

Abstract
This review summarizes recent insights in the cooperation of synaptic proteins that are central to synaptic vesicle fusion in presynaptic active zones, including SNAREs, synaptotagmin, complexin, Munc18, and Munc13. Structural and functional studies of the synaptic fusion machinery suggest new molecular models of synaptic vesicle priming and Ca2+-triggered fusion. These studies will be a stepping-stone towards answering the question of how the synaptic vesicle fusion machinery achieves such high speed and sensitivity.
Keywords: synaptic vesicle fusion, fusion protein, action potential, synaptic vesicle priming, Ca2+-triggering
1. Introduction
During synaptic transmission, Ca2+ influx into the presynaptic terminal triggers neurotransmitter release. This process involves sensing Ca2+, and subsequently fusing neurotransmitter-filled synaptic vesicles with the presynaptic membrane in less than a millisecond (122, 139). In the neuron, neurotransmitters are released “synchronously” upon action potential arrival in the synaptic terminal, “asynchronously” with a certain delay after an action potential, and “spontaneously” without an action potential; each of these processes likely involves specific sets of synaptic proteins (69). Many, if not most, of the key factors of the core synaptic fusion machinery have been identified, including SNAREs (Soluble N-ethylmaleimide sensitive factor Attachment protein REceptor), NSF (N-ethylmaleimide-sensitive factor), SNAP (soluble NSF adaptor protein), synaptotagmin (abbreviated as Syt), complexin (abbreviated as Cpx), Munc18 (mammalian uncoordinated-18) and Munc13 (mammalian uncoordinated-13). Yet, important questions remain: how is the process tightly regulated by Ca2+, and how is it triggered within less than a millisecond, much faster than any other biological membrane fusion process? We will first summarize the current knowledge about each individual factor in an encyclopedic fashion (Sections 1–7), and then discuss recent insights into the cooperation and interplay between these factors for neurotransmitter release (Sections 8–10). The reader may wish to skip to these sections first and consult the sections on the individual factors when more information is needed.
2. SNAREs
In this review, we focus on the neuronal SNAREs and their most extensively studied isoforms. Prior to membrane fusion, synaptobrevin-2 (also called VAMP2 – Vesicle Associated Membrane Protein 2) on the synaptic vesicle, and syntaxin-1A and SNAP-25A on the plasma membrane initially form a trans SNARE complex with the transmembrane domains of synaptobrevin-2 and syntaxin-1A in their respective membranes (Fig. 1). Moreover, individual syntaxin-1A molecules form clusters in the plasma membrane that are segregated from phosphatidylinositol-4,5-bisphosphate (PIP2) domains (149). During fusion, the SNARE complex completely zippers into the fully assembled cis SNARE complex where the soluble core consists of a parallel four α-helix bundle (144). The synaptobrevin-2 and syntaxin-1A helices of the cis SNARE complex extend into the merged membrane (136). The topology of the core of the SNARE complex is a left-handed four-helix coiled coil where the core consists of layers of primarily hydrophobic residues, except for an ionic layer at the center of the complex consisting of three glutamine amino acid residue and one arginine residue (referred to as ionic or zero layer) (39, 144). In addition to this parallel four α-helix bundle configuration, SNAREs can form a variety of non-canonical stoichiometries and configurations in vitro (reviewed in ref. (17)), and we will discuss potential physiological roles of these non-canonical assemblies in Section 10.
Figure 1. The roles of SNAREs in the synaptic vesicle cycle.
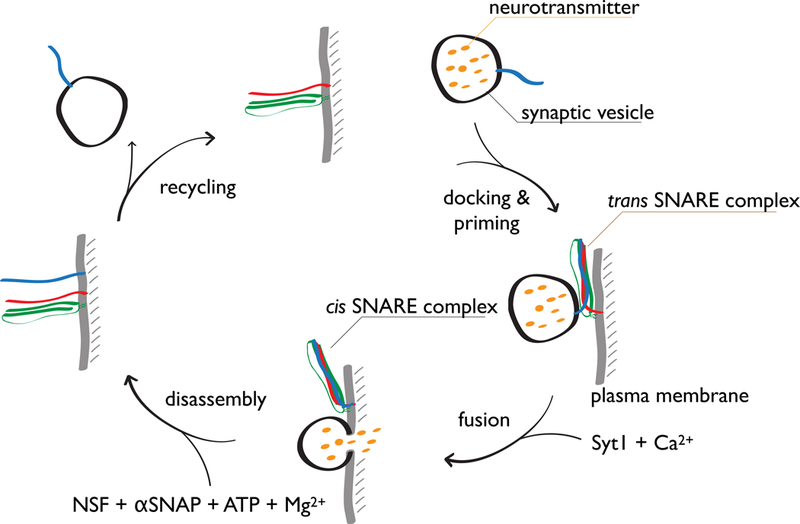
SNAREs form a trans complex that juxtaposes membranes after synaptic vesicle docking and priming. In combination with a Ca2+-sensor, evoked fusion occurs upon an action potential. During fusion, SNARE complexes are fully formed (cis complex). Cis SNARE complexes are disassembled by the ATPase NSF in conjunction with SNAPs and made available for another round of synaptic vesicle formation.
The assembly of the SNARE complex is thought to provide the energy necessary for membrane fusion (144, 158). Single-molecule optical and magnetic trap pulling experiments suggest that the free energy that is released by the zippering of one SNARE complex is approximately 36 kBT (42, 103), and that a half-zippered intermediate exists under “loaded” conditions (i.e., when the juxtamembrane regions of the SNARE complex are steadily pulled by a force apparatus), suggesting that the trans SNARE complex may consist of a partially zippered complex. However, we note that direct imaging of the trans SNARE complex has not yet been achieved. Nevertheless, the estimated free energy that is released by formation of one SNARE complex is somewhat less than the energy that is required to overcome the hydration-force barrier for the formation of a lipid stalk (i.e., the dehydration energy of ~40–90 kBT that is required to induce stalk formation) (2). Additional energy may be required for pore formation and expansion (28). Consistent with this notion, SNAREs mediate membrane exchange when reconstituted into proteoliposomes (158). As little as one SNARE complex (148) is sufficient for ensemble lipid mixing of proteoliposomes, but at least two synaptobrevin-2 molecules, and presumably two SNARE complexes, are required for fast Ca2+-triggered exocytosis (131). Although these lipid mixing experiments demonstrate that SNAREs mediate membrane exchange, content mixing is the proper correlation for neurotransmitter release since lipid mixing can occur in the absence of full fusion due to metastable hemifusion diaphragms (35, 54, 78). Moreover, dequenching of the fluorescent dyes used in lipid mixing assays may occur due to other phenomena (186). The first genuine content mixing experiment for precisely timed Ca2+-triggered fusion with synaptic proteins was reported by (78, 79) (Fig. 2) (for a review of other assays and more details see (18)). Over the years, this single-vesicle fusion assay has progressed to include more molecular components with the goal of a more complete physiological reconstitution (35, 80–82) (Section 10).
Figure 2. Single-vesicle fusion assay.
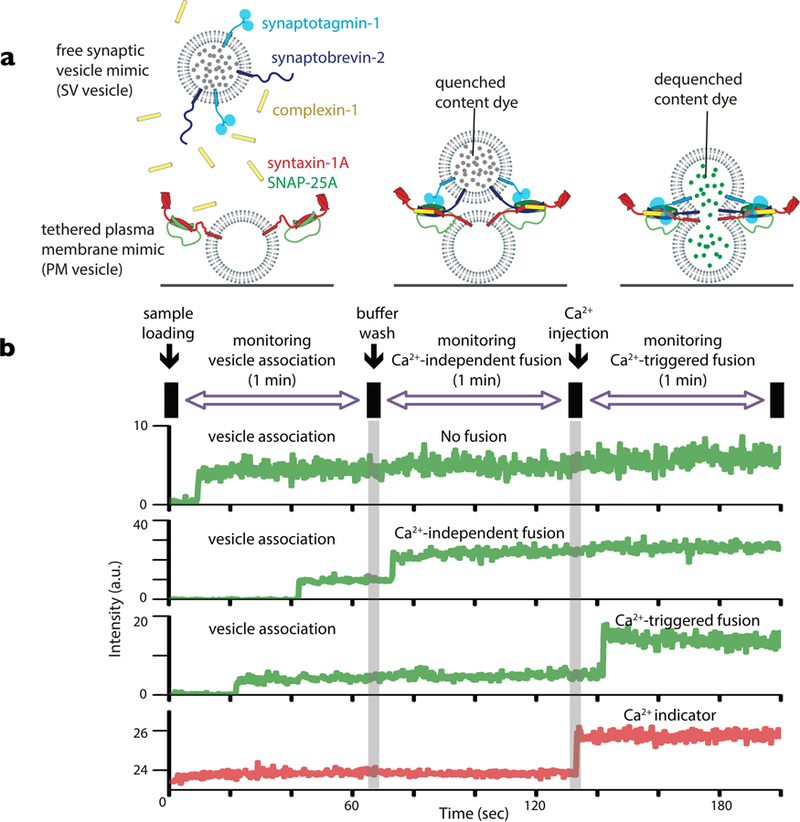
(a) The schema shows the initial setup with proteoliposomes with reconstituted synaptobrevin-2 and Syt1 that mimic synaptic vesicles (SV vesicles) and surface-tethered proteoliposomes with reconstituted syntaxin-1A and SNAP-25A that mimic the plasma membrane (PM vesicles). Cpx1 is included in solution. A soluble dye is encapsulated into the SV vesicles, and in some experiments, a lipid dye is incorporated into the SV vesicle membrane. These dyes are at high enough concentration to result in self-quenching of the fluorescence intensity. An increase in volume or surface area, respectively, will produce an increase in the respective fluorescence intensity. (b) Ca2+-independent fusion is observed for SV vesicles that are associated with tethered PM vesicles. (c) Ca2+-triggered fusion events are observed. For more details see references (35, 78–80, 82).
3. Synaptotagmin
In addition to SNAREs, synaptotagmins (Syts) constitute an evolutionary conserved family of proteins (105) that are composed of an N-terminal single transmembrane-spanning domain, a variable juxtamembrane linker and two C-terminal cytoplasmic C2 domains, termed C2A and C2B, respectively (112). In this review we focus on synaptotagmin-1 (Syt1), which resides on the synaptic vesicle and is vital for synchronous Ca2+-triggered synaptic vesicle fusion (41, 43).
Syt2 and Syt9 are also involved in Ca2+-triggered synchronous neurotransmitter release for different subsets of neurons (167), while Syt7 mediates asynchronous release (5, 160). In addition, Syt1 and Syt7 together act as redundant Ca2+ sensors for neuroendocrine exocytosis (125, 141), and they also participate in postsynaptic AMPA receptor exocytosis during long term potentiation (LTP) (164).
Syt1 interacts with anionic membranes and SNARE complexes in both Ca2+-dependent and Ca2+-independent manners (7, 15, 16, 22, 26, 31, 41, 73, 77, 111, 152, 156, 184). Syt1 function and membrane binding is specific to Ca2+, i.e., other divalent cations, such as Sr2+, only weakly trigger synaptic vesicle fusion (38). Under certain experimental in vitro conditions, (i.e., in the presence of Ca2+), the Syt1 C2B domain or the C2A-C2B fragment binds to curved membranes or favors deformation of the membrane (60, 93), along with membrane clustering (3). Computer simulations support the notion that both C2 domains cooperate to induce membrane bending (165). Moreover, the linker between the C2 domains is important for Syt1 function in neurons (6, 87), and both C2 domains can insert into the same membrane at the same time upon Ca2+-binding in vitro (59). Additionally, the juxtamembrane linker domain—the domain between the transmembrane domain and the C2 domains—is important for Syt1 function in fusion assays (83) and in neurons (85). Thus the relative locations of the C2 domains are critical for proper function, but the precise location of Syt C2 domains before and after Ca2+-triggering remains to be directly visualized.
While mutations in the Ca2+-binding sites of the Syt1 C2A domain can partially rescue Syt1-deficient mammalian neurons, mutations in the Ca2+-binding sites of the Syt1 C2B domain cannot (107, 130, 168). The mutations in the Syt1 C2B domain not only block Ca2+-evoked synchronous release in an intact synapse, but also dominantly inhibit the ability of endogenous wild-type Syt1 to trigger fusion, consequently increasing spontaneous miniature release when co-expressed (84, 164, 185).
SNAREs and Syt1 alone are sufficient to promote Ca2+-triggered proteoliposome lipid mixing (147). However, as mentioned above, lipid mixing does not necessarily correlate with neurotransmitter release since lipid mixing can occur without content mixing. While SNAREs and Syt1 can indeed promote full fusion (i.e., content mixing) upon Ca2+-triggering (35, 78) with a ratio of Ca2+-dependent fusion to Ca2+-independent fusion of ~ 10 (82), Ca2+-triggered fusion with this minimal system is relatively inefficient. As discussed in Section 10, a more complete reconstitution greatly increases the efficiency and synchrony of Ca2+-triggered fusion.
The first crystal structure of a Syt C2 domain (143) revealed that there are no large conformational changes upon Ca2+ binding. Rather, the C2 domain acts as an electrostatic switch where Ca2+-binding neutralizes the negative charges in that region as suggested by NMR experiments (127). Structures of complexes between Syts and the SNARE complex have been extremely difficult to obtain due to the multiple binding modes of the components of the complex (15, 26), and it had been questioned if SNARE-Syt interactions are functionally important (110). It is only recently that crystal structures of SNARE/Syt1 and SNARE/Cpx1/Syt1 complexes have been obtained and functionally validated (184, 185) (Section 9).
4. Complexin
The cytoplasmic protein complexin (Cpx) also plays critical roles in neurotransmitter release. Specifically, synchronous evoked neurotransmitter release depends on Cpx (99), and this activating role of Cpx is conserved across all species and different types of Ca2+-induced exocytosis studied to date (21, 55, 61, 67, 94, 97, 119, 173, 175, 177). Cpx also regulates spontaneous release, although this effect is less conserved among species and experimental conditions: for example, in Drosophila, spontaneous release increases with knockout of Cpx compared to wildtype neurons (65, 171). Likewise, knockdown in cultured cortical mouse neurons increases spontaneous release compared to wild-type neurons, although knockout of Cpx only affects spontaneous release depending on the particular neuronal cell type (67, 97, 146, 175).
We focus here on the homolog complexin-1 (Cpx1) of the mammalian Cpx family whose primary sequence is highly conserved (96%) in mouse, rat, and human. Cpx1 consists of four domains: the N-terminal domain is important for activation of synchronous Ca2+-triggered release in murine neurons (97, 170, 172) and in isolated chromaffin cells (33); the accessory domain regulates spontaneous release (97); the central domain is required for all functions of Cpx1 and it binds with high affinity (~10 nM) to the neuronal ternary SNARE complex (24, 99, 109); and the C-terminal domain is involved in vesicle priming and binds to anionic membranes in a curvature sensitive fashion (24, 47, 67, 97, 133).
Structurally, in isolation, both the N- and C-terminal domains of Cpx1 are largely unstructured, while the accessory and central domains have α-helical propensity (108). The central domain of Cpx1 binds to the groove formed by the synaptobrevin-2 and syntaxin-1A α-helices in the SNARE complex (14, 24). This interaction is anti-parallel, i.e., the direction of the α-helix of the accessory domain of Cpx1 is anti-parallel to the direction of the α-helices in the ternary SNARE complex.
The accessory domain of Cpx1 is not required for activation of Ca2+-triggered single-vesicle fusion with reconstituted neuronal SNAREs and Syt1 (81), and mutations of this domain have no effect on evoked release in neurons (176). However, mutations of the Cpx1 accessory domain affect spontaneous release (25, 176), and elimination of the accessory domain increases Ca2+-independent single-vesicle fusion (content mixing) (81) compared to wild-type control. In another study, introduction of an α-helix breaking mutation into the accessory domain of Cpx1 increased spontaneous release in C. elegans (117).
Functional studies, along with molecular modeling suggested that Cpx1 may inhibit full ternary SNARE complex formation by preventing the C-terminal part of synaptobrevin-2 from binding to the syntaxin-1A/SNAP-25A subcomplex of the ternary SNARE complex (45, 46). However, a subsequent crystal structure of a mutant of Cpx1 (D27L, E34F, R37A) in complex with a partially truncated SNARE complex (containing a truncated synaptobrevin-2 fragment), along with isothermal titration calorimetry (ITC) and light scattering experiments instead found that Cpx1 bridges two partially truncated SNARE complexes: one partial SNARE complex binds to the central domain of Cpx1 while another partial SNARE complex binds weakly to the accessory domain of the same Cpx1 molecule (PDB IDs 3RK3 and 3RL0) (75, 76). Although this weak interaction of the accessory domain is observable by ITC (75, 116), it was not observed by NMR (146), perhaps due to differences in constructs (75, 116). Finally, single-molecule experiments demonstrated that the accessory domain also weakly interacts with the syntaxin-1A/SNAP-25A (binary) complex (27). Moreover, these single-molecule studies showed that both binding modes of the Cpx1 accessory helix are possible, reconciling the previous results. However, the biological context of these weak interactions involving the accessory domain remains to be elucidated.
The C-terminal domain of Cpx1 binds to phospholipids (67, 126) and it is important for vesicle priming in neurons (33, 34, 67). Moreover, Cpx1 without the C-terminal domain does not reduce spontaneous release, but it still activates Ca2+-triggered release in neuronal cultures (67) and in a reconstituted system (82). The C-terminal domain is sensitive to membrane curvature, and it may thus localize Cpx1 to the synaptic membrane (47, 133, 163). Interestingly, the curvature sensing value of the C-terminal domain is comparable to that of α-synuclein (47, 63), i.e., a value that corresponds to the relatively high curvature of synaptic vesicles with a typical diameter of 40 nm. In support of this notion, replacement of the C-terminal domain with plasma membrane-localizing elements, results in increased spontaneous neurotransmission and a prolonged synchronous decay time-constant of NMDA-type glutamate receptor evoked postsynaptic currents in cultured neurons (47).
In conjunction with neuronal SNAREs and Syt1, Cpx1 increases the Ca2+-triggered amplitude and synchrony of proteoliposome fusion, and it also suppresses Ca2+-independent single vesicle fusion (content mixing) (82), resulting in a 50–100-fold ratio of Ca2+-triggered fusion to Ca2+-independent fusion. Moreover, the functions of the four domains of Cpx1 have been reproduced in vitro (81, 82). By varying the Cpx1 concentration, these single-vesicle studies suggest that the regulatory effect on Ca2+-independent fusion and the facilitating role on Ca2+-triggered fusion are governed by distinct molecular mechanisms involving different subsets of the four domains of Cpx1. Similarly, genetic experiments in Drosophila also suggest distinct mechanisms for activation of fast synchronous release and regulation of spontaneous release (25).
5. Munc18
Sec1/Munc18 (SM) proteins are also required components of all membrane trafficking pathways as exemplified by the block of synaptic vesicle fusion upon Munc18–1 knockout in mice (151). Following disassembly of SNARE complexes with NSF and SNAP, Munc18–1 captures syntaxin-1A, locking it into a “closed” conformation and preventing reassembly of the ternary SNARE complex (91). Munc18–1 binds tightly (Kd ~1–4 nM) to syntaxin-1A in which the N-terminal three helix bundle of syntaxin-1A (Habc domain) forms a cis conformation with the syntaxin-1A SNARE motif (H3 domain) (19, 23, 104), in turn hindering SNARE complex formation (114, 174).
Moreover, Munc18 co-localizes with syntaxin and SNAP-25 in fixed cultured neurons and forms nano-clusters in the plasma membrane (113). Interestingly, in neuroendocrine cells, transfection of syntaxin alone results in its mislocalization, whereas co-transfection of both Munc18 and syntaxin results in proper trafficking to the plasma membrane (100, 123). Thus, one function of Munc18 may be to stabilize or “chaperone” syntaxin, which also agrees with the aforementioned knockout studies of Munc18 in mice that showed a reduced level of syntaxin-1 in the plasma membrane (13). The crystal structure of the Munc18–1/syntaxin-1A complex revealed extensive interactions of syntaxin-1A in the closed conformation with the concave surface formed by domains 1 and 3 of Munc18–1 (104), explaining the inability of the syntaxin-1A H3 domain in this closed conformation to interact with SNAP-25A and synaptobrevin-2.
Binding of Munc18–1 to the closed conformation of syntaxin-1A is not always required, as the syntaxin-1A Habc domain is not essential for evoked synaptic vesicle fusion in cultured syntaxin-1A-deficient neurons, although it is required for spontaneous synaptic vesicle fusion (183). In marked contrast, the N-terminal residues of syntaxin-1A are required for binding of the ternary SNARE complex to Munc18–1, and may thereby serve as a recruiting factor for Munc18–1 (118). Munc18–1 binds to the assembled ternary SNARE complex via the syntaxin-1A N-terminal residues, with syntaxin-1A in an “open” conformation (128) (32, 58, 169). In this open conformation, the H3 domain of syntaxin-1A interacts with its partner SNARE motifs to form the SNARE complex (144), while the syntaxin-1A Habc domain is presumably flexibly linked to the four-helix core of the SNARE complex (51, 106).
6. Munc13
Munc13–1 and its homologues are primarily brain-specific, cytoplasmic proteins in the presynaptic terminal that are involved in synaptic vesicle priming and short-term synaptic plasticity (138, 139). Munc13–1 is expressed in all neurons of the rat central nervous system, while Munc13–2 and Munc13–3 are mostly restricted to different brain regions. The ubiquitously expressed splice variant ubMunc13–2 and the Munc13–4 isoform are predominantly expressed in non-neuronal cells (72). Munc13–4 only includes the C2B, MUN, and C2C domains (Domain functions will be detailed below). The UNC-13 and Dunc-13 homologs of Munc13–1 lack the N-terminal C2A domain, as do a brain-specific splice-variant of bMunc13–2 and the Munc13–3 isoform. Here we will focus on the Munc13–1 isoform.
Early studies found that neurons in mice lacking Munc13–1 exhibit severely limited activity-dependent as well as hypertonic sucrose-induced neurotransmitter release (4). Moreover, deletion of Munc13–1 results in substantially larger distances between synaptic vesicles and active zones as observed by electron microscopy (50, 62, 159). The priming phenotype of Munc13–1 knockout is much stronger than that of other priming factors, including Cpx1 (175), i.e., the size of the readily releasable pool of synaptic vesicles is much smaller in Munc13–1 knockout mice compared to Cpx1 knockout mice. Moreover, Munc13–1, together with Munc18–1, prevents NSF-dependent de-priming of synaptic vesicles (53). Finally, Munc13–1 and −2 double-knockout mice have essentially no release-competent synaptic vesicles (150) and exhibit a severe docking defect (62).
Below, we briefly summarize structural and functional studies for each domain of Munc13–1. Munc13–1 consists of a C1 domain (C1), a calmodulin-binding domain, two C2 domains (C2A, C2B), and a MUN domain, and another C2 domain (C2C). The crystal structure of the C1C2BMUN fragment of Munc13–1 revealed inter-domain interactions between the C1, C2B, and MUN domains and a folded linker region between the C1 and C2B domains (166). An electron cryo-tomography (cryoET) study of vesicles with bound C1C2BMUN revealed filamentous features that suggest hinge bending of this fragment, with the straight conformation being consistent with the crystal structure (44).
Functionally, the C2A domain of Munc13–1 interacts with the Rab3 effector RIM, forming a tripartite complex (36) which is important for the priming function of Munc13–1 and for the presynaptic localization in mice (68). Likewise, the C2A domain of UNC-13 in C. elegans is important for regulating both evoked and spontaneous release, as well as its precise localization to the active zone (182).
Munc13–1 and the ubiquitously expressed isoform Munc13–2 contain a conserved calmodulin-binding domain. When calmodulin binding is disrupted by mutation, evoked release upon a single action potential is unchanged, but short-term plasticity and Ca2+-dependent modulation of the readily-releasable pool is reduced (66).
The diacyglycerol (DAG)-phorbol C1 binding domain regulates Munc13–1 functions in short-term synaptic plasticity and priming of synaptic vesicles, although mutation of the site does not affect isolated evoked neurotransmitter release (120). The C2B domain binds to phospholipids in a Ca2+-dependent fashion, with particular specificity for phosphatidylinositol phosphate (PIP) and phosphatidylinositol bisphosphate (PIP2). Additionally, an interaction between the C2B domain and presynaptic voltage-gated calcium channels modulates use-dependence of the channels (20). Interestingly, deletion of the C2B domain enhances Ca2+-triggered exocytosis in C. elegans neurons, suggesting an auto-inhibitory function of this domain (102).
The MUN domain of Munc13–1 has two major roles in vesicle fusion: catalyzing the transit of syntaxin-1A from the syntaxin-1A/Munc18–1 complex into the ternary SNARE complex (10, 91, 178) and promoting the proper assembly of the SNARE complex, in particular the parallel configuration of syntaxin-1A and synaptobrevin-2 (80). Supporting this latter role, the so-called LE mutant of syntaxin-1A (syntaxin-1ALE) (37) bypasses the requirement of Munc13–1 for the transit of syntaxin-1A into the ternary SNARE complex in presence of SNAP-25A and synaptobrevin-2 in vitro (90). It accomplishes this by preferentially adopting an open conformation of syntaxin-1A (155). However, this open syntaxin-1A mutant can only partially restore neurotransmitter release in unc-13 mutants of C. elegans (50), because it does not promote the proper assembly of the SNARE complex, i.e., SNARE complexes with the syntaxin-1A LE mutant are scrambled (80); this observation explains the poor rescue of Munc13 deletion by this mutant. Earlier studies in C. elegans suggest a similar function of full-length unc-13 (121), although the role of the MUN domain has not been directly studied in that system.
Finally, the C2C domain of Munc13–1 also plays a functional role in evoked release in chromaffin granule secretion and neurotransmission in C. elegans (92, 137), and in evoked release in priming in mouse neurons (88). It also enhances ensemble lipid and single-vesicle content mixing experiments (80, 88).
Similar to Munc13, CAPS proteins are members of an extended Munc13/CAPS family of proteins that are also important for priming of synaptic vesicles (64). They contain one Munc13 homology domain (MHD) that comprises part of the MUN domain, and a pleckstrin homology domain that interacts with PIP2. It is likely that the MHD domain in CAPS proteins is partially responsible for their function in synaptic priming (30). Both Munc13s and CAPS proteins are probably jointly important for synaptic vesicle priming. In support of a partially redundant function of both molecules, the C1C2BMUN fragment of Munc13 can substitute for deletion of CAPS in Ca2+-triggered dense core vesicle fusion with a planar supported bilayer (74).
7. NSF and SNAPs
Historically, the ATPase NSF was first discovered as a key factor for inter-compartmental transport in eukaryotic cells and, subsequently, SNAREs were found to be substrates for NSF (9, 134). Independently, SNAREs were discovered to be important for neurotransmitter release (11, 12, 140). In concert with the adaptor protein, SNAP, the ATPase NSF disassembles the SNARE complex into individual proteins upon ATP hydrolysis (51, 98, 135). NSF is a member of the AAA+ family consisting of two ATPase rings (known as Type II AAA+), and an N-terminal domain. Most eukaryotic organisms encode only one NSF gene. By contrast, there are three homologues of SNAP, with αSNAP being the most widely studied. Although earlier work suggested that disruption of the SNARE complex by ATP hydrolysis drives fusion (135), subsequent work clarified that it is actually the SNARE complex formation that drives fusion (51, 98). For more structural details of NSF and SNAPs we refer to a recent review (180).
8. Cooperation between Syt1, Cpx1 and SNAREs for evoked release
SNARE/Cpx1/Syt1 synaptic fusion complex
As mentioned in sections 1–3, Syt1, Cpx1 and SNAREs are all important for evoked release upon action potential arrival in the synaptic terminal (97, 168). The structures of the neuronal SNARE/Cpx1 subcomplex (24) and of the SNARE/Syt1 subcomplex (184), along with functional studies, suggested that each of these binary interactions are essential for neurotransmitter release. For example, the 4M mutant of Cpx1 that disrupts binding to the ternary SNARE complex neither rescued deletion of wildtype Cpx1 in neuronal cultures (97), nor increased the Ca2+-triggered amplitude in single-vesicle fusion experiments with reconstituted SNAREs and Syt1 (82). Similarly, disruption of the primary SNARE/Syt1 complex interface by mutations abolished fast synchronous release in cultured neurons and greatly reduced the Ca2+-triggered amplitude in single-vesicle fusion experiments (184).
The primary SNARE/Syt1 interface is structurally conserved in very different crystal packing environments and conditions (Fig. 5) (184, 185), demonstrating that it is a genuine and specific interface. In the presence of anionic phospholipid membranes, the polybasic region of Syt1 C2B primarily interacts with the membrane (111), and this membrane interaction likely stabilizes the SNARE/Syt1 primary interface (184), lowering the dissociation constant by about an order of magnitude (156). Based on primary sequence alignment, the SNARE/Syt1 primary interface is specific for fast Ca2+ sensors (Syt1, Syt2, Syt9) (167, 184).
Figure 5. The primary SNARE/Syt1 interface is structurally conserved.
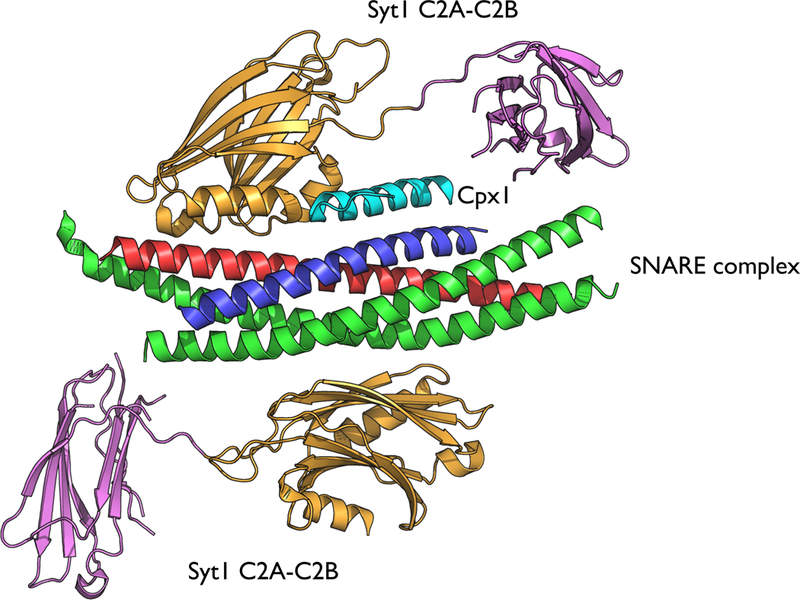
Superposition of the primary interface observed in crystal structures of the Ca2+ (3.5 Å resolution, PDB code 5CCG, color) and Mg2+ (4.1 Å resolution, PDB code 5CCI, gray/white) bound structures of the SNARE/Syt1 (184), and of the SNARE/Cpx1/Syt1 complex (1.8 Å resolution, PDB code 5W5C, black) (185). Note, that the crystal packing of the SNARE/Cpx1/Syt1 complex is very different from that of the SNARE/Syt1 complexes (185).
The binary SNARE/Syt1 and SNARE/Cpx1 interactions alone do not explain certain experimental results. For example, mutation of the Ca2+-binding region of the C2B domain of Syt1 has dominant negative effects on both evoked and spontaneous neurotransmitter release (84, 164) while deletion of Cpx1 abrogates these dominant negative phenotypes (185), suggesting a tripartite cooperation between SNAREs, Cpx1, and Syt1. The structure of the tripartite SNARE/Cpx1/Syt1 complex (Fig. 6) (185) explained these experimental results for the first time since it revealed a new interface between one Syt1 C2B domain and both the SNARE complex and Cpx1 (Fig. 6) (185). Simultaneously, a second Syt1 C2B domain interacts with the other side of the SNARE complex via the abovementioned SNARE/Syt1 primary interface (Fig. 6). Structure-guided mutagenesis, solution-binding studies by ITC, and electrophysiological recordings demonstrated that both Ca2+-triggered synaptic release and suppression of spontaneous release depend on both the SNARE/Cpx1/Syt1 tripartite and the SNARE/Syt1 primary interfaces (185).
Figure 6. Structure of the SNARE/Cpx1/Syt1 complex.
Shown are both the primary and the tripartite interface. For details see (185).
For the tripartite interface, the Syt1 C2B domain binds to the SNARE/Cpx1 subcomplex via interactions with both the SNARE and Cpx1 components (Fig. 6). Among the most striking structural features of this tripartite interface is the continuation of the Cpx1 central helix into the α-helix HA of Syt1. Since the α-helix HA is structurally conserved in C2B domains of all synaptotagmins, Doc2b, and Rabphilin, but not present in the Munc13–1 C2B domain and synaptotagmin C2A domains, the SNARE/Cpx1/Syt1 tripartite interface may be more general and involve other Syts. The different types of Syt-regulated exocytosis are mediated by similar Cpx1-dependent fusion mechanisms (Syt1, Syt2, Syt7, Syt9, and Syt10) (21, 49, 125, 167), so it is conceivable that these other Syts could participate in a tripartite interface while another Syt1, Syt2, or Syt9 molecule could be involved in the primary interface.
The tripartite interface involves the central α-helix, but not the accessory helix of Cpx1 or any other part of Cpx1. Indeed, the accessory domain can be entirely eliminated while maintaining the activating function of Cpx1 in single-vesicle fusion experiments with reconstituted SNAREs, Syt1, and Cpx1 (81). The structure of the SNARE/Cpx1/Syt1 complex now explains the functional requirement of the central α-helix of Cpx1 since it is an integral part of the tripartite interface (Fig. 6). On the other hand, the N-terminal domain of Cpx1 is important for activation of synchronous Ca2+-triggered release (33, 97, 170, 172), and it increases the Ca2+-triggered amplitude in single-vesicle fusion (content mixing) experiments when added as an independent fragment in addition to the Cpx1 central domain (81). One possibility is that the Cpx1 N-terminal domain may independently interact with the splayed open trans SNARE complex (27). Note that the membrane proximal parts of the trans SNARE complex were not included in the crystal structure of the SNARE/Cpx1/Syt1 complex, so it is not surprising that the Cpx1 N-terminal domain is not visible in that crystal structure.
Why did it take so many years since publication of the first crystal structure of the SNARE complex (144) to obtain structures of the SNARE/Syt1 and SNARE/Cpx1/Syt1 complexes? In retrospect, we now know that there are many weak or dynamic interactions between Syt1 and the SNARE complex (15, 26) which obscure the specific interface(s) in solution (185). In order to obtain the SNARE/Syt1 complex, covalently linked chimeras of the components of the complex were designed in order to stabilize the complex (184). Crystallization of the tripartite SNARE/Cpx1/Syt1 complex was achieved by truncation of the 23 C-terminal residues of the cytoplasmic domain of synaptobrevin-2 which prevented formation of the six membrane proximal layers of the SNARE complex, but left the binding interface to the central α-helix of Cpx1 intact (185). This truncated complex mimics a pre-fusion state of the trans SNARE complex where the membrane proximal part of the complex is not fully zippered. For crystallization, this feature of the truncated complex was essential since it prevented formation of inter-SNARE complex interactions that would otherwise interfere with formation of the SNARE/Cpx1/Syt1 complex. Moreover, this strategy alleviated the need for covalent linkers between the components.
Supramolecular arrangements of synaptic fusion complexes
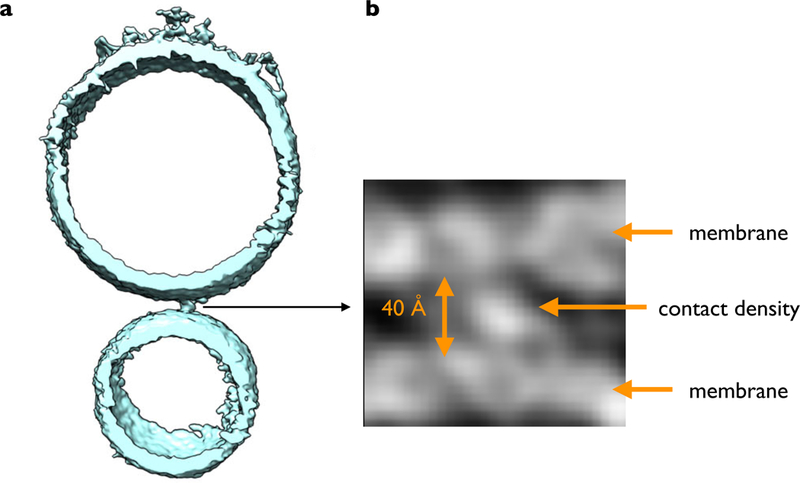
The structure of the SNARE/Cpx1/Syt1 complex likely represents the pre-fusion primed state of the system (185), one of likely two or more synaptic complexes involved in Ca2+-triggered fusion (131). These two or more synaptic complexes likely interact with each other. For example, one Syt1 C2B domain could bridge two SNARE complexes via the primary and tripartite interfaces and it could be sandwiched between the two membranes (185). CryoET images of the same proteoliposomes as used for the single-vesicle fusion experiments indeed revealed contacts with a variety of morphologies between the vesicle membranes with a preference for relatively compact point contacts (Fig. 7A) (44), although it remains to be established that it is these point contacts that undergo fast Ca2+-triggered fusion. These contacts are likely proteinaceous since the observed densities of these contacts are relatively high (Fig. 7B) (44) and the observed separation in the cryoET images between the membranes is 40 to 60 Å, substantially longer than the critical distance at which lipid stalks can form (< 9 Å) (2). Volumetric analysis suggested that the compact point contacts can accommodate two synaptic complexes while the lone contacts can accommodate more complexes (44). Thus, the juxtaposition of protein complexes between membranes prevents fusion until Ca2+-triggering, in remote analogy to the action of viral fusion proteins where fusion only happens when a sufficient number of neighboring viral fusion proteins switch to their fusogenic form (52).
Figure 7. Point contact between proteoliposomes with neuronal SNAREs, Cpx1, Syt1, and Munc13.
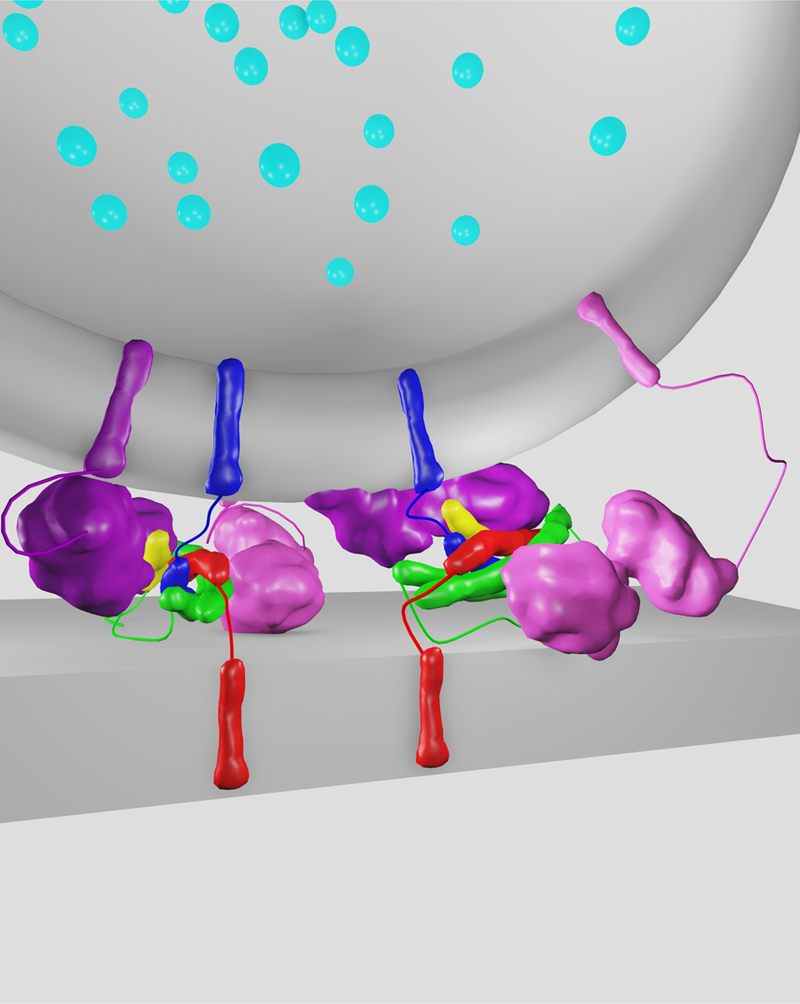
(a) Shown is a slice of an isosurface representation of a SV vesicle (bottom) and a PM vesicle (top) in the presence of the C1C2BMUN fragment of Munc13. (b) Close-up view of grey scale tomographic 2D-slices of the contact site between the two membranes. For more details see (44).
How does Cpx fit into this picture? Cpx1 has at least two conformations when bound to the ternary SNARE complex (27). In one conformation, the accessory domain cooperates with the N-terminal domain of Cpx1 to induce a conformational change at the membrane-proximal C-terminal end of the bound SNARE complex, likely making it similar to the splayed open trans SNARE complex. Conversely, if the SNARE complex is in the trans conformation, the interaction of the N-terminal domain of Cpx1 can occur, regardless of the accessory domain, consistent with the expendable nature of the accessory domain for evoked release. In the other Cpx1 conformation, Cpx1 can bridge two SNARE complexes, such as, a fully-assembled ternary SNARE complex and a binary SNARE complex, consisting of syntaxin-1A and SNAP-25A, requiring the presence of both the central and accessory domains (27). It has been proposed that Cpx1 could bridge many SNARE complexes in a “zigzag” arrangement (76). However, such an arrangement would be incompatible when Syt1 would bridge SNARE complexes via the primary and tripartite interfaces (185), although the abovementioned bridging of a ternary and a binary SNARE complex would be possible.
In addition to forming a complex with SNAREs and Cpx1, Syts are capable of forming ring-like homo-oligomeric assemblies on membranes containing acidic lipids (153) and in solution (154), although the functional importance of these assemblies has yet to be established. These rings are disrupted by Ca2+ when bound to acid phospholipid membranes (153, 179). Finally, synaptobrevin is capable of forming ring-like structures in complex with synaptophysin in vitro (1). In contrast to these studies, no rings are observed by electron cryo-tomography of slices of unstained, vitrified frozen-hydrated mouse synapses (40), or electron cryo-tomography of thin sections of high-pressure frozen hippocampal neuronal cultures (29). Moreover, cryoET studies of proteolipsomes with neuronal SNAREs, Syt1, Cpx1, and Munc13 revealed short and long contacts, but no ring-like features, i.e., regular point-group symmetry was not discernible in these cryoET studies (44). Clearly, the long contacts may represent higher order oligomeric, but asymmetric, assemblies. Perhaps multiple membrane contact morphologies co-exist in the neuron and are relevant in different contexts (e.g., fast synchronous release, asynchronous release, and spontaneous release).
Model of unlocking and Ca2+-triggering
Regardless of the supramolecular arrangement of the synaptic protein complexes in the observed contacts between membranes, they appear stably locked into a conformation from which fusion is unlikely considering the observed membrane separation of 40–60 Å (Fig. 7). Consequently, the pre-fusion SNARE/Cpx1/Syt1 complexes would be only partially zippered (Fig. 8). The readily releasable pool of synaptic vesicles requires both interfaces (185), suggesting that the crystal structure of the SNARE/Cpx1/Syt1 complex represents the primed state. Upon Ca2+-binding to the C2 domains of the primed complex, the SNARE/Cpx1/Syt1 tripartite interface must be unlocked or un-inhibited, allowing membranes to move closer and Ca2+-triggered fusion to commence. It is likely that upon Ca2+-binding, the Syt1 molecule that is involved in the SNARE/Cpx1/Syt1 tripartite interface is dislodged by interacting with the synaptic vesicle membrane (185). This Syt1 dissociation may also induce a conformational change of Cpx1 (27). However, full dislodging of Cpx1 (145) is unlikely considering the tight binding (Kd ≈ 10–100 nM) and the relatively slow off-rate of 0.1 s−1 between Cpx1 and the cis ternary SNARE complex (27). Upon unlocking the primed complex, the SNARE then may fully zipper (144, 158), and trigger fusion, likely in conjunction with the membrane-bending action of the Syt1 molecule (60, 93) that is involved in the primary interface (184).
Figure 8. Model of primed pre-fusion SNARE/Cpx1/Syt1 complexes.
In this model, two SNARE/Cpx1/Syt1 complexes (185) form a contact between a synaptic vesicle (top) and the plasma membrane (bottom). Blue: synaptobrevin-2, green: SNAP-25A, red: syntaxin-1A, yellow: Cpx1, light/dark magenta: Syt1s. The molecules and membranes are drawn to scale, with a ~ 40 Å separation between membranes as observed by cryoET (44). Note that the relative arrangement of the two SNARE/Cpx1/Syt1 complexes is unknown. Also, there can be more than two such complexes forming long membrane contacts (44).
A model of locked and primed SNARE/Cpx1/Syt1 complexes that juxtapose synaptic and plasma membranes (Fig. 8) explains the dominant negative effect of certain mutations of the Ca2+-binding region of the Syt1 C2B domain (84, 164, 185): the presence of permanently locked complexes would prevent membranes from approaching the critical distance to form lipid stalks (less than 9 Å) (2). As mentioned in Section 3, neuronal SNAREs and Syt1 alone are a minimal system for Ca2+-triggered membrane fusion (35, 78, 82, 147). Thus, the triggering mechanism in this even more minimal system may be similar to that of the unlocked SNARE/Cpx1/Syt1 system after dislodging of the Syt1 molecule that is involved in the tripartite interface. Clearly, high resolution images of the SNARE/Cpx1/Syt1 complexes between membranes, and monitoring the rapid conformational transitions of the complexes upon Ca2+-triggering will be important next steps.
9. Implications for spontaneous release
Syt1, Cpx1, and SNAREs are also important for spontaneous release (33, 65). More specifically, both the primary SNARE/Syt1 and tripartite SNARE/Cpx1/Syt1 interfaces are required for the suppression of spontaneous release in neurons, i.e., elimination of either interface resulted in an increase in mini frequency (185). Why is there an increase in spontaneous fusion frequency when either interface is disrupted? At a structural level, it could simply mean that membranes can get closer when one or more components of the SNARE/Cpx1/Syt1 complex are missing, consistent with the closer membrane separation distance observed by cryoET of proteoliposomes when Cpx1 is absent (44). However, there is an observation that counters this simple membrane separation distance argument: the probability of Ca2+-independent fusion did not increase when Syt1 is absent in single-vesicle fusion experiments with SNAREs and Cpx1 only (82). Despite this, these experiments should be repeated with full reconstitution (80), since in the absence of full reconstitution, improperly formed SNARE complexes may obscure the more subtle effect of suppression of Ca2+-independent fusion.
We would like to emphasize that Ca2+-independent fusion of single-vesicle experiments cannot strictly be compared with spontaneous release in neurons. First, Ca2+-independent fusion probabilities are normalized to the number of associated vesicles. Second, the frequency of spontaneous release measured in electrophysiological experiments depends on both the number of functional synapses as well as the number of synaptic vesicles that are capable of undergoing spontaneous fusion. Third, there is likely another Ca2+-sensor that competes with Syt1 in binding to the tripartite interface (142). Elimination of the primary interface or presence of the certain Syt1 C2B mutants would affect the binding equilibrium, possibly explaining the increase of spontaneous release in neurons (185).
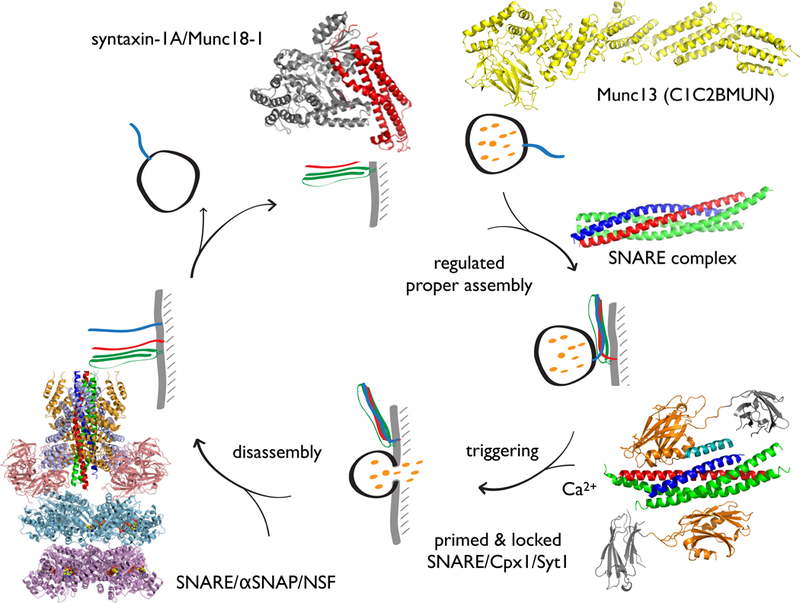
10. Cooperation between Munc18 and Munc13 for proper SNARE assembly
SNARE assembly is fuzzy
As mentioned above, the two interfaces in the SNARE/Cpx1/Syt1 complex are important for the readily releasable pool of synaptic vesicles (185). The readily releasable pool of synaptic vesicles in turn is established by a process that is referred to as priming, i.e., the association of synaptic vesicles with presynaptic proteins that enables them to undergo fast evoked fusion (70). There are several factors involved in priming, including Munc18 and Munc13, but the molecular basis of the priming function of these molecules had been unknown until recently. There is now evidence that at least one function of priming factors is to ensure the proper assembly of SNARE complexes (80). Why is it necessary to assist the proper assembly of the SNARE complex? When soluble fragments of SNARE proteins are simply mixed in solution, improper configurations may occur (27, 89, 124, 162). In particular, the existence of antiparallel SNARE complex configurations were directly probed by using FRET dye pairs (27, 162). As described (162), antiparallel configurations can be suppressed by extensive purification of the ternary SNARE complex, including a urea wash step. Another biochemical method to obtain more fusogenic complexes consists of incubating with C-terminal fragments of synaptobrevin-2 (101, 115) prior to assembling the SNARE complex. Single-molecule FRET experiments now revealed that these C-terminal synaptobrevin fragments promote proper SNARE complex formation (27). Clearly, urea treatments or incubation with synaptobrevin-2 fragments are not physiological mechanisms, but they illustrate the importance of proper SNARE complex assembly in establishing maximum fusogenicity.
Munc13 and Munc18 promote proper SNARE complex assembly
In search for a physiological mechanism that ensures proper SNARE complex formation, Lai et al. found that the MUN domain of Munc13–1 promotes the proper parallel subconfiguration between syntaxin-1A and synaptobrevin-2 when assembling the ternary SNARE complex when starting from the syntaxin-1A/SNAP-25A complex, i.e., the MUN domain prevented improper subconfigurations, and it increased the Ca2+-triggered amplitude in single-vesicle fusion (content mixing) experiments (80). Inclusion of the longer C1C2BMUN or C1C2BMUNC2C fragments of Munc13–1 in the fusion assay enhanced the Ca2+-triggered amplitude and fusion ratio in a manner similar to the MUN domain. However, these longer fragments were able to achieve these effects at twenty- and hundred-fold lower concentrations than the MUN domain, respectively.
In addition to its autonomous function of promoting the proper syntaxin-1A/synaptobrevin-2 subconfiguration, Munc13 also cooperates with Munc18–1 to promote the proper syntaxin-1A/SNAP-25A subconfiguration within the assembled ternary SNARE complex (80). Thus, while Munc13 alone is sufficient to promote the proper syntaxin-1A/synaptobrevin-2 subconfiguration starting from the syntaxin-1A/SNAP-25A complex and consequently increasing the Ca2+-triggered fusion amplitude, the proper syntaxin-1A/SNAP-25A subconfiguration is only achieved when starting from the syntaxin-1A/Munc18–1 complex. Moreover, near physiological Ca2+-sensitivity of the assay was achieved when both Munc13–1 and Munc18–1 were included (80). Taken together, Munc13–1 and Munc18–1 are factors for establishing the proper subconfigurations during trans ternary SNARE complex assembly (with parallel arrangements of the SNARE components and 1:1:1 stoichiometry) by suppressing improper subconfigurations, thus promoting proper ternary SNARE complex assembly. The cooperation of Munc18–1 and Munc13–1 in promoting the proper SNARE complex assembly suggests that Munc18–1 has an indirect but essential role in enabling high fusion efficiency, perhaps explaining the severe phenotype of deletion of Munc18–1 in neurons (151).
When some of the SNARE complexes around the contact site between the synaptic and plasma membranes are improperly assembled in the absence of Munc13, they have a large effect on morphologies of the contact sites between SV and PM vesicles as observed by cryoET of these vesicles (44). Upon inclusion of Munc13, the contacts are largely restricted to point contacts only, i.e., no long contacts are present and very few “dead-end” hemifusion diaphragms were observed prior to Ca2+-triggering. The ternary SNARE complex interacts with both Syt1 and Cpx1, forming the primed complex (185). These interactions in the SNARE/Cpx1/Syt1 complex may not occur if there are improper subconfigurations within the ternary SNARE complex. In particular, an anti-parallel subconfiguration between syntaxin-1A and synaptobrevin-2 would prevent formation of the SNARE/Cpx1/Syt1 tripartite interface (Fig. 6) (185). Moreover, an anti-parallel subconfiguration between SNAP-25A and syntaxin-1A would prevent formation of the primary SNARE/Syt1 interface since it involves the SNAP-25A and syntaxin-1A components of the SNARE complex (Fig. 5) (184).
Towards more complete reconstitution of synaptic vesicle fusion
Consistent with their molecular functions, Munc18 and Munc13 increased the efficiency of ensemble proteoliposome lipid and content mixing in the presence of SNAREs, Syt1, NSF and αSNAP (91). Moreover, when Munc13 and Munc18 were included, the Ca2+-triggered amplitude of single vesicle content mixing was four-fold higher for a reconstitution with SNAREs, Syt1, Cpx1, NSF, αSNAP (80). Remarkably, the Ca2+-sensitivity of this single-vesicle fusion assay was about 23 µM, which is close to the physiological range (Fig. 3). These improvements in amplitude and sensitivity are achieved by proper assembly of all components of the SNARE complex by Munc18 and Munc13 (Fig. 4) (80).
Figure 3. Reconstitution with neuronal SNAREs, Cpx1, Syt1, NSF, αSNAP, Munc18, Munc13.
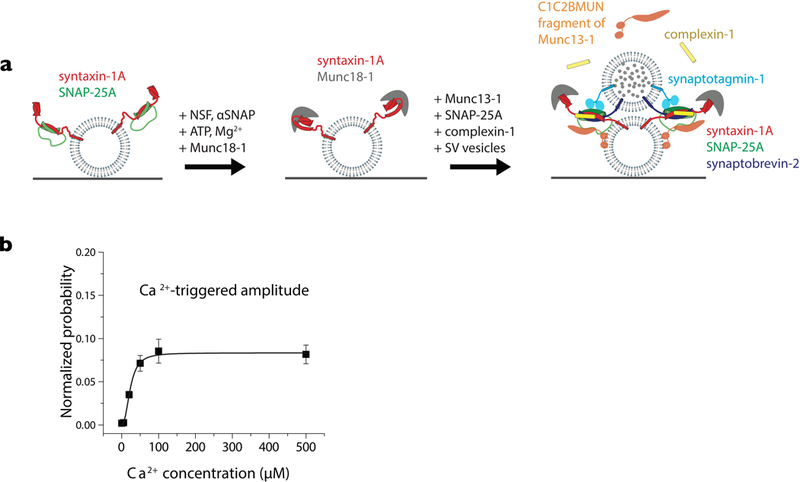
(a) Prior to the schema shown in Fig. 2a, a step is added that dissociates the syntaxin-1A/SNAP-25A complex by NSF, αSNAP, ATP, Mg2+ and concomitant formation of the syntaxin-1A/Munc18 complex. Incubation of the resulting syntaxin-1A/Munc18 vesicles is performed in the presence of Munc13–1, Cpx1, and SNAP-25A. (b) Ca2+-sensitivity of the fusion assay using the schema shown in panel a. For more details see (80).
Figure 4. Munc13 and Munc18 assist in proper SNARE complex formation.
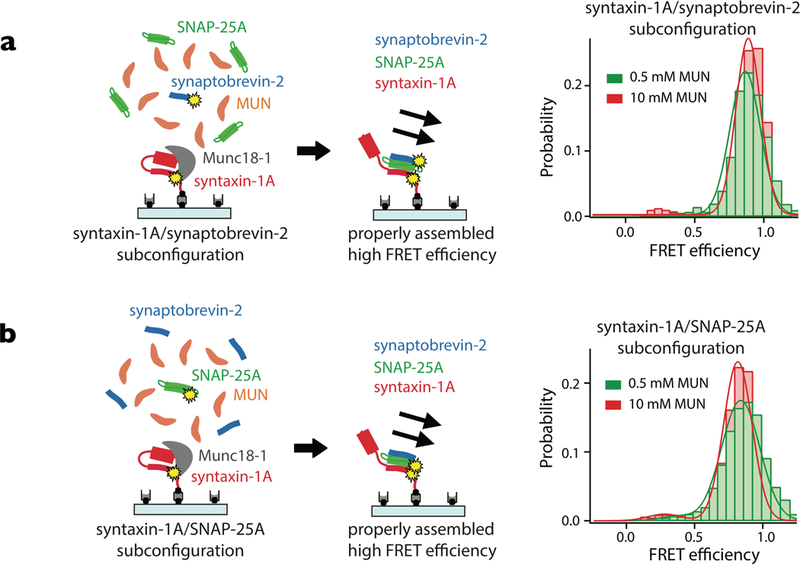
Starting from the syntaxin-1A/Munc18 complex, ternary SNARE complex is formed upon addition of the MUN domain of Munc13, SNAP-25A, and synaptobrevin-2. Single-molecule FRET measurements involving pairs of fluorescent dyes indicate that the components of the SNARE complex are parallel with respect to each other. For more details see (80).
How do Munc13 and Munc18 function?
The molecular mechanism by which Munc13 and Munc18 promote proper SNARE complex assembly is still a mystery, although there is some information about the molecular interactions. The MUN domain initially interacts with the Munc18–1/syntaxin-1A complex via a conserved hydrophobic pocket located at the midpoint of the MUN domain (90, 178). Moreover, two conserved residues R151 and I155 in the syntaxin-1 linker region are also critical for the function of Munc13–1, and they likely interact with the MUN domain (155). Single-molecule FRET experiments revealed that the MUN domain does not dissociate the closed Munc18–1/syntaxin-1A complex, but that it rather induces a conformational change in the syntaxin-1A linker region, thus enabling syntaxin-1A to transit into the ternary SNARE complex once synaptobrevin-2 and SNAP-25A are added. In addition to these interactions, the MUN domain weakly interacts with synaptobrevin-2 (80, 132), syntaxin-1A (80, 90), the syntaxin-1A/SNAP-25A complex (48, 161), and the ternary SNARE complex (48).
In some ways, assistance of proper ternary SNARE complex formation by Munc13–1 is reminiscent of certain ATP-independent chaperones such as Trigger Factor that assist protein folding of the nascent polypeptide chain upon synthesis by the ribosome (57). It has been proposed that Trigger Factor functions by shielding exposed hydrophobic residues during folding (71), inhibiting premature folding (96), unfolding preexisting structures and intermediate structures (56), or assisting protein assembly (95). While the MUN domain of Munc13–1 is structurally quite different than Trigger Factor, some of the properties of Trigger Factor also apply to the MUN domain in that both act through a series of weak and transient interactions to ensure a given conformation.
The existence of factors for proper assembly of ternary SNARE complex may be a general phenomenon. In the context of a different SNARE-mediated fusion process, the so-called homotypic fusion and protein sorting (HOPS) complex is essential in promoting maximum efficiency of homotypic vacuolar fusion (186), where the Munc18–1 homolog in the HOPS complex (Vps33) affects vacuolar SNARE-mediated membrane fusion (8). The crystal structures of a complex with the Munc18 homolog Vps33 and the synaptobrevin homolog Nyv1 as well as the complex of Vps33 and the syntaxin homolog Vam3 suggest that Vps33 regulates the proper assembly of Nyv1 and Vam3. Moreover, NMR experiments showed that synaptobrevin-2 interacts with Munc18–1 (132) in a similar fashion as suggested by the Vps33/Nyv1 complex. It is of course possible that the Vps33/Nyv1 interaction is promiscuous and may allow interactions with other SNAREs as well. Such a promiscuous interaction would be consistent with the cooperation of Munc18 and Munc13 in promoting the proper assembly of all components of the neuronal SNARE complex. Finally, there is no Munc13 homolog in homotypic vacuolar fusion and all Munc13 homologues are thought to be involved in Ca2+-triggered exocytosis (72, 86). Thus, the evolution of the Ca2+-triggered fusion machineries deviated from that of constitutive vesicle fusion.
Hypothesis: presynaptic plasticity may be a manifestation of regulation of proper SNARE complex assembly
The key roles of Munc13–1 in priming and short-term plasticity may be related to the role of the MUN domain in proper assembly of the SNARE complex (80). This function of the MUN domain which may in turn be subject to regulation by the other domains of Munc13–1. For example, Ca2+-binding to the C2B domain, the synergy between C1, C2B, and C2C domains (88), and the Ca2+-dependent interaction between calmodulin and Munc13 (66) may localize Munc13 to sites of docked synaptic vesicles and increase its ability to assist in proper SNARE complex assembly, as corroborated by the concentration dependence of the various Munc13 fragments in single-vesicle fusion experiments (80). Indeed, mutations of the Munc13–2 C2B domain affect neurotransmitter release upon a single action potential as well as short-term synaptic plasticity (129) and mutations in the interface regions between the C1, C2B, and MUN domains have differential effects on evoked release and the readily releasable pool (166), suggesting that the two molecular functions of Munc13 may be differentially regulated in the neuron. Taken together, regulation of presynaptic plasticity via Munc13 may be in part accomplished by its profound molecular function on ensuring proper assembly of the ternary SNARE complex. The starting point for the action by Munc18 and Munc13 is the disassembled SNARE complex mediated by NSF and αSNAP (Fig. 9). Thus, NSF, αSNAP, Munc18, and Munc13 constitute a quality control system that produces maximal fusogenic synaptic complexes.
11. Perspectives and Outlook
There are other important factors that were not reviewed here. In particular, factors that are involved in tethering, regulation through Rab G-proteins, and voltage gated Ca2+ channels (68, 157). Clearly, complexes with these factors also need to be imaged at near atomic resolution, and functionally reconstituted. Moreover, there are many proteins in synaptic vesicles with little known function that may deserve closer examination. Ultimately, studying the effects of disease and aging on the synaptic neurotransmitter release machinery and its regulations may provide clues for entirely new therapeutic developments.
Acknowledgements:
We thank Kristopher Ian White for stimulating discussions and critical reading of this review, and acknowledge support by the National Institutes of Health (R37MH63105 to A.T.B.).
References
- 1.Adams DJ, Arthur CP, Stowell MHB. 2015. Architecture of the Synaptophysin/Synaptobrevin Complex: Structural Evidence for an Entropic Clustering Function at the Synapse. Sci. Rep 5(1):13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aeffner S, Reusch T, Weinhausen B, Salditt T. 2012. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc. Natl. Acad. Sci 109(25):E1609–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, et al. 2006. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol 13(3):209–17 [DOI] [PubMed] [Google Scholar]
- 4.Augustin I, Rosenmund C, Südhof TC, Brose N. 1999. Munc13–1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400:457–61 [DOI] [PubMed] [Google Scholar]
- 5.Bacaj T, Wu D, Yang X, Morishita W, Zhou P, et al. 2013. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80(4):947–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai H, Xue R, Bao H, Zhang L, Yethiraj A, et al. 2016. Different states of synaptotagmin regulate evoked versus spontaneous release. Nat. Commun 7:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai J, Tucker WC, Chapman ER. 2004. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol 11(1):36–44 [DOI] [PubMed] [Google Scholar]
- 8.Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. 2015. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science (80-. ) 349(6252):1111–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balch WE, Glick BS, Rothman JE. 1984. Sequential intermediates in the pathway of intercompartmental transport in a cell-free system. Cell 39(3 Pt 2):525–36 [DOI] [PubMed] [Google Scholar]
- 10.Basu J, Shen N, Dulubova I, Lu J, Guan R, et al. 2005. A minimal domain responsible for Munc13 activity. Nat. Struct. Mol. Biol 12(11):1017–18 [DOI] [PubMed] [Google Scholar]
- 11.Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R. 1989. Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J 8(2):379–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett MK, Calakos N, Scheller RH. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Sci. (New York, NY) 257(5067):255–59 [DOI] [PubMed] [Google Scholar]
- 13.Bouwman J, Spijker S, Schut D, Wächtler B, Ylstra B, et al. 2006. Reduced expression of neuropeptide genes in a genome-wide screen of a secretion-deficient mouse. J. Neurochem 99(1):84–96 [DOI] [PubMed] [Google Scholar]
- 14.Bracher A, Kadlec J, Betz H, Weissenhorn W. 2002. X-ray structure of a neuronal complexin-SNARE complex from squid. J. Biol. Chem 277(29):26517–23 [DOI] [PubMed] [Google Scholar]
- 15.Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, et al. 2015. Dynamic binding mode of a Synaptotagmin-1–SNARE complex in solution. Nat. Struct. Mol. Biol 22(7):555–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brose N, Petrenko G a, Südhof TC, Jahn R. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256(5059):1021–25 [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT. 2006. Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys 38(1):1. [DOI] [PubMed] [Google Scholar]
- 18.Brunger AT, Cipriano DJ, Diao J. 2015. Towards reconstitution of membrane fusion mediated by SNAREs and other synaptic proteins. Crit. Rev. Biochem. Mol. Biol 0(0):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. 2008. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J 27(7):923–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calloway N, Gouzer G, Xue M, Ryan T a. 2015. The active-zone protein Munc13 controls the use-dependence of presynaptic voltage-gated calcium channels. Elife 4(July):1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao P, Yang X, Sudhof TC, Südhof TC. 2013. Complexin Activates Exocytosis of Distinct Secretory Vesicles Controlled by Different Synaptotagmins. J. Neurosci 33(4):1714–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman ERER, Davis AFAF. 1998. Direct Interaction of a Ca 2 + -binding Loop of Synaptotagmin with Lipid Bilayers. J. Biol. Chem 273(22):13995–1 [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Lu J, Dulubova I, Rizo J. 2008. NMR analysis of the closed conformation of syntaxin-1. J. Biomol. NMR 41(1):43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, et al. 2002. Three-Dimensional Structure of the Complexin/SNARE Complex. Neuron 33(3):397–409 [DOI] [PubMed] [Google Scholar]
- 25.Cho RW, Kümmel D, Li F, Baguley SW, Coleman J, et al. 2014. Genetic analysis of the Complexin trans-clamping model for cross-linking SNARE complexes in vivo. Proc. Natl. Acad. Sci. U. S. A 111(28):10317–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR. 2010. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol 17(3):318–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi UB, Zhao M, Zhang Y, Lai Y, Brunger AT. 2016. Complexin induces a conformational change at the membrane-proximal C-terminal end of the SNARE complex. Elife 5(9):e16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen FS, Melikyan GB. 2004. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol 199(1):1–14 [DOI] [PubMed] [Google Scholar]
- 29.Cole AA, Chen X, Reese TS. 2016. A Network of Three Types of Filaments Organizes Synaptic Vesicles for Storage, Mobilization, and Docking. J. Neurosci 36(11):3222–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daily NJ, Boswell KL, James DJ, Martin TFJ. 2010. Novel interactions of CAPS (Ca2+-dependent activator protein for secretion) with the three neuronal SNARE proteins required for vesicle fusion. J. Biol. Chem 285(46):35320–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davletov BA, Südhof TC. 1993. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J. Biol. Chem 268(35):26386–90 [PubMed] [Google Scholar]
- 32.Deák F, Xu Y, Chang W-P, Dulubova I, Khvotchev M, et al. 2009. Munc18–1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J. Cell Biol 184(5):751–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhara M, Yarzagaray A, Schwarz Y, Dutta S, Grabner C, et al. 2014. Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. J. Cell Biol 204(7):1123–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diao J, Cipriano DJ, Zhao M, Zhang Y, Shah S, et al. 2013. Complexin-1 enhances the on-rate of vesicle docking via simultaneous SNARE and membrane interactions. J. Am. Chem. Soc 135(41):15274–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, et al. 2012. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife 1:e00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dulubova I, Lou X, Lu J, Huryeva I, Alam A, et al. 2005. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24(16):2839–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, et al. 1999. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18(16):4372–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans CS, Ruhl DA, Chapman ER. 2015. An Engineered Metal Sensor Tunes the Kinetics of Synaptic Transmission. J. Neurosci 35(34):11769–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fasshauer D, Sutton RB, Brunger AT, Jahn R. 1998. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. U. S. A 95(26):15781–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Busnadiego R, Asano S, Oprisoreanu AM, Sakata E, Doengi M, et al. 2013. Cryo-electron tomography reveals a critical role of RIM1 α in synaptic vesicle tethering. J. Cell Biol 201(5):725–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Chacón R, Königstorfer A, Gerber SHH, García J, Matos MFF, et al. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410(6824):41–49 [DOI] [PubMed] [Google Scholar]
- 42.Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, et al. 2012. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337(6100):1340–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geppert M, Goda Y, Hammer RERERE, Li C, Rosahl TWTWTW, et al. 1994. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 79(4):717–27 [DOI] [PubMed] [Google Scholar]
- 44.Gipson P, Fukuda Y, Danev R, Lai Y, Chen D-H, et al. 2017. Morphologies of synaptic protein membrane fusion interfaces. Proc. Natl. Acad. Sci 114(34):9110–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giraudo CG, Eng WS, Melia TJ, Rothman JE. 2006. A clamping mechanism involved in SNARE-dependent exocytosis. Science 313(5787):676–80 [DOI] [PubMed] [Google Scholar]
- 46.Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, et al. 2009. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science 323(5913):512–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong J, Lai Y, Li X, Wang M, Leitz J, et al. 2016. C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proc. Natl. Acad. Sci 113(47):E7590–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guan R, Dai H, Rizo J. 2008. Binding of the Munc13–1 MUN domain to membrane-anchored SNARE complexes. Biochemistry 47(6):1474–81 [DOI] [PubMed] [Google Scholar]
- 49.Gustavsson N, Han W. 2009. Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci. Rep 29(4):245. [DOI] [PubMed] [Google Scholar]
- 50.Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, Jorgensen EM. 2007. Open syntaxin docks synaptic vesicles. PLoS Biol 5(8):e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. 1997. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90(3):523–35 [DOI] [PubMed] [Google Scholar]
- 52.Harrison SC. 2017. Pictures of the prologue to neurotransmitter release. Proc. Natl. Acad. Sci, p. 201712038. [DOI] [PMC free article] [PubMed]
- 53.He E, Wierda K, van Westen R, Broeke JH, Toonen RF, et al. 2017. Munc13–1 and Munc18–1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat. Commun 8(May):15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, et al. 2012. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336(6088):1581–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. 2011. Complexin maintains vesicles in the primed state in C. elegans. Curr. Biol 21(2):106–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffmann A, Becker AH, Zachmann-Brand B, Deuerling E, Bukau B, Kramer G. 2012. Concerted Action of the Ribosome and the Associated Chaperone Trigger Factor Confines Nascent Polypeptide Folding. Mol. Cell 48(1):63–74 [DOI] [PubMed] [Google Scholar]
- 57.Hoffmann A, Bukau B, Kramer G. 2010. Structure and function of the molecular chaperone Trigger Factor. Biochim. Biophys. Acta 1803(6):650–61 [DOI] [PubMed] [Google Scholar]
- 58.Hu S-H, Christie MP, Saez NJ, Latham CF, Jarrott R, et al. 2011. Possible roles for Munc18–1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc. Natl. Acad. Sci. U. S. A 108(3):1040–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hui E, Bai J, Chapman ER. 2006. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J 91(5):1767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. 2009. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell 138(4):709–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huntwork S, Littleton JT. 2007. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat. Neurosci 10(10):1235–37 [DOI] [PubMed] [Google Scholar]
- 62.Imig C, Min S-W, Krinner S, Arancillo M, Rosenmund C, et al. 2014. The Morphological and Molecular Nature of Synaptic Vesicle Priming at Presynaptic Active Zones. Neuron 84(2):416–31 [DOI] [PubMed] [Google Scholar]
- 63.Jensen MB, Bhatia VK, Jao CC, Rasmussen JE, Pedersen SL, et al. 2011. Membrane curvature sensing by amphipathic helices: a single liposome study using α-synuclein and annexin B12. J. Biol. Chem 286(49):42603–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jockusch WJ, Speidel D, Sigler A, Sørensen JB, Varoqueaux F, et al. 2007. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131(4):796–808 [DOI] [PubMed] [Google Scholar]
- 65.Jorquera R a, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT 2012. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J. Neurosci 32(50):18234–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Junge HJ, Rhee J, Jahn O, Varoqueaux F, Spiess J, et al. 2004. Calmodulin and Munc13 Form a Ca2+ Sensor/Effector Complex that Controls Short-Term Synaptic Plasticity. Cell 118(3):389–401 [DOI] [PubMed] [Google Scholar]
- 67.Kaeser-Woo YJ, Yang X, Südhof TC. 2012. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J. Neurosci 32(8):2877–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, et al. 2011. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144(2):282–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaeser PS, Regehr WG. 2014. Molecular Mechanisms for Synchronous, Asynchronous, and Spontaneous Neurotransmitter Release. Annu. Rev. Physiol 76(1):333–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaeser PS, Regehr WG. 2017. The readily releasable pool of synaptic vesicles. Curr. Opin. Neurobiol 43:63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaiser CM, Chang H-C, Agashe VR, Lakshmipathy SK, Etchells SA, et al. 2006. Real-time observation of trigger factor function on translating ribosomes. Nature 444(7118):455–60 [DOI] [PubMed] [Google Scholar]
- 72.Koch H, Hofmann K, Brose N. 2000. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem. J 349(Pt 1):247–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kochubey O, Schneggenburger R. 2011. Synaptotagmin Increases the Dynamic Range of Synapses by Driving Ca2+-Evoked Release and by Clamping a Near-Linear Remaining Ca2+ Sensor. Neuron 69(4):736–48 [DOI] [PubMed] [Google Scholar]
- 74.Kreutzberger AJB, Kiessling V, Liang B, Seelheim P, Jakhanwal S, et al. 2017. Reconstitution of calcium-mediated exocytosis of dense-core vesicles. Sci. Adv 3(7):e1603208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krishnakumar SS, Li F, Coleman J, Schauder CM, Kümmel D, et al. 2015. Re-visiting the trans insertion model for complexin clamping. Elife 4:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kümmel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, et al. 2011. Complexin cross-links prefusion SNAREs into a zigzag array. Nat. Struct. Mol. Biol 18(8):927–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuo W, Herrick DZ, Ellena JF, Cafiso DS. 2009. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J. Mol. Biol 387(2):284–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, et al. 2011. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc. Natl. Acad. Sci 108(29):E304–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kyoung M, Zhang Y, Diao J, Chu S, Brunger AT. 2012. Studying calcium-triggered vesicle fusion in a single vesicle-vesicle content and lipid-mixing system. Nat. Protoc 8(1):1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, et al. 2017. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95:591–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lai Y, Choi UB, Zhang Y, Zhao M, Pfuetzner RA, et al. 2016. N-terminal domain of complexin independently activates calcium-triggered fusion. Proc. Natl. Acad. Sci 113(32):E4698–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner R a, et al. 2014. Complexin inhibits spontaneous release and synchronizes Ca 2+ -triggered synaptic vesicle fusion by distinct mechanisms. Elife 3:e03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lai Y, Lou X, Jho Y, Yoon T-Y, Shin Y-K. 2013. The synaptotagmin 1 linker may function as an electrostatic zipper that opens for docking but closes for fusion pore opening. Biochem. J 456:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee J, Guan Z, Akbergenova Y, Littleton JT. 2013. Genetic Analysis of Synaptotagmin C2 Domain Specificity in Regulating Spontaneous and Evoked Neurotransmitter Release. J. Neurosci 33(1):187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee J, Littleton JT. 2015. Transmembrane tethering of synaptotagmin to synaptic vesicles controls multiple modes of neurotransmitter release. Proc. Natl. Acad. Sci 112(12):201420312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lipstein N, Schaks S, Dimova K, Kalkhof S, Ihling C, et al. 2012. Nonconserved Ca2+/Calmodulin Binding Sites in Munc13s Differentially Control Synaptic Short-Term Plasticity. Mol. Cell. Biol 32(22):4628–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu H, Bai H, Xue R, Takahashi H, Edwardson JM, Chapman ER. 2014. Linker mutations reveal the complexity of synaptotagmin 1 action during synaptic transmission. Nat. Neurosci 17(5):670–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu X, Seven AB, Camacho M, Esser V, Xu J, et al. 2016. Functional synergy between the Munc13 C-terminal C1 and C2 domains. Elife 5(MAY2016):1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lou X, Shin J, Yang Y, Kim J, Shin Y-K. 2015. Synaptotagmin-1 Is an Antagonist for Munc18–1 in SNARE Zippering. J. Biol. Chem [DOI] [PMC free article] [PubMed]
- 90.Ma C, Li W, Xu Y, Rizo J. 2011. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol 18(5):542–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma C, Su L, Seven AB, Xu Y, Rizo J. 2013. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339(6118):421–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Madison JM, Nurrish S, Kaplan JM. 2005. UNC-13 Interaction with Syntaxin Is Required for Synaptic Transmission. Curr. Biol 15(24):2236–42 [DOI] [PubMed] [Google Scholar]
- 93.Martens S, Kozlov MM, McMahon HT. 2007. How Synaptotagmin Promotes Membrane Fusion. Science (80-. ) 316(5828):1205–8 [DOI] [PubMed] [Google Scholar]
- 94.Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. 2011. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr. Biol 21(2):97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martinez-Hackert E, Hendrickson WA. 2009. Promiscuous Substrate Recognition in Folding and Assembly Activities of the Trigger Factor Chaperone. Cell 138(5):923–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mashaghi A, Kramer G, Bechtluft P, Zachmann-Brand B, Driessen AJM, et al. 2013. Reshaping of the conformational search of a protein by the chaperone trigger factor. Nature 500(7460):98–101 [DOI] [PubMed] [Google Scholar]
- 97.Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. 2009. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323(5913):516–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mayer A, Wickner W, Haas A. 1996. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85(1):83–94 [DOI] [PubMed] [Google Scholar]
- 99.McMahon HT, Missler M, Li C, Südhof TC. 1995. Complexins: Cytosolic proteins that regulate SNAP receptor function. Cell 83(1):111–19 [DOI] [PubMed] [Google Scholar]
- 100.Medine CN, Rickman C, Chamberlain LH, Duncan RR. 2007. Munc18–1 prevents the formation of ectopic SNARE complexes in living cells. J. Cell Sci 120(Pt 24):4407–15 [DOI] [PubMed] [Google Scholar]
- 101.Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, et al. 2002. Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J. Cell Biol 158(5):929–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michelassi F, Liu H, Hu Z, Dittman JS, Michelassi F, et al. 2017. Article Article A C1-C2 Module in Munc13 Inhibits Calcium-Dependent Neurotransmitter Release. Neuron 95(3):577–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Min D, Kim K, Hyeon C, Cho YH, Shin Y-K, Yoon T-Y. 2013. Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force-generating mechanism. Nat. Commun 4:1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Misura KM, Scheller RH, Weis WI. 2000. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404(6776):355–62 [DOI] [PubMed] [Google Scholar]
- 105.Mittelsteadt T, Seifert G, Alvárez-Barón E, Steinhäuser C, Becker AJ, Schoch S. 2009. Differential mRNA expression patterns of the synaptotagmin gene family in the rodent brain. J. Comp. Neurol 512(4):514–28 [DOI] [PubMed] [Google Scholar]
- 106.Nicholson KL, Munson M, Miller RB, Filip TJ, Fairman R, Hughson FM. 1998. Regulation of SNARE complex assembly by an N-terminal domain of the t- SNARE Sso1p. Nat. Struct. Biol 5(9):793–802 [DOI] [PubMed] [Google Scholar]
- 107.Nishiki T, Augustine GJ. 2004. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci 24(39):8542–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pabst S, Hazzard JW, Antonin W, Südhof TC, Jahn R, et al. 2000. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem 275(26):19808–18 [DOI] [PubMed] [Google Scholar]
- 109.Pabst S, Margittai M, Vainius D, Langen R, Jahn R, Fasshauer D. 2002. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J. Biol. Chem 277(10):7838–48 [DOI] [PubMed] [Google Scholar]
- 110.Park Y, Seo JB, Fraind A, Pérez-Lara A, Yavuz H, et al. 2015. Synaptotagmin-1 binds to PIP2-containing membrane but not to SNAREs at physiological ionic strength. Nat. Struct. Mol. Biol [DOI] [PMC free article] [PubMed]
- 111.Pérez-Lara Á, Thapa A, Nyenhuis SBB, Nyenhuis DAA, Halder P, et al. 2016. PtdInsP 2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. Elife 5:1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Perin MS, Johnston PA, Ozcelik T, Jahn R, Francke U, Südhof TC. 1991. Structural and functional conservation of synaptotagmin (p65) in Drosophila and humans. J. Biol. Chem 266(1):615–22 [PubMed] [Google Scholar]
- 113.Pertsinidis A, Mukherjee K, Sharma M, Pang ZP, Park SR, et al. 2013. Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc. Natl. Acad. Sci. U. S. A 110(30):E2812–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting E a, et al. 1994. Specificity and regulation of a synaptic vesicle docking complex. Neuron 13(2):353–61 [DOI] [PubMed] [Google Scholar]
- 115.Pobbati AV, Stein A, Fasshauer D. 2006. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science 313(5787):673–76 [DOI] [PubMed] [Google Scholar]
- 116.Prinslow EA, Brautigam CA, Rizo J. 2017. Reconciling isothermal titration calorimetry analyses of interactions between complexin and truncated SNARE complexes. Elife 6:e30286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Radoff DT, Dong Y, Snead D, Bai J, Eliezer D, Dittman JS. 2014. The accessory helix of complexin functions by stabilizing central helix secondary structure. Elife 3:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J. 2010. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc. Natl. Acad. Sci. U. S. A 107(52):22399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, et al. 2001. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104(1):71–81 [DOI] [PubMed] [Google Scholar]
- 120.Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, et al. 2002. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108(1):121–33 [DOI] [PubMed] [Google Scholar]
- 121.Richmond JE, Weimer RM, Jorgensen EM. 2001. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412(6844):338–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rothman JE. 2014. The Principle of Membrane Fusion in the Cell (Nobel Lecture). Angew. Chemie Int. Ed n/a-n/a [DOI] [PubMed]
- 123.Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, et al. 1999. Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J. Cell Sci 112 ( Pt 1:1865–77 [DOI] [PubMed] [Google Scholar]
- 124.Ryu J-K, Min D, Rah S-H, Kim SJ, Park Y, et al. 2015. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science (80-. ) 347(6229):1485–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schonn J-S, Maximov A, Lao Y, Sudhof TC, Sorensen JB. 2008. Synaptotagmin-1 and −7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc. Natl. Acad. Sci 105(10):3998–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Seiler F, Malsam J, Krause JM, Söllner TH. 2009. A role of complexin-lipid interactions in membrane fusion. FEBS Lett 583(14):2343–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shao X, Li C, Fernandez I, Zhang X, Südhof TC, Rizo J. 1997. Synaptotagmin-syntaxin interaction: The C2 domain as a Ca2+-dependent electrostatic switch. Neuron 18:133–42 [DOI] [PubMed] [Google Scholar]
- 128.Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. 2007. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128(1):183–95 [DOI] [PubMed] [Google Scholar]
- 129.Shin O-H, Lu J, Rhee J-S, Tomchick DR, Pang ZP, et al. 2010. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol 17(3):280–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shin O-H, Xu J, Rizo J, Südhof TC. 2009. Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. U. S. A 106(38):16469–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sinha R, Ahmed S, Jahn R, Klingauf J. 2011. Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc. Natl. Acad. Sci. U. S. A 108(34):14318–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sitarska E, Xu J, Park S, Liu X, Quade B, et al. 2017. Autoinhibition of Munc18–1 modulates synaptobrevin binding and helps to enable Munc13-dependent regulation of membrane fusion. Elife 6:618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Snead D, Wragg RT, Dittman JS, Eliezer D. 2014. Membrane curvature sensing by the C-terminal domain of complexin. Nat. Commun 5:4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, et al. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature 362(6418):318–24 [DOI] [PubMed] [Google Scholar]
- 135.Söllner TH, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. 1993. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75(3):409–18 [DOI] [PubMed] [Google Scholar]
- 136.Stein A, Weber G, Wahl MC, Jahn R. 2009. Helical extension of the neuronal SNARE complex into the membrane. Nature 460(7254):525–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stevens DR, Wu ZX, Matti U, Junge HJ, Schirra C, et al. 2005. Identification of the minimal protein domain required for priming activity of munc13–1. Curr. Biol 15(24):2243–48 [DOI] [PubMed] [Google Scholar]
- 138.Südhof TC. 2012. The presynaptic active zone. Neuron 75(1):11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Südhof TC. 2013. Neurotransmitter Release: The Last Millisecond in the Life of a Synaptic Vesicle. Neuron 80(3):675–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Südhof TC, Jahn R. 1991. Proteins of synaptic vesicles involved in exocytosis and membrane recycling. Neuron 6(5):665–77 [DOI] [PubMed] [Google Scholar]
- 141.Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, et al. 2001. Synaptotagmin VII as a Plasma Membrane Ca2+ Sensor in Exocytosis. Neuron 30(2):459–73 [DOI] [PubMed] [Google Scholar]
- 142.Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. 2007. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450(7170):676–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sutton RB, Davletov B a, Berghuis a M, Südhof TC, Sprang SR 1995. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80:929–38 [DOI] [PubMed] [Google Scholar]
- 144.Sutton RB, Fasshauer D, Jahn R, Brunger AT. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395(6700):347–53 [DOI] [PubMed] [Google Scholar]
- 145.Tang J, Maximov A, Shin O-H, Dai H, Rizo J, et al. 2006. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126(6):1175–87 [DOI] [PubMed] [Google Scholar]
- 146.Trimbuch T, Xu J, Flaherty D, Tomchick DR, Rizo J, Rosenmund C. 2014. Re-examining how complexin inhibits neurotransmitter release. Elife 3:e02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tucker WC, Weber T, Chapman ER. 2004. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304(5669):435–38 [DOI] [PubMed] [Google Scholar]
- 148.van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. 2010. One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol 17(3):358–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, et al. 2011. Membrane protein sequestering by ionic protein-lipid interactions. Nature 479(7374):552–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Varoqueaux F, Sigler A, Rhee J-S, Brose N, Enk C, et al. 2002. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U. S. A 99(13):9037–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, et al. 2000. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Sci. (New York, NY) 287(5454):864–69 [DOI] [PubMed] [Google Scholar]
- 152.Vrljic M, Strop P, Hill RCC, Hansen KCC, Chu S, Brunger ATT. 2011. Post-translational modifications and lipid binding profile of insect cell-expressed full-length mammalian synaptotagmin 1. Biochemistry 50(46):9998–10012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang J, Bello O, Auclair SM, Coleman J, Pincet F, et al. 2014. Calcium sensitive ring-like oligomers formed by synaptotagmin. Proc. Natl. Acad. Sci. U. S. A 111(38):13966–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang J, Li F, Bello OD, Sindelar CV, Pincet F, et al. 2017. Circular oligomerization is an intrinsic property of synaptotagmin. Elife 6:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang S, Choi UB, Gong J, Yang X, Li Y, et al. 2017. Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. EMBO J 36(6):816–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang S, Li Y, Ma C. 2016. Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. Elife 5(9):1689–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang SSH, Held RG, Wong MY, Liu C, Karakhanyan A, Kaeser PS. 2016. Fusion Competent Synaptic Vesicles Persist upon Active Zone Disruption and Loss of Vesicle Docking. Neuron 91(4):777–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Weber T, Zemelman B V, McNew JA, Westermann B, Gmachl M, et al. 1998. SNAREpins: minimal machinery for membrane fusion. Cell 92(6):759–72 [DOI] [PubMed] [Google Scholar]
- 159.Weimer RM, Gracheva EO, Meyrignac O, Miller KG, Richmond JE, Bessereau J. 2006. UNC-13 and UNC-10 / Rim Localize Synaptic Vesicles to Specific Membrane Domains. Analysis 26(31):8040–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wen H, Linhoff MW, McGinley MJ, Li G-L, Corson GM, et al. 2010. Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc. Natl. Acad. Sci 107(31):13906–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT. 2008. Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure 16(2):308–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Weninger K, Bowen ME, Chu S, Brunger AT. 2003. Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc. Natl. Acad. Sci. U. S. A 100(25):14800–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, et al. 2013. Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77(2):323–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, et al. 2017. Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544(7650):316–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wu Z, Schulten K. 2014. Synaptotagmin’s Role in Neurotransmitter Release Likely Involves Ca2+-induced Conformational Transition. Biophys. J 107(5):1156–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xu J, Camacho M, Xu Y, Esser V, Liu X, et al. 2017. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13–1 C 1 C 2 BMUN. Elife 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu J, Mashimo T, Südhof TC. 2007. Synaptotagmin-1, −2, and −9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54(4):567–81 [DOI] [PubMed] [Google Scholar]
- 168.Xu J, Pang ZP, Shin O-H, Südhof TC. 2009. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat. Publ. Gr 12(6):759–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xu Y, Su L, Rizo J. 2010. Binding of Munc18–1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry 49(8):1568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xue M, Craig TK, Xu J, Chao H-T, Rizo J, Rosenmund C. 2010. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat. Struct. Mol. Biol 17(5):568–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xue M, Lin YQ, Pan H, Reim K, Deng H, et al. 2009. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64(3):367–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xue M, Reim K, Chen X, Chao H-T, Deng H, et al. 2007. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat. Struct. Mol. Biol 14(10):949–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xue M, Stradomska A, Chen H, Brose N, Zhang W, et al. 2008. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc. Natl. Acad. Sci. U. S. A 105(22):7875–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yang B, Steegmaier M, Gonzalez LC, Scheller RH. 2000. nSec1 binds a closed conformation of syntaxin1A. J. Cell Biol 148(2):247–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yang X, Cao P, Sudhof TC. 2013. Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proc. Natl. Acad. Sci 110(51):20777–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC. 2010. Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory α helix. Neuron 68(5):907–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yang X, Pei J, Kaeser-woo YJ, Bacaj T, Grishin N V, Südhof TC. 2015. Evolutionary conservation of complexins : from choanoflagellates to mice. EMBO Rep 16(10):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang X, Wang S, Sheng Y, Zhang M, Zou W, et al. 2015. Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol 22(7):547–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zanetti MN, Bello OD, Wang J, Coleman J, Cai Y, et al. 2016. Ring-like oligomers of Synaptotagmins and related c2 domain proteins. Elife 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhao M, Brunger AT. 2016. Recent Advances in Deciphering the Structure and Molecular Mechanism of the AAA + ATPase N-Ethylmaleimide-Sensitive Factor (NSF). J. Mol. Biol 428(9):1912–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, et al. 2015. Mechanistic insights into the recycling machine of the SNARE complex. Nature 518:61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhou K, Stawicki TM, Goncharov A, Jin Y. 2013. Position of UNC-13 in the active zone regulates synaptic vesicle release probability and release kinetics. Elife 2:e01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, et al. 2013. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32(1):159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, et al. 2015. Architecture of the synaptotagmin–SNARE machinery for neuronal exocytosis. Nature 525(7567):62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, et al. 2017. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature in press: [DOI] [PMC free article] [PubMed]
- 186.Zick M, Wickner WT. 2014. A distinct tethering step is vital for vacuole membrane fusion. Elife 3:e03251. [DOI] [PMC free article] [PubMed] [Google Scholar]