point represents a replicate with 10 to 15 experimental and 20 to 30 control animals. Raw numbers are provided in table S2C. Error bars indicate SEM. DMSO, dimethyl sulfoxide.
Drug screening
We previously demonstrated that rescue from lethality can be used as a quantitative phenotypic readout for high-throughput drug screening. Targeting transgene expression to the developing hindgut epithelium can lead to broad transformation in the epithelium and organismal lethality; this lethality can be rescued by drugs mixed with the fly’s food. A drug’s ability to rescue Drosophila cancer models to pupariation or adulthood indicates that the drug is both effective and nontoxic.
For drug screening, we assembled a custom “focused FDA library” consisting of 121 drugs FDA-approved for (i) cancer, (ii) noncancer indications with reported antitumor effects, and (iii) noncancer indications with cancer-relevant targets. The first round of screening did not identify any drugs that provided significantly improved survival (see table S2A for raw data). This result is consistent with our previous work demonstrating that genetically complex cancer models are often resistant to single agents and may require drug combinations to be effective at addressing genetic complexity (27).
Given the presence of oncogenic RAS in the patient’s tumor, we focused on identifying effective drug combinations that included the MEK (mitogen activated protein kinase kinase) inhibitor trametinib. Trametinib is strongly effective against oncogenic RAS alone but not against highly multigenic CRC Drosophila models. A combination screen focusing on noncancer drugs in a focused FDA library was screened in the presence of trametinib; the bisphosphonate class drug ibandronate was identified as strongly effective in combination with trametinib (see table S2B for raw data). These results were confirmed in an independent experiment in which trametinib was tested in combination with three different doses of ibandronate (Fig. 2C and table S2C).
Bisphosphonates have been previously reported to have antitumor effects as single agents and in combination with different tyrosine kinase inhibitors. We therefore tested two additional bisphosphonates, pamidronate and zoledronate, in combination with trametinib. Zoledronate also proved to be an effective partner for trametinib (Fig. 2D and table S2C). Ibandronate synergized with trametinib at a wider range of doses and provided a more significant rescue than zoledronate. The reason for this difference is not clear; for example, it may reflect subtle toxicity that was not apparent in controls, differences in off-target activities that lead to toxicity at higher doses, or differences in drug stability/metabolism in Drosophila. A multidisciplinary tumor board that included pharmacists, oncologists, and clinical trial experts who reviewed our findings noted that oral ibandronate can cause esophagitis (36); intravenous administration of bisphosphonates would avoid esophagitis. Given the data supporting zoledronate as a potential anticancer agent (37–40), the tumor board recommended a combination of intravenous zoledronate and oral trametinib for the patient.
We also explored drug response at the molecular and phenotypic level in the patient model’s hindgut (Fig. 3). We first evaluated RAS/MAPK signaling pathway output using dually phosphorylated extracellular signal–regulated kinase (dpERK) in hindgut lysates from drug-treated experimental animals. Lysates from the patient model demonstrated significantly increased dpERK levels compared to control animals (Fig. 3A and fig. S2C). Trametinib significantly reduced dpERK levels in the patient model, while zoledronate had no detectable effect on MAPK signaling output. Combining trametinib with zoledronate led to a stronger reduction in dpERK levels than trametinib alone, indicating that zoledronate enhances the ability of trametinib to inhibit MAPK signaling. Regarding phenotypic changes, we found a statistically significant reduction in the expansion of the anterior portion of the hindgut in the patient model in response to each single agent; the trametinib/zoledronate combination directed a stronger rescue than either drug alone (Fig. 3, B and C). Notably, the observation that zoledronate (i) partially rescued the anterior portion of the hindgut but (ii) had no effect on MAPK signaling output (Fig. 3A) suggests a complex, pleiotropic mechanism of action for the combination.
Fig. 3. Secondary assays of drug response.
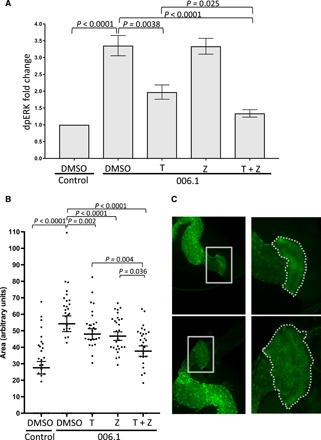
(A) Western blot analysis of MAPK signaling pathway output from control and drug-treated hindgut lysates using dpERK as a readout. Quantification represents two independent experiments with different sets of biological replicates. Each experiment was performed in triplicate with 10 hindguts per biological replicate (gel images are shown in fig. S2C). (B and C) Analysis of the expansion of the anterior hindgut in control and drug-treated animals. (B) Quantification of the anterior region of the hindgut. Data points indicate individual hindguts. (C) Two images representing the high and low ends of the size distribution observed in the assay. Quantified region of the hindgut is outlined by white dashed lines. T, 1 μM trametinib; Z, 0.7 μM zoledronate in the food. Statistical significance in (A) and (B) was determined using multiple t tests with Holm-Sidak correction for multiple hypotheses.
Patient treatment
Before beginning third-line therapy, the patient underwent an ophthalmologic exam and a cardiac multigated acquisition scan; both were in the normal range. A pretreatment baseline CT reported target lesions, including left axillary and para-aortic, aortocaval, and right external iliac adenopathy and a left upper lobe pulmonary nodule. The sum of the longest diameter for all target lesions was 74 mm. Pretreatment baseline CEA was 2.2 ng/ml.
Patient treatment was initiated with oral trametinib (2 mg daily) plus zoledronate (4 mg intravenously every 4 weeks). Within 2 weeks of starting therapy, the patient developed a grade 2 acneiform rash on his face, neck, and upper back, which was attributed to trametinib. The rashes progressed, and the patient was prescribed minocycline, topical clindamycin, and antihistamines. Despite these measures, the rash progressed to grade 3 in severity and the patient developed facial swelling without dyspnea or dysphonia by week 4 of therapy. Trametinib was suspended, and the patient was referred to a dermatologist, who confirmed the diagnosis of drug-induced dermatitis. His symptoms improved with the addition of prednisone. Zoledronate infusions continued every 4 weeks.
A CT of the chest, abdomen, and pelvis, performed 8 weeks from the initial start date of therapy, revealed that the sum of the target lesion diameters had decreased to 41 mm, representing a 45% decrease from baseline and partial response to treatment based on RECIST (Response Evaluation Criteria in Solid Tumors) 1.1 (Fig. 4, A and B, and table S3). The patient subsequently resumed trametinib a week later at a reduced dose of 0.5 mg every other day. Serum CEA at the time was 2.5 ng/ml. He tolerated the modified dose of trametinib well, except for grade 1 pruritis. A repeat CT scan performed 5 weeks after resuming trametinib demonstrated a sustained partial response in target lesions (sum of diameters, 41 mm). New peripancreatic and periportal adenopathy emerged, measuring 16 mm by 15 mm and 15 mm by 53 mm, respectively. On the basis of these results, the dose of trametinib was increased to 0.5 mg daily. Twelve weeks after resuming trametinib, another CT was performed, showing a 10% increase in the sum of target lesions (now 45 mm) from nadir but still 39% below baseline, indicative of a sustained partial response. The two new nontarget lesions were also slightly larger (19 mm by 16 mm and 21 mm by 65 mm), but there were no new lesions.
Fig. 4. Patient response.
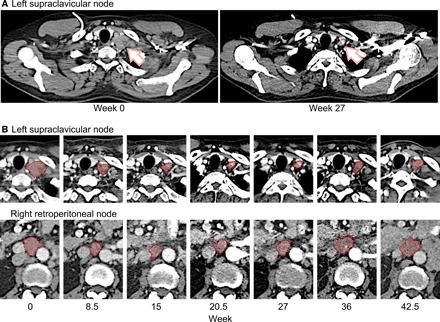
(A) Patient scans before treatment and 27 weeks after treatment. Arrow indicates an example of lesion in left supraclavicular node. (B) Two examples of target lesion shrinkage at indicated time points are highlighted by pink shading plus red dashed outline; the top panels provide detail to (A).
Given the tolerance of 0.5-mg daily trametinib without any new cutaneous toxicity, the dose was gradually increased to 1 mg daily. A further dose increase to trametinib (1.5 mg) was attempted, but the patient developed a pruritic rash after 1 week, causing the dose to be reduced back to 1 mg daily. A CT performed 18 weeks after resuming trametinib showed that the sum of target lesions was now 46 mm, constituting a 12% increase from nadir but still 38% lower than baseline measurements. In addition, the peripancreatic nodes had increased to 28 mm by 26 mm and the periportal nodes to 27 mm by 85 mm.
Following the CT scan, trametinib was held while a 10-day course of stereotactic radiation was initiated to the abdominal adenopathy. Trametinib was resumed 11 days later at a dose of 1 mg daily. Serum CEA was 3.0 ng/ml. At this dose of trametinib, the patient occasionally experienced mild exacerbations of the drug rash and/or skin dryness involving his face or arms, but these reactions remained grade 1 in severity. Although he still maintained a good performance status [ECOG 1 (Eastern Cooperative Oncology Group 1)], he reported increasing fatigue, occasional postprandial nausea without vomiting, and abdominal bloating. He stopped trametinib on his own for 4 days because of these symptoms and then resumed. Approximately 5 weeks after completing radiation, a new CT demonstrated that the sum of the target lesions (now 62 mm) had increased by 51% from nadir, and the total sum was now 16% below baseline. New nontarget lesions had also appeared: a left perirenal soft tissue nodule measuring 32 mm by 23 mm and an aortopulmonary window nodule measuring 15 mm by 18 mm. The irradiated periportal nodes were stable, but the peripancreatic nodes were slightly larger, measuring 28 mm by 26 mm. At this juncture, the decision was made to discontinue study therapy and switch to fourth-line therapy with regorafenib.
Overall, the patient was treated with trametinib plus zoledronate for approximately 11 months, exhibiting a maximum of 45% reduction in tumor burden. The primary toxicity was a severe rash controlled with antibiotics and antihistamines, permitting him to resume trametinib. The patient was eventually removed from treatment primarily because of emergence of previously unobserved lesions; the full genomic landscape of these lesions is unknown. We had the opportunity to explore the mutational profile of the treatment-resistant peripancreatic and periportal nodes using a specimen obtained from an endoscopic ultrasound-guided biopsy. The biopsy provided sufficient material for a targeted, high-coverage analysis using Oncomine Comprehensive Panel version 2. No new mutations were reported on the panel, ruling out most druggable targets and at least many of the mutations known to promote resistance. A similar analysis using circulating cell-free DNA identified a similar profile and also did not identify a specific resistance mechanism.
DISCUSSION
Here, we report a novel treatment approach for a patient with advanced KRAS-mutant mCRC. Before our personalized therapy, the patient had received but eventually failed multiple courses of chemotherapy. Anticipated response for this class of patients to third-line targeted therapy or chemotherapy is poor, with marginal improvement in overall survival (41–43). Instead, on the basis of extensive genomic analysis of the tumor, we developed a personalized Drosophila model as a whole-animal screening platform. A combination of trametinib plus a bisphosphonate reduced animal lethality. Treating the patient with trametinib/zoledronate led to a progression-free interval of 3 months overall but a partial response of target lesions lasting 8 months, including a maximum of 45% reduction in target lesions.
Our goal in creating this model was to capture tumor complexity: Drugs and lead compounds that have shown success in genetically less complex KRAS-mutant tumor models, including models that contain one to two additional targeted genes, have typically performed poorly in clinical trials. Genetically complex models can also identify candidate therapeutics that act through multiple targets, providing an opportunity for a “network approach” to address tumor complexity. Notably, regorafenib is approved as a multikinase inhibitor (44) that can extend survival of patients with mCRC by several weeks.
The model described here is one of the most genetically complex transgenic whole-animal disease models described to date. Still, we were able to capture only a small subset of genomic alterations observed in the patient’s tumor. Using functional prediction algorithms to prioritize those variants that are most likely to deleteriously affect protein function eliminated a substantial number of variants most likely to be passenger events. We then focused on variants in genes identified as recurrently mutated drivers of cancer and those with clear cancer-relevant functions; however, our exclusion criteria are necessarily incomplete, and a large number of candidate variants remained. Further expanding the multigenic platform technology described here would provide an opportunity to generate even more sophisticated models that can better capture the genomic complexity of tumor genomic landscapes.
Most tumor genome landscapes contain a combination of heterozygous and homozygous loss of genes. Knockdown of a large number of genes to the desired level is a technically challenging issue. Use of hairpin sequences based on their predicted efficacy introduces a degree of uncertainty regarding how well they would perform in vivo, particularly in these genetically complex backgrounds. Generating two models each with a different set of hairpins targeting the same genes has been a useful approach to increasing the likelihood of success. For instance, we did not find any significant knockdown of ft in model 006.1 and ft or shg in model 006.2. The knockdown profiles of the models would be further optimized by replacing the ineffective hairpins with improved version. However, building and validating additional models were not feasible in the time frame of the clinical study with our current approach.
Trametinib is a potent RAS pathway inhibitor, and its clinical failure to slow progression of most KRAS-mutant solid tumor types has been unexpected. Here, we demonstrate that trametinib can act on a nine-hit Drosophila model when dosed in combination with a bisphosphonate; this effectiveness translated into a partial response by the patient. The nature of zoledronate’s synergy with trametinib is not clear. Zoledronate has been previously demonstrated to inhibit RAS pathway signaling through direct inhibition of EGFR activity and inhibition of prenylation (37–40). In addition, zoledronate and related bisphosphonates are associated with strong protection against CRC: Women who took bisphosphonates to protect from excess bone resorption—patients with breast cancer and postmenopausal women—exhibited a 40 to 59% reduced incidence of CRC, leading to speculation of its use as a therapeutic for patients with CRC (45, 46). Whether any of these activities are related to zoledronate’s ability to synergize with trametinib is unclear.
Preclinical mammalian models, including patient-derived xenografts (PDXs) and organoid cultures, are key components of the drug discovery pipeline. We explored the possibility of incorporating these models into our pipeline. However, success rates for growing patient-derived tumor tissue in mice or in three-dimensional culture are generally low and highly variable, and the cost and time frame are generally prohibitive. Furthermore, these approaches require live tumor specimen from patients; our Drosophila pipeline on the other hand can be applied to any patient with isolated tumor tissue, including fixed [formalin-fixed, paraffin-embedded (FFPE)] tissue. As both Drosophila and mammalian technologies advance, an integrated approach that combines Drosophila and organoid/PDX studies may prove feasible, with the latter initiated in parallel to generate sufficient models to test the most promising candidates that emerge from the Drosophila patient model.
In this study, we present a new, highly personalized approach to treating CRC that can be adapted for use in other tumor types and other diseases that can be modeled in Drosophila. The advantage of Drosophila is the ability to alter a large number of genes in a single tissue and screen a large number of drugs and drug combinations in a whole-animal setting with a simple readout for efficacy and toxicity. Identifying an effective, unique drug combination, trametinib plus zoledronate, emphasizes the potential for moderately high-throughput screens that can be accomplished in a time frame that is useful for treating a patient. This approach may prove especially useful in tumors with challenging profiles, for example, KRAS-mutant tumor types. Outcome data from additional patients to evaluate success rates will be necessary to determine the feasibility of integrating this approach into clinical practice.
MATERIALS AND METHODS
Enrollment
The study was regulated by three separate protocols approved by the Mount Sinai Institutional Review Board: (i) a biorepository protocol that regulated inventory and processing of tumor and patient-specific normal control (whole blood in EDTA) specimens; (ii) a molecular analysis protocol that included genomic analysis, model building/validation, and drug screening pipelines; and (iii) a treatment protocol including a personalized treatment consent for the recommended therapy after the results were reviewed and approved by a multidisciplinary tumor board.
Sample processing and genome assays
Genomic analysis was performed on (i) FFPE primary tumor specimen and (ii) whole blood collected at the time of consent to serve as a patient-matched normal control. Detailed protocols for sample processing, next-generation sequencing assays, and data integration were described previously (47).
Variant selection and validation
WES of tumor and blood DNA identified 132 somatic and 965 rare germline variants. We focused our analysis on genes recurrently mutated in cancers including colorectal and those involved in cancer-relevant signaling pathways and cellular processes. To determine the likelihood that observed missense variants are deleterious (e.g., negatively affect protein function), two functional prediction algorithms were used: database for functional prediction and annotation of all potential non-synonymous single nucleotide variants in the human genome (dbNSFP) and Combined Annotation Dependent Depletion (CADD) (48, 49). Variants predicted to be benign (e.g., unlikely to affect protein function) by both methods were eliminated. The remaining variants were first manually reviewed by examining the raw sequence reads to exclude false positives from automated WES variant calling algorithms. In addition, each variant was independently assessed by a Pacific Biosciences sequencing platform for orthogonal validation using targeted amplicon circular consensus sequencing, as previously described (47, 50). Using this method, we confirmed the presence of each variant except for SMARCA4, which was inconclusive strictly because of technical reasons and was included in our final selection of variants for building the Drosophila model.
Immunohistochemical analysis
To confirm the findings of the gene expression analysis, we performed immunohistochemical assays on 5-μm FFPE primary tumor sections for both FLT1 (1:200; catalog no. ab9540, Abcam) and FLT3 (1:100; catalog no. ab150599, Abcam) with appropriate antibody controls (51). Immunohistochemical scoring was performed semiquantitatively with an H-score (i.e., “histo” score) with intensity of staining ranging from 0 to 3+ multiplied by the percentage of positive expressing cells with a final score ranging from 0 to 300. The sample was considered overexpressed on the basis of a discriminating threshold greater than or equal to an H-score of 150.
Model building
Patient-specific models were generated using a multigenic UAS vector modified from a previously reported Drosophila transformation vector (35). The modified vector contains three UAS cassettes, each with their own UAS promoter, SV40 terminator sequences, and unique multiple cloning sites (MCSs). Oncogenic Drosophila ras85D(G12V) was PCR-amplified from a previously validated transgenic construct using primers designed to append restriction sites for enzymes Fse I and Pac I to the 5′ and 3′ ends of the product and cloned into one of the MCSs (Fig. 1D).
Short hairpins for gene knockdown were selected using DSIR, a publicly available tool for designing shRNAs (52) following previously established hairpin selection criteria for Drosophila (35). Individual hairpins were separated by spacer sequences found 5′ to well-expressed Drosophila microRNAs. To help ensure that a personalized model with a desired knockdown profile was obtained, two independent clusters that target the same eight genes using different hairpin clusters were generated (006.1 and 006.2). Hairpin, spacer, and final cluster sequences are provided in table S1.
Hairpin clusters were generated by gene synthesis (GENEWIZ). Sequence-confirmed products were then cloned into the multigenic vector using Xba I (5′) and Not I (3′). Transgenic flies were generated by PhiC31-mediated targeted integration into the attp40 site on the second chromosome (BestGene) (53). To ensure strong knockdown for biallelically inactivated genes, previously validated transgenic RNAi knockdown lines for apc (VDRC) and ago (TRIP) were introduced by standard genetic crosses after transgenic flies were generated.
Model validation
Personalized models were validated by qPCR and Western blots. Experimental and control animals for validation were generated by crossing both models (006.1 and 006.2) to a tub-gal4 tub-gal80ts line to transiently and ubiquitously induce transgene expression for 3 days. Whole larvae with the genotypes (i) tub-gal4 tub-gal80ts > UAS-006.1; UAS agoRNAi UAS-apcRNAi; (ii) tub-gal4 tub-gal80ts > UAS-006.2; UAS agoRNAi UAS-apcRNAi; and (iii) tub-gal4 tub-gal80ts/+ as controls were collected (three biological replicates per genotype and six larvae per replicate).
For protein extraction, larvae were homogenized using a motorized pestle in ice-cold 100-μl radioimmunoprecipitation assay buffer (Sigma-Aldrich) with the Phosphatase Inhibitor Cocktail Set III (EMD Millipore) and Protease Inhibitor Cocktail (Roche). Lysates were centrifuged at 4°C for 10 min at 13,000 rpm, supernatants (70 μl) were transferred to a fresh tube, and 25 μl of 4× NuPAGE LDS Sample Buffer and 10 μl of NuPAGE 10× Reducing Agent (Invitrogen) were added. After a brief spin down, samples were boiled for 10 min, briefly spun down, and centrifuged at 4°C for 5 min at 13,000 rpm. Supernatant (80 μl) was transferred to new tubes and stored at −80°C. Western blots were performed as described previously (27) using the following primary and secondary antibodies: mouse anti-p53 [1:1000; Developmental Studies Hybridoma Bank (DSHB) Dmp53-H3], mouse anti-dpERK (1:1000; Sigma-Aldrich), mouse anti-syntaxin as loading control (1:1000; DSHB), and goat anti-mouse horseradish peroxidase secondary (1:10,000).
Larvae collected for RNA extraction were stored in 300-μl RNAlater (Life Technologies). RNA extraction was performed using the RNeasy Plus Kit with the RNase-free DNase Set for on-column DNA digestion (Qiagen) following the manufacturer’s instructions. RNA concentration was measured using Qubit. For qPCR analysis, 1 μg of RNA was converted to complementary DNA (cDNA) using the High-Capacity RNA-to-cDNA Kit (Life Technologies), and qPCR was performed using the PerfeCTa SYBR Green FastMix for IQ (VWR Scientific). A panel of four housekeeping genes (rpl32, cyp33, gapdh, and sdha) were first assayed to identify the best candidate, and cyp33 was selected as providing the most robust and consistent results. qPCR data were analyzed using the ΔΔC(t) method (54).
Model imaging
Whole guts were dissected from third instar byn-GAL4 tubulin-GAL80ts UAS-GFP/UAS-transgene larvae that were induced at 25°C for 4 days. Control and experimental animals were fixed with 4% paraformaldehyde, washed, and mounted. Images were taken at 5× (low magnification) and 10× in Fig. 2. Quantification of the anterior portions of hindguts from drug-treated animals was performed with ImageJ software using images captured at 10× magnification.
Drug screening
Drugs in our custom focused FDA library were purchased individually as powder from the following commercial sources: Selleck Chemicals, LC Laboratories, Tocris Bioscience, and MedChemExpress. Drugs were dissolved in 100% dimethyl sulfoxide (DMSO) or water on the basis of the solubility information provided by the manufacturers. For each drug, the highest possible dose (based on solubility) that did not lead to detectable toxicity on wild-type animals was selected for screening, and drugs were aliquoted into 384-well plates.
The library was screened at a single dose for each drug along with DMSO controls (eight replicates per condition) by diluting each drug in the library 1:1000, which brings the DMSO concentration in the food to 0.1%. Drug-food mixtures were made using an automated liquid handling workstation (PerkinElmer) by adding 0.7 μl of drug into round-bottom test tubes (12 mm by 75 mm; Sarstedt), followed by 700 μl of semidefined Drosophila medium (recipe obtained from the Bloomington Drosophila Stock Center) and mixing by pipetting.
After food was solidified, a mixture of experimental and control embryos (at a ratio of 1:2 based on expected Mendelian ratios) was aliquoted into each drug/food tube (15 μl per tube). Embryo suspensions were generated using the buffer that we designed to minimize embryo clumping and settling (15% glycerol, 1% bovine serum albumin, and 0.1% Tween 20 in water). Embryos for drug screening were generated from the following cross in cages: w/Y; UAS-006.1; UAS agoRNAi UAS-apcRNAi/Stub-gal80-T X w UAS-dicer2; +; byn-gal4 UAS-GFP tub-gal80ts/TM6, Hu, Tb. Embryos were obtained from each cage for four to five consecutive days by providing daily a fresh apple juice plate with yeast paste. Egg lays were performed at 22°C to minimize transgene expression during embryogenesis to prevent embryonic defects or lethality that could not potentially be rescued by drug feeding. After embryos were aliquoted, drug tubes were transferred to 25°C to induce transgene expression. After 2 weeks, the number of surviving experimental pupae (EP) was counted in each tube. Drugs that showed significantly higher numbers of experimental survivors compared to vehicle controls (multiple Student’s t tests corrected for multiple comparisons using the Holm-Sidak method, PRISM software) were considered hits.
Drug combination screens were performed by combining trametinib at its screening dose (1 μM in the food) with each drug in the library and mixing with Drosophila medium (eight replicates for each combination). DMSO and trametinib alone served as controls. Drug combinations identified as candidate hits were retested in an independent experiment by combining the screening dose of trametinib with three different doses of each partner drug (original screening dose and 10 and 1% of screening dose). Experimental and control pupae (EP and CP, respectively) were counted for each tube, and percentage of survival to pupal stage was calculated using the formula [(EP × 2/CP) × 100]. Statistical analysis was performed as described above.
Supplementary Material
Acknowledgments
We acknowledge the Cagan Laboratory for important discussions during the progression of the study and the Genomics Core Facility at the Icahn Institute and Department of Genetics and Genomic Sciences for technical support. Computational resources and staff expertise were provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai. We acknowledge support from the Novartis Patient Assistance Program. A patent application has been filed related to this work. Funding: This work was supported by funds from the Dean’s Office and by U54OD020353 from the NIH. Author contributions: E.B., P.S., C.H., N.G., W.J.Y., D.M., D.L., and R.L.C. were responsible for developing and screening the Drosophila models. E.B., A.V.U., A.G.T., Y.A., R.C., C.Y.L., R.S., E.W., M.D., E.E.S., and R.L.C. were responsible for the genomics and tissue analyses. C.A., I.S., H.R., A.S.M., P.T., A.B., M.S., S.K., K.Y., K.M., and M.R.P. were responsible for patient clinical interactions. E.B., C.A., K.M., M.R.P., and R.L.C. had the primary roles in writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data and accession numbers needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav6528/DC1
Fig. S1. Validation of patient’s personalized Drosophila model (see Materials and Methods for details).
Fig. S2. Validation of patient’s personalized Drosophila model and drug response from hindgut lysates (see Materials and Methods for details).
Table S1. Multi-hairpin cluster sequences for patient’s personalized model.
Table S2. Raw drug screening and testing data.
Table S3. Patient’s tumor measurements.
REFERENCES AND NOTES
- 1.Izzedine H., Rixe O., Billemont B., Baumelou A., Deray G., Angiogenesis inhibitor therapies: Focus on kidney toxicity and hypertension. Am. J. Kidney Dis. 50, 203–218 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Hayman S. R., Leung N., Grande J. P., Garovic V. D., VEGF inhibition, hypertension, and renal toxicity. Curr. Oncol. Rep. 14, 285–294 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Røed Skårderud M., Polk A., Kjeldgaard Vistisen K., Larsen F. O., Nielsen D. L., Efficacy and safety of regorafenib in the treatment of metastatic colorectal cancer: A systematic review. Cancer Treat. Rev. 62, 61–73 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Chang Y. Y., Lin P. C., Lin H. H., Lin J. K., Chen W. S., Jiang J. K., Yang S. H., Liang W. Y., Chang S. C., Mutation spectra of RAS gene family in colorectal cancer. Am. J. Surg. 212, 537–544.e3 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Valtorta E., Misale S., Sartore-Bianchi A., Nagtegaal I. D., Paraf F., Lauricella C., Dimartino V., Hobor S., Jacobs B., Ercolani C., Lamba S., Scala E., Veronese S., Laurent-Puig P., Siena S., Tejpar S., Mottolese M., Punt C. J. A., Gambacorta M., Bardelli A., Di Nicolantonio F., KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 133, 1259–1265 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Jones R. P., Sutton P. A., Evans J. P., Clifford R., McAvoy A., Lewis J., Rousseau A., Mountford R., McWhirter D., Malik H. Z., Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br. J. Cancer 116, 923–929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karagkounis G., Torbenson M. S., Daniel H. D., Azad N. S., Diaz L. A. Jr., Donehower R. C., Hirose K., Ahuja N., Pawlik T. M., Choti M. A., Incidence and prognostic impact of KRAS and BRAF mutation in patients undergoing liver surgery for colorectal metastases. Cancer 119, 4137–4144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H. S., Heo J. S., Lee J., Lee J. Y., Lee M.-Y., Lim S. H., Lee W. Y., Kim S. H., Park Y. A., Cho Y. B., Yun S. H., Kim S. T., Park J. O., Lim H. Y., Choi Y. S., Kwon W. I., Kim H. C., Park Y. S., The impact of KRAS mutations on prognosis in surgically resected colorectal cancer patients with liver and lung metastases: A retrospective analysis. BMC Cancer 16, 120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo A. L., Borger D. R., Szymonifka J., Ryan D. P., Wo J. Y., Blaszkowsky L. S., Kwak E. L., Allen J. N., Wadlow R. C., Zhu A. X., Murphy J. E., Faris J. E., Dias-Santagata D., Haigis K. M., Ellisen L. W., Iafrate A. J., Hong T. S., Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer 120, 1482–1490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umeda Y., Nagasaka T., Mori Y., Sadamori H., Sun D.-S., Shinoura S., Yoshida R., Satoh D., Nobuoka D., Utsumi M., Yoshida K., Yagi T., Fujiwara T., Poor prognosis of KRAS or BRAF mutant colorectal liver metastasis without microsatellite instability. J. Hepatobiliary Pancreat. Sci. 20, 223–233 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Andreyev H. J. N., Norman A. R., Cunningham D., Oates J., Dix B. R., Iacopetta B. J., Young J., Walsh T., Ward R., Hawkins N., Beranek M., Jandik P., Benamouzig R., Jullian E., Laurent-Puig P., Olschwang S., Muller O., Hoffmann I., Rabes H. M., Zietz C., Troungos C., Valavanis C., Yuen S. T., Ho J. W. C., Croke C. T., O’Donoghue D. P., Giaretti W., Rapallo A., Russo A., Bazan V., Tanaka M., Omura K., Azuma T., Ohkusa T., Fujimori T., Ono Y., Pauly M., Faber C., Glaesener R., Goeij A. F. P. M., Arends J. W., Andersen S. N., Lövig T., Breivik J., Gaudernack G., Clausen O. P. F., Angelis P. D., Meling G. I., Rognum T. O., Smith R., Goh H. S., Font A., Rosell R., Sun X. F., Zhang H., Benhattar J., Losi L., Lee J. Q., Wang S. T., Clarke P. A., Bell S., Quirke P., Bubb V. J., Piris J., Cruickshank N. R., Morton D., Fox J. C., al-Mulla F., Lees N., Hall C. N., Snary D., Wilkinson K., Dillon D., Costa J., Pricolo V. E., Finkelstein S. D., Thebo J. S., Senagore A. J., Halter S. A., Wadler S., Malik S., Krtolica K., Urosevic N., Kirsten ras mutations in patients with colorectal cancer: the 'RASCAL II' study. Br. J. Cancer 85, 692–696 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M., K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim S. M., Westover K. D., Ficarro S. B., Harrison R. A., Choi H. G., Pacold M. E., Carrasco M., Hunter J., Kim N. D., Xie T., Sim T., Jänne P. A., Meyerson M., Marto J. A., Engen J. R., Gray N. S., Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Eng. 53, 199–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmermann G., Papke B., Ismail S., Vartak N., Chandra A., Hoffmann M., Hahn S. A., Triola G., Wittinghofer A., Bastiaens P. I. H., Waldmann H., Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 497, 638–642 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D., Valtorta E., Schiavo R., Buscarino M., Siravegna G., Bencardino K., Cercek A., Chen C.-T., Veronese S., Zanon C., Sartore-Bianchi A., Gambacorta M., Gallicchio M., Vakiani E., Boscaro V., Medico E., Weiser M., Siena S., Nicolantonio F. D., Solit D., Bardelli A., Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nazarian R., Shi H., Wang Q., Kong X., Koya R. C., Lee H., Chen Z., Lee M.-K., Attar N., Sazegar H., Chodon T., Nelson S. F., McArthur G., Sosman J. A., Ribas A., Lo R. S., Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephen A. G., Esposito D., Bagni R. K., McCormick F., Dragging ras back in the ring. Cancer Cell 25, 272–281 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Falchook G. S., Lewis K. D., Infante J. R., Gordon M. S., Vogelzang N. J., DeMarini D. J., Sun P., Moy C., Szabo S. A., Roadcap L. T., Peddareddigari V. G., Lebowitz P. F., Le N. T., Burris H. A. III, Messersmith W. A., O’Dwyer P. J., Kim K. B., Flaherty K., Bendell J. C., Gonzalez R., Kurzrock R., Fecher L. A., Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 13, 782–789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Infante J. R., Fecher L. A., Falchook G. S., Nallapareddy S., Gordon M. S., Becerra C., DeMarini D. J., Cox D. S., Xu Y., Morris S. R., Peddareddigari V. G., Le N. T., Hart L., Bendell J. C., Eckhardt G., Kurzrock R., Flaherty K., Burris H. A. III, Messersmith W. A., Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 13, 773–781 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Lee M. S., Helms T. L., Feng N., Gay J., Chang Q. E., Tian F., Wu J. Y., Toniatti C., Heffernan T. P., Powis G., Kwong L. N., Kopetz S., Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 7, 39595–39608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinelli E., Morgillo F., Troiani T., Ciardiello F., Cancer resistance to therapies against the EGFR-RAS-RAF pathway: The role of MEK. Cancer Treat. Rev. 53, 61–69 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Benvenuti S., Sartore-Bianchi A., Di Nicolantonio F., Zanon C., Moroni M., Veronese S., Siena S., Bardelli A., Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 67, 2643–2648 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Di Nicolantonio F., Martini M., Molinari F., Sartore-Bianchi A., Arena S., De Dosso P. S., Mazzucchelli L., Frattini M., Siena S., Bardelli A., Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 26, 5705–5712 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Gong J., Cho M., Fakih M., RAS and BRAF in metastatic colorectal cancer management. J. Gastrointest. Oncol. 7, 687–704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lièvre A., Bachet J.-B., Corre D. L., Boige V., Landi B., Emile J.-F., Côté J.-F., Tomasic G., Penna C., Ducreux M., Rougier P., Penault-Llorca F., Laurent-Puig P., KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 66, 3992–3995 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Benson A. B. III, Venook A. P., Al-Hawary M. M., Cederquist L., Chen Y.-J., Ciombor K. K., Cohen S., Cooper H. S., Deming D., Engstrom P. F., Garrido-Laguna I., Grem J. L., Grothey A., Hochster H. S., Hoffe S., Hunt S., Kamel A., Kirilcuk N., Krishnamurthi S., Messersmith W. A., Meyerhardt J., Miller E. D., Mulcahy M. F., Murphy J. D., Nurkin S., Saltz L., Sharma S., Shibata D., Skibber J. M., Sofocleous C. T., Stoffel E. M., Stotsky-Himelfarb E., Willett C. G., Wuthrick E., Gregoryand K. M., Freedman-Cass D. A., NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Canc. Netw. 16, 359–369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bangi E., Murgia C., Teague A. G. S., Sansom O. J., Cagan R. L., Functional exploration of colorectal cancer genomes using Drosophila. Nat. Commun. 7, 13615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colucci G., Gebbia V., Paoletti G., Giuliani F., Caruso M., Cartenì N. G., Agostara B., Pezzella G., Manzione L., Borsellino N., Misino A., Romito S., Durini E., Cordio S., Seri M. D., Lopez M., Maiello E., Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: A multicenter study of the Gruppo Oncologico Dell'Italia Meridionale. J. Clin. Oncol. 23, 4866–4875 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Deng T., Zhang L., Liu X.-j., Xu J.-m., Bai Y.-x., Wang Y., Han Y., Li Y.-h., Ba Y., Bevacizumab plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as the second-line therapy for patients with metastatic colorectal cancer, a multicenter study. Med. Oncol. 30, 752 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Giantonio B. J., Catalano P. J., Meropol N. J., O'Dwyer P. J., Mitchell E. P., Alberts S. R., Schwartz M. A., Benson A. B. III; Eastern Cooperative Oncology Group Study E3200 , Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 25, 1539–1544 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Qu C.-Y., Zheng Y., Zhou M., Zhang Y., Shen F., Cao J., Xu L.-M., Value of bevacizumab in treatment of colorectal cancer: A meta-analysis. World J. Gastroenterol. 21, 5072–5080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saltz L. B., Clarke S., Díaz-Rubio E., Scheithauer W., Figer A., Wong R., Koski S., Lichinitser M., Yang T. S., Rivera F., Couture F., Sirzén F., Cassidy J., Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 26, 2013–2019 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Network , Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand A. H., Perrimon N., Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Ni J.-Q., Zhou R., Czech B., Liu L.-P., Holderbaum L., Yang-Zhou D., Shim H.-S., Tao R., Handler D., Karpowicz P., Binari R., Booker M., Brennecke J., Perkins L. A., Hannon G. J., Perrimon N., A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guay D. R., Ibandronate: A new oral bisphosphonate for postmenopausal osteoporosis. Consult. Pharm. 20, 1036–1055 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Konstantinopoulos P. A., Karamouzis M. V., Papavassiliou A. G., Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Mo H., Elson C. E., Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 229, 567–585 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Wong W. W.-L., Dimitroulakos J., Minden M. D., Penn L. Z., HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 16, 508–519 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Yuen T., Stachnik A., Iqbal J., Sgobba M., Gupta Y., Lu P., Colaianni G., Ji Y., Zhu L. L., Kim S. M., Li J., Liu P., Izadmehr S., Sangodkar J., Bailey J., Latif Y., Mujtaba S., Epstein S., Davies T. F., Bian Z., Zallone A., Aggarwal A. K., Haider S., New M. I., Sun L., Narla G., Zaidi M., Bisphosphonates inactivate human EGFRs to exert antitumor actions. Proc. Natl. Acad. Sci. U.S.A. 111, 17989–17994 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grothey A., Van Cutsem E., Sobrero A., Siena S., Falcone A., Ychou M., Humblet Y., Bouché O., Mineur L., Barone C., Adenis A., Tabernero J., Yoshino T., Lenz H. J., Goldberg R. M., Sargent D. J., Cihon F., Cupit L., Wagner A., Laurent D.; CORRECT Study Group , Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Li J., Qin S., Xu R., Yau T. C., Ma B., Pan H., Xu J., Bai Y., Chi Y., Wang L., Yeh K. H., Bi F., Cheng Y., Le A. T., Lin J. K., Liu T., Ma D., Kappeler C., Kalmus J., Kim T. W.; CONCUR Investigators , Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 16, 619–629 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Mayer R. J., Van Cutsem E., Falcone A., Yoshino T., Garcia-Carbonero R., Mizunuma N., Yamazaki K., Shimada Y., Tabernero J., Komatsu Y., Sobrero A., Boucher E., Peeters M., Tran B., Lenz H. J., Zaniboni A., Hochster H., Cleary J. M., Prenen H., Benedetti F., Mizuguchi H., Makris L., Ito M., Ohtsu A.; RECOURSE Study Group , Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl. J. Med. 372, 1909–1919 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Wilhelm S. M., Dumas J., Adnane L., Lynch M., Carter C. A., Schütz G., Thierauch K.-H., Zopf D., Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 129, 245–255 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Pazianas M., Abrahamsen B., Eiken P. A., Eastell R., Russell R. G., Reduced colon cancer incidence and mortality in postmenopausal women treated with an oral bisphosphonate—Danish National Register Based Cohort Study. Osteoporos. Int. 23, 2693–2701 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Rennert G., Pinchev M., Rennert H. S., Gruber S. B., Use of bisphosphonates and reduced risk of colorectal cancer. J. Clin. Oncol. 29, 1146–1150 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uzilov A. V., Ding W., Fink M. Y., Antipin Y., Brohl A. S., Davis C., Lau C. Y., Pandya C., Shah H., Kasai Y., Powell J., Micchelli M., Castellanos R., Zhang Z., Linderman M., Kinoshita Y., Zweig M., Raustad K., Cheung K., Castillo D., Wooten M., Bourzgui I., Newman L. C., Deikus G., Mathew B., Zhu J., Glicksberg B. S., Moe A. S., Liao J., Edelmann L., Dudley J. T., Maki R. G., Kasarskis A., Holcombe R. F., Mahajan M., Hao K., Reva B., Longtine J., Starcevic D., Sebra R., Donovan M. J., Li S., Schadt E. E., Chen R., Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med. 8, 62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kircher M., Witten D. M., Jain P., O’Roak B. J., Cooper G. M., Shendure J., A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X., Wu C., Li C., Boerwinkle E., dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uzilov A. V., Cheesman K. C., Fink M. Y., Newman L. C., Pandya C., Lalazar Y., Hefti M., Fowkes M., Deikus G., Lau C. Y., Moe A. S., Kinoshita Y., Kasai Y., Zweig M., Gupta A., Starcevic D., Mahajan M., Schadt E. E., Post K. D., Donovan M. J., Sebra R., Chen R., Geer E. B., Identification of a novel RASD1 somatic mutation in a USP8-mutated corticotroph adenoma. Cold Spring Harb. Mol. Case Stud. 3, a001602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donovan M. J., Kotsianti A., Bayer-Zubek V., Verbel D., Teverovskiy M., Cordon-Cardo C., Costa J., Greco F. A., Hainsworth J. D., Parums D. V., A systems pathology model for predicting overall survival in patients with refractory, advanced non-small-cell lung cancer treated with gefitinib. Eur. J. Cancer 45, 1518–1526 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Vert J.-P., Foveau N., Lajaunie C., Vandenbrouck Y., An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics 7, 520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bischof J., Maeda R. K., Hediger M., Karch F., Basler K., An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc. Natl. Acad. Sci. U.S.A. 104, 3312–3317 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sopko R., Foos M., Vinayagam A., Zhai B., Binari R., Hu Y., Randklev S., Perkins L. A., Gygi S. P., Perrimon N., Combining genetic perturbations and proteomics to examine kinase-phosphatase networks in Drosophila embryos. Dev. Cell 31, 114–127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav6528/DC1
Fig. S1. Validation of patient’s personalized Drosophila model (see Materials and Methods for details).
Fig. S2. Validation of patient’s personalized Drosophila model and drug response from hindgut lysates (see Materials and Methods for details).
Table S1. Multi-hairpin cluster sequences for patient’s personalized model.
Table S2. Raw drug screening and testing data.
Table S3. Patient’s tumor measurements.