

Abstract
Inositol pyrophosphates (IPPs) are present in organisms ranging from plants, slime moulds and fungi to mammals. Distinct classes of kinases generate different forms of energetic diphosphate-containing IPPs from inositol phosphates (IPs). Conversely, polyphosphate phosphohydrolase enzymes dephosphorylate IPPs to regenerate the respective IPs. IPPs and/or their metabolizing enzymes regulate various cell biological processes by modulating many proteins via diverse mechanisms. In the last decade, extensive research has been conducted in mammalian systems, particularly in knockout mouse models of relevant enzymes. Results obtained from these studies suggest impacts of the IPP pathway on organ development, especially of brain and testis. Conversely, deletion of specific enzymes in the pathway protects mice from various diseases such as diet-induced obesity (DIO), type-2 diabetes (T2D), fatty liver, bacterial infection, thromboembolism, cancer metastasis and aging. Furthermore, pharmacological inhibition of the same class of enzymes in mice validates the therapeutic importance of this pathway in cardio-metabolic diseases. This review critically analyses these findings and summarizes the significance of the IPP pathway in mammalian health and diseases. It also evaluates benefits and risks of targeting this pathway in disease therapies. Finally, future directions of mammalian IPP research are discussed.
Keywords: inositol pyrophosphate, IP6K, PPIP5K, DIPP, TNP, development, obesity, diabetes, cardiovascular disease, cancer, aging
I. INTRODUCTION
Myo-inositol and its various soluble and lipid-associated inositol phosphate (IP) derivatives play important biological functions in some bacteria, and in fungi, higher plants, and mammals (Michell, 1975; Holub, 1986). In the early 20th century, the inositol hexakisphosphate or phytic acid [I(1,2,3,4,5,6)P6 or IP6] was identified as the principal phosphate-storage molecule in plants (Plimmer & Page, 1913; Vucenik & Shamsuddin, 2003). In the mid-1980s, the discovery of inositol trisphosphate [I(1,4,5)P3 or IP3]-mediated release of intracellular calcium accelerated interest in the biological functions of IPs (Streb et al., 1983; Berridge, Lipp & Bootman, 2000). IP derivatives with energetic di(β)phosphates of IP6 were identified in the early 1990s (Europe-Finner, Gammon & Newell, 1991; Stephens et al., 1991; Oliver et al., 1992; Wong et al., 1992). These molecules were termed ‘inositol pyrophosphates (IPPs)’, to distinguish them from the monoester-based IPs (Glennon & Shears, 1993; Menniti et al., 1993; Stephens et al., 1993). Due to a rapid turnover rate, this class of molecules was predicted to function as signalling modulators (Glennon & Shears, 1993).
IPP nomenclature has been discussed elsewhere (Barker et al., 2009; Saiardi, 2012; Wundenberg & Mayr, 2012; Shears, 2015). Four well-characterized mammalian IPPs, 5-diphosphoinositol (1,3,4,6)-tetrakisphosphate (5PP-IP4), 1-diphosphoinositol (2,3,4,5,6) pentakisphosphate (1PP-IP5 or 1-IP7), 5-diphosphoinositol (1,2,3,4,6) pentakisphosphate (5PP-IP5 or 5-IP7) and 1,5-bisdiphosphoinositol (2,3,4,6) tetrakisphosphate (1,5PP2-IP4 or 1,5-IP8) are discussed here. When 5-IP7 and 1-IP7 are indistinguishable, the term IP7 is used. PPP-IP5, PP-IP3, (PP)2-IP3 and (PP)2-IP2 (Draskovic et al., 2008; Wundenberg & Mayr, 2012; Thota & Bhandari, 2015) are not characterized in mammals and thus, are omitted from further discussion.
Studies in lower organisms establish that IPPs regulate various biological processes. A slime mould-specific IP7 regulates chemotaxis (Luo et al., 2003). In fungi, the pathway modulates telomere length (Saiardi et al., 2005; York et al., 2005), DNA hyper-recombination (Luo et al., 2002), vacuole biogenesis (Saiardi et al., 2000), endocytosis and call wall integrity (Saiardi et al., 2002), ribosomal RNA (rRNA) transcription (Thota et al., 2015), environmental stress response (Worley, Luo & Capaldi, 2013), adaptation to the host environment and pathogenicity (Lev et al., 2015), virulence (Li et al., 2016), phosphate homeostasis (Lee et al., 2007) and cell death (Onnebo & Saiardi, 2009). In fruit flies Drosophila melanogaster, it is associated with insulin signalling (Williams et al., 2015). In zebrafish Danio rerio, the IPP pathway acts as an effector of the hedgehog signalling pathway, which modulates development of craniofacial structures and neural crest cells (Sarmah & Wente, 2010).
A major question is ‘what is the significance of the IPP pathway in mammalian health?’ To address this, extensive research has been carried out in mammalian systems, including detection of IPPs (Menniti et al., 1993; Stephens et al., 1993), and cloning and characterization of relevant enzymes (Nagase et al., 1996; Voglmaier et al., 1996; Saiardi et al., 1999, 2001; Yang, Safrany & Shears, 1999; Caffrey et al., 2000; Choi et al., 2007; Fridy et al., 2007). Impacts of the IPP pathway on mammalian cells were also discovered (Barker et al., 2009; Chakraborty, Kim & Snyder, 2011; Wundenberg & Mayr, 2012; Wilson, Livermore & Saiardi, 2013; Thota & Bhandari, 2015) and generation of knockout (KO) mouse models established the in vivo significance of the IPP pathway (Bhandari et al., 2008; Morrison et al., 2009; Rao et al., 2014; Fu et al., 2015; Moritoh et al., 2016; Zhu et al., 2016). The beneficial effects of pharmacological inhibition of this pathway in various rodent disease models demonstrated its therapeutic significance. This review encompasses impacts of the IPP pathway in health and diseases.
II. IPP-METABOLIZING ENZYMES IN MAMMALS
The intricacies of the IPP metabolic pathway have been discussed elsewhere (Irvine & Schell, 2001; Shears, 2004; Bennett et al., 2006; Barker et al., 2009; Chakraborty et al., 2011; Wilson et al., 2013; Thomas & Potter, 2014; Thota & Bhandari, 2015; Shears et al., 2016; Shah et al., 2017) and thus are only briefly presented here (Figs 1 and 2). In mammals, hormone- or growth factor-stimulated enzyme phospholipase C (PLC) cleaves the phosphoinositide phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate diacylglycerol (DAG) and IP3. Inositol polyphosphate multikinase (IPMK) and IP3 3-kinase (IP3K) phosphorylate IP3 to generate I(1,3,4,5)P4 (IP4) (Takazawa et al., 1990; Saiardi et al., 1999) that is further phosphorylated to I(1,3,4,5,6)P5 (IP5) by IPMK. Alternatively, the inositol polyphosphate 5-phosphatase [INPP5; primarily type I, although type II and SH2-domain containing inositol phosphatase-1 (SHIP1) also possess this activity] dephosphorylates IP4 to I(1,3,4)P3 (IP3*) (Erneux et al., 1998). Inositol tetrakisphosphate 1-kinase (ITPK1) synthesizes I(1,3,4,6)P4 (IP4*) from IP3*, which is converted to IP5 by IPMK (Fig. 1). It is not known to what extent these enzymes contribute in vivo to IP5 level.
Fig. 1.
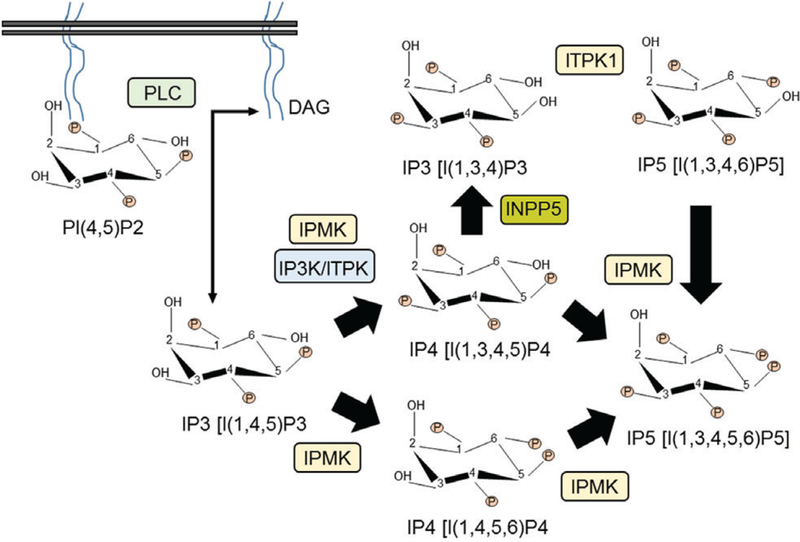
Generation of inositol phosphate IP5 from the lipid phosphoinositide phosphatidylinositol (4,5)-bisphosphate (PIP2) in mammals. Only major enzymes are denoted. DAG, diacylglycerol; INPP5, inositol polyphosphate 5-phosphatase; IP3K, IP3 3-kinase; IPMK, inositol polyphosphate multikinase; ITPK1, inositol tetrakisphosphate 1-kinase; PLC, phospholipase C.
Fig. 2.
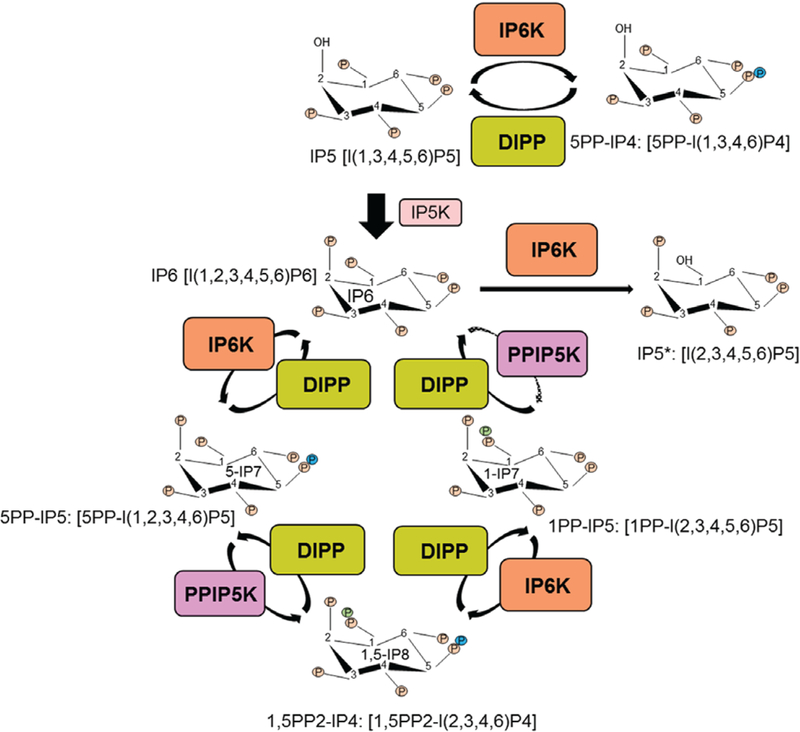
Synthesis of inositol phosphate IP6 and inositol pyrophosphates. Inositol hexakisphosphate kinases (IP6Ks) convert IP5, IP6 and 1-IP7 to generate 5PP-IP4, 5-IP7 and 1,5-IP8 respectively. Diphosphoinositol pentakisphosphate kinases (PPIP5Ks) primarily convert 5-IP7 to 1,5-IP8. PPIP5Ks also generate 1-IP7 from IP6, albeit to a lesser extent (dotted arrow). Diphosphoinositol polyphosphate phosphohydrolases (DIPPs) hydrolyse inositol pyrophosphates to regenerate the respective inositol phosphates. At a lower ATP/ADP ratio, IP6Ks dephosphorylate IP6 to IP5*. Only well-characterized enzymes are depicted.
Inositol pentakisphosphate 2-kinase (IP5K) converts IP5 to IP6. A family of three inositol hexakisphosphate kinases (IP6Ks) synthesizes 5PP-IP4, 5-IP7 and 1,5-IP8 from IP5, IP6 and 1-IP7 respectively. IP6Ks can also dephosphorylate IP6 to I(2,3,4,5,6)P5 (IP5*) at a reduced ATP/ADP ratio (Wundenberg et al., 2014). Diphosphoinositol pentakisphosphate (PPIP5) kinases (PPIP5Ks) are a distinct family of enzymes which primarily produce 1,5-IP8 from 5-IP7 and to a lesser extent 1-IP7 from IP6 (Shears et al., 2016). Diphosphoinositol polyphosphate phosphohydrolases (DIPPs) hydrolyse IPPs to their respective IPs (Fig. 2). The genes involved in IPP metabolism are not only evolutionarily conserved, but also are amplified in mammals, which indicates advanced isoform-selective functions in higher organisms.
(1). Inositol hexakisphosphate kinase (IP6K)
The three human IP6K (E.C.2.7.4.21) proteins range from 410 to 441 amino acids (Saiardi et al., 1999, 2001; Thomas & Potter, 2014). Critical amino acids in and around the catalytic domain are largely conserved in IP kinases including IP6K1/2/3 (Fig. 3, cyan – catalytic) (Saiardi et al., 2001; Holmes & Jogl, 2006), although isoform-specific structural details are lacking. Substantial homology is also observed in some other regions (Fig. 3, cyan). Conversely, distinct isoform-specific sequences also exist (Fig. 3, white). These regions are involved in protein–protein interactions and post-translational modifications. These events cause specificity of IP6K’s functions in vivo by regulating activity, stability, subcellular distribution and target-protein identification of the enzymes (Barker et al., 2009; Chakraborty et al., 2011,b; Ghoshal et al., 2016) (see Sections IV–VI).
Fig. 3.
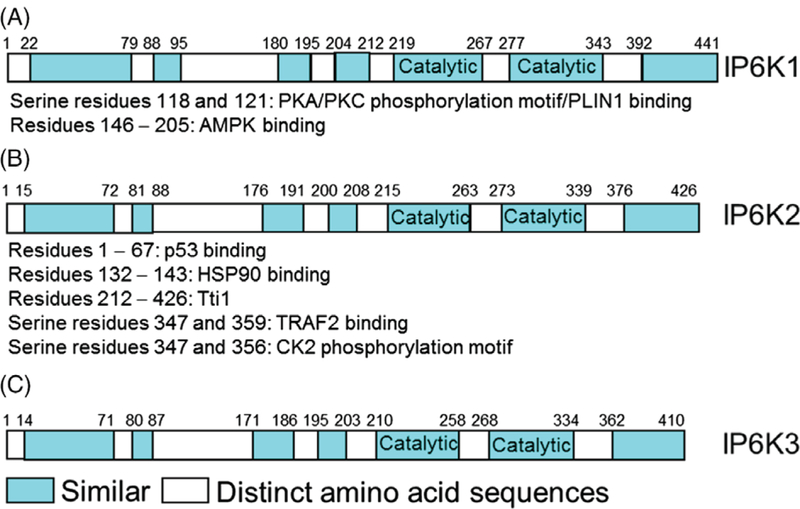
A–C. Schematic presentation of the human inositol hexakisphosphate kinase (IP6K) isoforms. In addition to the highly conserved catalytic domain (cyan: catalytic), various other regions display substantial similarities (cyan). Conversely, certain (white) regions are isoform-specific. IP6Ks interact with various protein targets in an isoform-selective manner. A few representative protein-interactors are shown for IP6K1 and IP6K2, for which the target binding sites were mapped. Some isoform-specific interactions are partly explained by substantial dissimilarity in the binding regions. IP6Ks also bind to certain targets [perilipin-1 (PLIN1) for IP6K1 and TNF receptor-associated factor-2 (TRAF2) for IP6K2] via phosphorylation of isoform-specific residues. Details of these and other interactions are presented in Sections IV–VI. AMPK, AMP-activated protein kinase; CK2, casein kinase 2; HSP90, heat shock protein 90; p53, tumour suppressor protein 53; PKA/C, protein kinase A/C; Tti1, telomere length regulation protein 2 (Tel2)-interacting proteins 1.
IP6Ks also differ in their tissue expression patterns. IP6K1 is abundant in the majority of murine tissues, with highest expression in brain and testis (Saiardi et al., 1999; Moritoh et al., 2016). Conversely, in humans, IP6K1 and IP6K2 are equally abundant, although IP6K2 is slightly higher in mammary gland, thymus, colon, adipose tissue, testis, prostate and smooth muscle. IP6K3 is minimally expressed in murine tissues with exceptions in heart, skeletal muscle and brain (Fu et al., 2015; Moritoh et al., 2016). However, in humans, IP6K3 is expressed at similar levels to IP6K1/2 in the thyroid, whereas it is the primary form in the heart. IP6K3 is the major form in murine and human skeletal muscles (Moritoh et al., 2016). However, unlike mouse, the human samples in that study were pooled from subjects of different ages, which means that age-dependent variations could not be ruled out. The above comparisons were based on messenger RNA (mRNA) expression, and should be confirmed at the levels of protein and isoform-specific activities. Thus, despite their fairly ubiquitous nature, differential isoform-specific expression patterns of IP6Ks are observed in mammalian tissues.
(2). Diphosphoinositol pentakisphosphate kinase (PPIP5K)
Soon after the discovery of the 1-IP7 synthesizing enzyme VIP1 in Saccharomyces cerevisiae, (Mulugu et al., 2007; Lin et al., 2009; Wang et al., 2012), its mammalian orthologs PPIP5K1 and PPIP5K2 were cloned (Choi et al., 2007; Fridy et al., 2007). Compared to IP6Ks, PPIP5Ks (E.C. 2.7.4.24) are relatively large proteins (PPIP5K1: 160 kDa; PPIP5K2: 140 kDa) (Choi et al., 2007; Fridy et al., 2007). The kinase domain of PPIP5Ks resides at the N-terminus (Fig. 4) (Shears et al., 2016). The substrate-binding pocket of the human PPIP5K2-kinase domain is composed of two near-parallel grooves that accommodate the inositol with six phosphate/pyrophosphate groups (Wang et al., 2012). PPIP5Ks also host a phosphatase domain (Fig. 4) (Fridy et al., 2007). Although the amino-acid sequence of the phosphatase domain resembles the phytase subgroup of histidine acid phosphatases, PPIP5Ks hydrolyse the 1-β-phosphate of 1-IP7 and 1,5-IP8 (Wang et al., 2015). PPIP5K1 is widely expressed, although higher expression levels are observed in skeletal muscle, heart and brain (Fridy et al., 2007).
Fig. 4.
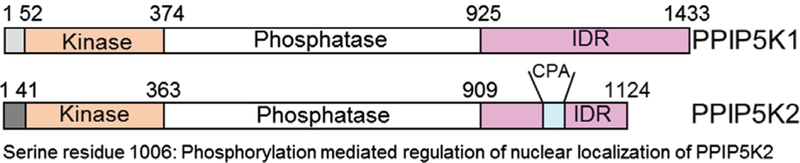
Domain analyses of human diphosphoinositol pentaphosphate kinases PPIP5K1 and PPIP5K2 (redrawn from Shears et al., 2016). PPIP5Ks contain a kinase and a phosphatase domain, which share 86 and 77% homology between the isoforms. This group also contains an intrinsically disordered domain (IDR), essential for protein– protein interaction, which shares 3% identity. PPIP5K2 contains a penta-arginine (CPA) domain that serves as a nuclear-localization signal (NLS). Phosphorylation of the serine 1006 residue, adjacent to the NLS regulates the nuclear localization of PPIP5K2.
(3). Diphosphoinositol polyphosphate phosphohydrolase (DIPP)
The IPP-hydrolysing DIPP (E.C. 3.6.1.52) enzymes belong to the NUDT gene family (McLennan, 2006). Nudix enzymes hydrolyse a wide range of organic pyrophosphates, including nucleoside di- and triphosphates, dinucleoside and diphosphoinositol polyphosphates, inorganic polyphosphates (polyP), nucleotide sugars and RNA caps, with varying degrees of substrate specificity (Yang et al., 1999; Fisher et al., 2002; McLennan, 2006; Lonetti et al., 2011). DIPP enzymes range from 164 to 181 amino acids (Leslie, McLennan & Safrany, 2002). In mammals, NUDT3 and NUDT4 encode DIPP1 and DIPP2 respectively, of which, the latter has two alternatively spliced isoforms, DIPP2α and DIPP2β (Safrany et al., 1998; Caffrey et al., 2000; Caffrey & Shears, 2001; Fisher et al., 2002). DIPP2α and DIPP2β differ by one amino acid, yet, it causes a significant difference in their catalytic constant (Kcat) values on IPPs (Caffrey et al., 2000). The products of NUDT10 and NUDT11 are DIPP3α and DIPP3β, which also differ by a single amino acid (Fisher et al., 2002;Hidaka et al., 2002; Leslie et al., 2002). In general, DIPPs have lower substrate concentration at half maximum velocity (Km) values for diphosphoinositol polyphosphates (0.004–0.088 μM) than for diadenosine polyphosphates (5.9–43 μM) (McLennan, 2006). However, substrate preference and utilization vary with divalent ions or redox conditions in vitro suggesting possible switching in their catalytic activities in vivo (Leslie et al., 2002; Hua et al., 2003). The ranking of Kcat values of DIPPs are in the order 1-IP7 > 5-IP7 = 1,5,-IP8, and DIPP1 is 10- to 60-fold more active than DIPP2α/β and DIPP3α/β (Kilari et al., 2013). DIPP1 expression is higher in brain, heart, pancreas and liver (Safrany et al., 1998). DIPP2 is expressed in the heart, and to a lower extent in skeletal muscle, pancreas, and kidney (Caffrey et al., 2000; Hidaka et al., 2002). DIPP3α expression is greater in the brain and liver (Leslie et al., 2002). DIPP3β expression is higher in brain, pancreas and testis (Hidaka et al., 2002; Leslie et al., 2002). The significance of DIPP in vivo is yet to be understood.
III. REGULATION OF IPPS
Intracellular IPPs are measured using a standard technique of labelling cells with [3H]myo-inositol followed by high-performance liquid chromatography (HPLC)-based separation [Azevedo & Saiardi, 2006 and references therein]. In mammalian cells, about 50% of IP6 and 20% of IP5 are converted to pyrophosphates per hour (Menniti et al., 1993). The IP7 pool can turn over around 10 times in every 40 min (Glennon & Shears, 1993). 5-IP7 and IP6 concentrations are 0.5–5 and 10–100 μM, respectively (Albert et al., 1997; Barker et al., 2004; Illies et al., 2007; Lin et al., 2009; Wundenberg & Mayr, 2012). Thus, intracellular 5-IP7 concentration is about 1–5% of IP6 levels. 1-IP7 comprises 2–10% of total intracellular IP7 levels (Padmanabhan et al., 2009; Gu et al., 2016; Shears et al., 2016). 1,5-IP8 levels may be undetectable under basal conditions, although it can reach up to 10–20% of total IP7, depending on cell type and conditions [see Gu et al. (2016), Thota & Bhandari (2015), Wundenberg & Mayr (2012) and references therein].
Intracellular IP7 levels respond to diverse conditions such as metabolic, apoptotic and aging. In mouse embryonic fibroblasts (MEFs) or human hepatocellular carcinoma (HepG2) cells, overnight serum starvation decreases IP7 levels, which are restored by insulin-like growth factor-1 (IGF-1) or insulin (Chakraborty et al., 2010). Insulin-induced adipogenesis in 3T3L1 cells also enhances the IP7 level (Chakraborty et al., 2010). Cellular ATP/ADP ratio is a major determinant of IP6K activity (due to its higher Km toward ATP), which may explain differential levels of IP7 in anabolic versus catabolic conditions (Wundenberg et al., 2014). For the same reason, inhibitors of phosphoinositide 3-kinase (PI3K) (wortmannin and LY294002) or insulin receptor tyrosine kinase [hydroxy-2-naphthalenylmethylphosphonic acid (HNMPA)] that reduce the ATP/ADP ratio, also diminish IP7 levels in an insulin-secreting HIT-T15 cell line. The PI4K and tyrosine phosphatase inhibitor phenylarsine oxide (PAO) and the PLC inhibitor 1-[6-((17β−3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole −2,5-dione (U73122) also reduce the ATP/ADP ratio and IP7 levels. However, they also directly inhibit IP6K1 (Rajasekaran et al., 2017).
Apoptosis inducers, such as the anticancer drug cisplatin, the heat shock protein 90 (HSP90) inhibitor novobiocin, the broadspectrum kinase inhibitor staurosporine and the cytokine interferon-β (IFNβ) increase IP7 concentrations in various mammalian cell lines (see Section VI.4a) (Morrison et al., 2001; Nagata et al., 2005; Chakraborty et al., 2008, 2011). Acute or prolonged treatments of certain stress/apoptosis inducers differentially influence IP7 levels. For example, overnight treatment of the endoplasmic reticulum (ER) Ca2+-ATPase inhibitor thapsigargin, which induces ER stress and apoptosis (Janssen et al., 2009) increases IP7 levels in the smooth muscle cell line DDT1- MF2 (Safrany, 2004), although its short-term (40 min) treatment has the opposite effect in hepatocytes (Glennon & Shears, 1993). Similarly, an apoptotic dose of the casein kinase-2 (CK2) inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB, 50 μM, 16 h) enhances IP7 level in IP6K2-overexpressing osteosarcoma (U2OS) cells (Chakraborty et al., 2011), whereas a lower dose (10 μM, 30 min) reduces IP7 levels and ATP/ADP ratio in IP6K1-predominated HIT-T15 cells (Rajasekaran et al., 2017).
IP7 levels also alter in diseases. Pancreatic islets of leptin-deficient obese and hyperinsulinemic ob/ob mice display ~50% higher 5-IP7 levels than lean mice (Section VI.3a) (Illies et al., 2007). IP7 is higher in older (10-month old) primary hepatocytes than younger (2-month old) controls (Section VI.3i) (Chakraborty et al., 2010). Aged (18-month old) murine bone marrow mesenchymal stem cells (BM-MSCs) also exhibit an approximately twofold increase in IP7, compared to young (2-month old) cells (Section VII.1d) (Zhang et al., 2014). IP7 level is higher in lymphoblasts of Huntington’s Disease patients (Nagata et al., 2011). The chemotactic bacterial peptide formyl-met-leu-phe (fMLP) depletes IP7 in neutrophils (human promyelocytic leukemia cell line HL60) (Prasad et al., 2011), whereas nicotine-mediated reduction of IP7 decelerates apoptosis in aged neutrophils (Section VI.3h) (Xu et al., 2013). The cytokine oncostatin-M (OSM), which alleviates cardiac ischaemic/reperfusion (I/R) injury by inhibiting cardiomyocyte apoptosis, also reduces IP7 levels. This study measured IP7 levels in the border zone of myocardial infarction (MI) by injecting [3H] myoinositol in the MI region (Section VII.1c) (Sun et al., 2015).
Intracellular 1,5-IP8 level is also sensitive to diverse factors. Experiments were conducted in Syrian hamster vas deferens smooth muscle DDT1-MF2 cells. Acute heat shock at 42°C increases 1,5-IP8 levels in these cells, although it decreases IP7 level (Choi, Mollapour & Shears, 2005). Hyperosmotic-stress-inducing agents such as sorbitol and sucrose significantly raise 1,5-IP8 levels (but slightly lower IP7 levels) in the above cell line (Safrany, 2004). Endothelial growth factor (EGF) treatment of DDT1-MF2 cells causes a modest increase in 1,5-IP8 without altering IP7 levels (Pesesse et al., 2004), whereas platelet-derived growth factor (PDGF) treatment does not influence IPP levels in NIH3T3 cells (Gokhale et al., 2013). The compound genistein, but not herbimycin decreases 1,5-IP8 level (Safrany, 2004). These compounds are broad-spectrum tyrosine kinase inhibitors, yet they also target oestrogen receptor, DNA topoisomerase, α-glucosidase, and HSP90, often more potently than tyrosine kinases, which presumably explains their differential effects. The ATP synthase inhibitor oligomycin, which increases cellular AMP levels, reduces 1,5-IP8 levels without directly affecting levels of PPIP5Ks (Choi et al., 2008). The AMP-mimetic aminoimidazol-carboxamide-ribonucleoside (AICAR) decreases 1,5-IP8 levels via an adenosine monophosphate-activated protein kinase (AMPK)-independent mechanism (Choi et al., 2008). Activation of the cyclic AMP– protein kinase A (cAMP– PKA) pathway by 3-isobutyl-1-methylxanthine (IBMX) or the beta-adrenergic receptor (β-AR) agonist isoproterenol reduces IP8 level (Safrany & Shears, 1998).
IV. REGULATION OF IPP-METABOLIZING ENZYMES
IPP-metabolizing enzymes are regulated at various levels by diverse factors. Cold exposure reduces IP6K1 expression in white adipose tissue (WAT) (Zhu et al., 2016). Protein kinases PKA/PKC specifically phosphorylate IP6K1 at serine residues 118 and 121, which modulate its interaction with the lipolytic regulator perilipin-1 (PLIN1) (Figs 3A and 5A and Section VI.3j) (Ghoshal et al., 2016). IP6K2 is also regulated by phosphorylation. CK2 selectively phosphorylates IP6K2 at serine residues S347 and S356 within the degradation-specific PEST motif, which enhances its ubiquitination and degradation (Figs 3B and 5B and Section VI.4a) (Chakraborty et al., 2011). IP6K1 is also phosphorylated at serine 151 (Dephoure et al., 2008), although the functional significance of this is currently unknown. A number of other modifications of unidentified function are also specific to IP6K1 (A. Chakraborty, unpublished observation). Conversely, catalytic activity of IP6K is regulated by protein interaction. DNA damage binding protein-1 (DDB1) and HSP90 inhibit IP6K1 and IP6K2, respectively (Figs 3B, 5C, D and Section VI.4a) (Chakraborty et al., 2008; Rao et al., 2014).
Fig. 5.
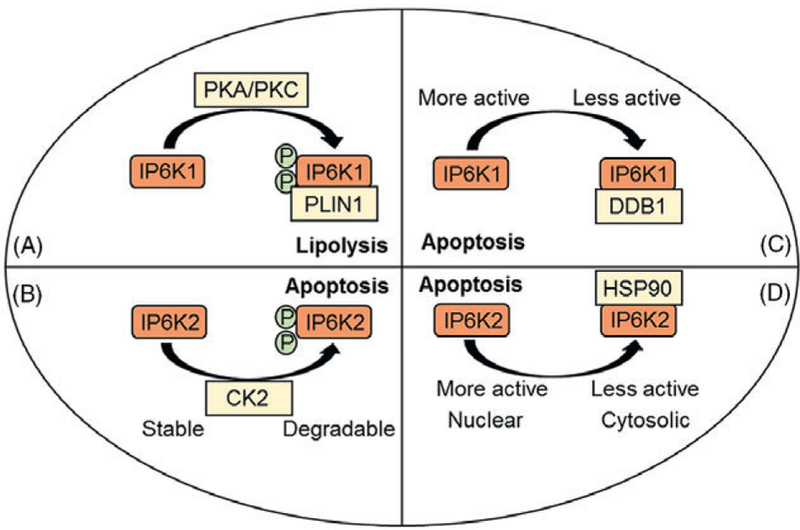
Isoform-specific post-translational modifications regulate inositol hexakisphosphate kinase (IP6K) stability or interaction with other proteins. Moreover, isoform-selective interactions with certain proteins regulate the catalytic activity of IP6K. (A) Protein kinase A/C (PKA/PKC)-mediated phosphorylation of IP6K1 mediates its interaction with the lipolytic modulator perilipin 1 (PLIN1). (B) Casein kinase-2 (CK2) phosphorylation facilitates degradation of IP6K2. (C) DNA damage binding protein-1 (DDB1) inhibits IP6K1 catalytic activity. (D) Heat shock protein 90 (HSP90) binds to inhibit the catalytic activity of IP6K2.
PPIP5Ks contain a polyphosphoinositide binding domain (PBD), within their phosphatase domain, which mediates stimulus-dependent translocation of cytosolic PPIP5K1 to the plasma membrane (Fig. 4 and Section VI.2b) (Gokhale, Zaremba & Shears, 2011). PPIP5Ks also contain an intrinsically disordered domain (IDR) at the C-terminus, which mediates interactions with proteins of vesicular trafficking, cytoskeleton, and lipid metabolism, although the functional significance of these interactions is unknown (Machkalyan et al., 2016; Shears et al., 2016). PPIP5K2 contains a penta-arginine (CPA) nuclear-localization sequence in the IDR domain (Fig. 4) (Yong et al., 2015). Overexpressed green fluorescent protein (GFP)-tagged CPA domain is nuclear, and phosphorylation of a serine residue adjacent to the domain regulates its nuclear localization (Yong et al., 2015). The impacts of phosphorylation of DIPP3α at multiple C-terminal residues are unknown (Gauci et al., 2009).
V. MECHANISMS BY WHICH IPP-METABOLIZING ENZYMES REGULATE CELLULAR PROCESSES
IPPs and their metabolizing enzymes regulate various cellular processes by modulating proteins via diverse mechanisms (Fig. 6). IPPs can regulate proteins by binding or by pyrophosphorylation. The mechanism of action of 5-IP7 is the best characterized. Most, but not all, actions of IPPs depend on the catalytic activity of their metabolizing enzymes. IP6Ks can interact directly with proteins to facilitate 5-IP7-mediated effects, although this is not always essential. Protein interaction is involved in scaffolding actions of IP6Ks, which do not require their catalytic activity. The physiological significance of these mechanisms is discussed in Section VI.
Fig. 6.
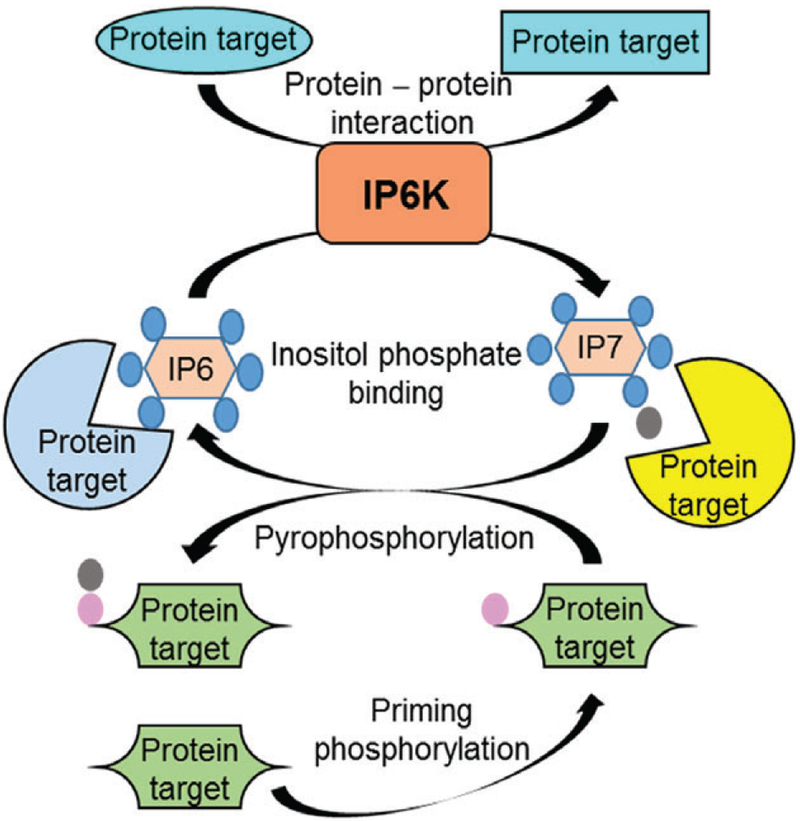
Mechanisms by which inositol pyrophosphate biosynthetic enzymes regulate protein targets. The model is based primarily on inositol hexakisphosphate kinases (IP6Ks). IP6Ks generate inositol phosphate IP7 from IP6. IP7 modulates protein (yellow) targets by direct binding. This activity also diminishes the effects of IP6 on its protein (blue) targets. Moreover, IP7 donates its β-phosphate (grey) to the serine residue of a protein (green), which is already phosphorylated (pink) by a priming protein kinase. This event is called pyrophosphorylation. IP6 is released by the process, which may enhance its interaction with protein targets. In addition, IP6Ks bind to certain protein targets (cyan), to alter their conformations and functions. This does not require catalytic activity of IP6Ks. The catalytic activity-mediated functions of IP6K may also require protein interaction. Diphosphoinositol pentakisphosphate kinase (PPIP5K) may display similar activities. See Section V for further details.
(1). Interaction of IPPs with proteins
(a). Pleckstrin homology (PH)-domain interaction
PH-domains in proteins bind phosphoinositides (Lemmon, 2007). Growth factor-stimulated receptor tyrosine kinases promote class I PI3K-mediated conversion of membrane-bound PIP2 to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). The protein kinase B (PKB or Akt) and phosphoinositide-dependent kinase-1 (PDK1) bind to PIP3 via their PH-domains. PDK1 then phosphorylates Akt at the threonine 308 residue, which activates the kinase. Studies have established that 5-IP7 reduces the phosphorylation, membrane translocation and activation of Akt (Luo et al., 2003; Chakraborty et al., 2010). 5-IP7 inhibits PDK1-mediated phosphorylation of Akt with half-maximal inhibitory concentration (IC50) values of ~20–200 nM in vitro (Chakraborty et al., 2010; Wu et al., 2013). A stable version of 5-IP7, 5PCP-IP5 has an IC50 of 129 nM (Wu et al., 2013). 1-IP7 and its analog 1PCP-IP5 also potently inhibit Akt phosphorylation (IC50 of ~55 and 105 nM, respectively) (Wu et al., 2014).
PDK1-mediated phosphorylation of Akt is increased by PIP3 (Calleja et al., 2007). In the presence of 1 μM PIP3, 5-IP7 prevents phosphorylation of Akt (IC50 ~1 μM), whereas IP6 shows a 10- to 15-fold lower efficiency. This inhibitory effect is observed when 5-IP7 and Akt are pre-incubated prior to PIP3 addition. When PIP3 is added before 5-IP7, the pyrophosphate is less effective (Downes et al., 2005; Chakraborty et al., 2010). Accordingly, IC50 values for 5-IP7 and 1-IP7 analogs (5PCP-IP5 and 1PCP-IP5, respectively) on Akt phosphorylation are 129 and 105 nM, respectively, in the absence of PIP3, but increase to 4.5 and 3.6 μM if PIP3 is added simultaneously with the analogs (Wu et al., 2014). 5-IP7, IP6, 1-IP7 and 1,5-IP8 bind the Akt-PH-domain with IC50 values of 7, 14, 45 and >50 μM, respectively (Gokhale et al., 2013). Thus, binding affinities of IPPs are higher than their inhibitory potencies, presumably due to differences in assay conditions.
In vivo, Akt activity is higher in IP6K1-deleted or IP6K-inhibited mice (Chakraborty et al., 2010; Prasad et al., 2011; Zhang et al., 2014; Sun et al., 2015; Zhu et al., 2016; Ghoshal et al., 2016). Furthermore, photouncaging-induced enhancement in intracellular 5-IP7 rapidly excludes the PH-domain of Akt from the plasma membrane (Pavlovic et al., 2016), whereas IP6 is ineffective (H. J. Jessen, personal communication). Although the precise molecular and structural details of this mechanism are yet to be understood, it is conceivable that ligand-induced conformational alterations of the PH-domain of Akt may contribute to its differential regulation by phosphoinositides and IPPs. In fact, the inactive (PH-in) conformation of Akt is inaccessible to PDK1. PIP3 binding converts Akt to the PDK1-accesible PH-out conformation (Calleja et al., 2007). 5-IP7 appears to stabilize PH-in. IPPs also interact with other mammalian PH-domain-containing proteins such as PI3-kinase enhancer (PIKE), T-lymphoma invasion and metastasis-inducing protein (TIAM), general receptor for phosphoinositides-1 (GRP1) and stress-activated protein kinase-interaction protein-1 (SIN1) in vitro (Luo et al., 2003; Gokhale et al., 2013), although the in vivo significance of these interactions is not known.
(b). C2-domain interaction
C2-domains, comprising about 130 residues, are found in various eukaryotic proteins. In classical PKCs, C2-domains are responsible for Ca2+-dependent membrane binding (Corbalan-Garcia & Gomez-Fernandez, 2014). The other class of C2-domain, containing the Ca2+-sensor protein synaptotagmin (Syt), regulates vesicle fusion and neurotransmitter release in the pre-synaptic terminal (Chapman, 2008). IPs such as IP4 and IP6 are known binders of the C2B domain of Syt (Mikoshiba et al., 1999; Yang et al., 2012). A recent study demonstrated that 5-IP7 displays a 45-fold higher binding affinity to Syt1 over IP6 (Lee et al., 2016). Molecular details of 5-IP7 and C2B domain interaction are not yet known.
(c). Other interactions that require catalytic activity and target-binding of IPP-metabolizing enzymes
IP6Ks interact with diverse proteins or protein complexes in an isoform-specific manner, followed by its catalytic activity-dependent modulation of the target proteins. For example, IP6K2/5-IP7 regulates the interaction of CK2 with telomere length regulation protein 2 (Tel2)-interacting proteins 1 and 2 (Tti1 and Tti2) complex (TTT) (Rao et al., 2014), tumour suppressor protein p53 (Koldobskiy et al., 2010) and liver kinase B1 (LKB1) (Rao et al., 2015), IP6K1/5-IP7 modulates Cullin-ring finger ligase-4 (CRL4) complex (Rao et al., 2014), α-actinin (Fu et al., 2017) and jumonji domain-containing 2C protein (JMJD2C) (Burton et al., 2013), whereas IP6K1/IP6 controls AMPK (Zhu et al., 2016). Details are described in Section VI.
(2). IPP-mediated pyrophosphorylation of target proteins
Protein phosphorylation is a common post-translational event, which regulates every aspect of cell growth, metabolism and survival (Manning et al., 2002; Ubersax & Ferrell, 2007; Derouiche, Cousin & Mijakovic, 2012). A distinct post-translational modification, termed ‘protein pyrophosphorylation’, by IPPs further modulates protein phosphorylation-mediated regulation of cellular functions (Wilson et al., 2013; Thota & Bhandari, 2015). Because of steric hindrance caused by the negatively charged bulky phosphate groups, the phosphoanhydride bond of IPP has a high energy of hydrolysis (Stephens et al., 1993; Laussmann et al., 1996; Hand & Honek, 2007; Wilson et al., 2013; Shears, 2017). Therefore, transfer of β-phosphate from 5-IP7 or 1,5-IP8 is considered thermodynamically favourable (Hand & Honek, 2007). In fact, the β-phosphate of 5-IP7 is transferred to ADP by IP6K1 in vitro (Voglmaier et al., 1996). This information suggests that IPPs are phosphorylating agents (Voglmaier et al., 1996). Indeed, IPPs donate their β-phosphate to the prephosphorylated serine residue in a non-enzymatic fashion (Saiardi et al., 2004; Bhandari et al., 2007). All IPPs are capable of pyrophosphorylation (Thota & Bhandari, 2015). The pyrophosphates in proteins are acid labile, but are resistant to protein phosphatases (Bhandari et al., 2007). Longer incubations of synthetic pyrophosphopeptides with alkaline phosphatases can remove the β-phosphate group on pyrophosphoserine-containing peptides (Yates & Fiedler, 2015), indicating that protein pyrophosphorylation is a reversible process (Thota & Bhandari, 2015). CK2 primarily acts as the priming kinase for the pyrophosphorylation event. However, direct in vivo evidence of pyrophosphorylation is lacking.
(3). Maintenance of intracellular inorganic polyphosphate (polyP) levels by IPPs
PolyPs are linear polymers of orthophosphates that are linked by phosphoanhydride bonds, as observed in ATP. These polymers, with various chain lengths, are found in all organisms (Rao, Gomez-Garcia & Kornberg, 2009). The idea that IP6K/5-IP7 regulates cellular polyP levels in mammals arose from studies in yeast, in which deletion of the KCS1 (IP6K1 ortholog), but not the PPIP5K ortholog, reduces polyP levels. Moreover, complementation of active but not inactive mouse IP6K1 restores polyP levels in KCS1-null yeast (Auesukaree et al., 2005; Lonetti et al., 2011). PolyPs are critical in coagulation of platelets. Accordingly, IP6K1-KO platelets display a reduction in polyP levels and in platelet coagulation (Section VI.3d) (Ghosh et al., 2013).
(4). Modulation of targets via protein–protein interactions
The above mechanisms imply that IPP-metabolizing enzymes require their catalytic activity to modulate protein targets. However, phylogenetic analysis predicts that IP6Ks evolved prior to the upstream IP-kinases, which generate substrates for IP6Ks (Bennett et al., 2006). What was IP6K doing in the absence of its substrates? Perhaps it was involved in scaffolding functions, which do not require involvement of IPPs. A number of studies suggest that IP6Ks can function as a scaffold to modulate protein targets, for which their catalytic activities are not required. Binding of IP6K1 to the guanine nucleotide exchange factor for Rab3A (GRAB) (Luo et al., 2001), the protein kinase glycogen synthase kinase (GSK3) (Chakraborty et al., 2014) and the lipolytic regulator PLIN1 (Ghoshal et al., 2016), interaction of IP6K2 with TNF receptor-associated factor-2 (TRAF2) (Morrison et al., 2007) and binding of IP6K3 to spectrin and adducin (Fu et al., 2015) are examples of such functions. Details of these interactions and their functional significance are described in Section VI.
VI. FUNCTIONS OF IPP-METABOLIZING ENZYMES IN MAMMALS
Pleiotropic effects of the IPP pathway modulate various cellular processes via diverse mechanisms, thus affecting the animal’s phenotype. Below, the functional significance of IPP enzymes is discussed. The mechanisms (where known) associated with each function are also mentioned.
(1). DIPP
(a). Extracelluar signal-regulated kinase (ERK) signalling
The Ras family of GTP-binding proteins transduces signals from several receptor tyrosine kinases, activating mitogen-activated protein kinases (MAPKs) like ERK. Ectopic expression of murine DIPP1 inhibits the ERK1/2 pathway. However, phosphohydrolase activity-deficient DIPP1 blocks ERK1/2 signalling with a greater efficiency, indicating that phosphohydrolase activity is not required by this process (Chu et al., 2004). The exact mechanism is not known.
(b). Cell migration
The 5′7-methyloguanosine cap and 3′-terminal poly(A) tract regulate the synthesis, translation, and degradation of mRNA. DIPP1 possesses mRNA-decapping activity, which modulates migration of the breast cancer cell line MCF-7. A reduction in DIPP1 level promotes migration of these cells, due to enhanced filopodial extension. The phenotype is reversed by complementation with the wild type, but not by the DIPP1 mutant that lacks mRNA-decapping activity. The impact of the inositol pyrophosphatase activity of DIPP1 is unknown (Grudzien-Nogalska et al., 2016).
(2). PPIP5K
(a). Type-1 interferon response
The retinoic acid-inducible gene-I (RIG-I) receptor facilitates type-I interferon production. A human genome-wide RNA interference (RNAi) screen identified PPIP5Ks to be positive regulators of this process, which is involved in cellular infection by Sendai and influenza A viruses (Pulloor et al., 2014). Overexpression of catalytically active IP5K, IP6Ks and PPIP5Ks induces interferon induction. Conversely, silencing of IP5K and PPIP5Ks but not IP6Ks has the opposite effect, indicating involvement of 1-IP7 in this process. PPIP5Ks mediate phosphorylation and activation of the interferon regulatory transcription factor 3 (IRF3) that promotes expression of type-1 interferon. The addition of purified IPPs to a cell-free reconstituted RIG-I signalling assay also showed 1-IP7 to be essential for IRF3 activation. 1-IP7 is thought to act by pyrophosphorylation, since its action was not mimicked by a synthetic phosphonoacetate analog, although no experimental evidence was presented (Pulloor et al., 2014). Intracellular 1-IP7 level is low and at times undetectable (Gu et al., 2016), so determining the concentration and regulation of 1-IP7 in primary macrophages and/or other relevant cells will be critical to evaluate the physiological relevance of this interesting finding.
(b). PIP3-mediated mammalian target of rapamycin complex-2 (mTORC2) signalling
In L6 myotubes, knockdown of PPIP5K1 impairs insulin-induced phosphorylation of S473 but not of T308 of Akt (Gokhale et al., 2013). The mTORC2 complex phosphorylates Akt at S473 and PPIP5K is proposed to activate this pathway. PPIP5K substrates (IP6/5-IP7) but not products (1-IP7/1,5-IP8) bind to the PH-domain of the mTORC2 regulator SIN1 in vitro (Section V.1a). In unstimulated cells, the PH-domain of SIN1 binds to and suppresses mTORC2 activity. Upon PI3K activation, SIN1 binds to PIP3 at the membrane, which activates mTORC2 (Yuan & Guan, 2015). Similar stimulation also promotes the binding of PPIP5K1 to PIP3 (Gokhale et al., 2011). Presumably, the catalytic activity of PPIP5K1 activates mTORC2 by a local reduction in IP6/5-IP7 level, which leads to increased SIN1–PIP3 and reduced SIN1–mTORC2 binding. PPIP5K1 activity seems specifically to regulate SIN1–PIP3 but not Akt–PIP3 binding, although the reason is unknown. Influences of various kinase, phosphatase and PBD mutants of PPIP5K1 on Akt phosphorylation should be assessed to determine the precise cellular mechanism involved. It is clear, however, that the IPP pathway regulates PI3K signalling at various levels.
(c). Cell growth and metabolism
Clustered regularly interspaced short palindromic repeats and its associated protein 9 (CRISPR-Cas9)-based genetic deletion of both PPIP5K1 and PPIP5K2 revealed impacts of 1,5-IP8 depletion on various phenotypes of the HCT116 colon cancer cell line. PPIP5K deletion abolishes 1,5-IP8, which causes a threefold increase in 5-IP7 levels in these cells. PPIP6K-KO cells display growth-inhibited and hypermetabolic phenotypes (Gu et al., 2017). Phenotypes of PPIP5K-KO cells are discussed and compared with IP6K1-or IP6K2-deleted cells in Sections VI.3 and 4.
(3). IP6K1
(a). Vesicular trafficking
Vesicular trafficking includes endocytosis and exocytosis, by which vesicles transport materials among different cellular compartments and between a cell and its environment. Various studies demonstrate that IP6K1 exerts both stimulatory and inhibitory effects on vesicular trafficking.
IP6K1 stimulates insulin exocytosis. Pancreatic β-cells express IP6K1 and IP6K2 and maintain relatively high levels (~6 μM) of 5-IP7, which is further increased in hyperinsulinemic ob/ob mice (Section III) (Illies et al., 2007). Exogenous IPPs (5-IP7, 1-IP7, 3-IP7 and 4-IP7) enhance exocytosis in mouse β-cells. Overexpression of catalytically active IP6K1 or IP6K2 enhances exocytosis, whereas silencing of IP6K1 but not IP6K2 inhibits the process. Depletion of IP6K1 or IP6K2 in β-cells causes ~50 or 25% reduction in 5-IP7 levels, respectively. Presumably, an optimal concentration of IP6K1- but not IP6K2-generated 5-IP7 is critical for insulin exocytosis (Illies et al., 2007). A reduced serum insulin level in IP6K1-KO but not in IP6K2-KO mice supports this possibility (Bhandari et al., 2008; Morrison et al., 2009). The precise mechanism by which IP6K1/5-IP7 mediates insulin release is not known.
IP6K1 regulates neuroexocytosis via both catalytic activity-dependent and independent mechanisms. The nucleotide exchange factor GRAB inhibits exocytosis via its action on the GTPase Rab3A. Both catalytically active and inactive versions of IP6K1 interact (Section V.4) and compete with GRAB for binding to Rab3A, which stimulates dopamine or growth hormone release in PC12 cells (Luo et al., 2001). Conversely, IP6K1-generated 5-IP7 reduces Ca2+-mediated neuroexocytosis in PC12 and in primary hippocampal neuronal cells by interacting with the C2-domain of Syt1 (Section V.1b) (Lee et al., 2016). Overexpression of IP6K1 suppresses, whereas its depletion promotes, exocytosis. 5-IP7 inhibits vesicle fusion in vitro, at a 10-fold higher affinity than IP6 or 1-IP7 (Lee et al., 2016). 5-IP7-dependent reduction of synaptic vesicle fusion was abolished by increasing Ca2+ levels, suggesting competition between them. Ca2+-bound Syt directs vesicular fusion via macromolecular interactions of lipid membranes and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins. IP6K1/5-IP7-dependent inhibition of synaptic vesicle fusion was abolished by increasing Ca2+ levels or by expressing the Ca2+-binding C2B domain of Syt1. Thus, binding of 5-IP7 to Syt1 interferes with the fusogenic activity of Ca2+. Whether 5-IP7 regulates other C2 domain-containing proteins, is not known.
Two independent studies suggest that 5-IP7 reduces or facilitates trafficking by pyrophoshorylation (Section V.2) of distinct cytoskeletal proteins. Long-range intracellular transport occurs on the microtubule cytoskeleton and is driven by two classes of motor proteins: kinesins and cytoplasmic dynein (Vallee, McKenney & Ori-McKenney, 2012). In MEFs, 5-IP7 inhibits kinesin-induced exocytosis of viral particles (Azevedo et al., 2009). Interaction of AP3β1, a clathrin-associated protein complex with the kinesin family motor protein Kif3A is required for release of viral particles. Catalytically active IP6K1 or IP6K2 reduces AP3β1– Kif3A interaction and subsequent release of virus particles (Azevedo et al., 2009). Direct in vitro and indirect in vivo (back phosphorylation) studies demonstrated that 5-IP7 pyrophosphorylates AP3β1, which may regulate this process.
On the other hand, 5-IP7 facilitates dynein-mediated trafficking. One study employed the transferrin protein as a model to show that IP6K1-deleted MEFs display defective dynein-dependent trafficking pathways, including endosomal sorting, vesicle movement, and Golgi maintenance. Complementation of catalytically active IP6K1 reverses the phenotype. Accordingly, phagosomal motility is reduced in IP6K1-KO macrophages (see Section VI.3h for neutrophil phagocytosis). The interaction between dynein intermediate chain (DIC) and the p150Glued subunit of dynactin is critical for the transport, which is reduced in IP6K1-KO cells. Direct in vitro and indirect in vivo (back phosphorylation) pyrophosphorylation, together with mutation studies, suggests that 5-IP7-mediated pyrophosphorylation of the serine 51 residue of DIC regulates its interaction with p150Glued and presumably the transport process (Chanduri et al., 2016). Acquiring further evidence such as in vivo detection of pyrophosphorylation and impacts of pyrophosphorylation-deficient mutants on trafficking are critical to determine the molecular details of the mechanisms involved. IP6K1/5-IP7 regulates vesicle trafficking via diverse mechanisms, which can diminish or stimulate exocytosis depending on the cell/tissue and/or physiological context. The mechanisms and physiological relevance of insulin and neurotransmitter release are distinct, which explains differential role of IP6K1 in these processes. The activity-independent stimulatory and activity-dependent inhibitory actions of IP6K1 on neurotransmitter release require further study. Transgenic mouse models of active and inactive IP6K1 are necessary to distinguish these functions in vivo.
(b). Chromatin remodelling
Eukaryotic DNA is packaged in a complex manner, which hinders transcription and DNA repair. Coordinated remodelling of chromatin at specific sites is critical to initiate such processes. Various modifications including DNA and histone methylation regulate chromatin remodelling, and are modulated by IP6K1. DNA methylation leads to long-term gene repression (Cedar & Bergman, 2009; Rose & Klose, 2014). Nuclear-localized IP6K1 enhances DNA methylation in a catalytic activity-dependent manner, which inhibits expression of the inositol biosynthetic gene mIno1 (Yu, Ye & Greenberg, 2016).
Conversely, histone methylation can either activate or inhibit the transcription process. For histone H3, methylation of the lysine 4 residue activates, whereas similar modification at the lysine 9 residue blocks the transcription process (Kouzarides, 2002; Rice et al., 2003). The interaction of IP6K1 with the histone lysine demethylase JMJD2C dissociates the demethylase from the chromatin and causes a corresponding increase in trimethyl-histone H3 lysine 9 (H3K9me3) levels. Accordingly, IP6K1-KO MEFs display increased association of JMJD2C with the chromatin and reduced levels of H3K9me3, which is reversed by expression of catalytically active but not inactive IP6K1. Thus, IP6K1 inhibits transcription of the JMJD2C target gene, the E3 ubiquitin ligase MDM2 (Burton et al., 2013). The molecular mechanism by which 5-IP7 regulates DNA or histone methylation is not understood.
(c). DNA damage and repair
IP6K1-KO MEFs show decreased viability and reduced recovery after induction of DNA damage. Although homologous recombination repair is initiated, it is not completed in knockout cells. Expression of catalytically active but not inactive IP6K1 restores repair in these cells, implying a requirement for 5-IP7 in this process (Jadav et al., 2013), although the precise mechanism is not known.
A response to DNA damage (DDR)-induced stress is regulated by various pathways including the ubiquitin proteasome system. Cullin complex (CRL4) is a multi-protein complex primarily composed of DDB1, DDB2 (XPE), cullin-4A (Cul4A), regulator of Cullins-1 E3 (Roc1-E3) and the Cop9 de-ubiquitination signalosome (CSN) (Bergink, Jaspers & Vermeulen, 2007; Scrima et al., 2011). Following ultraviolet (UV) radiation (and other DNA-damaging agents), CSN is dissociated from the complex, leading to activation of the E3 ligase, which ubiquitinates several components of the nucleotide excision repair (NER) machinery, which is required to initiate NER. In MEF cells, IP6K1 forms a ternary complex with CSN and CRL4, in which both IP6K1 and CRL4 are inactive under basal conditions. DDB1 present in the complex inhibits IP6K1 (Section IV). UV dissociates IP6K1 from the complex, which activates the kinase. IP6K1-generated 5-IP7 disrupts the CSN–CRL4 complex, which activates CRL4-mediated NER. Accordingly, IP6K1-KO MEFs display enhanced UV-induced CRL4 activation, NER and reduced apoptosis. Thus, IP6K1 mediates UV-induced disassembly of the CRL4–CSN complex, which regulates NER and apoptosis (Rao et al., 2014). The molecular details of the process by which 5-IP7 dissociates the signalosome–Cullin complex are unclear.
(d). Haemostasis
Haemostasis is a physiological response to prevent blood loss following vascular injury. In aberrant haemostasis, the timing of blood clotting is altered. Slow blood clotting leads to diathesis, which is a condition of susceptibility to bleeding. Conversely, abnormally fast clotting leads to thromboembolism, which can cause fatal pathological conditions such as stroke, pulmonary embolism, deep vein thrombosis, and myocardial infarction. 5-IP7 is a key player in maintaining haemostasis (Ghosh et al., 2013). IP6K1-KO mice display a significant delay in platelet aggregation, lengthening plasma clotting time (Ghosh et al., 2013) and hence a longer tail bleeding time. As a result, IP6K1-KO mice are protected from ADP-induced pulmonary thromboembolism. Mechanistic studies reveal that levels of polyP, a critical clotting factor, are reduced in the platelets of IP6K1-KO mice. Addition of polyP but not 5-IP7 rescues clotting in sera derived from IP6K1-KO mice, indicating that 5-IP7 modulates the process in vivo indirectly by regulating polyP levels (Ghosh et al., 2013) (Section V.3).
(e). Spermatogenesis
IP6K1-KO male mice are sterile, with few advanced spermatids in the testes, and no sperm in the epididymis (Bhandari et al., 2008). IP6K1 is highly expressed in round spermatids and is enriched in a perinuclear ribonucleoprotein complex called the chromatoid body. Deletion of IP6K1 impairs formation of the chromatoid body, which causes defects in elongation and condensation of sperm nuclei that subsequently block differentiation of spermatids. During spermiogenesis, nucleosomal histones are replaced by transition proteins 1 and 2 (TNP1 and TNP2) and protamines (PRM1 and PRM2). Premature expression of these proteins is associated with defective spermatid formation and infertility in mice. Juvenile IP6K1-KO mice display premature synthesis of TNP2 and PRM2 in round spermatids, which correlates with this phenotype (Malla & Bhandari, 2017). Thus, IP6K1 mediates the post-transcriptional regulatory machinery of testicular germ cells via a hitherto unknown mechanism. However, pharmacological inhibtion of the relevant IPP pathway does not cause sterility (Ghoshal et al., 2016) (Section VII.2). The molecular details by which IP6K1 regulates TNP2 and PRM2 are not known.
(f). Cell migration-mediated neuronal development and cancer metastasis
Cell migration is critical in many physiological and pathological motility processes such as development, immunity and cancer metastasis. Two independent studies demonstrated that IP6K1 regulates cell migration. IP6K1-KO MEFs display impaired cell spreading and migration. The knockout cells also display reduced tyrosine phosphorylation and activity of the focal adhesion kinase (FAK), which diminishes cell migration (Jadav et al., 2016). Expression of catalytically active IP6K1 reverses migration defects in these cells suggesting involvement of 5-IP7 in this process. Actin cytoskeleton remodelling and cell migration support the ability of cancer cells to achieve their oncogenic potential. Accordingly, cancer cells like HeLa and HCT116 with depleted IP6K1 levels, display reduced migration, invasion, and anchorage-independent growth (Jadav et al., 2016). Subcutaneous injections of control and IP6K1-depleted HCT116 cells in nude mice form similar-sized tumours, yet, the invasive property of IP6K1-depleted cells is significantly less. Accordingly, IP6K1-KO mice fed the oral carcinogen 4-nitroquinoline-1-oxide (4-NQO) show reduced progression of invasive carcinoma (Jadav et al., 2016). This is discussed further in Sections VI.4b,c in the context of IP6K2.
IP6K1/5-IP7-mediated cell migration is also crucial for brain development. IP6K1-KO mice display abnormal brain development due to impaired neuronal migration. IP6K1 localizes to focal adhesion and interacts with the microfilament protein α-actinin. Experiments in MEF and HeLa cells revealed that IP6K1 binds α-actinin, which promotes its tyrosine phosphorylation by FAK, intracellular localization and function. These events are critical for stress fibre formation, cell migration and spreading. Catalytic activity of IP6K1 is not required for binding, but is indispensable in FAK-mediated phosphorylation of α-actinin. Accordingly, 5-IP7 promotes FAK autophosphorylation-mediated activation in vitro (Fu et al., 2017), which demonstrates the mechanism by which IP6K1/5-IP7 promotes cell migration (Jadav et al., 2016; Fu et al., 2017). The molecular details of 5-IP7-mediated FAK phosphorylation are unknown.
(g). Behavioural responses
IP6K1-KO mice display certain behavioural alterations such as reduced locomotor activity in novel environments and impaired social interactions (Chakraborty et al., 2014). Behavioural responses are results of complex biological actions and thus, may arise due to multiple mechanistic changes in the knockout mice. One such change is altered Akt/GSK3 signalling in the brain. IP6K1 activates the protein kinase GSK3 by: (i) 5-IP7-mediated inhibition of its negative regulator Akt (Chakraborty et al., 2010); and (ii) by direct protein interaction (Sections V.1a and 4) (Chakraborty et al., 2014). Consequently, IP6K1-KO mice display reduced GSK3 activity in diverse brain regions. However, the extent to which the catalytic or protein-interaction activity of IP6K1 contributes in vivo remains unclear. Defective brain development due to impaired neuronal migration (Section VI.3f) may also cause these alterations. Generation of brain-specific transgenic mice with catalytically active and inactive versions of IP6K1 may provide answers, but the above studies at least suggest that IP6K1 is essential for brain development. Conversely, targeting IP6K1 in the brain may have beneficial effects in psychiatric and neurodegenerative diseases, where abnormalities in insulin signalling are demonstrated (Chakraborty et al., 2014).
(h). Neutrophil functions
Neutrophils are critical players in innate immunity and host defence. However, excessive accumulation or hyper-responsiveness of neutrophils can be detrimental to the host, so neutrophils are under stringent regulation. IPPs are major regulators of neutrophil function in infection and inflammation (Luo & Mondal, 2015). PIP3-induced Akt activation plays a critical role in regulating neutrophil chemotaxis, phagocytosis, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-mediated superoxide production. IP6K1 regulates neutrophil function through 5-IP7-mediated Akt inhibition (Section V.1a). The chemoattractant fMLP stimulates Akt activity in neutrophils by reducing 5-IP7 levels (Section III). Accordingly, fMLP-induced Akt activation is significantly higher in IP6K1-KO neutrophils. Moreover, IP6K1-KO neutrophils display enhanced Akt-mediated transient superoxide production and bactericidal activity. However, unlike HCT116 or neuron cells, IP6K1-KO neutrophils do not display alterations in cell adhesion or migration (Prasad et al., 2011).
Abnormal accumulation of neutrophils in the lungs generates reactive oxygen species (ROS), which augments pathological phenotypes in chronic obstructive pulmonary disease (COPD) (Barnes, 2003). A 5-IP7-mediated decrease in Akt activity promotes spontaneous neutrophil death, which protects mice from nicotine-induced lung inflammation. Cigarette smoke extract or nicotine reduces 5-IP7 in aging neutrophils, which delays spontaneous neutrophil death and thus contributes to the pathogenesis of COPD. Accordingly, deletion of IP6K1 augments cigarette smoke-induced neutrophil accumulation and lung damage (Xu et al., 2013). Thus, 5-IP7-mediated Akt inhibition and neutrophil death exert differential effects on immunity and COPD. IP6K1-KO macrophages display reduced phagosomal motility (Chanduri et al., 2016), which may reduce phagocytosis. Genrally, neutrophils respond to acute, whereas macrophages function during chronic inflammation. Thus, IP6K1 may differentially regulate inflammation.
(i). Insulin signalling
Insulin induces 5-IP7, which in turn inhibits the hormone’s stimulatory action on Akt (Sections III and V.1a) (Manning, 2010; Chakraborty et al., 2011) implying that IP6K1 deletion will enhance insulin sensitivity. Indeed, mild insulin sensitivity is observed in young, chow-fed IP6K1-KO mice (Bhandari et al., 2008), and significant sensitivity in older (10-month old) knockouts (Chakraborty et al., 2010). In gastrocnemius muscle, epididymal WAT (EWAT) and primary hepatocytes of the older knockouts, insulin-induced Akt activity is markedly increased. These results, together with higher IP7 level in older wild-type hepatocytes compared to younger controls (Section III), suggest that age-induced insulin resistance is caused, at least in part, by 5-IP7-mediated Akt inhibition (Chakraborty et al., 2010). IP6K1 regulates insulin signalling in the adipose tissue, which in turn modulates global insulin sensitivity. As a result, both global (IP6K1-KO) and adipocyte-specific IP6K1-KO (AdKO) mice are also protected from high fat diet (HFD)-induced insulin resistance (Chakraborty et al., 2010; Zhu et al., 2016). HFD-fed AdKO mice display higher Akt activity in the EWAT, liver and gastrocnemius muscle. Thus, adipocyte-specific IP6K1 deletion alters expression/secretion of certain adipokines, enhancing insulin sensitivity in other tissues. Indeed, the plasma level of the insulin-sensitizing adipokine adiponectin (ADIPOQ) is higher in HFD-fed AdKO mice (Zhu et al., 2016). Serum insulin level is lower in HFD-fed AdKO mice compared to the corresponding wild-type, which suggests that adipocyte-specific IP6K1 deletion-mediated insulin hypersensitivity reduces insulin secretion indirectly (Zhu et al., 2016). Thus, IP6K1 regulates insulin secretion and signalling via pleiotropic mechanisms, which can be targeted in type-2 diabetes (T2D) (Chakraborty et al., 2011; Boucher, Kleinridders & Kahn, 2014; MacKenzie & Elliott, 2014; Zhang, Liu & Liu, 2017).
(j). Energy metabolism
IPPs are proposed to be metabolic messengers (Shears, 2009). Yet, the suggestion that IP6K1 inhibits energy expenditure (EE) emerged from the observation that IP6K1-KO mice are protected from HFD-induced weight gain despite eating the same amount of food (Chakraborty et al., 2010; Zhu et al., 2017). Furthermore, cold exposure, which enhances adipose tissue browning-mediated thermogenesis, reduces IP6K1 expression. These results suggest that IP6K1 deletion reduces weight gain by enhancing EE in adipocytes. Protection of AdKO mice from HFD-induced weight gain further supports this possibility (Zhu et al., 2016). Stored energy is expended by coupled respiration that generates ATP and/or by uncoupling protein-1 (UCP1)-mediated uncoupled respiration that releases heat. Brown adipocytes in brown adipose tissue (BAT) or beige adipocytes in WAT expend energy primarily by thermogenesis. Therefore, enhanced browning of WAT facilitates thermogenesis-mediated fat loss in rodents and humans (van Dam et al., 2015; Sidossis & Kajimura, 2015, and references therein). Indeed, both IP6K1-KO and AdKO mice expend more energy, at least in part, due to enhanced thermogenic EE (Chakraborty et al., 2010; Zhu et al., 2016, 2017). Stromal vascular fractions isolated from WAT display higher beige adipogenesis, further evidenced by enhanced expression of the EE machinery. Accordingly, mitochondrial oxygen consumption rate (OCR) is higher in AdKO beige adipocytes, whereas glycolysis is unaltered.
The protein kinase AMPK enhances EE (Zhu et al., 2016, and references therein). IP6K1 deletion augments EE by stimulating AMPK via a novel mechanism. IP6 stimulates LKB1-mediated stimulatory phosphorylation of the AMPKα (catalytic) subunit at the threonine 172 residue, whereas IP7 is ineffective. Catalytically active IP6K1 interferes with IP6-mediated AMPK activation. The amino acid residues 146– 205 in IP6K1 are required for its interaction with AMPKα (Fig. 3A). Presumably, IP6K1 interferes with IP6-mediated AMPKα activation by converting IP6 to 5-IP7 (or IP5*) (Fig. 2). Thus, dynamic alterations in IP6/5-IP7 ratio in a microenvironment also regulate protein targets (Zhu et al., 2016). However, the molecular mechanism by which IP6 enhances AMPK phosphorylation is not understood. IP6K1 may also indirectly inhibit intracellular AMPK. Similar to IP6K2 (Rao et al., 2015; Section VI.4c), IP6K1-generated 5-IP7 may promote nuclear localization of LKB1, which is known to perturb AMPK activation (Xie et al., 2009). Nevertheless, IP6K1-mediated inhibition of AMPK reduces adipocyte EE, promoting weight gain (Zhu et al., 2016). AMPK also displays insulin-sensitizing effects, which may contribute to insulin hypersensitivity in IP6K1-deleted mice. Although AMPK and Akt display synergistic effects on insulin sensitivity, they have differential roles in cell proliferation and survival. In general, Akt stimulates but AMPK inhibits these processes. Thus, in certain tissues/conditions, if IP6K1 inhibits one kinase, it may activate the other.
Moreover, IP6K1 regulates EE by modulating lipolysis via protein– protein interaction, which does not require catalytic activity of IP6K1 (Ghoshal et al., 2016). In lipolysis, lipases hydrolyse stored triglycerides (TAGs) that release free fatty acids (FFA) and glycerol. These products are utilized at various stages of the EE pathway. PLIN and comparative gene identification-58 (CGI-58) proteins regulate lipolysis by modulating activities of various lipases. PLIN1 binds and perturbs the stimulatory action of CGI-58 on adipose triglyceride lipase (ATGL) and thus, inhibits basal lipolysis (Ghoshal et al., 2016). Basal lipolysis is enhanced in IP6K1-KO mice. IP6K1 is a major PLIN1-interacting protein in the adipose tissue. Furthermore, PKA/PKC phosphorylation of IP6K1 at serine residues 118 and 121 (Fig. 3A) facilitates its binding to PLIN1, which modulates lipolysis (Section IV) (Ghoshal et al., 2016). Thus, IP6K1 regulates lipolysis and thermogenesis to promote fat accumulation.
IP6K1 also inhibits EE in MEFs, albeit via reducing glycolysis (Szijgyarto et al., 2011). Under basal conditions, IP6K1-KO MEFs display higher ATP levels due to increased glycolysis, but mitochondrial activity is compromised. The mechanism of this phenotype is known in S. cerevisiae, which exhibit similar phenotypes in the absence of the IP6K ortholog KCS1. The interaction between glycolytic genes transcriptional activators 1 and 2 (GCR1 and GCR2) is essential for transcription of the glycolytic genes. The KCS1-KO strain displays increased GCR1–GCR2 interaction, which stimulates expression of glycolytic genes such as GAPDH and PGK1. In vitro pyrophosphorylation and mutation studies suggest that 5-IP7 pyrophosphorylates (Section V.2) GCR1 and destabilizes its interaction with GCR2, which subsequently reduces glycolysis (Szijgyarto et al., 2011).
Experiments in HCT116 cells suggest that the IPP 1,5-IP8 plays a role in regulating ATP level, although the precise mechanism by which 1,5-IP8 regulates this process is unknown. In these cells, PPIP5K deletion eliminates 1,5-IP8 (but increases 5-IP7) and elevates ATP levels. PPIP5K-KO cells accumulate more ATP due to increased EE caused by enhanced glycolysis and oxidative phosphorylation. Overexpressed active but not kinase-inactive PPIP5K1 restores 1,5-IP8 level and returns ATP to normal levels, whereas reduction of 5-IP7 does not (Gu et al., 2017). IP6K1 may also regulate EE partly via 1,5-IP8. Although the IPP pathway inhibits EE, the precise mechanism may vary depending on the specific enzyme and cell type/conditions as energy metabolism varies among cell types, conditions and proliferative nature (Bereiter-Hahn, Munnich & Woiteneck, 1998).
(k). Bone marrow-derived mesenchymal stem cell (BM-MSC) fitness and skeletal involution
During skeletal aging, oxidative stress reduces bone mineral density and increases marrow adiposity due to impaired osteogenesis and enhanced adipogenesis of bone marrow-resident MSCs. For this reason, obese and/or diabetic patients have an increased risk of fracture. Therefore, targeting pathways that prevent these dysfunctions have therapeutic potential in bone diseases. IP6K1-KO mice display an increased yield of BM-MSCs. Moreover, IP6K1-KO MSCs exhibit enhanced growth, survival, osteogenesis and haematopoiesis. Conversely, they show reduced adipogenesis, which further explains reduced adipose mass in these animals (Chakraborty et al., 2010). Unlike adipocytes (Section VI.3j), IP6K1-KO MSCs display unaltered OCR, yet a reduced ROS level. Oxidative stress induces p53-mediated growth arrest and apoptosis in murine MSCs. Accordingly, p53 protein level is reduced and its E3 ubiquitin ligase mouse double minute-2 homolog (MDM2) is increased in IP6K1-KO MSCs. IP6K1 inhibits the transcription of MDM2 by inhibiting the demethylase JMJD2C in MEFs (Section VI.3b), although this mechanism has not been tested in MSCs. In summary, IP6K1-mediated regulation of MSCs promotes fat accumulation but reduce bone formation (Boregowda et al., 2017). The therapeutic significance of this finding in osteoporosis is discussed in Section VII.1b.
IP6K1, which is abundant in brain and testis (Section II.1), is essential for development of these organs. Conversely, its role in metabolic and other tissues is regulatory, representing a possible therapeutic targeted in diet- or age-induced diseases.
(4). IP6K2
(a). Apoptosis
IP7 level is increased during apoptosis (Section III). Several studies suggest an essential role of IP6K2-generated 5-IP7 in this process. An antisense screening study in OVCAR3 cells first identified IP6K2 as a mediator of IFNβ-induced apoptosis (Morrison et al., 2001). In addition to OVCAR3 cells, IP6K2 also sensitizes various other cells including HeLa, HEK293, PC12, Jurkat and HL60 to apoptotic actions of cisplatin, etoposide, hydrogen peroxide, staurosporine, hypoxia, γ-irradiation and mutant huntingtin expression (Morrison et al., 2002; Nagata et al., 2005, 2011). Consequently, IP6K2-KO mice display a better survival rate following total body irradiation (8–10 Gy; 1 Gy = 1J radiation/kg) (Morrison et al., 2002, 2009).
Catalytically active IP6K2 regulates the transcriptional function of p53 (Koldobskiy et al., 2010). In HEK293 cells, the chaperone HSP90 binds IP6K2 at the amino acid region 132– 143, which inhibits the kinase (Fig. 3B). Disruption of IP6K2–HSP90 binding promotes the activation and nuclear localization of IP6K2 (Chakraborty et al., 2008; Koldobskiy et al., 2010). Nuclear IP6K2 binds p53 via amino acids 1–67 (Fig. 3B), which suppresses p53-mediated transcription of the cell cycle arrest regulator p21. As a result, DNA repair is hampered, and the cells undergo apoptosis. Conversely, IP6K2 enhances the expression of one of p53’s apoptotic targets, phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1 or NOXA). Thus, IP6K2 reduces the cell cycle arrest functions of p53, whereas it enhances its apoptotic actions (Koldobskiy et al., 2010). Subsequent studies provided further details about the mechanism involved (Rao et al., 2014). The phosphoinositide-3-kinase-related kinases (PIKKs) DNA dependent protein kinase (DNA-PK) and ataxia–telangiectasia-mutated protein kinase (ATM) phosphorylate and activate p53. The TTT co-chaperone family of proteins stabilises PIKKs. The C-terminus of IP6K2 (amino acids 212–426, Fig. 3B) binds to the TTT complex through direct interactions with Tti1, which facilitates 5-IP7’s binding-mediated activation of CK2. 5-IP7 binds more potently than IP6 (IC50 of 5-IP7 ~ 0.4 μM; IC50 of IP6 ~14 μM) to the lysine-rich cluster of CK2α, which facilitates access of CK2’s substrate to the kinase. Activated CK2 stimulates phosphorylation of the TTT complex, thereby enhancing the ability of DNA-PK/ATM to activate p53. Thus, in IP6K2-KO HCT116 cells, DNA damage (etoposide)-mediated induction of p53 is reduced, attenuating expression of its apoptotic targets PUMA and NOXA. Even a reduced level of p53 is sufficient to elevate p21 in IP6K2-KO cells (Rao et al., 2014). Studies in PPIP5K-KO HCT116 cells further demonstrate the impacts of IPPs on cell proliferation. PPIP5K deletion increases levels of 5-IP7 (opposite effect to IP6K2-KO), decreases 1,5-IP8 (similar to IP6K2-KO), enhances p53 (opposite to IP6K2-KO) and p21 (similar to IP6K2-KO) and reduces proliferation (similar to IP6K2-KO) (Gu et al., 2017). Thus, although IP6K2 or PPIP5K deletion reduces cell proliferation, the mechanisms cannot be attributed solely to differences in IPPs. IP6K2 and PPIP5K may have distinct targets in this process.
Catalytic activity-independent apoptotic action of IP6K2 has also been reported. IP6K2 interacts with TRAF2, which interferes with transforming growth factor beta-activated kinase-1 (TAK1) phosphorylation, nuclear factor kappa B (NF-κB) signalling, and apoptosis. Overexpression of the TRAF2-binding deficient IP6K2 (S347A/S359A) (Fig. 3B) mutant in OVCAR3 cells enhances NF-κB signalling, which partly impairs tumour necrosis factor alpha (TNFα)-induced apoptosis (Morrison et al., 2007). On the other hand, CK2, which mediates cell survival, degrades IP6K2 by phosphorylating it at residues S347 and S356 (Fig. 3B) (Section IV) (Chakraborty et al., 2011). Further studies are required to determine the precise effects of these IP6K2 residues in stressor-mediated apoptosis. Although the apoptotic function of IP6K2 is established, to what extent its subcellular distribution regulates this process remains unclear. Besides a nuclear role (Morrison et al., 2005; Koldobskiy et al., 2010), apoptotic functions of mitochondrial (Nagata et al., 2005) and cytosolic (Nagata et al., 2011, 2016; Moriya et al., 2017) IP6K2 have also been reported, which require elucidation.
(b). Chemical-induced carcinogenesis
IP6K2-KO mice display normal embryogenesis, development, growth, blood parameters, serum insulin level and fertility (Morrison et al., 2009). Although these mice do not develop spontaneous tumours, they do show accelerated development of aerodigestive tract carcinoma when exposed to the UV-mimetic chemical carcinogen 4-NQO (Morrison et al., 2009). Enhanced susceptibility of IP6K2-KO mice to this chemical carcinogen is associated with upregulation of the transcription termination factor 1 (TTF1) and the twist family of basic helix-loop-helix transcription factor neighbour protein (TWISTNB) that may have implications in cancer (Morrison et al., 2009). Conversely, downregulation of putative tumor suppressor genes like DUSP16 and EXT2 are observed in IP6K2-KO mice. IP6K2 promotes the apoptosis actions of p53 (Koldobskiy et al., 2010; Rao et al., 2014). Transgenic mice with a dominant negative p53 mutation are also susceptible to 4-NQO-induced oral cancer (Zhang et al., 2006). Therefore, susceptibility of IP6K2-KO mice to 4-NQO-induced cancer may involve p53 activity. IP6K2, but not IP6K1, exhibits apoptotic functions, which may partly explain why IP6K1 deletion protects, whereas IP6K2 deletion augments chemical carcinogenesis.
(c). Cell migration and cancer metastasis
IP6K2 enhances cell–matrix adhesion, but reduces cell–cell adhesion (Rao et al., 2015). IP6K2-KO HCT116 cells exhibit delayed growth, reduced cell–matrix adhesion, spreading and tyrosine phosphorylation of FAK (see Section VI.3f). Conversely, cell–cell adhesion and level of the adhesion-promoting protein zona occludin-1 (Zo-1) are increased in knockout cells. As a result, these cells display reduced anchorage-independent growth and migration. In vitro tumour invasion, monitored by tumour growth in a collagen invasion assay, is greatly reduced by IP6K2 deletion. Moreover, subcutaneous xenograft tumours grown from IP6K2-KO HCT116 cells are reduced in size. Knockout xenografts, when implanted into the caecum of nude mice, display reduced growth and diminished metastasis to liver. Overexpression of catalytically active but not inactive IP6K2 restores adhesion and associated features in the knockout cells. A complex mechanism is suggested. The tumour suppressor LKB1 inhibits cell migration and metastasis via activation of cytosolic tyrosine phosphatases that dephosphorylate FAK. The catalytic activity of IP6K2 is not required for binding, but is critical for nuclear sequestration of LKB1, which inhibits the phosphatases. Accordingly, IP6K2-KO cells accumulate cytosolic LKB1 and display higher tyrosine phosphatase activity, which facilitates FAK dephosphorylation and cell migration. However, direct evidence of enhanced LKB1 phosphorylation-dependent activation of phosphatases in IP6K2-KO cells is lacking. Nevertheless, results obtained in two different studies indicate that IP6K1- or IP6K2-generated 5-IP7 promotes cell migration via: (i) direct stimulation of FAK autophosphorylation (Fu et al., 2017); and/or (ii) LKB1–tyrosine phosphatase-mediated indirect regulation of FAK (Rao et al., 2015).
Based on the studies discussed here and in Sections VI.1b and 3f, it is clear that deletion of IP6K1 or IP6K2 reduces cell migration, whereas a reduction of DIPP1 level has the opposite effects (Grudzien-Nogalska et al., 2016). Conversely, deletion of IP6K2 but not IP6K1 reduces tumour volume. This phenotypic variation may arise due to: (i) distinct methods of disruption of these genes; (ii) differential expression patterns of these isoforms; or (iii) additional, yet to be identified mechanisms. Understandably, 5-IP7 levels in these two systems are different. IP6K2-KO HCT116 cells display an almost total loss, whereas IP6K1-depleted HCT116 cells show a modest decrease in IPP levels (Koldobskiy et al., 2010; Jadav et al., 2016). Perhaps cell migration but not tumour growth is susceptible to a smaller decrease in IPP levels (Jadav et al., 2016). Tissue-specific IP6K-KO mice or isoform-selective inhibitors may allow the roles of these isoforms in the initiation, progression and metastasis of cancer to be clarified.
(5). IP6K3
(a). Synapse formation and motor function
Although IP6K3-KO mice do not display gross abnormalities in brain structure, their motor learning and coordination are disturbed, which suggests Purkinje cell dysfunction. IP6K3 is highly expressed in Purkinje cells. Moreover, IP6K3-KO mice display alterations in Purkinje cell structure with withered dendritic trees. The cell size and spine density of mutant Purkinje cells are also decreased in these knockouts. Both inhibitory symmetric and excitatory asymmetric synapses in the cerebellar molecular layer of IP6K3-KO mice are reduced. IP6K3-KO cerebella display reduced levels of the γ-aminobutyric acid (GABA)-synthesizing enzyme glutamic acid decarboxylase (GAD65), which is a marker of GABAnergic inhibitory neurons. Whether IP6K3 directly influences GAD65, is not known. The shape of Purkinje cell dendritic spines is determined by the arrangements and attachments of their cytoskeletal proteins such as adducin and spectrin. IP6K3 binds to the calponin homology (CH) domain of adducin and spectrin, whose mutual interactions are perturbed in IP6K3-KO mice. Catalytic activity of IP6K3 is not required to maintain β-adducin–β2-spectrin binding (Fu et al., 2015).
(b). Metabolism and aging
IP6K3 is highly expressed in both mouse and human myotubes and muscle tissues (Moritoh et al., 2016). Furthermore, IP6K3 expression is elevated in skeletal muscle under diabetic, fasting, and disuse conditions. Chow-fed, aged (1.5-year old) IP6K3-KO mice display reduced body mass and accumulate less fat, although they eat a similar amount of food. Moreover, aged IP6K3-KO mice exhibit lower blood glucose and reduced circulating insulin levels. These mice also exhibit increased plasma lactate levels, suggesting that glycolysis is increased in the knockouts. IP6K3-KO mice (15-weeks old) also display enhanced glucose removal during glucose- and insulin-tolerance tests. The knockouts exhibit reduced expression of the muscle pyruvate dehydrogenase kinase-4 (PDK4) enzyme. PDK4 inhibits pyruvate to acetyl coenzyme A (acetyl-CoA) conversion and thus inhibits glucose oxidation. Thus, reduced PDK4 expression may also increase glucose oxidation, which may reduce lipogenesis-mediated fat accumulation in these mice. Published results (Chakraborty et al., 2010) and recent experiments (S. Ghoshal & A. Chakraborty, unpublished data) suggest that IP6K1-KO mice are also protected from age-induced weight gain and insulin resistance. However, unlike IP6K1-KO, IP6K3-KO mice are not protected against diet-induced obesity (DIO) (Moritoh et al., 2016).
IP6K3-KO mice exhibit an increased survival rate, although the lifespan experiment was not conducted to completion (Moritoh et al., 2016). Phosphorylation of the mTORC1 effector S6 ribosomal protein is reduced in heart but not in skeletal muscle of IP6K3-KO mice, although stimulatory phosphorylation of the direct mTORC1 target S6 kinase-1 (S6K1) and the mTORC2 target Akt are unchanged (Moritoh et al., 2016). Although mTOR activity is associated with aging (Kennedy & Lamming, 2016), it is not clear whether the same mechanism is operative in IP6K3-KO mice. Although the impact of IP6K1 on mTOR pathway in the heart is unknown, in various IP6K1-KO cells and in the skeletal muscle of IP6K1-KO HFD-fed mice, it is upregulated along with the Akt pathway (Chakraborty et al., 2010). Further studies are needed to distinguish tissue-specific functions of IP6K1 and IP6K3 in metabolism and aging.
VII. PHARMACOLOGICAL TARGETING OF IP6KS
(1). Characterization of the pan-IP6K inhibitor TNP
The above studies indicate that pharmacological targeting of the IPP pathway may have beneficial effects in diseases (Chakraborty et al., 2011; MacKenzie & Elliott, 2014; Shears, 2016). The compound N2-(m-trifluorobenzyl)N6-(p-nitrobenzyl)purine (TNP) was originally discovered as an IP3–3K inhibitor (Chang et al., 2002), but was later found to be more potent on IP6Ks. TNP is fairly specific to IP6Ks; it does not inhibit PPIP5Ks or ~70 other kinases (Padmanabhan et al., 2009). TNP reduces 5-IP7 level in various cells (Padmanabhan et al., 2009; Chakraborty et al., 2010; Prasad et al., 2011; Wundenberg et al., 2014; Zhang et al., 2014; Sun et al., 2015). Moreover,TNP inhibits both kinase and phosphatase activities of IP6K on IP6 (Wundenberg et al., 2014). Accordingly, TNP regulates IP6K-mediated cellular processes. For example, TNP hampers the tyrosine phosphorylation and membrane localization of α-actinin, which reduces migration of HeLa cells (Fu et al., 2017). TNP substantially increases Cul4A–CSN binding and attenuates the UV-elicited dissociation of the Cul4A–CSN complex, which conforms with the role of 5-IP7 in this process (Rao et al., 2014). Under various conditions, TNP increases Akt activity in HEK293 cells, MEFs, neutrophils, aged MSCs and diabetic cardiomyocytes (Chakraborty et al., 2010; Prasad et al., 2011; Xu et al., 2013; Zhang et al., 2014; Sun et al., 2015), whereas it enhances AMPK activity in adipocytes and diabetic cardiomyocytes (Sun et al., 2015; Zhu et al., 2017). TNP enhances EE, whereas it inhibits fatty acid biosynthesis in 3T3L1 cells (Zhu et al., 2017). TNP is also bioavailable. For example, 6 h post-administration of a single intraperitoneal dose, TNP accumulated at ~10–25 μM in liver, skeletal muscle, and adipose tissue, although to a much lesser extent (0.63 μM) in the brain. Plasma protein binding (PPB) of TNP is 90–93% which is similar to or better than a number of FDA-approved drugs (Ghoshal et al., 2016).
(a). TNP ameliorates DIO and insulin resistance
Similar to IP6K1 deletion (Sections VI.3i,j) (Chakraborty et al., 2010; Zhu et al., 2016, 2017), TNP protects mice from DIO and associated metabolic abnormalities (Ghoshal et al., 2016). Both short-term (2 weeks) and long-term (10 weeks) TNP treatments (10–20 mg/kg daily, intraperitoneal) reduce fat mass, reduce fatty liver, enhance insulin sensitivity and restore metabolic homeostasis in DIO mice without altering food intake. TNP decelerates both initiation and progression of DIO and insulin resistance in mice (Zhu et al., 2016; Ghoshal et al., 2016). These effects are abolished in IP6K1-KO mice, indicating that at least part of the beneficial effects of TNP on metabolism act via inhibition of IP6K1. TNP enhances insulin sensitivity in DIO mice via Akt activation (Ghoshal et al., 2016). Moreover, TNP reduces weight gain by enhancing AMPK-mediated browning and EE in the adipose tissue and adipocytes (Zhu et al., 2016). Thus, TNP has strong anti-obesity and anti-diabetic effects.
(b). TNP attenuates HFD-induced loss of trabecular bone volume
IP6K1-KO mice display increased MSC fitness and osteogenesis (Section VI.3k). Accordingly, long-term TNP treatment (Ghoshal et al., 2016) increases yield and colony-formation capacity of MSCs in DIO mice (Boregowda et al., 2017). TNP conserved trabecular bone, but decreased marrow adiposity. Therefore, in addition to its beneficial effects on obesity and T2D, TNP also protects mice from HFD-induced MSC dysfunctions in the marrow (Boregowda et al., 2017). Insulin sensitizers like peroxisome proliferator-activated receptor gamma (PPARγ) antagonists increase obesity and fracture risk. Thus, IP6K1 inhibitors may have greater acceptability as anti-obesity and anti-diabetic drugs due to their beneficial effects on bone properties.
(c). TNP protects against cardiac I/R injury in leptin receptor mutant db/db mice
The cytokine OSM, which works via activation of the OSM receptor, alleviates cardiac ischaemic/reperfusion (I/R) injury by inhibiting cardiomyocyte apoptosis in leptin receptor-deficient obese and diabetic db/db mice (Sun et al., 2015). OSM enhances mitochondrial biogenesis and function, whereas it reduces IP7 levels in db/db mice subjected to cardiac I/R injury (Section III). Conversely, OSM receptor knockout increases IP7, impairs mitochondrial biogenesis, glucose homeostasis and insulin sensitivity and augments cardiomyocyte apoptosis, together aggravating cardiac I/R injury. Short-term TNP treatment (10 mg/kg daily, tail vein injection) mimics the effects of OSM in db/db mice (Sun et al., 2015). OSM exerts pleiotropic signalling effects, which may regulate 5-IP7 production, although the precise mechanism is not clear.
(d). TNP improves aging effects of BM-MSCs and their therapeutic efficacy for myocardial infarction (MI)
Decreased viability and impaired function of aged BM-MSCs hamper their therapeutic efficacy. Aged (18-month old) BM-MSCs exhibit higher 5-IP7 levels compared to young cells (Section III). In addition, Akt activity is decreased, whereas apoptosis is increased in aged BM-MSCs. TNP treatment (10 μM, 2 h) reduces 5-IP7 level and apoptosis, whereas it enhances Akt activity in aged BM-MSCs. Levels of angiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), IGF-1 and hepatocyte growth factor (HGF), which are decreased in aged BM-MSCs, are restored by TNP treatment (Zhang et al., 2014). Moreover, TNP-treated BM-MSCs, when transplanted into infarcted hearts, display enhanced survival, which promotes their anti-apoptotic and pro-angiogenic efficacy in vivo. Thus, TNP-treated BM-MSC therapy decreases fibrosis and preserves heart function (Zhang et al., 2014).
(2). Potential risks of IP6K inhibition
From the above discussion, it is clear that the IPP metabolic pathway regulates major cellular processes (Fig. 7), via diverse mechanisms (Fig. 6), modulating various phenotypes in mice (Fig. 8). Although targeting this pathway is beneficial in certain disease models, it may also have potential risks (Fig. 8). For example, IP6K1-KO mice exhibit premature death, impaired social interaction and male sterility (Bhandari et al., 2008; Chakraborty et al., 2014; Fu et al., 2017; Malla & Bhandari, 2017), whereas IP6K3-KO mice display reduced motor learning and coordination (Fu et al., 2015). IP6K1 deletion also aggravates lung inflammation in nicotine-exposed mice (Xu et al., 2013). Moreover, platelet aggregation is delayed in these knockouts, which may cause excess bleeding. IP6K2-KO mice are sensitive to chemical carcinogenesis (Morrison et al., 2009). Therefore, it is critical to determine whether pharmacological inhibition of IP6Ks can cause these aberrations. Chronic TNP treatment (15 weeks, 10 mg/kg daily) does not lead to neuronal or reproductive anomalies (Ghoshal et al., 2016). TNP does not cross the blood– brain barrier (BBB) efficiently. Functioning in a similar way to the BBB, the blood– testes barrier (BTB) blocks entry of drugs in the apical compartment (Cheng & Mruk, 2012), which may explain the ineffectiveness of TNP in these organs. Thus, developmental defects caused by germ-line deletion of IP6K1 are not observed following its pharmacological inhibition in adult mice. Studies involving very long-term treatment should be conducted to rule out such concerns.
Fig. 7.

The inositol pyrophosphate (IPP) metabolic pathway regulates various cellular processes in mammals.
Fig. 8.
Summary of benefits and potential risks of perturbation of the inositol pyrophosphate (IPP) pathway. Studies using IP6K1-, IP6K2- and IP6K3-knockout mouse models and pan-pharmacological inhibition reveal that disruption of the pathway is beneficial in several diseases (highlighted in green). However, the knockout mice also display isoform-specific aberrations (highlighted in pink). Thus, although the IPP pathway is a novel and encouraging therapeutic target, precautions must be taken during drug development to minimize potential risks. Development of isoform-selective inhibitors would be beneficial. 4-NQO, 4-nitroquinoline-1-oxide; BM-MSC, bone marrow-derived mesenchymal stem cell; I/R, ischaemia/reperfusion; MI, myocardial infarction; MSC, mesenchymal stem cell.
Does Akt activation represent a potential cancer risk in patients treated with IP6K inhibitors? Gene amplification or constitutive hyperactivation of Akt kinase (~10- to 50-fold) (Cheng et al., 1996; Vincent et al., 2011) causes tumorigenesis (Mundi et al., 2016). However, tumours are not formed if Akt hyperactivation is not achieved (Mundi et al., 2016). IP6K1 deletion only causes a ~2- to 3-fold increase in phosphorylation stimulated by Akt in insulin-treated mice (Chakraborty et al., 2010). DIO mice with IP6K1 deletion exhibit normal Akt activity, unlike DIO mice with intact IP6K1 in which Akt activity is reduced (Zhu et al., 2016). Therefore, it is unlikely that IP6K inhibition will result in Akt-mediated tumorigenesis. IP6Ks regulate various other pathways and thus have differential effects on cell proliferation and survival (Chakraborty et al., 2011; Wilson et al., 2013; Thota & Bhandari, 2015; Zhu et al., 2016). IP6K1 or IP6K3 knockout mice do not display tumorigenesis (Chakraborty et al., 2010; Jadav et al., 2016; Moritoh et al., 2016). In fact, IP6K1-KO mice are protected from chemical carcinogenesis and colon cancer metastasis (Jadav et al., 2016). IP6K2 deletion, on the other hand, sensitizes mice to chemical carcinogenesis (Morrison et al., 2009), although it protects them from cancer metastasis (Rao et al., 2015). However, TNP does not cause tumorigenesis. To summarize, the therapeutic significance of IP6K inhibition in cancer is yet to be determined.
(3). Development of safe and isoform-selective IP6K inhibitors is necessary
TNP inhibits all three IP6K isoforms. It also inhibits human cytochrome P450s (Ghoshal et al., 2016), which may interfere with the metabolism of other drugs. Moreover, TNP slightly elevates Ca2+ levels in human promyelocytic leukemia (HL60) cells (Chang et al., 2002) and cytosolic Zn2+ levels in cortical neurons (Stork & Li, 2010) and thus, may cause signalling alterations. At higher concentrations, TNP inhibits IP3–3K which enhances neurite outgrowth in PC12 cells (Eva et al., 2012). TNP also enhances the stimulatory phosphorylation of ERK in PC12 cells (Eva et al., 2012), but inhibits the same process in endothelial cells (Sekar et al., 2014). TNP has a half-life (T1/2) of 32 and 9 min in human and mouse hepatic microsomal stability assays (Ghoshal et al., 2016). The development of an IP6K1 inhibitor with greater metabolic stability is required to achieve oral efficacy in patients. Thus, although TNP is an exciting proposition, further improvements are required before it advances to the next level. To minimize undesirable effects, isoform-selective and safer inhibitors are needed. BBB- and BTB-impermeable IP6K1 (or IP6K1 + IP6K3) selective inhibitors are desirable for the treatment of diet- or age-induced metabolic diseases. Furthermore, the therapeutic window for each disease should be determined.
VIII. THE IPP PATHWAY IN HUMAN HEALTH
Pathways regulated by IPP enzymes are highly conserved in mice and humans. Thus, pharmacological targeting of IPP enzymes is expected to work similarly in human subjects. Yet, murine and human IP6Ks do exhibit certain differences. For example, although IP6K isoforms are expressed in human tissues, isoform-specific expression may vary between mice and humans (Moritoh et al., 2016) (Section II). In addition, regulatory domains of IPP enzymes display minor variations in amino acid sequences, which may alter certain in vivo functions.
Limited information is available on gene expression and polymorphism profiles of IPP enzymes in diseases. An epigenome-wide association study identified higher CpG methylation upstream of IP6K1 in the saliva of heavy (BMI = 24.1) adolescent Finnish girls compared to lean (BMI = 14.4) girls (Rounge et al., 2016), suggesting diminished expression of the gene in obesity. It is conceivable that IP6K1 expression is reduced in a natural negative-feedback mechanism. Disruption of IP6K1 at intron 1 was observed in a single Japanese family with type-2 diabetes (Kamimura et al., 2004). However, this anomaly was not found in 405 unrelated patients, indicating the disrupted IP6KI is either family-specific or a chance association. An intron-specific NUDT3 single nucleotide polymorphism (SNP) (rs206936) is associated with increased BMI and obesity but not metabolic disorders in Japanese women. The rs206936 polymorphism may enhance gene expression, although confirmation is required (Kitamoto et al., 2013).
IP6K2 is located in chromosome 3p21, with an array of other tumour-suppressor genes including NPRL2, TUSC2, RASSF1A, SEMA3B, and SEMA3F (Shames & Minna, 2008). A missense mutation in IP6K2 (R325C; arginine to cysteine) was reported in patients with low-grade serous carcinoma (LGSC) (Jones et al., 2012). The arginine residue is adjacent to the FYSSSLL motif of the enzyme, which is critical for its catalytic activity. Loss of the IP6K2 gene, among others, was reported in B-cell chronic lymphocytic leukaemia (CLL) patients (Kay et al., 2010).
A genetic variability study identified IP6K3 polymorphism in Alzheimer’s disease. The SNPs rs28607030 and rs10947435, located in the 5j-flanking region of IP6K3, are associated with late-onset alzheimer’s disease (LOAD) risk (Crocco et al., 2016). The ‘G’ allele of SNP rs28607030 in the promoter region of IP6K3 forms an intact binding site for the cellular myelocytomatosis viral oncogene homolog (Myc) transcription factor and thus shows higher transcriptional activity than the corresponding ‘A’ allele in a luciferase assay (Crocco et al., 2016). The impact of this SNP on endogenous IP6K3 is not known. The above studies are interesting, albeit inadequate, as in many cases mRNA expression does not correlate with protein or activity level (Tian et al., 2004). Further studies are required to determine activity, post-translational modifications, localization and protein interactions of these enzymes in human tissues.
IX. CURRENT AND FUTURE DIRECTIONS OF IPP RESEARCH IN MAMMALS
(1). Complete characterization of IP6K-KO mice and generation of other models
Although IP6K-KO mice are fairly well characterized, further details are necessary. Transgenic and inducible mouse models of IP6K, PPIP5K and DIPP are required to determine the IPP-dependent and independent functions of these enzymes. Knockout cell lines of PPIP5K reveal that these enzymes are also critical in modulating cell proliferation and metabolism (Gu et al., 2017). Therefore, genetic-knockout mouse models of PPIP5K and DIPP are needed to study their roles in vivo. Appropriate tissue-specific mouse models are also required. Most importantly, impacts of the pathway in human health and diseases should be assessed.
(2). Design high-throughput assays and obtain structural information for IPP-metabolizing enzymes
The only available IP6K inhibitor TNP is not an ideal lead compound (Section VIII.3). Although PAO and U73122 inhibit IP6K1 activity in vitro (Rajasekaran et al., 2017) (Section III), they are not exciting lead compounds. The toxic arsenic atom in PAO (Ralph, 2008) and off-target effects of U73122 (Huang et al., 2013) limit their use. Thus, chemical libraries should be screened to discover new inhibitors. A rate-limiting step in this approach is lack of a high-throughput assay. Although radiolabelled-HPLC or non-radiolabelled sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and mass-spectrometry-based methods (Azevedo & Saiardi, 2006; Draskovic et al., 2008; Losito et al., 2009) are useful for estimating IP6K activity and cellular IPP levels, they are not suitable for high-throughput screening of thousands of compounds. A recent paper addressed this issue by designing a high-throughput assay for IP6Ks (Wormald et al., 2017). In addition, crystallization of human enzymes will provide an insight into active sites and regulatory regions, which is critical for structure-based drug development. To date, crystal structures of IP6K from the parasite Entamoeba histolytica and PPIP5Ks from humans (Wang et al., 2012, 2014) are known. Similarly, structures of human IP6Ks should be resolved.
(3). Develop new chemical tools for IPP research
Lack of appropriate chemical tools hampers IPP research. Encouragingly, various laboratories have initiated efforts to design novel tools such as methods for the cellular delivery of IPPs (Pavlovic et al., 2016), non-hydrolysable IPP analogs (Wu et al., 2013, 2014), isolation and detection of IPPs from tissue samples (Wilson et al., 2015), modification of current isolation protocols to differentiate 5-IP7 and 1-IP7 (Gu et al., 2016), synthesis of IP- and IPP-based affinity reagents (Wu et al., 2016), 5-IP7 analogs for traceable phosphoryl transfer (Brown, Marmelstein & Fiedler, 2016), synthetic peptides to establish pyrophosphate ester stability (Yates & Fiedler, 2015), an affinity reagent for the recognition of pyrophosphorylated peptides (Conway & Fiedler, 2015), in-gel detection of pyrophosphorylated proteins (Williams & Fiedler, 2015) and stabilized mimics of IPPs (Williams & Fiedler, 2015). A recently reported mass-spectrometry approach detected peptide pyrophosphorylation (Penkert et al., 2017), which hopefully can be utilized in vivo. A reliable method for quantitative estimation of IPPs from biological samples is required to measure their levels in healthy and diseased subjects.
X. CONCLUSIONS
(1). IPPs are soluble IP derivatives with diphosphate moieties, which are metabolized by distinct classes of enzymes.
(2). Post-translational modifications or protein interactions regulate IP6Ks.
(3). IPP-metabolizing enzymes regulate proteins via diverse mechanisms.
(4). The IPP pathway regulates various cellular functions.
(5). Genetic deletion of IP6K s has detrimental effects on development, but beneficial impacts on several diseases.
(6). Pharmacological inhibition of IP6Ks in mice reveals that this pathway has great potential to become a target in the treatment of various diseases.
XI. ACKNOWLEDGEMENTS
I thank Solomon Snyder, Hongbo Luo and Bahaa Elgendy for providing critical comments and Henning J. Jessen for sharing unpublished data. I also thank Molee Chakraborty and Sarbani Ghoshal for proofreading. I sincerely apologize to any authors whose research I failed to cite. This work was supported by NIHR01DK103746.
XII. REFERENCES
- ALBERT C, SAFRANY ST, BEMBENEK ME, REDDY KM, REDDY K, FALCK J, BROCKER M, SHEARS SB & MAYR GW (1997). Biological variability in the structures of diphosphoinositol polyphosphates in Dictyostelium discoideum and mammalian cells. Biochemical Journal 327, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AUESUKAREE C, TOCHIO H, SHIRAKAWA M, KANEKO Y & HARASHIMA S (2005). Plc1p, Arg82p, and Kcs1p, enzymes involved in inositol pyrophosphate synthesis, are essential for phosphate regulation and polyphosphate accumulation in Saccharomyces cerevisiae. Journal of Biological Chemistry 280, 25127–25133. [DOI] [PubMed] [Google Scholar]
- AZEVEDO C, BURTON A, RUIZ-MATEOS E, MARSH M & SAIARDI A (2009). Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proceedings of the National Academy of Sciences of the United States of America 106, 21161–21166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZEVEDO C & SAIARDI A (2006). Extraction and analysis of soluble inositol polyphosphates from yeast. Nature Protocols 1, 2416–2422. [DOI] [PubMed] [Google Scholar]
- BARKER CJ, ILLIES C, GABOARDI GC & BERGGREN PO (2009). Inositol pyrophosphates: structure, enzymology and function. Cellular and Molecular Life Sciences 66, 3851–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARKER CJ, WRIGHT J, HUGHES PJ, KIRK CJ & MICHELL RH (2004). Complex changes in cellular inositol phosphate complement accompany transit through the cell cycle. Biochemical Journal 380, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNES PJ (2003). New concepts in chronic obstructive pulmonary disease. Annual Review of Medicine 54, 113–129. [DOI] [PubMed] [Google Scholar]
- BENNETT M, ONNEBO SM, AZEVEDO C & SAIARDI A (2006). Inositol pyrophosphates: metabolism and signaling. Cellular and Molecular Life Sciences 63, 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEREITER-HAHN J, MUNNICH A & WOITENECK P (1998). Dependence of energy metabolism on the density of cells in culture. Cell Structure and Function 23, 85–93. [DOI] [PubMed] [Google Scholar]
- BERGINK S, JASPERS NG & VERMEULEN W (2007). Regulation of UV-induced DNA damage response by ubiquitylation. DNA Repair 6, 1231–1242. [DOI] [PubMed] [Google Scholar]
- BERRIDGE MJ, LIPP P & BOOTMAN MD (2000). The versatility and universality of calcium signalling. Nature Reviews Molecular Cell Biology 1, 11–21. [DOI] [PubMed] [Google Scholar]
- BHANDARI R, JULURI KR, RESNICK AC & SNYDER SH (2008). Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proceedings of the National Academy of Sciences of the United States of America 105, 2349–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHANDARI R, SAIARDI A, AHMADIBENI Y, SNOWMAN AM, RESNICK AC, KRISTIANSEN TZ, MOLINA H, PANDEY A, WERNER JK Jr., JULURI KR, XU Y, PRESTWICH GD, PARANG K & SNYDER SH (2007). Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proceedings of the National Academy of Sciences of the United States of America 104, 15305–15310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOREGOWDA SV, GHOSHAL S, BOOKER CN, KRISHNAPPA V, CHAKRABORTY A & PHINNEY DG (2017). IP6K1 reduces mesenchymal stem/stromal cell fitness and potentiates high fat diet-induced skeletal involution. Stem Cells 35, 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUCHER J, KLEINRIDDERS A & KAHN CR (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harbor Perspectives in Biology 6, a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN NW, MARMELSTEIN AM & FIEDLER D (2016). Chemical tools for interrogating inositol pyrophosphate structure and function. Chemical Society Reviews 45, 6311–6326. [DOI] [PubMed] [Google Scholar]
- BURTON A, AZEVEDO C, ANDREASSI C, RICCIO A & SAIARDI A (2013). Inositol pyrophosphates regulate JMJD2C-dependent histone demethylation. Proceedings of the National Academy of Sciences of the United States of America 110, 18970–18975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAFFREY JJ, SAFRANY ST, YANG X & SHEARS SB (2000). Discovery of molecular and catalytic diversity among human diphosphoinositol-polyphosphate phosphohydrolases. An expanding Nudt family. Journal of Biological Chemistry 275, 12730–12736. [DOI] [PubMed] [Google Scholar]
- CAFFREY JJ & SHEARS SB (2001). Genetic rationale for microheterogeneity of human diphosphoinositol polyphosphate phosphohydrolase type 2. Gene 269, 53–60. [DOI] [PubMed] [Google Scholar]
- CALLEJA V, ALCOR D, LAGUERRE M, PARK J, VOJNOVIC B, HEMMINGS BA, DOWNWARD J, PARKER PJ & LARIJANI B (2007). Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biology 5, e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CEDAR H & BERGMAN Y (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nature Reviews Genetics 10, 295–304. [DOI] [PubMed] [Google Scholar]
- CHAKRABORTY A, KIM S & SNYDER SH (2011). Inositol pyrophosphates as mammalian cell signals. Science Signaling 4, re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABORTY A, KOLDOBSKIY MA, BELLO NT, MAXWELL M, POTTER JJ, JULURI KR, MAAG D, KIM S, HUANG AS, DAILEY MJ, SALEH M, SNOWMAN AM, MORAN TH, MEZEY E & SNYDER SH (2010). Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 143, 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABORTY A, KOLDOBSKIY MA, SIXT KM, JULURI KR, MUSTAFA AK, SNOWMAN AM, VAN ROSSUM DB, PATTERSON RL & SNYDER SH (2008). HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proceedings of the National Academy of Sciences of the United States of America 105, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABORTY A, LATAPY C, XU J, SNYDER SH & BEAULIEU JM (2014). Inositol hexakisphosphate kinase-1 regulates behavioral responses via GSK3 signaling pathways. Molecular Psychiatry 19, 284–293. [DOI] [PubMed] [Google Scholar]
- CHAKRABORTY A, WERNER JK Jr., KOLDOBSKIY MA, MUSTAFA AK, JULURI KR, PIETROPAOLI J, SNOWMAN AM & SNYDER SH (2011). Casein kinase-2 mediates cell survival through phosphorylation and degradation of inositol hexakisphosphate kinase-2. Proceedings of the National Academy of Sciences of the United States of America 108, 2205–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDURI M, RAI A, MALLA AB, WU M, FIEDLER D, MALLIK R & BHANDARI R (2016). Inositol hexakisphosphate kinase 1 (IP6K1) activity is required for cytoplasmic dynein-driven transport. Biochemical Journal 473, 3031–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG YT, CHOI G, BAE YS, BURDETT M, MOON HS, LEE JW, GRAY NS, SCHULTZ PG, MEIJER L, CHUNG SK, CHOI KY, SUH PG & RYU SH (2002). Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem 3, 897–901. [DOI] [PubMed] [Google Scholar]
- CHAPMAN ER (2008). How does synaptotagmin trigger neurotransmitter release? Annual Review of Biochemistry 77, 615–641. [DOI] [PubMed] [Google Scholar]
- CHENG CY & MRUK DD (2012). The blood-testis barrier and its implications for male contraception. Pharmacological Reviews 64, 16–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG JQ, RUGGERI B, KLEIN WM, SONODA G, ALTOMARE DA, WATSON DK & TESTA JR (1996). Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proceedings of the National Academy of Sciences of the United States of America 93, 3636–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI JH, WILLIAMS J, CHO J, FALCK JR & SHEARS SB (2007). Purification, sequencing, and molecular identification of a mammalian PP-InsP5 kinase that is activated when cells are exposed to hyperosmotic stress. Journal of Biological Chemistry 282, 30763–30775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K, MOLLAPOUR E, CHOI JH & SHEARS SB (2008). Cellular energetic status supervises the synthesis of bis-diphosphoinositol tetrakisphosphate independently of AMP-activated protein kinase. Molecular Pharmacology 74, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K, MOLLAPOUR E & SHEARS SB (2005). Signal transduction during environmental stress: InsP8 operates within highly restricted contexts. Cellular Signalling 17, 1533–1541. [DOI] [PubMed] [Google Scholar]
- CHU C, ALAPAT D, WEN X, TIMO K, BURSTEIN D, LISANTI M, SHEARS S & KOHTZ DS (2004). Ectopic expression of murine diphosphoinositol polyphosphate phosphohydrolase 1 attenuates signaling through the ERK1/2 pathway. Cellular Signalling 16, 1045–1059. [DOI] [PubMed] [Google Scholar]
- CONWAY JH & FIEDLER D (2015). An affinity reagent for the recognition of pyrophosphorylated peptides. Angewandte Chemie International Edition in English 54, 3941–3945. [DOI] [PubMed] [Google Scholar]
- CORBALAN-GARCIA S & GOMEZ-FERNANDEZ JC (2014). Signaling through C2 domains: more than one lipid target. Biochimica et Biophysica Acta 1838, 1536–1547. [DOI] [PubMed] [Google Scholar]
- CROCCO P, SAIARDI A, WILSON MS, MALETTA R, BRUNI AC, PASSARINO G & ROSE G (2016). Contribution of polymorphic variation of inositol hexakisphosphate kinase 3 (IP6K3) gene promoter to the susceptibility to late onset alzheimer’s disease. Biochimica et Biophysica Acta 1862, 1766–1773. [DOI] [PubMed] [Google Scholar]
- VAN DAM AD, KOOIJMAN S, SCHILPEROORT M, RENSEN PC & BOON MR (2015). Regulation of brown fat by AMP-activated protein kinase. Trends in Molecular Medicine 21, 571–579. [DOI] [PubMed] [Google Scholar]
- DEPHOURE N, ZHOU C, VILLEN J, BEAUSOLEIL SA, BAKALARSKI CE, ELLEDGE SJ & GYGI SP (2008). A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEROUICHE A, COUSIN C & MIJAKOVIC I (2012). Protein phosphorylation from the perspective of systems biology. Current Opinion in Biotechnology 23, 585–590. [DOI] [PubMed] [Google Scholar]
- DOWNES CP, GRAY A, FAIRSERVICE A, SAFRANY ST, BATTY IH & FLEMING I (2005). The regulation of membrane to cytosol partitioning of signalling proteins by phosphoinositides and their soluble headgroups. Biochemical Society Transactions 33, 1303–1307. [DOI] [PubMed] [Google Scholar]
- DRASKOVIC P, SAIARDI A, BHANDARI R, BURTON A, ILC G, KOVACEVIC M, SNYDER SH & PODOBNIK M (2008). Inositol hexakisphosphate kinase products contain diphosphate and triphosphate groups. Chemistry & Biology 15, 274–286. [DOI] [PubMed] [Google Scholar]
- ERNEUX C, GOVAERTS C, COMMUNI D & PESESSE X (1998). The diversity and possible functions of the inositol polyphosphate 5-phosphatases. Biochimica et Biophysica Acta 1436, 185–199. [DOI] [PubMed] [Google Scholar]
- EUROPE-FINNER GN, GAMMON B & NEWELL PC (1991). Accumulation of [3H]-inositol into inositol polyphosphates during development of Dictyostelium. Biochemical and Biophysical Research Communications 181, 191–196. [DOI] [PubMed] [Google Scholar]
- EVA R, BOUYOUCEF-CHERCHALLI D, PATEL K, CULLEN PJ & BANTING G (2012). IP3 3-kinase opposes NGF driven neurite outgrowth. PLoS One 7, e32386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISHER DI, SAFRANY ST, STRIKE P, MCLENNAN AG & CARTWRIGHT JL (2002). Nudix hydrolases that degrade dinucleoside and diphosphoinositol polyphosphates also have 5-phosphoribosyl 1-pyrophosphate (PRPP) pyrophosphatase activity that generates the glycolytic activator ribose 1,5-bisphosphate. Journal of Biological Chemistry 277, 47313–47317. [DOI] [PubMed] [Google Scholar]
- FRIDY PC, OTTO JC, DOLLINS DE & YORK JD (2007). Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. Journal of Biological Chemistry 282, 30754–30762. [DOI] [PubMed] [Google Scholar]
- FU C, XU J, CHENG W, ROJAS T, CHIN AC, SNOWMAN AM, HARRAZ MM & SNYDER SH (2017). Neuronal migration is mediated by inositol hexakisphosphate kinase 1 via alpha-actinin and focal adhesion kinase. Proceedings of the National Academy of Sciences of the United States of America 114, 2036–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FU C, XU J, LI RJ, CRAWFORD JA, KHAN AB, MA TM, CHA JY, SNOWMAN AM, PLETNIKOV MV & SNYDER SH (2015). Inositol hexakisphosphate kinase-3 regulates the morphology and synapse formation of cerebellar Purkinje cells via spectrin/adducin. Journal of Neuroscience 35, 11056–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUCI S, HELBIG AO, SLIJPER M, KRIJGSVELD J, HECK AJ & MOHAMMED S (2009). Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Analytical Chemistry 81, 4493–4501. [DOI] [PubMed] [Google Scholar]
- GHOSH S, SHUKLA D, SUMAN K, LAKSHMI BJ, MANORAMA R, KUMAR S & BHANDARI R (2013). Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood 122, 1478–1486. [DOI] [PubMed] [Google Scholar]
- GHOSHAL S, TYAGI R, ZHU Q & CHAKRABORTY A (2016). Inositol hexakisphosphate kinase-1 interacts with perilipin1 to modulate lipolysis. The International Journal of Biochemistry & Cell Biology 78, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHOSHAL S, ZHU Q, ASTEIAN A, LIN H, XU H, ERNST G, BARROW JC, XU B, CAMERON MD, KAMENECKA TM & CHAKRABORTY A (2016). TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Molecular Metabolism 5, 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLENNON MC & SHEARS SB (1993). Turnover of inositol pentakisphosphates, inositol hexakisphosphate and diphosphoinositol polyphosphates in primary cultured hepatocytes. Biochemical Journal 293, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOKHALE NA, ZAREMBA A, JANOSHAZI AK, WEAVER JD & SHEARS SB (2013). PPIP5K1 modulates ligand competition between diphosphoinositol polyphosphates and PtdIns(3,4,5)P3 for polyphosphoinositide-binding domains. Biochemical Journal 453, 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOKHALE NA, ZAREMBA A & SHEARS SB (2011). Receptor-dependent compartmentalization of PPIP5K1, a kinase with a cryptic polyphosphoinositide binding domain. Biochemical Journal 434, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRUDZIEN-NOGALSKA E, JIAO X, SONG MG, HART RP & KILEDJIAN M (2016). Nudt3 is an mRNA decapping enzyme that modulates cell migration. RNA 22, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU C, NGUYEN H-N, GANINI D, CHEN Z, JESSEN HJ, GU Z, WANG H & SHEARS SB (2017). KO of 5-InsP7 kinase activity transforms the HCT116 colon cancer cell line into a hypermetabolic, growth-inhibited phenotype. Proceedings of the National Academy of Sciences of the United States of America 114, 11968–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU C, WILSON MS, JESSEN HJ, SAIARDI A & SHEARS SB (2016). Inositol pyrophosphate profiling of two HCT116 cell lines uncovers variation in InsP8 levels. PLoS One 11, e0165286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAND CE & HONEK JF (2007). Phosphate transfer from inositol pyrophosphates InsP5PP and InsP4(PP)2: a semi-empirical investigation. Bioorganic & Medicinal Chemistry Letters 17, 183–188. [DOI] [PubMed] [Google Scholar]
- HIDAKA K, CAFFREY JJ, HUA L, ZHANG T, FALCK JR, NICKEL GC, CARREL L, BARNES LD & SHEARS SB (2002). An adjacent pair of human NUDT genes on chromosome X are preferentially expressed in testis and encode two new isoforms of diphosphoinositol polyphosphate phosphohydrolase. Journal of Biological Chemistry 277, 32730–32738. [DOI] [PubMed] [Google Scholar]
- HOLMES W & JOGL G (2006). Crystal structure of inositol phosphate multikinase 2 and implications for substrate specificity. Journal of Biological Chemistry 281, 38109–38116. [DOI] [PubMed] [Google Scholar]
- HOLUB BJ (1986). Metabolism and function of myo-inositol and inositol phospholipids. Annual Review of Nutrition 6, 563–597. [DOI] [PubMed] [Google Scholar]
- HUA LV, HIDAKA K, PESESSE X, BARNES LD & SHEARS SB (2003). Paralogous murine Nudt10 and Nudt11 genes have differential expression patterns but encode identical proteins that are physiologically competent diphosphoinositol polyphosphate phosphohydrolases. Biochemical Journal 373, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG W, BARRETT M, HAJICEK N, HICKS S, HARDEN TK, SONDEK J & ZHANG Q (2013). Small molecule inhibitors of phospholipase C from a novel high-throughput screen. Journal of Biological Chemistry 288, 5840–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ILLIES C, GROMADA J, FIUME R, LEIBIGER B, YU J, JUHL K, YANG SN, BARMA DK, FALCK JR, SAIARDI A, BARKER CJ & BERGGREN PO (2007). Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science 318, 1299–1302. [DOI] [PubMed] [Google Scholar]
- IRVINE RF & SCHELL MJ (2001). Back in the water: the return of the inositol phosphates. Nature Reviews Molecular Cell Biology 2, 327–338. [DOI] [PubMed] [Google Scholar]
- JADAV RS, CHANDURI MV, SENGUPTA S & BHANDARI R (2013). Inositol pyrophosphate synthesis by inositol hexakisphosphate kinase 1 is required for homologous recombination repair. Journal of Biological Chemistry 288, 3312–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JADAV RS, KUMAR D, BUWA N, GANGULI S, THAMPATTY SR, BALASUBRAMANIAN N & BHANDARI R (2016). Deletion of inositol hexakisphosphate kinase 1 (IP6K1) reduces cell migration and invasion, conferring protection from aerodigestive tract carcinoma in mice. Cellular Signalling 28, 1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANSSEN K, HORN S, NIEMANN MT, DANIEL PT, SCHULZE-OSTHOFF K & FISCHER U (2009). Inhibition of the ER Ca2+ pump forces multidrug-resistant cells deficient in Bak and Bax into necrosis. Journal of Cell Science 122, 4481–4491. [DOI] [PubMed] [Google Scholar]
- JONES S, WANG TL, KURMAN RJ, NAKAYAMA K, VELCULESCU VE, VOGELSTEIN B, KINZLER KW, PAPADOPOULOS N & SHIH IEM (2012). Low-grade serous carcinomas of the ovary contain very few point mutations. The Journal of Pathology 226, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMIMURA J, WAKUI K, KADOWAKI H, WATANABE Y, MIYAKE K, HARADA N, SAKAMOTO M, KINOSHITA A, YOSHIURA K, OHTA T, KISHINO T, ISHIKAWA M, KASUGA M, FUKUSHIMA Y, NIIKAWA N & MATSUMOTO N (2004). The IHPK1 gene is disrupted at the 3p21.31 breakpoint of t(3;9) in a family with type 2 diabetes mellitus. Journal of Human Genetics 49, 360–365. [DOI] [PubMed] [Google Scholar]
- KAY NE, ECKEL-PASSOW JE, BRAGGIO E, VANWIER S, SHANAFELT TD, VAN DYKE DL, JELINEK DF, TSCHUMPER RC, KIPPS T, BYRD JC & FONSECA R (2010). Progressive but previously untreated CLL patients with greater array CGH complexity exhibit a less durable response to chemoimmunotherapy. Cancer Genetics 203, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNEDY BK & LAMMING DW (2016). The mechanistic target of rapamycin: the grand conducTOR of metabolism and aging. Cell Metabolism 23, 990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KILARI RS, WEAVER JD, SHEARS SB & SAFRANY ST (2013). Understanding inositol pyrophosphate metabolism and function: kinetic characterization of the DIPPs. FEBS Letters 587, 3464–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAMOTO A, KITAMOTO T, MIZUSAWA S, TERANISHI H, SO R, MATSUO T, NAKATA Y, HYOGO H, OCHI H, NAKAMURA T, KAMOHARA S, MIYATAKE N, KOTANI K, KOMATSU R & ITOH N (2013). NUDT3 rs206936 is associated with body mass index in obese Japanese women. Endocrine Journal 60, 991–1000. [DOI] [PubMed] [Google Scholar]
- KOLDOBSKIY MA, CHAKRABORTY A, WERNER JK Jr., SNOWMAN AM, JULURI KR, VANDIVER MS, KIM S, HELETZ S & SNYDER SH (2010). p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proceedings of the National Academy of Sciences of the United States of America 107, 20947–20951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOUZARIDES T (2002). Histone methylation in transcriptional control. Current Opinion in Genetics & Development 12, 198–209. [DOI] [PubMed] [Google Scholar]
- LAUSSMANN T, EUJEN R, WEISSHUHN CM, THIEL U & VOGEL G (1996). Structures of diphospho-myo-inositol pentakisphosphate and bisdiphospho-myo-inositol tetrakisphosphate from Dictyostelium resolved by NMR analysis. Biochemical Journal 315, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE TS, LEE JY, KYUNG JW, YANG Y, PARK SJ, LEE S, PAVLOVIC I, KONG B, JHO YS, JESSEN HJ, KWEON DH, SHIN YK, KIM SH, YOON TY & KIM S (2016). Inositol pyrophosphates inhibit synaptotagmin-dependent exocytosis. Proceedings of the National Academy of Sciences of the United States of America 113, 8314–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE YS, MULUGU S, YORK JD & O’SHEA EK (2007). Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science 316, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEMMON MA (2007). Pleckstrin homology (PH) domains and phosphoinositides. Biochemical Society Symposium 74, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESLIE NR, MCLENNAN AG & SAFRANY ST (2002). Cloning and characteri-sation of hAps1 and hAps2, human diadenosine polyphosphate-metabolising Nudix hydrolases. BMC Biochemistry 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEV S, LI C., DESMARINI D, SAIARDI A, FEWINGS NL, SCHIBECI SD, SHARMA R, SORRELL TC & DJORDJEVIC JT (2015). Fungal inositol pyrophosphate IP7 is crucial for metabolic adaptation to the host environment and pathogenicity. MBio 6, e00531–e00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI C, LEV S, SAIARDI A, DESMARINI D, SORRELL TC & DJORDJEVIC JT (2016). Identification of a major IP5 kinase in Cryptococcus neoformans confirms that PP-IP5/IP7, not IP6, is essential for virulence. Scientific Reports 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN H, FRIDY PC, RIBEIRO AA, CHOI JH, BARMA DK, VOGEL G, FALCK JR, SHEARS SB, YORK JD & MAYR GW (2009). Structural analysis and detection of biological inositol pyrophosphates reveal that the family of VIP/diphosphoinositol pentakisphosphate kinases are 1/3-kinases. Journal of Biological Chemistry 284, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONETTI A, SZIJGYARTO Z, BOSCH D, LOSS O, AZEVEDO C & SAIARDI A (2011). Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. Journal of Biological Chemistry 286, 31966–31974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOSITO O, SZIJGYARTO Z, RESNICK AC & SAIARDI A (2009). Inositol pyrophosphates and their unique metabolic complexity: analysis by gel electrophoresis. PLoS One 4, e5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO HR, HUANG YE, CHEN JC, SAIARDI A, IIJIMA M, YE K, HUANG Y, NAGATA E, DEVREOTES P & SNYDER SH (2003). Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell 114, 559–572. [DOI] [PubMed] [Google Scholar]
- LUO HR & MONDAL S (2015). Molecular control of PtdIns(3,4,5)P3 signaling in neutrophils. EMBO Reports 16, 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO HR, SAIARDI A, NAGATA E, YE K, YU H, JUNG TS, LUO X, JAIN S, SAWA A & SNYDER SH (2001). GRAB: a physiologic guanine nucleotide exchange factor for Rab3A, which interacts with inositol hexakisphosphate kinase. Neuron 31, 439–451. [DOI] [PubMed] [Google Scholar]
- LUO HR, SAIARDI A, YU H, NAGATA E, YE K & SNYDER SH (2002). Inositol pyrophosphates are required for DNA hyperrecombination in protein kinase c1 mutant yeast. Biochemistry 41, 2509–2515. [DOI] [PubMed] [Google Scholar]
- MACHKALYAN G, TRIEU P, PETRIN D, HEBERT TE & MILLER GJ (2016). PPIP5K1 interacts with the exocyst complex through a C-terminal intrinsically disordered domain and regulates cell motility. Cellular Signalling 28, 401–411. [DOI] [PubMed] [Google Scholar]
- MACKENZIE RW & ELLIOTT BT (2014). Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metabolic Syndrome and Obesity 7, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALLA AB & BHANDARI R (2017). IP6K1 is essential for chromatoid body formation and temporal regulation of TNP2 and PRM2 expression in mouse spermatids. Journal of Cell Science 130, 2854–2866. [DOI] [PubMed] [Google Scholar]
- MANNING BD (2010). Insulin signaling: inositol phosphates get into the Akt. Cell 143, 861–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANNING G, PLOWMAN GD, HUNTER T & SUDARSANAM S (2002). Evolution of protein kinase signaling from yeast to man. Trends in Biochemical Sciences 27, 514–520. [DOI] [PubMed] [Google Scholar]
- MCLENNAN AG (2006). The Nudix hydrolase superfamily. Cellular and Molecular Life Sciences 63, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENNITI FS, MILLER RN, PUTNEY JW Jr. & SHEARS SB (1993). Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. Journal of Biological Chemistry 268, 3850–3856. [PubMed] [Google Scholar]
- MICHELL RH (1975). Inositol phospholipids and cell surface receptor function. Biochimica et Biophysica Acta 415, 81–147. [DOI] [PubMed] [Google Scholar]
- MIKOSHIBA K, FUKUDA M, IBATA K, KABAYAMA H & MIZUTANI A (1999). Role of synaptotagmin, a Ca2+ and inositol polyphosphate binding protein, in neurotransmitter release and neurite outgrowth. Chemistry and Physics of Lipids 98, 59–67. [DOI] [PubMed] [Google Scholar]
- MORITOH Y, OKA M, YASUHARA Y, HOZUMI H, IWACHIDOW K, FUSE H & TOZAWA R (2016). Inositol hexakisphosphate kinase 3 regulates metabolism and lifespan in mice. Scientific Reports 6, 32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORIYA Y, NAGATA E, FUJII N, SATOH T, OGAWA H, HADANO S & TAKIZAWA S (2017). Inositol hexakisphosphate kinase 2 is a presymptomatic biomarker for amyotrophic lateral sclerosis. The Tokai Journal of Experimental and Clinical Medicine 42, 13–18. [PubMed] [Google Scholar]
- MORRISON BH, BAUER JA, HU J, GRANE RW, OZDEMIR AM, CHAWLA-SARKAR M, GONG B, ALMASAN A, KALVAKOLANU DV & LINDNER DJ (2002). Inositol hexakisphosphate kinase 2 sensitizes ovarian carcinoma cells to multiple cancer therapeutics. Oncogene 21, 1882–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRISON BH, BAUER JA, KALVAKOLANU DV & LINDNER DJ (2001). Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. Journal of Biological Chemistry 276, 24965–24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRISON BH, BAUER JA, LUPICA JA, TANG Z, SCHMIDT H, DIDONATO JA & LINDNER DJ (2007). Effect of inositol hexakisphosphate kinase 2 on transforming growth factor beta-activated kinase 1 and NF-kappaB activation. Journal of Biological Chemistry 282, 15349–15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRISON BH, HANEY R, LAMARRE E, DRAZBA J, PRESTWICH GD & LINDNER DJ (2009). Gene deletion of inositol hexakisphosphate kinase 2 predisposes to aerodigestive tract carcinoma. Oncogene 28, 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRISON BH, TANG Z, JACOBS BS, BAUER JA & LINDNER DJ (2005). Apo2L/TRAIL induction and nuclear translocation of inositol hexakisphosphate kinase 2 during IFN-beta-induced apoptosis in ovarian carcinoma. Biochemical Journal 385, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULUGU S, BAI W, FRIDY PC, BASTIDAS RJ, OTTO JC, DOLLINS DE, HAYSTEAD TA, RIBEIRO AA & YORK JD (2007). A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 316, 106–109. [DOI] [PubMed] [Google Scholar]
- MUNDI PS, SACHDEV J, MCCOURT C & KALINSKY K (2016). AKT in cancer: new molecular insights and advances in drug development. British Journal of Clinical Pharmacology 82, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGASE T, SEKI N, ISHIKAWA K, OHIRA M, KAWARABAYASI Y, OHARA O, TANAKA A, KOTANI H, MIYAJIMA N & NOMURA N (1996). Prediction of the coding sequences of unidentified human genes. VI. The coding sequences of 80 new genes (KIAA0201-KIAA0280) deduced by analysis of cDNA clones from cell line KG-1 and brain. DNA Research 3, 321–329 341–354. [DOI] [PubMed] [Google Scholar]
- NAGATA E, LUO HR, SAIARDI A, BAE BI, SUZUKI N & SNYDER SH (2005). Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. Journal of Biological Chemistry 280, 1634–1640. [DOI] [PubMed] [Google Scholar]
- NAGATA E, NONAKA T, MORIYA Y, FUJII N, OKADA Y, TSUKAMOTO H, ITOH J, OKADA C, SATOH T, ARAI T, HASEGAWA M & TAKIZAWA S (2016). Inositol hexakisphosphate kinase 2 promotes cell death in cells with cytoplasmic TDP-43 aggregation. Molecular Neurobiology 53, 5377–5383. [DOI] [PubMed] [Google Scholar]
- NAGATA E, SAIARDI A, TSUKAMOTO H, OKADA Y, ITOH Y, SATOH T, ITOH J, MARGOLIS RL, TAKIZAWA S, SAWA A & TAKAGI S (2011). Inositol hexakisphosphate kinases induce cell death in Huntington disease. Journal of Biological Chemistry 286, 26680–26686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVER KG, PUTNEY JW Jr., OBIE JF & SHEARS SB (1992). The interconversion of inositol 1,3,4,5,6-pentakisphosphate and inositol tetrakisphosphates in AR4–2J cells. Journal of Biological Chemistry 267, 21528–21534. [PubMed] [Google Scholar]
- ONNEBO SM & SAIARDI A (2009). Inositol pyrophosphates modulate hydrogen peroxide signalling. Biochemical Journal 423, 109–118. [DOI] [PubMed] [Google Scholar]
- PADMANABHAN U, DOLLINS DE, FRIDY PC, YORK JD & DOWNES CP (2009). Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. Journal of Biological Chemistry 284, 10571–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAVLOVIC I, THAKOR DT, VARGAS JR, MCKINLAY CJ, HAUKE S, ANSTAETT P, CAMUNA RC, BIGLER L, GASSER G, SCHULTZ C, WENDER PA & JESSEN HJ (2016). Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nature Communications 7, 10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENKERT M, YATES LM, SCHUMANN M, PERLMAN D, FIEDLER D & KRAUSE E (2017). Unambiguous identification of serine and threonine pyrophosphorylation using neutral-loss-triggered electron-transfer/higher-energy collision dissociation. Analytical Chemistry 89, 3672–3680. [DOI] [PubMed] [Google Scholar]
- PESESSE X, CHOI K, ZHANG T & SHEARS SB (2004). Signaling by higher inositol polyphosphates. Synthesis of bisdiphosphoinositol tetrakisphosphate (“InsP8”) is selectively activated by hyperosmotic stress. Journal of Biological Chemistry 279, 43378–43381. [DOI] [PubMed] [Google Scholar]
- PLIMMER RH & PAGE HJ (1913). An investigation of Phytin. Biochemical Journal 7, 157–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRASAD A, JIA Y, CHAKRABORTY A, LI Y, JAIN SK, ZHONG J, ROY SG, LOISON F, MONDAL S, SAKAI J, BLANCHARD C, SNYDER SH & LUO HR (2011). Inositol hexakisphosphate kinase 1 regulates neutrophil function in innate immunity by inhibiting phosphatidylinositol-(3,4,5)-trisphosphate signaling. Nature Immunology 12, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PULLOOR NK, NAIR S, MCCAFFREY K, KOSTIC AD, BIST P, WEAVER JD, RILEY AM, TYAGI R, UCHIL PD, YORK JD, SNYDER SH, GARCÍA-SASTRE A, POTTER BV, LIN R, SHEARS SB, et al. (2014). Human genome-wide RNAi screen identifies an essential role for inositol pyrophosphates in Type-I interferon response. PLoS Pathogens 10, e1003981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAJASEKARAN SS, ILLIES C, SHEARS SB, WANG H, AYALA TS, MARTINS JO, DARE E, BERGGREN PO & BARKER CJ (2017). Protein kinase- and lipase inhibitors of inositide metabolism deplete IP7 indirectly in pancreatic beta-cells: off-target effects on cellular bioenergetics and direct effects on IP6K activity. Cellular Signalling 42, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RALPH SJ (2008). Arsenic-based antineoplastic drugs and their mechanisms of action. Metal-Based Drugs 2008, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO F, CHA J, XU J, XU R, VANDIVER MS, TYAGI R, TOKHUNTS R, KOLDOBSKIY MA, FU C, BARROW R, WU M, FIEDLER D, BARROW JC & SNYDER SH (2014). Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Molecular Cell 54, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO F, XU J, FU C, CHA JY, GADALLA MM, XU R, BARROW JC & SNYDER SH (2015). Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proceedings of the National Academy of Sciences of the United States of America 112, 1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO F, XU J, KHAN AB, GADALLA MM, CHA JY, XU R, TYAGI R, DANG Y, CHAKRABORTY A & SNYDER SH (2014). Inositol hexakisphosphate kinase-1 mediates assembly/disassembly of the CRL4-signalosome complex to regulate DNA repair and cell death. Proceedings of the National Academy of Sciences of the United States of America 111, 16005–16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO NN, GOMEZ-GARCIA MR & KORNBERG A (2009). Inorganic polyphosphate: essential for growth and survival. Annual Review of Biochemistry 78, 605–647. [DOI] [PubMed] [Google Scholar]
- RICE JC, BRIGGS SD, UEBERHEIDE B, BARBER CM, SHABANOWITZ J, HUNT DF, SHINKAI Y & ALLIS CD (2003). Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Molecular Cell 12, 1591–1598. [DOI] [PubMed] [Google Scholar]
- ROSE NR & KLOSE RJ (2014). Understanding the relationship between DNA methylation and histone lysine methylation. Biochimica et Biophysica Acta 1839, 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROUNGE TB, PAGE CM, LEPISTO M, ELLONEN P, ANDREASSEN BK & WEIDERPASS E (2016). Genome-wide DNA methylation in saliva and body size of adolescent girls. Epigenomics 8, 1495–1505. [DOI] [PubMed] [Google Scholar]
- SAFRANY ST (2004). Protocols for regulation and study of diphosphoinositol polyphosphates. Molecular Pharmacology 66, 1585–1591. [DOI] [PubMed] [Google Scholar]
- SAFRANY ST, CAFFREY JJ, YANG X, BEMBENEK ME, MOYER MB, BURKHART WA & SHEARS SB (1998). A novel context for the ‘MutT’ module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. The EMBO Journal 17, 6599–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAFRANY ST & SHEARS SB (1998). Turnover of bis-diphosphoinositol tetrakisphosphate in a smooth muscle cell line is regulated by beta2-adrenergic receptors through a cAMP-mediated, A-kinase-independent mechanism. The EMBO Journal 17, 1710–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIARDI A (2012). Cell signalling by inositol pyrophosphates. Sub-Cellular Biochemistry 59, 413–443. [DOI] [PubMed] [Google Scholar]
- SAIARDI A, BHANDARI R, RESNICK AC, SNOWMAN AM & SNYDER SH (2004). Phosphorylation of proteins by inositol pyrophosphates. Science 306, 2101–2105. [DOI] [PubMed] [Google Scholar]
- SAIARDI A, CAFFREY JJ, SNYDER SH & SHEARS SB (2000). The inositol hexakisphosphate kinase family. Catalytic flexibility and function in yeast vacuole biogenesis. Journal of Biological Chemistry 275, 24686–24692. [DOI] [PubMed] [Google Scholar]
- SAIARDI A, ERDJUMENT-BROMAGE H, SNOWMAN AM, TEMPST P & SNYDER SH (1999). Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Current Biology 9, 1323–1326. [DOI] [PubMed] [Google Scholar]
- SAIARDI A, NAGATA E, LUO HR, SNOWMAN AM & SNYDER SH (2001). Identification and characterization of a novel inositol hexakisphosphate kinase. Journal of Biological Chemistry 276, 39179–39185. [DOI] [PubMed] [Google Scholar]
- SAIARDI A, RESNICK AC, SNOWMAN AM, WENDLAND B & SNYDER SH (2005). Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proceedings of the National Academy of Sciences of the United States of America 102, 1911–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIARDI A, SCIAMBI C, MCCAFFERY JM, WENDLAND B & SNYDER SH (2002). Inositol pyrophosphates regulate endocytic trafficking. Proceedings of the National Academy of Sciences of the United States of America 99, 14206–14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARMAH B & WENTE SR (2010). Inositol hexakisphosphate kinase-2 acts as an effector of the vertebrate Hedgehog pathway. Proceedings of the National Academy of Sciences of the United States of America 107, 19921–19926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCRIMA A, FISCHER ES, LINGARAJU GM, BOHM K, CAVADINI S & THOMA NH (2011). Detecting UV-lesions in the genome: the modular CRL4 ubiquitin ligase does it best! FEBS Letters 585, 2818–2825. [DOI] [PubMed] [Google Scholar]
- SEKAR MC, SHAHIWALA K, LELOUP L & WELLS A (2014). Modulation of epidermal growth factor stimulated ERK phosphorylation and cell motility by inositol trisphosphate kinase. Journal of Pharmaceutical Sciences and Pharmacology 1, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAH A, GANGULI S, SEN J & BHANDARI R (2017). Inositol pyrophosphates: energetic, omnipresent and versatile signalling molecules. Journal of the Indian Institute of Science 97, 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAMES DS & MINNA JD (2008). IP6K2 is a client for HSP90 and a target for cancer therapeutics development. Proceedings of the National Academy of Sciences of the United States of America 105, 1389–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB (2004). How versatile are inositol phosphate kinases? Biochemical Journal 377, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB (2009). Diphosphoinositol polyphosphates: metabolic messengers? Molecular Pharmacology 76, 236–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB (2015). Inositol pyrophosphates: why so many phosphates? Advances in Biological Regulation 57, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB (2016). Towards pharmacological intervention in inositol pyrophosphate signalling. Biochemical Society Transactions 44, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB (2017). Intimate connections: inositol pyrophosphates at the interface of metabolic regulation and cell-signaling. Journal of Cellular Physiology 233, 1897–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARS SB, BAUGHMAN BM, GU C, NAIR VS & WANG H (2016). The significance of the 1-kinase/1-phosphatase activities of the PPIP5K family. Advances in Biological. Regulation 63, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIDOSSIS L & KAJIMURA S (2015). Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. The Journal of Clinical Investigation 125, 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPHENS L, RADENBERG T, THIEL U, VOGEL G, KHOO KH, DELL A, JACKSON TR, HAWKINS PT & MAYR GW (1993). The detection, purification, structural characterization, and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). Journal of Biological Chemistry 268, 4009–4015. [PubMed] [Google Scholar]
- STEPHENS LR, HAWKINS PT, STANLEY AF, MOORE T, POYNER DR, MORRIS PJ, HANLEY MR, KAY RR & IRVINE RF (1991). myo-inositol pentakisphosphates. Structure, biological occurrence and phosphorylation to myo-inositol hexakisphosphate. Biochemical Journal 275, 485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STORK CJ & LI YV (2010). Zinc release from thapsigargin/IP3-sensitive stores in cultured cortical neurons. Journal of Molecular Signaling 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STREB H, IRVINE RF, BERRIDGE MJ & SCHULZ I (1983). Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306, 67–69. [DOI] [PubMed] [Google Scholar]
- SUN D, LI S, WU H, ZHANG M, ZHANG X, WEI L, QIN X & GAO E (2015). Oncostatin M (OSM) protects against cardiac ischaemia/reperfusion injury in diabetic mice by regulating apoptosis, mitochondrial biogenesis and insulin sensitivity. Journal of Cellular and Molecular Medicine 19, 1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZIJGYARTO Z, GAREDEW A, AZEVEDO C & SAIARDI A (2011). Influence of inositol pyrophosphates on cellular energy dynamics. Science 334, 802–805. [DOI] [PubMed] [Google Scholar]
- TAKAZAWA K, LEMOS M, DELVAUX A, LEJEUNE C, DUMONT JE & ERNEUX C (1990). Rat brain inositol 1,4,5-trisphosphate 3-kinase. Ca2(+)-sensitivity, purification and antibody production. Biochemical Journal 268, 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS MP & POTTER BV (2014). The enzymes of human diphosphoinositol polyphosphate metabolism. FEBS Journal 281, 14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOTA SG & BHANDARI R (2015). The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. Journal of Biosciences 40, 593–605. [DOI] [PubMed] [Google Scholar]
- THOTA SG, UNNIKANNAN CP, THAMPATTY SR, MANORAMA R & BHANDARI R (2015). Inositol pyrophosphates regulate RNA polymerase I-mediated rRNA transcription in Saccharomyces cerevisiae. Biochemical Journal 466, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIAN Q, STEPANIANTS SB, MAO M, WENG L, FEETHAM MC, DOYLE MJ, YI EC, DAI H, THORSSON V, ENG J, GOODLETT D, BERGER JP, GUNTER B, LINSELEY PS, STOUGHTON RB, et al. (2004). Integrated genomic and proteomic analyses of gene expression in mammalian cells. Molecular & Cellular Proteomics 3, 960–969. [DOI] [PubMed] [Google Scholar]
- UBERSAX JA & FERRELL JE Jr. (2007). Mechanisms of specificity in protein phosphorylation. Nature Reviews Molecular Cell Biology 8, 530–541. [DOI] [PubMed] [Google Scholar]
- VALLEE RB, MCKENNEY RJ & ORI-MCKENNEY KM (2012). Multiple modes of cytoplasmic dynein regulation. Nature Cell Biology 14, 224–230. [DOI] [PubMed] [Google Scholar]
- VINCENT EE, ELDER DJ, THOMAS EC, PHILLIPS L, MORGAN C, PAWADE J, SOHAIL M, MAY MT, HETZEL MR & TAVARE JM (2011). Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. British Journal of Cancer 104, 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOGLMAIER SM, BEMBENEK ME, KAPLIN AI, DORMAN G, OLSZEWSKI JD, PRESTWICH GD & SNYDER SH (1996). Purified inositol hexakisphosphate kinase is an ATP synthase: diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proceedings of the National Academy of Sciences of the United States of America 93, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VUCENIK I & SHAMSUDDIN AM (2003). Cancer inhibition by inositol hexaphosphate (IP6) and inositol: from laboratory to clinic. The Journal of Nutrition 133, 3778S–3784S. [DOI] [PubMed] [Google Scholar]
- WANG H, DEROSE EF, LONDON RE & SHEARS SB (2014). IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nature Communications 5, 4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, FALCK JR, HALL TM & SHEARS SB (2012). Structural basis for an inositol pyrophosphate kinase surmounting phosphate crowding. Nature Chemical Biology 8, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, NAIR VS, HOLLAND AA, CAPOLICCHIO S, JESSEN HJ, JOHNSON MK & SHEARS SB (2015). Asp1 from Schizosaccharomyces pombe binds a [2Fe-2S](2+) cluster which inhibits inositol pyrophosphate 1-phosphatase activity. Biochemistry 54, 6462–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS FJ & FIEDLER D (2015). A fluorescent sensor and gel stain for detection of pyrophosphorylated proteins. ACS Chemical Biology 10, 1958–1963. [DOI] [PubMed] [Google Scholar]
- WILLIAMS MJ, ERIKSSON A, SHAIK M, VOISIN S, YAMSKOVA O, PAULSSON J, THOMBARE K, FREDRIKSSON R & SCHIOTH HB (2015). The obesity-linked gene Nudt3 Drosophila homolog Aps is associated with insulin signaling. Molecular Endocrinology 29, 1303–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON MS, BULLEY SJ, PISANI F, IRVINE RF & SAIARDI A (2015). A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biology 5, 150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON MS, LIVERMORE TM & SAIARDI A (2013). Inositol pyrophosphates: between signalling and metabolism. Biochemical Journal 452, 369–379. [DOI] [PubMed] [Google Scholar]
- WONG NS, BARKER CJ, MORRIS AJ, CRAXTON A, KIRK CJ & MICHELL RH (1992). The inositol phosphates in WRK1 rat mammary tumour cells. Biochemical Journal 286, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WORLEY J, LUO X & CAPALDI AP (2013). Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell Reports 3, 1476–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WORMALD M, LIAO G, KIMOS M, BARROW J, WEI H (2017). Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS One 12, e0188852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU M, CHONG LS, CAPOLICCHIO S, JESSEN HJ, RESNICK AC & FIEDLER D (2014). Elucidating diphosphoinositol polyphosphate function with nonhydrolyzable analogues. Angewandte Chemie International Edition in English 53, 7192–7197. [DOI] [PubMed] [Google Scholar]
- WU M, CHONG LS, PERLMAN DH, RESNICK AC & FIEDLER D (2016). Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America 113, E6757–E6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU M, DUL BE, TREVISAN AJ & FIEDLER D (2013). Synthesis and characterization of non-hydrolysable diphosphoinositol polyphosphate second messengers. Chemical Science 4, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WUNDENBERG T, GRABINSKI N, LIN H & MAYR GW (2014). Discovery of InsP6-kinases as InsP6-dephosphorylating enzymes provides a new mechanism of cytosolic Ins P6 degradation driven by the cellular ATP/ADP ratio. Biochemical Journal 462, 173–184. [DOI] [PubMed] [Google Scholar]
- WUNDENBERG T & MAYR GW (2012). Synthesis and biological actions of diphosp hoinositol phosphates (inositol pyrophosphates), regulators of cell homeostasis. Biological Chemistry 393, 979–998. [DOI] [PubMed] [Google Scholar]
- XIE Z, DONG Y, ZHANG J, SCHOLZ R, NEUMANN D & ZOU MH (2009). Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Molecular and Cellular Biology 29, 3582–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU Y, LI H, BAJRAMI B, KWAK H, CAO S, LIU P, ZHOU J, ZHOU Y, ZHU H, YE K & LUO HR (2013). Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proceedings of the National Academy of Sciences of the United States of America 110, 7726–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG SN, SHI Y, YANG G, LI Y, YU L, SHIN OH, BACAJ T, SUDHOF TC, YU J & BERGGREN PO (2012). Inositol hexakisphosphate suppresses excitatory neurotransmission via synaptotagmin-1 C2B domain in the hippocampal neuron. Proceedings of the National Academy of Sciences of the United States of America 109, 12183–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X, SAFRANY ST & SHEARS SB (1999). Site-directed muta-genesis of diphosphoinositol polyphosphate phosphohydrolase, a dual specificity NUDT enzyme that attacks diadenosine polyphosphates and diphosphoinositol polyphosphates. Journal of Biological Chemistry 274, 35434–35440. [DOI] [PubMed] [Google Scholar]
- YATES LM & FIEDLER D (2015). Establishing the stability and reversibility of protein pyrophosphorylation with synthetic peptides. ChemBioChem 16, 415–423. [DOI] [PubMed] [Google Scholar]
- YONG ST, NGUYEN HN, CHOI JH, BORTNER CD, WILLIAMS J, PULLOOR NK, KRISHNAN MN & SHEARS SB (2015). Identification of a functional nuclear translocation sequence in hPPIP5K2. BMC Cell Biology 16, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YORK SJ, ARMBRUSTER BN, GREENWELL P, PETES TD & YORK JD (2005). Inositol diphosphate signaling regulates telomere length. Journal of Biological Chemistry 280, 4264–4269. [DOI] [PubMed] [Google Scholar]
- YU W, YE C & GREENBERG ML (2016). Inositol hexakisphosphate kinase 1 (IP6K1) regulates inositol synthesis in mammalian cells. Journal of Biological Chemistry 291, 10437–10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUAN HX & GUAN KL (2015). The SIN1-PH domain connects mTORC2 to PI3K. Cancer Discovery 5, 1127–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Z, LIANG D, GAO X, ZHAO C, QIN X, XU Y, SU T, SUN D, LI W, WANG H, LIU B & CAO F (2014). Selective inhibition of inositol hexakisphosphate kinases (IP6Ks) enhances mesenchymal stem cell engraftment and improves therapeutic efficacy for myocardial infarction. Basic Research in Cardiology 109, 417. [DOI] [PubMed] [Google Scholar]
- ZHANG Z, LIU H & LIU J (2017). Akt activation: a potential strategy to ameliorate insulin resistance. Diabetes Research and Clinical Practice pii S0168–8227(17)30315–7. [DOI] [PubMed] [Google Scholar]
- ZHANG Z, WANG Y, YAO R, LI J, LUBET RA & YOU M (2006). p53 Transgenic mice are highly susceptible to 4-nitroquinoline-1-oxide-induced oral cancer. Molecular Cancer Research 4, 401–410. [DOI] [PubMed] [Google Scholar]
- ZHANG Z, ZHAO C, LIU B, LIANG D, QIN X, LI X, ZHANG R, LI C, WANG H, SUN D & CAO F (2014). Inositol pyrophosphates mediate the effects of aging on bone marrow mesenchymal stem cells by inhibiting Akt signaling. Stem Cell Research & Therapy 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Q, GHOSHAL S, RODRIGUES A, GAO S, ASTERIAN A, KAMENECKA TM, BARROW JC & CHAKRABORTY A (2016). Adipocyte-specific deletion of Ip6k1 reduces diet-induced obesity by enhancing AMPK-mediated thermogenesis. The Journal of Clinical Investigation 126, 4273–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Q, GHOSHAL S, TYAGI R & CHAKRABORTY A (2017). Global IP6K1 deletion enhances temperature modulated energy expenditure which reduces carbohydrate and fat induced weight gain. Molecular Metabolism 6, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]