

Abstract
In a somewhat narrow diagnostic lens, Alzheimer’s Disease (AD) has been considered a brain specific disease characterized by the presence of β-Amyloid plaques and tau neural fibrillary tangles and neural inflammation; these pathologies lead to neuronal death and consequently clinical symptoms such as memory loss, confusion, and impaired cognitive function. However, for decades researchers have noticed a link between various cardiovascular abnormalities and AD - such as heart failure (HF), coronary artery disease (CAD), atrial fibrillation (AF), and vasculopathy. A considerable volume of work has pointed at this head to heart connection, focusing mainly on associations between cerebral hypoperfusion and neuronal degradation. However, new evidence of a possible systemic or metastatic profile to AD calls for further analysis of this connection. β amyloid aggregations - biochemically and structurally akin to those found in the typical AD pathology - are now known to be present in the hearts of individuals with idiopathic dilated cardiomyopathy (iDCM) as well as the hearts of patients with AD. These findings suggest a potential systemic profile of proteinopathies, and a new hypothesis for the link between peripheral and central symptoms of HF and AD. Herein, we provide an overview of the cardiovascular links to Alzheimer’s disease.
Keywords: Alzheimer’s Disease, heart, inflammation, reactive oxygen species
Introduction
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder which accounts for about 70% of all dementia cases1-3 and is projected to affect 13 million individuals in the US by 20503. It is characterized by the formation of senile plaques composed of aggregated β-Amyloid (Aβ) fibers and neurofibrillary tangles (NT) formed by hyperphosphorylated tau protein, with associated neuronal inflammation, oxidative stress, and widespread degeneration of neurons. These physiologic disruptions are accompanied by physical symptoms: memory loss, impaired cognitive function, personality and judgment disorder, speech abnormalities, and apraxia1.
A similar epidemiological profile characterizes Heart Failure (HF), a widespread public health problem, affecting 5.8 million individuals in the US alone and nearly 23 million worldwide4. Obesity, sex, history of diabetes and especially age are risk factors for both HF and AD4-7. Genetic factors, such as the presence of the Apoliprotein E4 (ApoE4) allele, and variants in the Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2) genes are associated with the development of AD 8-10 and the same, as well as novel genetic variants of the PSEN genes are associated with dilated cardiomyopathy 11, 12. Despite those similarities, much of the current understanding is centralized around the pathogenesis of AD via vascular factors and has failed to be extended to cardiac contributions. Vascular factors, such as macro- and micro-infarcts, white matter hyperintensities, atherosclerosis, and hypertension that lead to decreased cerebral blood flow prior to β-amyloid deposition would accelerate AD progression13.
Here, we will review the current knowledge on the vascular component and explore the new “head to heart” connection that could help defining cardiac abnormalities occurring in concert with the development of AD.
Vascular Role in Alzheimer’s Disease
For years, the role of the vasculature in the progression of Alzheimer’s Disease has prevailed as a main comorbidity contributing to AD. Vascular cognitive impairment (VCI) remains a common risk factor of dementia as over half of all patients with VCI will advance to dementia14, 15. Thus, when attempting to explain the pathophysiology of AD, a vascular hypothesis arose in opposition to the amyloid cascade hypothesis16. The amyloid cascade hypothesis articulates that the pathology behind AD is driven by the deposition of the Aβ peptide in the brain17. However, supporters of the vascular hypothesis propose that AD is a vascular disorder, in which the pathology is induced via cerebral microvascular abnormalities18. In 1993, it was discovered that cerebral microvascular abnormalities led to a decrease in cerebral blood flow (CBF), glucose metabolism, and oxygen consumption in AD patients19. De la Torre et. al established an inversely proportional relationship between these vascular abnormality symptoms and the severity of AD19. In this study, they noted multiple different risk factors such as aging, atherosclerosis, hyper/hypotension, and micro-vessel pathology; all of which are vascular risk factors that impair proper cerebral perfusion18.
Brain hypoperfusion in the pathogenesis of AD
It is commonly known that the brain relies on adequate blood supply (and thus oxygen) in order to maintain proper function20. It has been proposed that hypoxia and ischemia resulting from cerebral hypoperfusion is a direct contributor to the pathogenesis and development of AD20-22. Roher et al. observed total cerebral blood flow to be 20% lower in the AD group compared to the non-demented control group suggesting an association between brain hypoperfusion and dementia of AD22. The same study found a decrease in pulse pressure in AD patients, further indicating a decrease in cerebral blood flow, and consequently, a decrease in cognitive measures22. Nishimura et al. more specifically observed decreased cerebral blood flow to the frontal lobe in correlation to both reduced cognitive function and progression of AD23. The reduced cerebral perfusion causes a metabolic energy crisis due to the reduced oxygen exposure. This hypoxic state induces oxidative stress and acidosis eventually culminating in neuronal degradation24, 25.
Oxidative stress in AD
Contrary to popular belief, it has been discussed that a balanced reactive oxygen species (ROS) response actually promotes tissue repair via disinfection of existing tissue and stimulation of healthy tissue turnover26. However, the imbalanced metabolism of oxygen induced by this hypoxic state, leads to excess ROS, resulting in deleterious effects27. Furthermore, the mitochondria of the vascular wall cells have been identified as the primary target of oxidative stress prior to development and progression of AD28.
However, it is believed that the accumulation of ROS, and thereby oxidative stress, on neural tissue is a result of hypoperfusion in combination with Aβ proteotoxicity29. An increase in Aβ production has been linked to the progression of oxidative stress which can induce mitochondrial dysfunction. Work by Matsuoka et al.30 using transgenic mice carrying mutant APP (amyloid precursor protein) and PSEN1 illustrates the key role of oxidative stress in the AD model. Additionally, 3-nitrotyrosine (protein oxidative stress marker), and 4-hydroxy-2-noneal (lipid oxidative stress marker) have been found to be increased with progressing levels of fibrillary Aβ30. While most Aβ aggregations are found in extracellular regions, some collections of Aβ have been found in the mitochondria31, 32 of individuals with AD. This phenomenon may diminish mitochondrial function, specifically respiration and thereby increase the levels of ROS inside and potentially outside of the cell. Oxidative stress not only can induce further Aβ production, but triggers the production of tau protein, another critical component to the pathology of this disease31. Furthermore, it has been demonstrated that individuals with AD have decreased antioxidant levels- allowing higher levels of ROS to accumulate in the local environment29. This evidence therefore suggests a cyclic effect between Aβ aggregation and oxidative stress, driving the progression of AD and its physical symptoms.
Inflammation contributes to vascular and neurodegeneration in AD
Another key aspect of the pathogenesis of AD as a result of reduced blood flow is inflammation. Changes in the vasculature either as a result of AD or part of an overall vasculopathy are associated with the release of multiple inflammatory factors, with cerebral micro vessels secreting higher levels of TNF-α, IL-1β, IL-6, and leukocyte adhesion molecules than non-AD controls33. However, despite the vast amount of literature focused on neural inflammation and its role in AD, no clear evidence has illuminated whether inflammation is a contributing factor towards the etiology of AD, part of its pathology, or a secondary phenomenon34. However, it is widely accepted that the negative consequences of inflammation contribute towards the progression of AD, including neurodegeneration and thereby diminished cognitive function. It is also understood that microglia, the resident macrophage in the brain, play a key role in the activation of the inflammatory response. Studies have illustrated that microglia are increased in individuals with AD and in transgenic mouse models Although they exhibit heterogeneous phenotypes, it is understood that they are involved in Aβ maintenance35. Studies in vitro suggest that cytokine signaling and secretion from microglia can greatly impact the cerebral microenvironment and function of neurons. These cytokines - specifically IL-1β, IL-6, TNF-α, INF-γ - and various chemokines can strengthen the inflammatory response35. More recently, cytokine signaling in AD mouse models can have significant effects on amyloidosis, neurodegeneration and cognition35, potentially disrupting the blood brain barrier in neurodegenerative disorders36. In AD pathology, the integrity of the blood brain barrier (BBB) is reduced37, 38 as well. Whether as a result of the chronic cerebral inflammation or part of a larger systemic pathology, this defective BBB is believed to play a role in the pathogenesis of AD.
Structural consequences of hypoperfusion in the AD brain
Furthermore, this same cerebral hypoperfusion has been found to breakdown the neurovascular unit (NVU), enabling progression into neurodegenerative disorders like AD39. The NVU encompasses many different types of brain cells (endothelial, pericytes and vascular smooth muscle cells), which in turn control the blood brain barrier (BBB)39. Breakdown of the NVU would result in a dysfunctional BBB. Many recent studies have now correlated BBB dysfunction with the accrual of vasculotoxic molecules and hypoxia resulting from a decrease in CBF39.
Amyloid directly contributes to cerebral angiopathy
Changes in peripheral perfusion also have the potential to induce cerebral amyloid angiopathy (CAA) - fibrillary amyloid deposition in small cerebral vessels. However, the exact mechanism whereby CAA contributes towards the pathogenesis of AD remains unknown. It has been reported that CAA can affect cell viability, induce apoptosis or oxidative stress, and trigger an inflammatory response40. Furthermore, CAA can create vascular dysfunction through hemorrhagic complications or blocking blood flow (resulting in cerebral ischemia). These events, either alone or in combination, may contribute towards the progression of AD40.
Atherosclerosis, hypertension and ischemia cascade effects in AD
Additionally, for many decades, the role of decreased cerebral blood flow in the pathogenesis of Alzheimer’s relied on the impairment of vasculature through the lens of atherosclerosis14, 41. Similarities between AD and atherosclerosis have sparked investigation considering that both are found in conjunction with vascular wall thickening and blood vessel occlusion42. After examination of the cerebral arteries in AD patients, Roher et al found cases of significantly increased cerebral artery occlusion in comparison to control groups42, 43. Additionally, a positive correlation was determined between arterial stenosis and NFT43, which is a hallmark of AD. Other studies by Roher et. al found more widespread intracranial atherosclerosis in AD patients versus nondemented patients 44. Cognitive dysfunction was also found to be exacerbated in AD patients with cerebral atherosclerosis compared to non-atherosclerotic AD patients, regardless of intracranial or extracranial localization45. Furthermore, the Nun Study of Aging and Alzheimer’s Disease indicated that patients with multiple brain infarctions have lower cognitive function and higher prevalence of dementia46, 47.
Hypertension has been independently identified as a risk factor for AD, but many have reasoned that this is due to hypertension leading to other diseases, which then result in the onset of dementia46. Throughout time, there have been multiple intuitions about the exact mechanism of hypertension leading to AD: (1) vascular alterations leading to infarcts in the brain, (2) adverse effects on neuronal health leading to deposition of Aβ in the brain, and lastly (3) development of cardiovascular disease giving rise to AD46. Hypertension has also been proposed to evoke AD via ischemia, oxidative stress, inflammation and small-vessel disease48,49. More recently, hypertension has been largely noted as the most detrimental vascular risk in the progression of AD50. This designation is due to its causation of small-vessel disease, which later results in lacunar infarcts, white matter lesions/hyperintensities, and microinfarcts51; all of which are indicated in AD and accelerate the reduction in CBF52. Hypertension on the other hand is an independent risk factor for cardiovascular diseases including heart failure53 indirectly affecting AD through the old lens of cerebral hypoperfusion and the new one of the common pathogenesis of AD and HF as age-related proteinopathies.
One of the main hallmarks of cerebrovascular disease is found in the microvascular structural changes, namely white matter hyperintensities54, a manifestation of small-vessel disease55. White matter hyperintensities (WMH) are a commonality with age progression and a causative agent behind normal cognitive decline56. However, as imaging and other diagnostic techniques have progressed, it has been shown that small-vessel disease and WMHs in the brain both play a role in the development of AD57. Population studies using MRI have also designated a high occurrence of small-vessel disease being associated with higher risk of stroke and dementia20. These white matter hyperintensities are indicative of vascular lesions and have been found to lower the threshold for the clinical diagnosis of AD20. Tosto et. al established that WMH distributions in the brain can progress to AD both directly via neurodegenerative changes and indirectly via aggravation of the tau effect on clinical transformation56. This connection has enabled vascular risk factors to be common targets when testing novel therapeutic approaches against AD, such as hypertension and cholesterol treatments58. However, these studies have been preliminary and there is much more to be investigated.
In summary, the role of the vasculature in Alzheimer’s Disease progression is one that is implicated primarily via decreased cerebral blood flow. This remains the main talking point for the vascular hypothesis of AD, which, as seen in Figure 1, claims that many vascular risk factors exert their pathology through cerebral hypoperfusion59. However, there is a multitude of contributing vascular risk factors, some of which include atherosclerosis, hypertension, small-vessel disease, and BBB dysfunction, all contributing to reduced cerebral blood flow and the progression of AD, as shown in Figure 1. Much of the discussed literature has focused solely on the vasculature and the brain; however, it was recently proposed that there is a closer link between the brain and the heart than originally expected 60.
Figure 1.
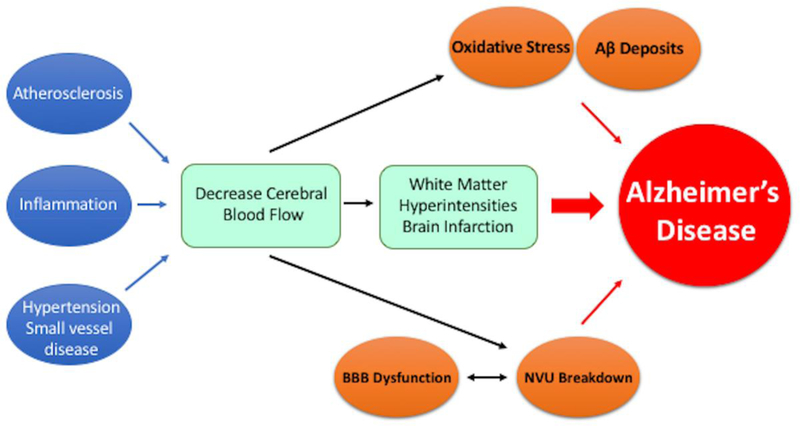
The vascular hypothesis of AD proposes that multiple vascular risk factors (atherosclerosis, inflammation, hypertension and small-vessel disease, etc.) result in decreased CBF. The reduced CBF proceeds to induce white matter hyperintensities and cerebral microinfarctions that ultimately contribute to the progression of AD. Furthermore, it has been proposed that oxidative stress, resultant of reduced CBF, can contribute to AD progression in conjunction with Aβ deposits.
A Newly Discovered Link Between HF and AD
Until recently, the only clear link between HF and AD was based on epidemiological data indicating that both of these debilitating conditions have a high incidence of co-existing, especially among older patients. Furthermore, both of these conditions share risk factors; some of which include obesity, sex, high cholesterol, and more importantly age. As described above, a link to cognitive impairment in HF was also recognized in cerebral hypoperfusion and anoxic state2, 61, 62. This theory follows a cascade effect: HF leads to decreased cerebral blood flow (CBF), causing a metabolic energy crisis. This, in turn causes acidosis and oxidative stress in multiple regions of the brain, eventually culminating in neuronal degradation24, 63, 64. Notably, the severity of HF has been shown to positively correlate with the degree of cognitive decline65, 66 and neuroimaging studies have demonstrated a link between HF and structural changes to the brain64, 67. Individuals with HF often exhibit regional brain atrophy and demyelination, as well as impaired axonal circuit functionality64, 67, 68.
However, cognitive impairment in HF may have a more complex pathogenic background. In fact, in a more general scope, misfolded protein disease is not limited to the brain60, 69.
Amylin cardiomyopathy: a direct link between heart and brain failure
Recent studies focusing on hyperamylinemia, a common disorder in diabetic patients, have revealed a more complex systemic pathogenesis of aggregating amylin. Previous work has found evidence of amylin/amyloid aggregations in the hearts of patients with diabetic cardiomyopathy70. Work by Jackson et al.71, 72 sought to examine the cerebrovascular tissue of diabetic patients with vascular dementia of AD for amyloid deposits wherein they found amylin oligomers and plaques in the temporal grey matter from diabetic patients. Furthermore, researchers found amylin deposition in the brain vasculature and parenchyma of individuals in a specific test group with late onset AD and no apparent diabetes 71. Jackson et al.71 also found some instances where amylin and Aβ depositions were mixed. Expanding on this finding, it has been suggested that amylin and Aβ are connected in terms of a wider net pathophysiology. This hypothesis is supported by the fact that both Aβ and amylin can form similar functioning toxic aggregates that can induce inflammation, oxidative stress, and changes in the microvasculature of brain parenchyma70, 71, 73. These findings suggest that amylin deposition and thereby its negative effects on the vasculature and parenchyma of the brain, may participate in the progression of AD.
Common genetic profiles between AD and HF
The concept of AD affecting both the head and heart is reinforced when looking at genetic profiles between connected disease states. Research has shown that there are similar genetic profiles between HF and AD. Work by Li et al.11 found that in the familial forms, these two conditions share variations in the PSEN1 or PSEN2 genes. In this specific study, a PSEN1 missense mutation (Asp333Gly) and a PSEN2 missense mutation (Ser130Leu) were associated with both DCM and HF11. Similarly, Gianni et al identified the same missense mutations in the PSEN1 and PSEN2 genes associated with AD in sporadic cases of iDCM and described new genetic variants of the promoter region of the genes affecting the expression levels of the protein12. In this study Gianni et al. discovered plaque-like amyloid deposits in the hearts of patients with iDCM. These aggregations of proteins hold a considerable degree of proteotoxicity, inducing cell death 74. At the micro scale, Demuro and colleagues investigated the biochemical mechanisms by which aggregations of soluble amyloid proteins have a neurotoxic effect75. Their findings suggest that oligomer amyloid aggregations can disrupt calcium flux homeostasis and thereby cell membrane function75. Similar to this proteotoxic effect in neurons, oligomeric aggregates exercise the same effect on cardiomyocyte Ca2+ homeostasis12. The toxic role of amyloid deposition as a causal agent for AD was challenged by the failure of some clinical trials targeting amyloid plaques for efficacy of clinical symptoms (the immunoglobulin/albumin combination Flebogamma/Albutein, and small molecule targeting the beta-secretase 1 cleaving enzyme Verubecestat) or causing side effects such as encephalopathy (the humanized anti-Aß mAb Ganatenerumab). An initial explanation for the failure of the trials, and therefore the failure of the “amyloid theory” altogether, included late administration of the drug when the amyloid had triggered neuronal cell death and other terminal changes. However, more recent trials targeting multiple forms of Aß, such as oligomers in addition to the insoluble fibrils (the mAb Crenezumab, Aducanumab) did not cause side effects and provided some evidence of clearance of plaques and decline of clinical symptoms. While waiting for the ongoing phase III clinical trial results (e.g for Crenezumab, Aducanumab, the BACE inhibitors Lanabecestatand Elenbecestat) in vitro studies from the del Monte laboratory using atomic force microscopy suggested that a possible explanation for the failure of some of the drugs may reside in targeting fibers vs. the more toxic oligomeric species. The study indicated that while dissolving fibers may result in increased release of toxic species, targeting the latter may at least slow the progression of the disease by removing the source of neuronal poison76.
Additional common protein profiles between AD and HF
Focusing on the heart, the del Monte laboratory analyzed the composition of the aggregates in the myocardium of iDCM patients and identified that cofilin-2 - along with its substrate actin and competing protein MLCII - was sequestered in the aggregates77. Cofilin is an actin-depolymerization protein that regulates the turnover of actin in contractile cells and the structural integrity of the cell. In a defective and sequestered state, this protein is also known to play a role in multiple neurodegenerative diseases such as corticobasal degeneration, William’s syndrome, fragile X syndrome, and spinal muscular atrophy12, 60, 77 as well as other cardiac disorders such as myocardial ischemia. Recently, the Salloum laboratory described similar changes in cofilin activity, as described in iDCM patients with end stage ischemic cardiomyopathy78. This new evidence further supports the link between neurodegenerative disease and various cardiovascular diseases leading to HF.
Following this work, Dr. del Monte’s group analyzed the complex pathology of proteinopathies and sought to discover a closer link between HF and AD by examining the hearts of individuals with AD diagnosis. In this work, Troncone et al. analyzed the hearts and brains of patients with AD. Upon investigation, they discovered, in the heart, Aβ (both Aβ40 and Aβ42) structurally akin to those found in the brain60 and, in a retrospective analysis, found that AD patients present with myocardial diastolic dysfunction. Thus, like in traditional cardiac amyloidosis, the pathophysiology of diastolic dysfunction in AD can be described by the accumulation of misfolded proteins in the heart 79.
The concept of peripheral accumulation of Aβ in patients with AD is not restricted to the heart. In fact, an early study by Joachim, Mori, & Selkoe80 analyzed Aβ deposition in non-neuronal tissue - most notably, skin and intestine. Eight of the samples from test subjects with AD showed clear and definite evidence of Aβ deposition in these peripheral tissues. Similarly, Aβ40 and Aβ42 aggregates were identified in skeletal muscle of AD individuals using fast performance liquid chromatographic (FPLC) size exclusion chromatography. Researchers discovered elevated levels of Aβ in the temporalis muscles of AD individuals, indicating a possible contributor to elevated concentrations of Aβ plasma levels and potentially indirectly contributing to Aβ deposits in cerebral blood vessels and brain parenchyma81. Furthermore, these varying levels of Aβ suggest an alteration in APP and/or Aβ metabolism in peripheral tissues outside of the CNS81.
Viewing Alzheimer’s disease with a new lens as a potential multi-organ disease60, as shown in Figure 2, it is possible that an inflamed and acidic cerebral microenvironment potentially contributes to a defective BBB (a common symptom of AD). This impaired BBB allows permeability of Aβ plaques into the bloodstream. This spreading event in combination with a defective production/clearance homeostasis of misfolded proteins would cause a combination of central nervous system (CNS) and peripheral symptoms. In the case of HF, the involvement of the heart would accelerate the progression of the disease by contributing to the oxidative stress and acidosis via brain hypoperfusion. On the other hand, there might be a possible specific genetic profile- including a miRNA signature - that accounts for the systemic link between the head and the peripheral organs.
Figure 2.
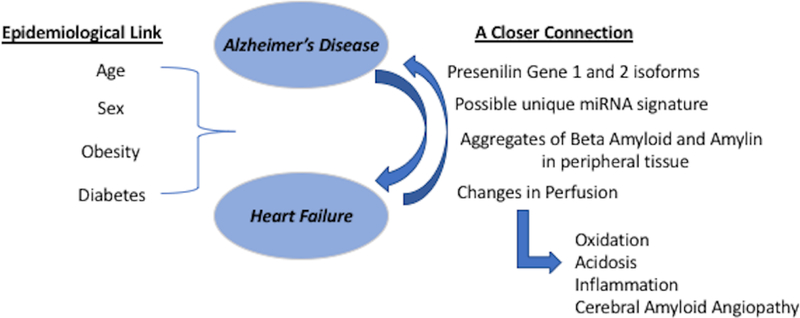
Overall schematic summary of the new findings on the head to heart connection in AD bringing attention to either a more systemic profile or a metastatic condition. This closer connection has some main tenants: 1. A shared genetic profile with PSEN1 and PSEN2 isoforms; 2. Discovery of aggregations of β-Amyloid and Amylin in peripheral tissue and their proteotoxic effects.
Conclusion
With recent studies discovering pathogenic mechanisms and possible links between AD and iDCM, including genetic and environmental background, and that AD individuals had significant amounts of Aβ plaques in peripheral organs, it is critical to understand the contribution of the peripheral organs to the overall clinical picture of proteostatic diseases. The failing heart, by reducing cerebral blood flow to peripheral organs, but also by sustaining the spread of the pathological fragments together with other peripheral organs, activates or aggravates aggregate pathology in the brain. Thus, within this new framework, understanding the disease in its entirety may help in discovering new approaches to delay or reverse proteinopathies involving these two vital organs.
Table 1:
Cardiovascular Problems Linked to AD
| Heart Failure | • Similar epidemiological profile to AD, with linked risk factors: obesity, sex, and age4-7. • Specifically in the case of patients with iDCM, there exists a common genetic (PSEN1 and PSEN2 genetic variants) and physiological link regarding the aggregation of amyloid protein in heart/brain12,58,. |
| Atrial Fibrillation | • Similar epidemiological profile to AD. • Chronic AF may lead to hypoperfusion and thereby induce acceleration of beta amyloid plaque formation82. |
| Hypertension | • Hypertension has been linked as risk factor for AD but the mechanism is poorly understood. • It is believed to either occur through: 1) vascular altercations leading to infarcts in the brain 2) development of other cardiovascular disease 3) adverse effects on neuronal health leading to beta amyloid deposition in brain45,47,48. |
| Vasculopathy | • Macro and micro infarcts, white matter hyperintensities, atherosclerosis, and hypertension → leading to decreased cerebral blood flow can accelerate AD pathogenesis and neurodegenerative symptoms. |
Acknowledgments
Sources of Funding
The authors are supported by NIH grants AG057046 (to FdM, CKC and LEW), HL139348, NR012618 and ES019923 (to LEW) and AHA grant 17CSA33620007 (to FdM).
Non-standard Abbreviations and Acronyms
- Aβ
beta-Amyloid
- AD
Alzheimer’s Disease
- ApoE4
Apoliprotein E4
- APP
Amyloid Precursor Protein
- BBB
Blood Brain Barrier
- CAA
Cerebral Amyloid Angiopathy
- CBF
Cerebral Blood Flow
- CNS
Central Nervous System
- NT/NFT
Neurofibrillary Tangles
- FPLC
Fast Performance Liquid Chromatography
- HF
Heart Failure
- iDCM
Idiopathic Dilated Cardiomyopathy
- NVU
Neurovascular Unit
- PSEN1
Presenilin 1
- PSEN2
Presenilin 2
- ROS
Reactive Oxygen Species
- VCI
Vascular Cognitive Impairment
- WMH
White Matter Hyperintensities
Footnotes
Disclosures
None
References
- 1.Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST, Ferris SH, Finkel SI, Gwyther LP, Khachaturian ZS, Lebowitz BD, McRae TD, Morris JC, Oakley F, Schneider LS, Streim JE, Sunderland T, Teri LA and Tune LE. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA. 1997;278:1363–1371. [PubMed] [Google Scholar]
- 2.Fratiglioni L, De Ronchi D and Agüero-Torres H. Worldwide prevalence and incidence of dementia. Drugs & Aging. 1999;15:365–375. [DOI] [PubMed] [Google Scholar]
- 3.Castellani RJ, Rolston RK and Smith MA. Alzheimer Disease. Disease-a-month : DM. 2010;56:484–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roger VL. Epidemiology of Heart Failure. Circulation research. 2013;113:646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viña J and Lloret A. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. Journal of Alzheimer’s disease: JAD. 2010;20 Suppl 2:S527–533. [DOI] [PubMed] [Google Scholar]
- 6.Hölscher C Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochemical Society Transactions. 2011;39:891–897. [DOI] [PubMed] [Google Scholar]
- 7.Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kåreholt I, Winblad B, Helkala E-L, Tuomilehto J, Soininen H and Nissinen A. Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Archives of Neurology. 2005;62:1556–1560. [DOI] [PubMed] [Google Scholar]
- 8.Van Uden E, Kang DE, Koo EH and Masliah E . LDL receptor-related protein (LRP) in Alzheimer’s disease: Towards a unified theory of pathogenesis. Microscopy Research and Technique. 2000;50:268–272. [DOI] [PubMed] [Google Scholar]
- 9.Pang CP and Baum L. Lipoproteins and related molecules in Alzheimer’s disease. Microscopy Research and Technique. 2000;50:259–260. [DOI] [PubMed] [Google Scholar]
- 10.Iadecola C The Pathobiology of Vascular Dementia. Neuron. 2013;80:844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M and Hershberger RE. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. The American Journal of Human Genetics. 2006;79:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gianni D, Li A, Tesco G, McKay KM, Moore J, Raygor K, Rota M, Gwathmey JK, Dec GW, Aretz T, Leri A, Semigran MJ, Anversa P, Macgillivray TE, Tanzi RE and Monte Fd. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation. 2010;121:1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, Schneider JA, Wellington C, Wilcock DM, Zipfel GJ, Zlokovic B, Bain LJ, Bosetti F, Galis ZS, Koroshetz W and Carrillo MC. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & Dementia. 2015;11:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wentzel C, Rockwood K, MacKnight C, Hachinski V, Hogan DB, Feldman H, Østbye T, Wolfson C, Gauthier S, Verreault R and McDowell I. Progression of impairment in patients with vascular cognitive impairment without dementia. Neurology. 2001;57:714–716. [DOI] [PubMed] [Google Scholar]
- 15.Jellinger KA. Understanding the pathology of vascular cognitive impairment. Journal of the Neurological Sciences. 2005;229–230:57-63. [DOI] [PubMed] [Google Scholar]
- 16.Rius-Pérez S, Tormos AM, Pérez S and Taléns-Visconti R. Vascular pathology: Cause or effect in Alzheimer disease? Neurologia (Barcelona, Spain). 2018;33:112–120. [DOI] [PubMed] [Google Scholar]
- 17.Karran E, Mercken M and De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10:698–712. [DOI] [PubMed] [Google Scholar]
- 18.de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–1162. [DOI] [PubMed] [Google Scholar]
- 19.de la Torre JC and Mussivand T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurological Research. 1993;15:146–153. [DOI] [PubMed] [Google Scholar]
- 20.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S and American Heart Association Stroke Council CoEaPCoCNCoCRaIaCoCSaA. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Torre JC. Cerebrovascular and cardiovascular pathology in Alzheimer’s disease. International Review of Neurobiology. 2009;84:35–48. [DOI] [PubMed] [Google Scholar]
- 22.Roher AE, Debbins JP, Malek-Ahmadi M, Chen K, Pipe JG, Maze S, Belden C, Maarouf CL, Thiyyagura P, Mo H, Hunter JM, Kokjohn TA, Walker DG, Kruchowsky JC, Belohlavek M, Sabbagh MN and Beach TG. Cerebral blood flow in Alzheimer’s disease. Vascular Health and Risk Management. 2012;8:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishimura T, Hashikawa K, Fukuyama H, Kubota T, Kitamura S, Matsuda H, Hanyu H, Nabatame H, Oku N, Tanabe H, Kuwabara Y, Jinnouchi S and Kubol A. Decreased cerebral blood flow and prognosis of Alzheimer’s disease: a multicenter HMPAO-SPECT study. Annals of Nuclear Medicine. 2007;21:15–23. [DOI] [PubMed] [Google Scholar]
- 24.Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ and Perry G. Oxidative stress and neurodegeneration. Annals of the New York Academy of Sciences. 2005;1043:545–552. [DOI] [PubMed] [Google Scholar]
- 25.Cermakova P, Eriksdotter M, Lund LH, Winblad B, Religa P and Religa D. Heart failure and Alzheimer′s disease. Journal of Internal Medicine. 2015;277:406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S and Hunt JA. Reactive oxygen species (ROS)--a family of fate deciding molecules pivotal in constructive inflammation and wound healing. European Cells & Materials. 2012;24:249–265. [DOI] [PubMed] [Google Scholar]
- 27.Uttara B, Singh AV, Zamboni P and Mahajan RT. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Current Neuropharmacology. 2009;7:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Smith MA, Honda K, Aliev G, Moreira PI, Nunomura A, Casadesus G, Harris PLR, Siedlak SL and Perry G. Vascular oxidative stress in Alzheimer disease. Journal of the Neurological Sciences. 2007;257:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim GH, Kim JE, Rhie SJ and Yoon S. The Role of Oxidative Stress in Neurodegenerative Diseases. Experimental Neurobiology. 2015;24:325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuoka Y, Picciano M, La Francois J and Duff K. Fibrillar beta-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience. 2001;104:609–613. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y and Zhao B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxidative Medicine and Cellular Longevity. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de la Monte SM and Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2006;9:167–181. [DOI] [PubMed] [Google Scholar]
- 33.Rius-Pérez S, Tormos AM, Pérez S and Taléns-Visconti R. Vascular pathology: Cause or effect in Alzheimer disease? Neurología (English Edition). 2018;33:112–120. [DOI] [PubMed] [Google Scholar]
- 34.Wyss-Coray T and Rogers J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harbor Perspectives in Medicine. 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Combs CK. Inflammation and microglia actions in Alzheimer’s disease. Journal of Neuroimmune Pharmacology: The Official Journal of the Society on NeuroImmune Pharmacology. 2009;4:380–388. [DOI] [PubMed] [Google Scholar]
- 36.Varatharaj A and Galea I. The blood-brain barrier in systemic inflammation. Brain, Behavior, and Immunity. 2017;60:1–12. [DOI] [PubMed] [Google Scholar]
- 37.Ujiie M, Dickstein DL, Carlow DA and Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation (New York, NY: 1994). 2003;10:463–470. [DOI] [PubMed] [Google Scholar]
- 38.Clifford PM, Zarrabi S, Siu G, Kinsler KJ, Kosciuk MC, Venkataraman V, D’Andrea MR, Dinsmore S and Nagele RG. Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Research. 2007;1142:223–236. [DOI] [PubMed] [Google Scholar]
- 39.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature Reviews Neuroscience. 2011;12:723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghiso J, Tomidokoro Y, Revesz T, Frangione B and Rostagno A. CEREBRAL AMYLOID ANGIOPATHY AND ALZHEIMER’S DISEASE. Hirosaki igaku = Hirosaki medical journal. 2010;61:S111–S124. [PMC free article] [PubMed] [Google Scholar]
- 41.Iadecola C The pathobiology of vascular dementia. Neuron. 2013;80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lathe R, Sapronova A and Kotelevtsev Y. Atherosclerosis and Alzheimer - diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatrics. 2014;14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI and Beach TG. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:2055–2062. [DOI] [PubMed] [Google Scholar]
- 44.Roher AE, Tyas SL, Maarouf CL, Daugs ID, Kokjohn TA, Emmerling MR, Garami Z, Belohlavek M, Sabbagh MN, Sue LI and Beach TG. Intracranial atherosclerosis as a contributing factor to Alzheimer’s disease dementia. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim T-W, Song I- U, Jeong D- S and Lee K- S. Clinical effect of cerebrovascular atherosclerosis on cognition in Alzheimer’s disease. Archives of Gerontology and Geriatrics. 2016;63:55–58. [DOI] [PubMed] [Google Scholar]
- 46.Dickstein DL, Walsh J, Brautigam H, Stockton SD, Gandy S and Hof PR. Role of Vascular Risk Factors and Vascular Dysfunction in Alzheimer’s Disease. The Mount Sinai Journal of Medicine, New York. 2010;77:82–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA and Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 48.Leszek J, Sochocka M and Gąsiorowski K. Vascular factors and epigenetic modifications in the pathogenesis of Alzheimer’s disease. Journal of the Neurological Sciences. 2012;323:25–32. [DOI] [PubMed] [Google Scholar]
- 49.Prins ND, van Dijk EJ, den Heijer T, Vermeer SE, Jolles J, Koudstaal PJ, Hofman A and Breteler MMB. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain: A Journal of Neurology. 2005;128:2034–2041. [DOI] [PubMed] [Google Scholar]
- 50.Hypertension Thorin E. and Alzheimer disease: another brick in the wall of awareness. Hypertension (Dallas, Tex: 1979). 2015;65:36–38. [DOI] [PubMed] [Google Scholar]
- 51.Faraco G and Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension (Dallas, Tex: 1979). 2013;62:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de la Torre JC. Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2012;32:553–567. [DOI] [PubMed] [Google Scholar]
- 53.Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D’Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D and Framingham Heart S. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. [DOI] [PubMed] [Google Scholar]
- 54.Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, Raman MR, Machulda MM, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Rocca WA, Roberts RO, Petersen RC and Jack CR. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain: A Journal of Neurology. 2015;138:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tuladhar AM, van Norden AGW, de Laat KF, Zwiers MP, van Dijk EJ, Norris DG and de Leeuw F-E. White matter integrity in small vessel disease is related to cognition. NeuroImage Clinical. 2015;7:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tosto G, Zimmerman ME, Hamilton JL, Carmichael OT, Brickman AM and Alzheimer’s Disease Neuroimaging I. The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2015;11:1510–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai Z, Wang C, He W, Tu H, Tang Z, Xiao M and Yan L-J. Cerebral small vessel disease and Alzheimer’s disease. Clinical Interventions in Aging. 2015;10:1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Brien JT and Markus HS. Vascular risk factors and Alzheimer’s disease. BMC medicine. 2014;12:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neuro-Degenerative Diseases. 2010;7:116–121. [DOI] [PubMed] [Google Scholar]
- 60.Troncone L, Luciani M, Coggins M, Wilker EH, Ho C-Y, Codispoti KE, Frosch MP, Kayed R and Del Monte F. Aβ Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. Journal of the American College of Cardiology. 2016;68:2395–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Odén A and Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet (London, England). 1996;347:1141–1145. [DOI] [PubMed] [Google Scholar]
- 62.Jagust W Untangling vascular dementia. The Lancet. 2001;358:2097–2098. [DOI] [PubMed] [Google Scholar]
- 63.Pirchl M and Humpel C. [Does acidosis in brain play a role in Alzheimer’s disease?]. Neuropsychiatrie: Klinik, Diagnostik, Therapie Und Rehabilitation: Organ Der Gesellschaft Osterreichischer Nervenarzte Und Psychiater. 2009;23:187–192. [PubMed] [Google Scholar]
- 64.Cermakova P, Eriksdotter M, Lund LH, Winblad B, Religa P and Religa D. Heart failure and Alzheimer’s disease. Journal of Internal Medicine. 2015;277:406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoth KF, Poppas A, Moser DJ, Paul RH and Cohen RA. Cardiac dysfunction and cognition in older adults with heart failure. Cognitive and Behavioral Neurology: Official Journal of the Society for Behavioral and Cognitive Neurology. 2008;21:65–72. [DOI] [PubMed] [Google Scholar]
- 66.Cohen MB and Mather PJ. A review of the association between congestive heart failure and cognitive impairment. The American Journal of Geriatric Cardiology. 2007;16:171–174. [DOI] [PubMed] [Google Scholar]
- 67.Vogels RLC, van der Flier WM, van Harten B, Gouw AA, Scheltens P, Schroeder-Tanka JM and Weinstein HC. Brain magnetic resonance imaging abnormalities in patients with heart failure. European Journal of Heart Failure. 2007;9:1003–1009. [DOI] [PubMed] [Google Scholar]
- 68.Kumar R, Woo MA, Macey PM, Fonarow GC, Hamilton MA and Harper RM. Brain Axonal and Myelin Evaluation in Heart Failure. Journal of the neurological sciences. 2011;307:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang J, Gu BJ, Masters CL and Wang Y-J. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nature Reviews Neurology. 2017;13:612–623. [DOI] [PubMed] [Google Scholar]
- 70.Despa F and DeCarli C. Amylin: what might be its role in Alzheimer’s disease and how could this affect therapy? Expert review of proteomics. 2013;10:403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jackson K, Barisone GA, Diaz E, Jin L-w, DeCarli C and Despa F. Amylin deposition in the brain: a second amyloid in Alzheimer’s disease? Annals of neurology. 2013;74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jackson G, Gibbs CR, Davies MK and Lip GYH. Pathophysiology. BMJ : British Medical Journal. 2000;320:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kawahara M, Kuroda Y, Arispe N and Rojas E. Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. The Journal of Biological Chemistry. 2000;275:14077–14083. [DOI] [PubMed] [Google Scholar]
- 74.Parry TL, Melehani JH, Ranek MJ and Willis MS. Functional Amyloid Signaling via the Inflammasome, Necrosome, and Signalosome: New Therapeutic Targets in Heart Failure. Frontiers in Cardiovascular Medicine. 2015;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demuro A, Mina E, Kayed R, Milton SC, Parker I and Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. The Journal of Biological Chemistry. 2005;280:17294–17300. [DOI] [PubMed] [Google Scholar]
- 76.Bernini F, Malferrari D, Pignataro M, Bortolotti CA, Di Rocco G, Lancellotti L, Brigatti MF, Kayed R, Borsari M, Del Monte F and Castellini E. Pre-amyloid oligomers budding:a metastatic mechanism of proteotoxicity. Scientific Reports. 2016;6:35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subramanian K, Gianni D, Balla C, Assenza GE, Joshi M, Semigran MJ, Macgillivray TE, Van Eyk JE, Agnetti G, Paolocci N, Bamburg JR, Agrawal PB and del Monte F. Cofilin-2 Phosphorylation and Sequestration In Myocardial Aggregates: Novel Pathogenetic Mechanisms For Idiopathic Dilated Cardiomyopathy. Journal of the American College of Cardiology. 2015;65:1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chau VQ, Devarakonda T, Romeo F, Balan B, Cain C, Toldo S, Tang D, Shah K, Kasirajan V and Salloum FN. Abstract 16744: Cardiac Expression Profiles of Cofilin-2 and H2S-Producing Enzyme 3-Mercaptopyruvate Sulfurtransferase in Patients With End-Stage Ischemic Cardiomyopathy. Circulation. 2017. [Google Scholar]
- 79.Willis MS and Patterson C. Proteotoxicity and Cardiac Dysfunction — Alzheimer’s Disease of the Heart? New England Journal of Medicine. 2013;368:455–464. [DOI] [PubMed] [Google Scholar]
- 80.Joachim CL, Mori H and Selkoe DJ. Amyloid β-protein deposition in tissues other than brain in Alzheimer’s disease. Nature. 1989;341:226–230. [DOI] [PubMed] [Google Scholar]
- 81.Kuo Y-M, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, Sue LI, Kalback WM, Emmerling MR, Beach TG and Roher AE. Elevated Aβ42 in Skeletal Muscle of Alzheimer Disease Patients Suggests Peripheral Alterations of AβPP Metabolism. The American Journal of Pathology. 2000;156:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ihara M and Washida K. Linking Atrial Fibrillation with Alzheimer’s Disease: Epidemiological, Pathological, and Mechanistic Evidence. Journal of Alzheimer’s disease: JAD. 2018;62:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]