

ABSTRACT
Macroautophagy (hereafter autophagy) is a lysosomal degradation pathway critical for maintaining cellular homeostasis and viability, and is predominantly regarded as a rapid and dynamic cytoplasmic process. To increase our understanding of the transcriptional and epigenetic events associated with autophagy, we performed extensive genome-wide transcriptomic and epigenomic profiling after nutrient deprivation in human autophagy-proficient and autophagy-deficient cells. We observed that nutrient deprivation leads to the transcriptional induction of numerous autophagy-associated genes. These transcriptional changes are reflected at the epigenetic level (H3K4me3, H3K27ac, and H3K56ac) and are independent of autophagic flux. As a proof of principle that this resource can be used to identify novel autophagy regulators, we followed up on one identified target: EGR1 (early growth response 1), which indeed appears to be a central transcriptional regulator of autophagy by affecting autophagy-associated gene expression and autophagic flux. Taken together, these data stress the relevance of transcriptional and epigenetic regulation of autophagy and can be used as a resource to identify (novel) factors involved in autophagy regulation.
KEYWORDS: Autophagy, ChIP-seq, EGR1, nutrient-deprivation, RNA-seq
Introduction
Macroautophagy (hereafter referred to as autophagy) is a highly conserved catabolic mechanism, involving the sequestration of bulk cytoplasmic components by transient double-membrane compartments called phagophores; these mature into autophagosomes, which allow subsequent delivery of the cargo into lysosomes for degradation [1,2]. Autophagy is essential for maintaining cellular homeostasis by removal of damaged or unnecessary proteins and organelles, and is important for cell viability by maintaining the energy balance upon cellular stresses, such as nutrient starvation [3].
Because autophagy is a rapid, dynamic process that constantly requires adaptation to environmental changes, research has often focused on cytoplasmic post-translational modifications of autophagy-associated genes [4]. In fact, for a long time autophagy has been viewed as mainly a cytoplasmic process, especially because enucleated cells are still capable of undergoing autophagy [5]. Recently it is becoming apparent that transcriptional and epigenetic events are also involved in regulating autophagy [6]. One of the first transcription factors identified to be involved in autophagy regulation under amino acid and serum starvation is TFEB (transcription factor EB). Besides its role in regulating lysosomal biogenesis, TFEB is involved in autophagy initiation because its overexpression can induce autophagy [7]. This is in part established by direct binding to the promotor of a set of autophagy-associated genes and thereby increasing their gene expression [7]. Transcription factors that have been implicated in the regulation of specific autophagy-associated gene expression under various starvation conditions can have an enhancing effect, such as the FOXO (forkhead box O) family of transcription factors [reviewed in 8], or a suppressing effect, such as ZKSCAN3 (zinc finger protein with KRAB and SCAN domains 3) [9]. NFKB (nuclear factor kappa B) is a transcription factor with a dual effect on autophagy-associated gene expression, by inhibiting BNIP3 (BCL2 interacting protein 3) transcription [10] and inducing BECN1 (beclin 1) [11], SQSTM1 (sequestosome 1) [12], and BCL2 [13] expression. While these studies have shed light on the transcriptional regulation of autophagy, it is still incompletely understood which transcription factors are involved in autophagy modulation and whether autophagy itself has a feedback regulation on its transcriptional regulation.
In addition to transcriptional regulation, there is limited evidence demonstrating whether autophagy is epigenetically regulated. EHMT2/G9a (euchromatic histone lysine methyltransferase 2) [14] and EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) [15] have both been implicated in autophagy repression under serum starvation by increasing H3K9me2 and H3K27me3 histone mark levels, respectively, of certain autophagy-associated genes. Furthermore, autophagy induction has been demonstrated to affect total H3R17me2, H4K16ac, and H2BK120ub levels through CARM1 (coactivator associated arginine methyltransferase 1) [16], KAT8/hMOF (lysine acetyltransferase 8) [17], and the deubiquitinase USP44 (ubiquitin specific peptidase 44) [18], respectively. These alterations affect transcription of genes involved in (the regulation of) autophagy and therefore function as an epigenetic switch in autophagy regulation under various starvation conditions and upon MTOR (mechanistic target of rapamycin kinase) inhibition. For example, autophagy induction downregulates KAT8, thereby decreasing H4K16 acetylation of autophagy-associated genes, which results in decreased gene expression. This reduces autophagy, thereby providing a feedback mechanism to control the amount of autophagy [17]. Furthermore, global changes in H4K20me3 [19], H3K4me3 [17], and H3K56ac [20] have been associated with autophagy induction, but whether and how this affects autophagy remains to be determined [17,19,20]. Importantly, extensive studies which assess and combine genome-wide transcriptomic and epigenomic events underlying autophagy are lacking. Taken together, further research is required to understand how, and which, epigenetic modifications contribute to the regulation of autophagy.
Here, we performed in-depth genome-wide transcriptional and epigenetic profiling to improve our understanding of the transcriptional and epigenetic events associated with amino acid and serum starvation-induced autophagy. RNA and chromatin immunoprecipitation (ChIP) sequencing of human cells revealed that nutrient deprivation leads to the transcriptional induction of many autophagy-associated genes. A similar induction was observed in autophagy-deficient cell lines, demonstrating that the induction of transcription of autophagy-associated genes is an autophagy-independent process in the cells used in this study. These transcriptional changes are reflected by POLR2/RNA polymerase 2 occupancy, and at the epigenetic level by H3K4me3, H3K27ac, and H3K56ac, indicating that the epigenome is involved in autophagy regulation. Our unbiased analyses identified EGR1 as a transcriptional regulator of many autophagy-associated genes, thereby affecting autophagy. This proof of principle demonstrates that these databases can function as a resource to further characterize the transcriptional and epigenetic events associated with autophagy, thereby facilitating the identification of (novel) mediators regulating autophagy in the future.
Results
Increased expression of autophagy-associated genes upon nutrient deprivation
For a better understanding of the transcriptional changes initiated by starvation, cells were deprived of amino acids and serum for 6 h in EBSS (Earle’s balanced salt solution; culture media without amino acids, serum and a low amount of glucose [21]), a common manner to starve cells and induce autophagy, and RNA-sequencing was performed. Nutrient deprivation of 6 h was chosen as this is long enough to allow for the detection of changes in the transcriptome and yet short enough to prevent interference of secondary modulators of transcriptional responses. We utilized the near-haploid human HAP1 cell line [22] in which autophagy genes can be readily manipulated, allowing us to study the effect of the autophagic flux on the transcriptome. Nutrient deprivation led to the induction of autophagy, as demonstrated by an increased autophagic flux as assessed by determining the levels of lipidated MAP1LC3B (microtubule associated protein 1 light chain 3 beta; hereafter referred to as LC3-II) in the presence or absence of bafilomycin A1 (Figure 1(a)), an increase in autolysosomal structures (Figure 1(b)), and an increase in the number of mCherry+ EGFP+ (yellow) and mCherry+ (red) dots (Figure 1(c)). Cell viability was not significantly affected at this time point (Figure S1A). Starvation had a profound effect on the transcriptome of these cells as many genes were significantly differentially expressed (Figure 1(d,e)). Analysis of genes affected by nutrient deprivation revealed that autophagy-associated genes were enriched within the genes upregulated upon nutrient deprivation (Figure 1(f); Table S1). The genes upregulated upon starvation also included the majority of the key genes regulating mammalian autophagosome formation, as defined by Mizushima et al. [23] (Figure 1(g)). The increased expression of genes associated with autophagy or involved in autophagosome formation, observed with RNA sequencing, was confirmed by qRT-PCR (Figure S1B). Thus, nutrient deprivation induces autophagy, and in parallel induces the expression of autophagy-associated genes in HAP1 cells.
Figure 1.
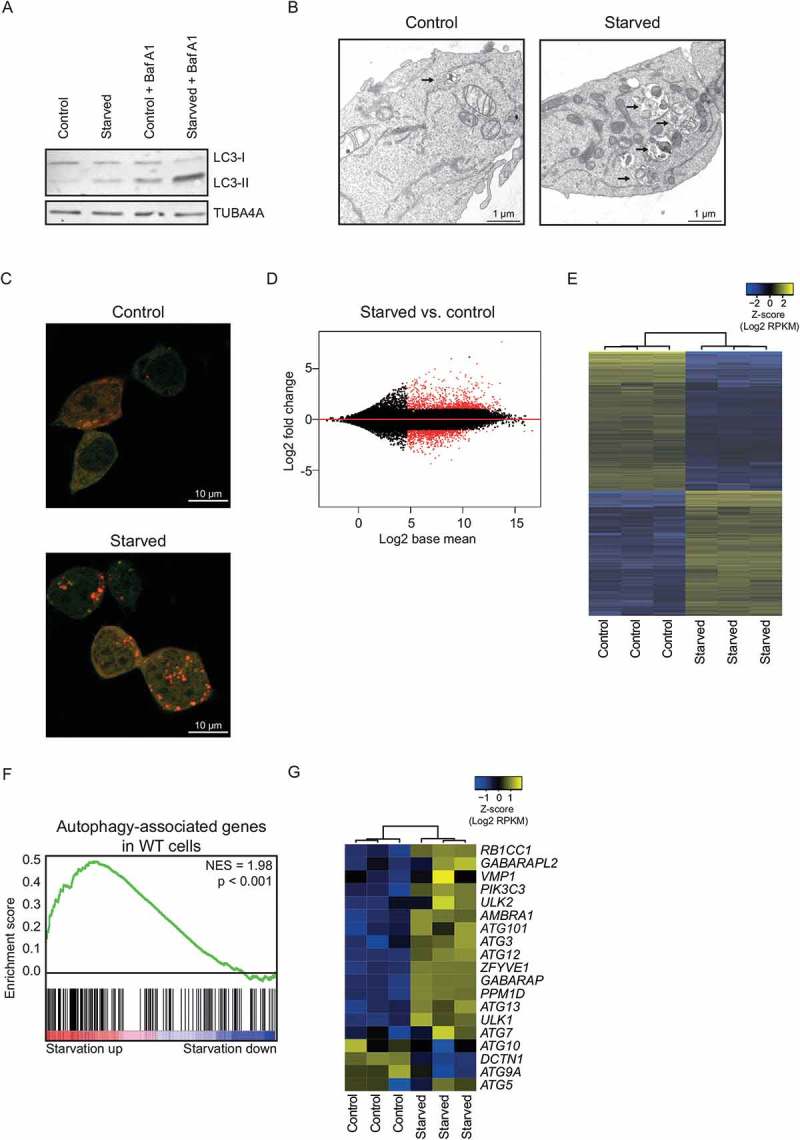
Increased expression of autophagy-associated genes after nutrient deprivation. (a) Western Blot of HAP1 cells in control and starved (6 h EBSS) condition, with and without bafilomycin A1 (40 nM). Representative blot is shown (n = 4). (b) Representative EM images of HAP1 cells in control and starved (6 h EBSS) condition, treated with bafilomycin A1. Autolysosomal structures are indicated by arrows. (c) Representative images of HAP1 cells transfected with a plasmid encoding mCherry-EGFP-LC3B in control and starved (6 h EBSS) condition. mCherry+ EGFP+ dots (yellow) are autophagosomes and mCherry+ dots (red) are autolysosomes. (d) MA plot of HAP1 cells upon 6 h starvation with EBSS, displaying all expressed genes. Red dots indicate genes with a FDR < 0.05. (e) Heatmap of genes differentially expressed in HAP1 cells after 6 h starvation with EBSS. (f) Gene set enrichment analysis for autophagy-associated genes in HAP1 cells upon starvation (6 h EBSS). (g) Heatmap depicting expression of key autophagy proteins upon starvation (6 h EBSS) of HAP1 cells. See also Figure S1.
Expression of autophagy-associated genes is independent of autophagic flux
To determine whether autophagy is required for the increased expression of autophagy-associated genes upon starvation, CRISPR/Cas9-mediated ATG7 (autophagy related 7)- and RB1CC1/FIP200 (RB1 inducible coiled-coil 1)-deficient HAP1 cells were utilized (ATG7 KO and RB1CC1 KO), 2 genes belonging to 2 different ATG protein functional clusters [2] (Figure S2A and S2B). These cells were unable to undergo normal autophagy, as demonstrated by the lack of LC3-II formation (Figure 2(a)) and the reduced formation of autophagosomal and autolysosomal structures (Figure 2(b,c)). Comparison of the transcriptomic changes of autophagy-deficient and wild-type (WT) cells upon nutrient deprivation demonstrated that both cell lines responded in a similar fashion (Figure 2(d,e)). This observation was supported by the analysis of differentially expressed genes upon starvation between WT and ATG7 KO or RB1CC1 KO cells, which revealed that only a few genes have a significantly different change in expression in RB1CC1 KO cells upon nutrient deprivation compared to their change in WT cells (Figure 2(f)). Importantly, autophagy-associated genes were significantly enriched in genes increased in both autophagy-deficient cell lines (Figure 2(g)). Furthermore, expression of autophagy-associated genes was affected similarly in autophagy-deficient cells compared to WT cells upon starvation (Figure 2(d), indicated by dark blue dots, and Figure 2(h)). These data demonstrate that the increased expression of autophagy-associated genes is not per se dependent on autophagic flux.
Figure 2.
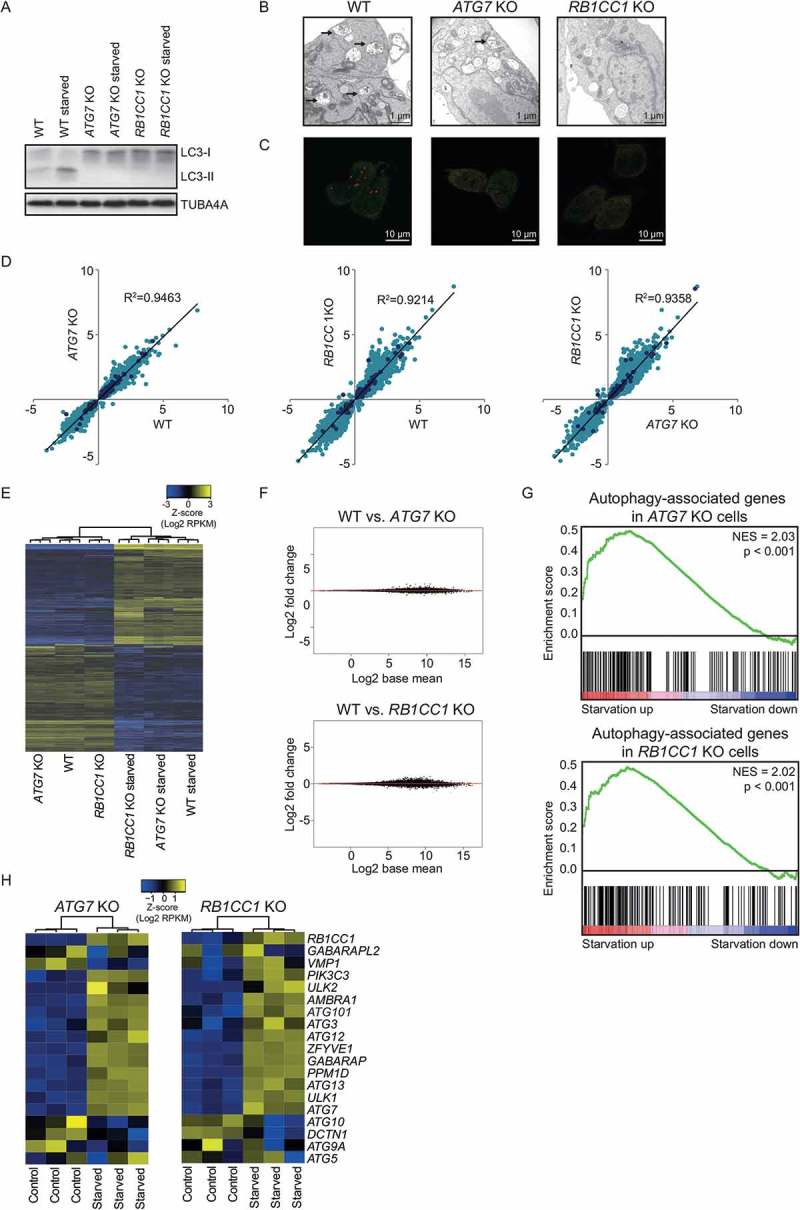
Increased expression of autophagy-associated genes upon nutrient deprivation in ATG7 KO and RB1CC1 KO cells. (a) Western Blot of WT, ATG7 KO, and RB1CC1 KO HAP1 cells in control and starved (3 h EBSS) condition, treated with bafilomycin A1 (40 nM). Representative blot is shown (n = 4). (b) Representative EM images of WT, ATG7 KO, and RB1CC1 KO HAP1 cells in starved (6 h EBSS) condition, treated with bafilomycin A1. Autolysosomes are indicated by arrows. (c) Representative images of HAP1 WT, ATG7 KO, and RB1CC1 KO cells transfected with a plasmid encoding mCherry-EGFP-LC3B in starved (6 h EBSS) condition. mCherry+ EGFP+ dots (yellow) are autophagosomes and mCherry+ dots (red) are autolysosomes. (d) Fold change of significantly differentially expressed genes in either WT, ATG7 KO and/or RB1CC1 KO HAP1 cells. Blue dots represent autophagy-associated genes. (e) Heatmap of WT, ATG7 KO, and RB1CC1 KO HAP1 cells upon starvation (6 h EBSS) displaying genes significantly different in one of the cell lines. (f) MA plot of genes differentially expressed between WT and ATG7 KO or RB1CC1 KO HAP1 cells upon starvation (6 h EBSS). Red dots indicate genes with a FDR < 0.05. (g) Gene set enrichment analysis of autophagy-associated genes in ATG7 KO and RB1CC1 KO HAP1 cells upon starvation (6 h EBSS). (h) Heatmap depicting expression of key autophagy proteins upon starvation (6 h EBSS) of ATG7 KO and RB1CC1 KO cells. See also Figure S2.
Increased transcription of autophagy-associated genes contributes to increased expression of autophagy-associated genes
To investigate whether the increased mRNA expression of autophagy-associated genes upon nutrient deprivation is the direct result of increased transcription, and to rule out that these differences are not only the result of increased mRNA stability, ChIP-sequencing for POLR2/Pol II (RNA polymerase Il) was performed. mRNA expression as defined by RNA-sequencing directly correlated with POLR2 signal, indicating active transcription (Figure 3(a,b)). Moreover, genes identified based on RNA-sequencing as upregulated after nutrient deprivation showed indeed an increased POLR2 signal after starvation, and genes defined as downregulated displayed a decrease in POLR2 signal, demonstrating that transcription indeed contributed to the changes in gene expression (Figure 3(c)). Similarly, the POLR2 signal was increased for the majority of key genes involved in autophagosome formation upon starvation (Figure 3(d-f); Figure S3). Collectively these POLR2 ChIP-seq data demonstrate that increased transcription directly contributes to the increased expression of autophagy-associated genes in HAP1 cells.
Figure 3.
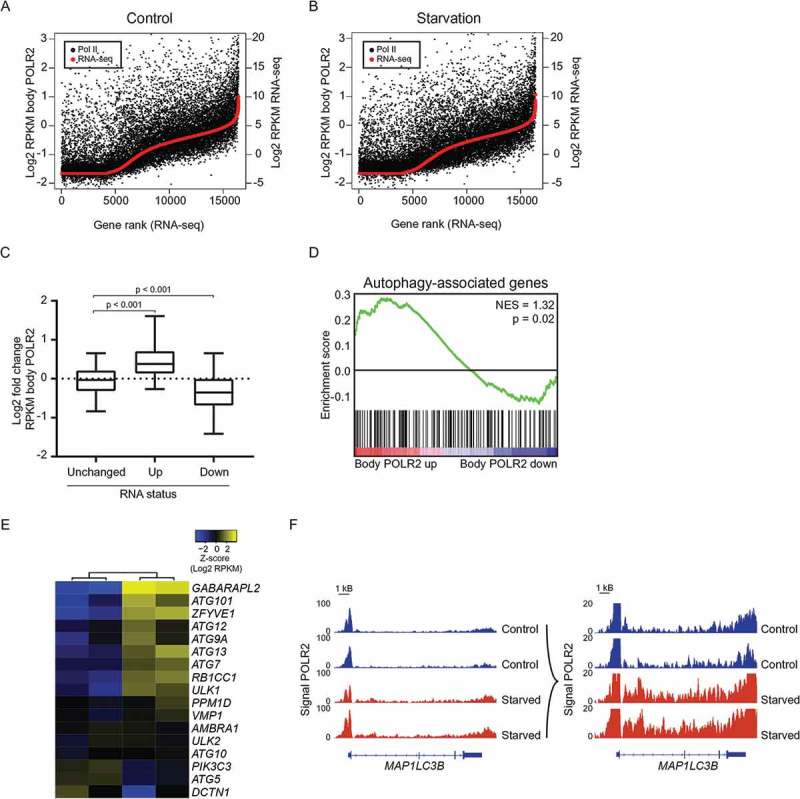
Increased transcription of autophagy-associated genes contributes to increased expression of autophagy-associated genes. Rank analysis of gene body POLR2 occupancy and RNA-sequencing signal in control (a) condition and after starvation (b). Genes are ranked according to RNA-sequencing data. (c) Boxplots with 5%-95% whiskers displaying log2 fold change of body POLR2 signal for genes unchanged, increased, and decreased ≥ log1 based on RNA-sequencing. (d) Gene set enrichment analysis of autophagy-associated genes for genes associated with an alteration of body POLR2 signal upon starvation (3 h EBSS). (e) Heatmap depicting body POLR2 signal for key autophagy genes upon starvation (3 h EBSS). (f) Gene track for MAP1LC3B displaying ChIP-seq signals for POLR2. See also Figure S3.
Increased transcription of autophagy-associated genes is reflected at the epigenetic level
To determine whether epigenetic modifications could contribute to the transcriptional changes upon nutrient deprivation, global levels of various histone marks (H4K16ac, H4K20me3, H3K9me2, H3K4me3, H3K27ac, and H3K56ac) were first assessed by western blotting 3 h after starvation. We did not observe an effect of nutrient-deprivation on global expression of these histone marks (Figure S4). Next, to evaluate whether nutrient deprivation may result in a more specific redistribution of chromatin marks, ChIP-sequencing was performed for histone marks associated with active transcription (H3K4me3 [24], H3K27ac [25], and H3K56ac [26]). Short-term nutrient deprivation resulted in alterations in all 3 histone marks (Figure 4(a)). As expected, the most pronounced effect was observed on the histone acetylation status, which has been demonstrated to be more dynamically regulated than methylation [27]. Increased H3K4me3, H3K27ac, and H3K56ac alterations directly correlated with increased mRNA expression (Figure 4(b)). Furthermore, autophagy-associated genes were enriched within the genes associated with an increase in H3K4me3, H3K27ac, or H3K56ac (Figure 4(c,d)). These data indicate that epigenetic alterations correlate with increased gene expression of autophagy-associated genes observed upon nutrient deprivation in HAP1 cells.
Figure 4.
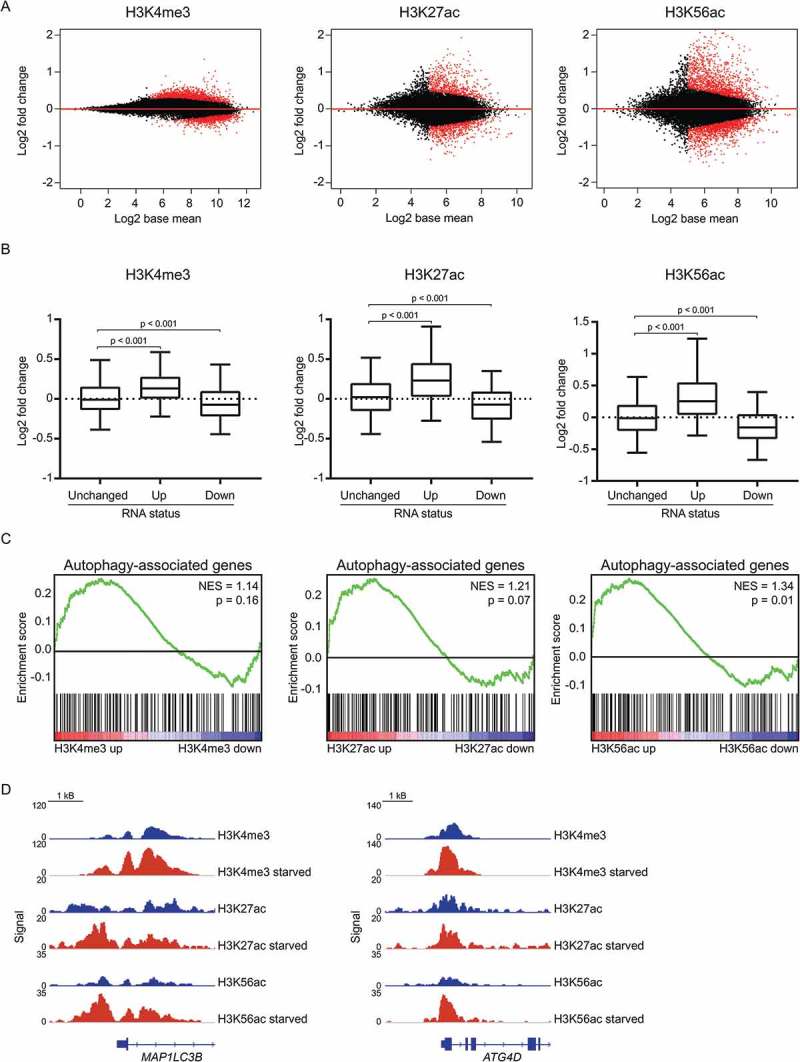
Increased transcription of autophagy-associated genes is reflected at the epigenetic level. (a) MA plots of H3K4me3, H3K27ac and H3K56ac signal upon starvation (3 h EBSS). Red dots indicate genes with a FDR < 0.05. (b) Boxplots with 5%-95% whiskers displaying log2 fold change in H3K4me3, H3K27ac or H3K56ac signal for genes unchanged, increased, and decreased ≥ log1 based on RNA-sequencing. (c) Gene set enrichment analysis of autophagy-associated genes for genes associated with an alteration of H3K4me3, H3K27ac or H3K56ac signal upon starvation (3 h EBSS). (d) Gene tracks for MAP1LC3B and ATG4D displaying ChIP-seq signals for H3K4me3, H3K27ac, and H3K56ac with and without starvation (3 h EBSS). See also Figure S4.
Epigenetic and transcriptomic analyses identify EGR1 as a candidate transcriptional regulator of autophagy
We next explored whether our epigenetic and transcriptomic datasets could be utilized to identify novel regulators of autophagy as a proof-of-principle exercise. To identify which transcription factor(s) could be involved in the increased transcription of autophagy-associated genes upon starvation, enrichment of transcription factor binding motifs in autophagy-associated genes was analyzed in silico. More specifically, open chromatin, indicated by H3K27ac, H3K56ac, or H3K4me3 peaks, associated with autophagy-associated genes with increased expression upon nutrient deprivation was combined with DNAse hypersensitivity data and analyzed for enrichment of transcription factor binding motifs (Figure 5(a)). For the 10 binding motifs with the highest enrichment, expression and induction of the corresponding transcription factors was assessed upon nutrient deprivation (Figure 5(b,c)). This analysis identified EGR1 as the transcription factor with the highest (increase in) expression under these conditions. Correspondingly, nutrient deprivation induced a strong increase in the POLR2 signal for EGR1, and EGR1 protein levels were demonstrated to increase upon starvation in both HAP1 and U2OS cell lines (Figure 5(d,e)). Serum or amino acid deprivation alone did not significantly affect EGR1 expression (Figure S5). To further validate the link between EGR1 and autophagy, we examined publically available EGR1 ChIP-sequencing data from 2 different lymphocytic cell lines, which indeed confirmed binding of EGR1 in the promotor region of many autophagy-related genes, including MAP1LC3B (Figure 5(f)). Furthermore, we identified the presence of 3 EGR1 motifs within the promoter region of MAP1LC3B corresponding to open chromatin regions in HAP1 cells. Altogether, these data identify EGR1 as a candidate transcriptional regulator of autophagy.
Figure 5.
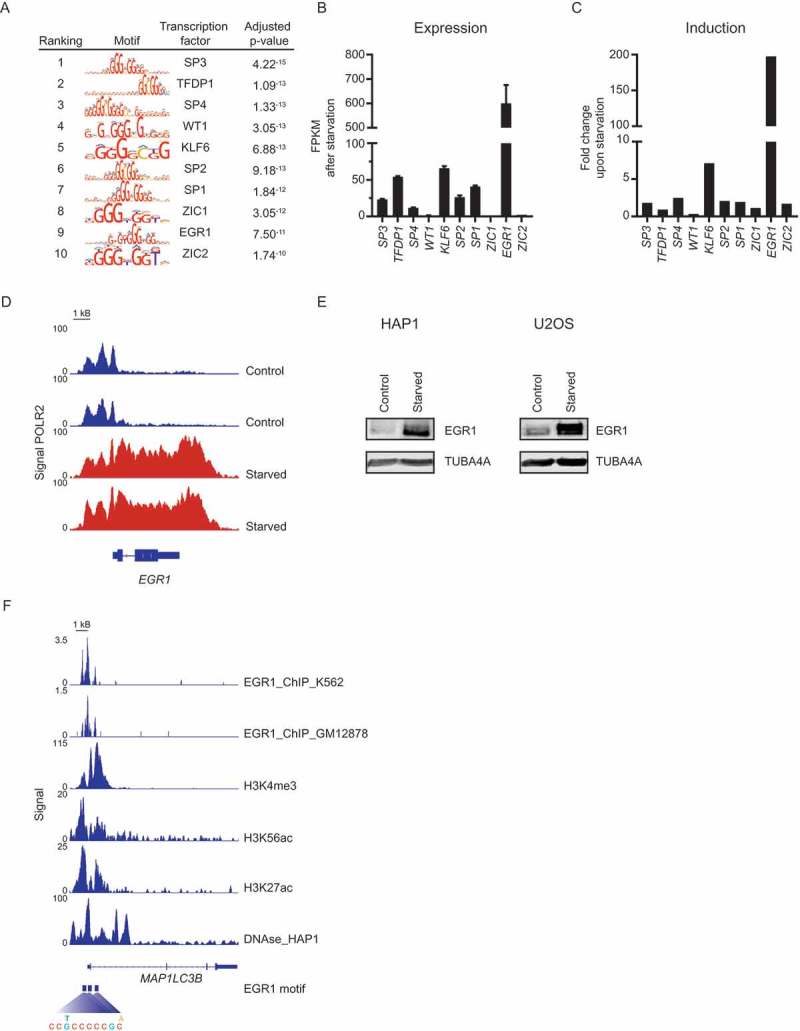
Epigenetic and transcriptomic analysis identifies EGR1 as a candidate transcriptional regulator of autophagy. (a) Top 10 transcription factor binding motifs enriched in autophagy-associated genes. Log2 FPKM (b) and fold change (c) after starvation (6 h EBSS) of transcription factors with binding motifs enriched in autophagy-associated genes. (d) Gene track for EGR1 displaying ChIP-seq signal for POLR2 with and without starvation (3 h EBSS). (e) EGR1 and TUBA4A/tubulin expression in HAP1 and U2OS cells upon starvation (6 h EBSS). Representative blots are shown (n = 3). (f) Gene track for MAP1LC3B displaying ChIP-seq signals for EGR1 (obtained from GEO GSE32465, samples GSM803414 and GSM803434), H3K4me3, H3K27ac, H3K56ac, DNAse I hypersensitivity sites, and EGR1 motif.
EGR1 acts as a transcriptional regulator of autophagy
To investigate whether EGR1 affects transcriptional regulation of autophagy, its expression levels were manipulated and autophagy-associated gene expression was analyzed. Upon nutrient deprivation, EGR1 knockdown resulted in a significant decrease in the transcription of the majority of autophagy-associated genes tested (Figure 6(a) and Figure S6A and S6B). Overexpression of EGR1 had a modest effect on the transcription of autophagy-associated genes (Figure 6(b) and Figure S6C and S6D). To substantiate these findings, we utilized CRISPR/Cas9-mediated EGR1-deficient HAP1 cells (EGR1 KO) and assessed autophagy-associated gene expression upon nutrient deprivation. In agreement with knockdown of EGR1, the expression of the majority of autophagy-associated genes was decreased in EGR1 KO cells compared to WT cells (Figure 6(c) and Figure S6E and S6F). Altogether, this demonstrates that EGR1 transcriptionally regulates autophagy-associated genes. To determine whether the decreased expression of autophagy-associated genes affects the autophagic flux, the LC3-II:LC3-I ratio was analyzed in EGR1 KO cells after 3 and 6 h of nutrient deprivation, in the presence or absence of bafilomycin A1. Indeed, autophagy was reduced in the absence of EGR1, as indicated by the decreased LC3-II:LC3-I ratio compared to WT cells (Figure 6(d)). In contrast, overexpression of EGR1 in either HAP1 cells or HEK293 cells resulted in an increase in the autophagic flux, observed after 3 as well as 6 h of EBSS treatment (Figure 6(e,f)). To validate the findings obtained with western blot, we transfected EGR1 KO cells with an mCherry-EGFP-LC3B construct and quantified the amount of autophagosomes (yellow) and autolysosomes (red) after 6 h of nutrient deprivation (Figure 6(g)). In agreement with the decrease in LC3-II:LC3-I observed with western blot, the ratio of red:yellow dots and the total amount of dots was decreased in EGR1 KO cells, suggesting a reduced autophagic flux compared to WT cells (Figure 6(h)). Altogether, nutrient deprivation induces EGR1 expression, which can subsequently induce autophagy through transcriptional control of numerous autophagy-associated genes, indicating that our datasets can indeed be utilized to identify (novel) regulators of autophagy.
Figure 6.
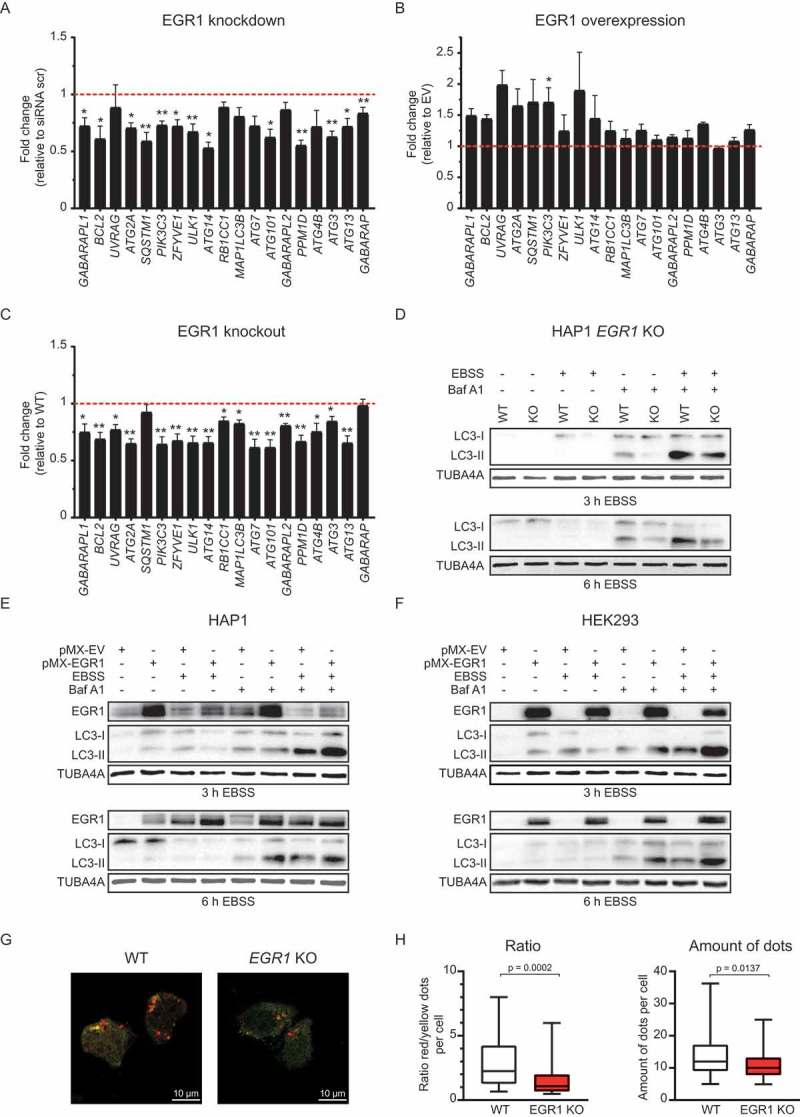
EGR1 acts as a transcriptional regulator of autophagy. (a) Expression of autophagy-associated genes in HAP1 cells with and without EGR1 knockdown starved for 6 h with EBSS. Fold change relative to cells transfected with scrambled siRNA is shown. Data are represented as mean ± SEM (n = 6–9). (b) Expression of autophagy-associated genes in HAP1 cells with and without EGR1 overexpression starved for 6 h with EBSS. Fold change relative to empty vector (EV)-transfected cells is shown. Data are represented as mean ± SEM (n = 3–6). (c) Expression of autophagy-associated genes in HAP1 EGR1 KO cells starved for 6 h with EBSS. Fold change relative to WT HAP1 starved for 6 h is shown. Data are represented as mean ± SEM (n = 6). (d) LC3 and TUBA4A expression in WT and EGR1 KO HAP1 cells in control and starved (3 h and 6 h EBSS) condition, treated with bafilomycin A1 (40 nM). Representative blot is shown (3 h: n = 4; 6 h: n = 6). (e) EGR1, LC3, and TUBA4A expression in WT HAP1 cells with and without EGR1 overexpression in control and starved (3 h and 6 h EBSS) condition, treated with bafilomycin A1 (40 nM). Representative blot is shown (3 h: n = 3; 6 h: n = 3). (f) LC3, EGR1, and TUBA4A expression in HEK293 cells with and without EGR1 overexpression in control and starved (3 h and 6 h EBSS) condition, treated with bafilomycin A1 (40 nM). Representative blot is shown (3 h: n = 3; 6 h: n = 6). (g) Representative images of HAP1 WT and EGR1 KO cells transfected with a plasmid encoding mCherry-EGFP-LC3B in starved (6 h EBSS) condition. mCherry+ EGFP+ dots (yellow) are autophagosomes and mCherry+ dots (red) are autolysosomes. (h) Boxplots with 5%-95% whiskers displaying the ratio red vs. yellow dots per cell and the total amount of dots per cell (75 cells were counted within 2 independent experiments). * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Discussion
Here, we generated an extensive transcriptomic and epigenomic database of human cells undergoing autophagy upon nutrient deprivation. We observed that nutrient deprivation induces an increase in expression of multiple autophagy-associated genes. This is in agreement with other studies, which analyzed the expression of a subset of proteins involved in autophagy under different starvation conditions, such as serum and amino acid deprivation [28] or glucose starvation [16]. Furthermore, we demonstrated that the expression of autophagy-associated genes was accompanied by an increase in POLR2 signal for these genes, validating that increased transcription contributes to the increased expression. We observed that nutrient deprivation had a similar effect on the transcriptome of ATG7 and RB1CC1 knockout cells compared to WT cells, including increased expression of autophagy-associated gene expression. This demonstrated that the transcriptional changes observed upon autophagy induction are not dependent on autophagic flux, but are rather the direct result of sensing nutrient deprivation [29]. This also indicates that within 6 h there is either limited feedback by autophagy itself on the transcriptional level, or that feedback still takes place in the autophagy-deficient cell lines, suggesting that autophagic flux is not itself necessary for feedback.
In atg5−/- mouse embryonic fibroblasts (MEFs), in contrast to WT MEFs, autophagy induction via rapamycin treatment does not lead to a decrease in H4K16ac, which is associated with downregulation of autophagy-associated genes and considered to be a feedback mechanisms [17]. This suggest that feedback at the transcriptional levels starts to become relevant after 6 h, or that these feedback loops are indirect, or that this is caused by differences between mice and men. Indeed, analysis of certain key autophagy genes in zebrafish has demonstrated that the increase in gene expression that is observed after 12 h of amino acid and serum starvation is absent after 24 h [28]. Additionally, our results indicate that the expression of autophagy-associated genes is not a measure of autophagic flux, because increased gene expression can still be observed in the absence of ATG7 and RB1CC1, when no autophagic flux is present. In contrast to other reports, we did not observe an effect of autophagy induction on global H4K16ac, H3K4me3, and H3K56ac levels. This discrepancy could be due to different methods to induce autophagy, for example nutrient deprivation versus MTOR inhibition, differences in timing, and differences in the type or species of the employed cells. For example, rapamycin-induced downregulation of H3K4me3 in MEFs is not observed after glucose starvation [16,17].
As a proof of principle we used our transcriptional and epigenetic datasets and identified EGR1 as a potential transcriptional regulator of autophagy, because the EGR1 binding site is enriched within open chromatin regions of autophagy-associated genes, and EGR1 expression increases dramatically upon nutrient deprivation. EGR1 is an immediate-early response gene, of which its expression can be induced within minutes after stimulation [30]. Mitogens [31,32], growth factors [33], and stress stimuli, such as cigarette smoke [34–36], hypoxia [37,38], and nutrient deprivation [39] regulate EGR1. For example, in agreement with our data, glucose restriction rapidly increases EGR1 protein levels in multiple cell lines [39]. The transcription factor EGR1 has been implicated in numerous processes, for example apoptosis, angiogenesis, proliferation, cell differentiation, and migration [40,41]. EGR1 has been linked to cigarette smoke, hypoxia, and irradiation-induced autophagy, by induction of ATG4B [32] and LC3B protein or gene expression [35,38]. Additionally, egr1−/- mice are more resistant to the pro-autophagic effects of chronic cigarette smoke exposure [35]. However, there are also indications that EGR1 might act as a negative regulator of autophagy, either by affecting ATG12–ATG5 conjugation with ATG16L1 [42] or by transcriptionally regulating the miRNA MIR152, which inhibits ATG14 and thereby decreases autophagy [43]. Our results are in line with a transcriptional activating role for EGR1 in autophagy and demonstrate that its transcriptional activity does not solely apply to ATG4B and MAP1LC3B, but to numerous autophagy-associated genes. SQSTM1 and GABARAP (GABA type A receptor-associated protein) expression was not significantly affected in EGR1 KO cells, whereas expression was decreased upon EGR1 knockdown. This discrepancy could be caused by adaptation of EGR1 KO cells to the long-term absence of EGR1, as knockdown of EGR1 is transient. The modest effect of EGR1 overexpression on autophagy-associated gene expression compared to EGR1 knockdown or knockout could be caused by the starvation conditions under which these experiments were performed. Upon starvation, EGR1 expression is already high, therefore a knockdown/knockout approach is more likely to have a more pronounced effect on the expression of EGR1 and autophagy-associated genes. Overall, the fact that EGR1 was unbiasedly identified as a transcriptional regulator of autophagy in our transcriptomic and epigenetic analyses, indicates that our data can facilitate the identification of additional transcription factors involved in the regulation of autophagy.
Various signaling pathways have been reported to be involved in the transcriptional regulation of EGR1 [44]. Reactive oxygen species (ROS), which can be induced by EBSS treatment, are a known inducer of autophagy [45]. Additionally, ROS production has been demonstrated to induce EGR1 expression in a MAPK/JNK- and MAPK/ERK-dependent manner [46]. Together, this suggests that nutrient deprivation may induce EGR1 expression through MAPK/JNKs and MAPK/ERKs. AMP-activated protein kinase (AMPK), a key energy sensor regulating cellular energy homeostasis, can induce autophagy through inactivation of MTOR complex 1 [47] and phosphorylation of ULK1 (unc-51 like autophagy activating kinase 1), a rapid and cytoplasmic process [48]. Recently, also a nuclear role for AMPK has also been described; upon prolonged glucose starvation, nuclear AMPK expression and activation is increased, leading to initiation of the FOXO3-SKP2 (S-phase kinase associated protein 2)-CARM1 axis, which can transcriptionally regulate specific autophagy-associated genes [16].AMPK activation has been demonstrated to induce EGR1 protein expression within 30 min [49,50]. These data indicate that the signaling pathway regulating the role of EGR1 in autophagy might involve AMPK. Because nuclear AMPK expression was only observed upon prolonged glucose starvation and the increase of EGR1 after starvation is rapid, it remains to be investigated whether nuclear AMPK is involved in the initial EGR1 induction or whether it is more important for maintaining EGR1 expression upon starvation.
In conclusion, our global transcriptomic and epigenomic profiling has demonstrated that nutrient deprivation regulates the transcriptional induction of autophagy-associated genes. This increase in autophagy-associated gene expression is accompanied by changes in chromatin remodeling and is not regulated by the autophagic flux. Furthermore, as a proof of principle, our data identified EGR1 as a transcriptional regulator of serum and amino acid starvation-induced autophagy. Taken together, these data increase our understanding of the molecular pathways regulating autophagy and can be used as a resource to identify (novel) factors involved in autophagy regulation. Because autophagy has been implicated in numerous diseases, a better understanding of the molecular pathways and transcription factors regulating autophagy might lead to the development of novel strategies aimed at restoring autophagy levels in the context of disease, for example therapies targeting EGR1 expression [51].
Materials and methods
Cell culture
HAP1 WT (C631), ATG7 KO (HZGHC000302c022), RB1CC1 KO (HZGHC000567c007), and EGR1 KO (HZGHC1958) cells were obtained from Horizon Genomics and cultured in Iscove’s Modified Dulbecco’s Medium (Gibco, 21,980,032; IMDM). U2OS (HTB-96) and HEK293 cells (CRL-1573) were obtained from ATCC and were cultured in Dulbecco’s Modified Eagle Medium (Gibco, 31966021l; DMEM). Both IMDM and DMEM were supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin (Gibco, 15,070–063) and 10% heat-inactivated fetal calf serum (Sigma-Aldrich, F7524) and all cells were cultured at 37°C in 5% CO2. For nutrient deprivation, cells were cultured with Earle’s Balanced Salt Solution (Sigma-Aldrich, E288; EBSS). For overexpression, cells were transfected with 2 µg DNA using polyethylenimine (Polysciences, 23,966–1). After 18 h, cells were washed with PBS (Sigma-Aldrich, D8537) and cultured for 24 h. For knockdown, cells were transfected with 25 pmol siRNA using Lipofectamine RNAiMAX transfection reagent (Invitrogen, 13,778,150). After 18 h, cells were washed with PBS and cultured for 6 h.
Antibodies and reagents
The following antibodies were used: mouse anti-MAP1LC3B (Nanotools, 0231–100/LC3-5F10), rabbit anti-ATG7 (Cell Signaling Technology, 2631S), rabbit anti-RB1CC1/FIP200 (ITK diagnostics, A301-536A), rabbit anti-EGR1 (Cell Signaling Technology, 4154S), mouse anti-RPB1 (Euromedex; PB-7C2), rabbit anti-histone H3 acetyl K27 (Abcam, ab4729), rabbit anti-histone H3 acetyl K56 (Active Motif, 39,281), rabbit anti-histone H3 trimethyl K4 (Active Motif, 39,159), mouse anti-histone H3 (Active Motif, 39,763), mouse anti-TUBA4A/tubulin (Sigma-Aldrich, T9026). pMXs-hs-EGR1 was a gift from Shinya Yamanaka (Addgene, 52,724) [52]. For EGR1 knockdown, human SMARTpool EGR1 siRNA (Dharmacon, M-006526–01-0005) was used. pBABE-puro-mCherry-EFGP-LC3B was a gift from Jayanta Debnath (Addgene, 22,418) [53]. To increase the intensity of the fluorescence, the EEF1A1/EF1α promoter was cloned into the construct using NaeI restriction sites. bafilomycin A1 was obtained from Sigma-Aldrich (B1793). Hydroxychloroquine (HCQ) was obtained from Acros Organics (263,010,250).
Western blot
Western blot was performed as described previously [54]. In short, cells were lysed in Laemmli buffer (0.12 M Tris–HCl, pH 6.8, 4% SDS, 20% glycerol, 0.05 µg/µl bromophenol blue, 35 mM β-mercaptoethanol). Samples were separated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane (Merck, IPFL00010), probed with the indicated antibodies and analyzed using enhanced chemiluminescence (Thermo Fisher Scientific, 34,075) or an Odyssey Sa Infrared Imaging System (LI-COR, Lincoln, NE, USA).
Electron microscopy
Cells were fixed with 50% karnovsky fixative (2.5% glutaraldehyde [Merck, 104,239], 2% paraformaldehyde [Sigma-Aldrich, 441,244], 0.1 M phosphate buffer, pH 7.4 (0.019 M NaH2PO4.1H20, 0.081 M Na2HPO4.2H2O), 0.25 mM CaCl2, 0.5 mM MgCl2) by adding equal amount of fixative to the medium and incubating for 10 min. Then, fixative was replaced for fresh 50% karnovsky fixative and incubated for 2 h at room temperature. Cells were washed 3 × 10 min with 0.1 M phosphate buffer, scraped and pelleted. Pellets were resuspended quickly in 2% low melting point agarose (Sigma-Aldrich, A9414-25G) at 63°C and immediately pelleted again. Pellets were incubated on ice for 30 min and cut into blocks, after which blocks were incubated in postfix solution (0.1 M phosphate buffer, pH 7.4, 1% OsO4 [Electron Microscopy Science, 19,110], 1.5% K3[Fe{CN}6] [Merck, 104,984]) for 2 h, 4°C. Samples were washed 3x with distilled water and incubated in 0.5% uranyl acetate (Electron Microscopy Science, 041209AB) for 1 h, 4°C in the dark. Afterwards, samples were rinsed with distilled water and incubated in 70% acetone overnight. Dehydration was done with increasing amounts of pure acetone with a final step of 100% acetone (Merck, 1,002,991,001). Epon infiltration (glycid ether 100 [Serva, 21,045], 2-dodecenyl succinic anhydride [Serva 20,755], methylnadic anhydride [Serva, 29,452], N-benzyldimethylamine [Electron Microscopy Science, 11,400–25] was done with acetone-epon mixtures with increasing amounts of epon and a final step of 100% epon. After the last step of 100% epon, fresh epon was added and polymerized for 3 days at 60°C. Cutting of ultra-thin sections was done on a Leica UCT/FCS (Leica, Wetzlar, Germany). Staining was done with a Leica AC20 (Leica, Wetzlar, Germany) with 45 min 0.5% uranyl acetate (Laurylab, 705,631,095) at 20°C and 5 min Reynolds lead citrate (Leica, D151214) at 20°C. The samples were examined with a Jeol101 electron microscope (Jeol Europe, Nieuw-Vennep, The Netherlands).
Confocal microscopy
Autophagy was analyzed using the pBABE-puro-Ef1alpha-mCherry-EFGP-LC3B construct (see section ‘antibodies and reagents’ how construct was created). Cells were seeded in 8-well µ-slides (Ibidi, 80,826) and cultured for 24 h, transfected with 0.2 µg DNA using polyethylenimine. After 18 h, cells were washed with PBS and cultured for 24 h. Next, cells were cultured for 6 h in either full IMDM or EBSS for 6 h, and 40 µM bafilomycin A1 was added after 5.5 h. Cells were washed twice with PBS, fixed with 1% formaldehyde (Merck Millipore, 104,003) and visualized with a Zeiss LSM 710 microscope (Carl Zeiss, Oberkochen, Germany) using the 63x objective.
Apoptosis measurements
Apoptosis was analyzed using the Annexin V Apoptosis Detection Kit (BD Biosciences, 556,547) according to the manufacturer’s protocol. Living cells were defined as ANXA5− 7-AAD−, early apoptotic cells as ANXA5+ 7-AAD−, and late apoptotic cells as ANXA5+ 7-AAD+.
Quantitative RT-PCR
Total RNA was extracted using the RNeasy kit (Qiagen, 74,106) and cDNA synthesis was performed using the iScript cDNA synthesis kit (Bio-Rad, 1,708,891). cDNA samples were amplified with SYBR Select mastermix (Life Technologies, 4,472,919) in a QuantStudio 12k flex (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. A list of primers used in this study can be found in Table S2.
RNA-sequencing and analysis
Cells were cultured till 80% confluence in 6w plates (Thermo Fisher Scientific, 140,675) and cultured for 6 h in full IMDM or EBSS. Total RNA was extracted from 3 independent biological replicates using the RNeasy kit. mRNA was isolated using NEXTflex®Poly(A) Beads (Bio Scientific, NOVA-512,980), libraries were prepared using the NEXTflex®Rapid Directional RNA-Seq Kit (Bio Scientific, NOVA-513,808) and samples were sequenced 75 base pair single-end on Illumina NextSeq500 (Illumina Inc., San Diego, CA, USA; Utrecht DNA Sequencing Facility). Reads were aligned to the human reference genome GRCh37 using STAR version 2.4.2a. Picard’s AddOrReplaceReadGroups (v1.98) was used to add read groups to the BAM files, which were sorted with Sambamba v0.4.5 and transcript abundances were quantified with HTSeq-count version 0.6.1p1 using the union mode. Subsequently, reads per kilobase million sequenced (RPKMs) were calculated with edgeR’s RPKM function. Differentially expressed genes were identified using the DESeq2 package with standard settings. Genes with absolute padj< 0.05 were considered as differentially expressed.
ChIP-sequencing and analysis
Cells were cultured until 80% confluence in 15-cm dishes (Corning, 430,599) and cultured for 3 h in full IMDM or EBSS. Cells from 2 independent biological replicates were crosslinked in 1% formaldehyde, 5 mM HEPES-KOH, pH 7.5, 10 mM NaCl, 0.1 mM EDTA, 50 µM EGTA and after 10 min crosslinking was stopped by adding 0.1 M glycine. Nuclei were isolated in 50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40 (Sigma-Aldrich, 56,741), 1% Triton X-100 (Sigma-Aldrich, T8787) and lysed in 20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.3% SDS. Lysates were resuspended in 20 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 and sonicated using Covaris (Covaris, Woburn, MA, USA) for 8 min, maximum output. Sheared DNA was incubated overnight with the indicated antibodies pre-coupled to protein A/G magnetic beads (Pierce, 88,802). Cells were washed and crosslinking was reversed by adding 1% SDS, 100 mM NaHCO3, 200 mM NaCl, 300 µg/ml proteinase K (Invitrogen, 25,530–015). DNA was purified using ChIP DNA Clean & Concentrator kit (Zymo Research, D5205), and end repair, A-tailing, and ligation of sequence adaptors was done using Truseq nano DNA sample preparation kit (Illumina, 20,015,965). Samples were PCR amplified, and barcoded libraries were sequenced 75 base pair single-end on Illumina NextSeq500. Peaks were called using Cisgenome 2.0 [55] (–e 150 -maxgap 200 – minlen 200). Peak coordinates were stretched to at least 2000 base pairs and collapsed into a single list. Overlapping peaks were merged based on their outermost coordinates. Only peaks identified by at least 2 independent datasets were further analyzed. Peaks with differential H3K27ac, H3K56ac or H3K4me3 occupancy were identified using DESeq (padj< 0.05) [56].
Motif enrichment analysis
H3K27ac, H3K56ac, and H3K4me3 peaks associated with autophagy-associated genes with log2FoldChange ≥ 0.5 upon starvation were overlapped with DNAse hypersensitivity sites, based on online DNAse-seq data in HAP1 cells (GEO GSE90371 [57]). The overlapping peaks were analyzed for motif enrichment using the AME tool of MEME Suite [58].
Gene set enrichment analysis (GSEA)
GSEA was performed using autophagy-associated genes that were identified via the human autophagy database (available at http://autophagy.lu/and see Table S1) [59]. Significance of the enrichment was calculated based on 1000 cycles of permutations.
Statistical analysis
For ChIP-seq and RNA-seq analysis, p-values were adjusted with the Benjamini-Hochberg procedure and a false discovery rate (FDR) ≤ 0.05 was considered significant. Cell viability was analyzed using two-way ANOVA with Sidak correction for multiple testing. Correlation between ChIP-seq and RNA-seq data and EGR1 induction by serum and/or nutrient deprivation was determined using an ordinary one-way ANOVA with Dunnett’s post-test. Starvation-induced changes of autophagy-associated genes, the POLR2 signal for key autophagy genes, and EGR1 knockdown, knockout, and overexpression were analyzed using Wilcoxon-matched pairs singed rank test (paired samples) or the Mann-Whitney U test (unpaired samples). Autophagic flux measurement using mCherry-EGFP-LC3B was analyzed using an unpaired t test with Welch’s correction. All analyses were performed using GraphPad Prism (GraphPad Software).
Data availability
The RNA-sequencing and ChIP-sequencing data from this publication have been deposited in the NCBI GEO database and together assigned the identifier GSE107603 (RNA-sequencing: GSE107600; ChIP-sequencing: GSE107599).
Funding Statement
This work was supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek [022.004.018];Nederlandse Organisatie voor Wetenschappelijk Onderzoek [91615113];Wilhelmina Children’s Hospital;Marie Skłodowska-Curie Cofund [713660];Marie Skłodowska-Curie ITN [765912];Reumafonds [14-3-201];ZonMw [016.130.606].
Abbreviations
- ATG12
autophagy related 12
- ATG14
autophagy related 14
- ATG16
autophagy related 16
- ATG4B
autophagy related 4B cysteine peptidase
- ATG5
autophagy related 5
- ATG7
autophagy related 7
- BECN1
beclin 1
- BNIP3
BCL2 interacting protein 3
- CARM1
coactivator associated arginine methyltransferase 1
- ChIP
chromatin immunoprecipitation
- EBSS
Earle’s balanced salt solution
- EGR1
early growth response 1
- EHMT2/G9a
euchromatic histone lysine methyltransferase 2
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- FOXO
forkhead box O
- KAT8/hMOF
lysine acetyltransferase 8
- MAP1LC3B
microtubule associated protein 1 light chain 3 beta
- MEF
mouse embryonic fibroblast
- MTOR
mechanistic target of rapamycin kinase
- POLR2/Pol II RNA
polymerase Il
- RB1CC1
RB1 inducible coiled-coil 1
- ROS
reactive oxygen species
- SKP2
S-phase kinase associated protein 2
- SQSTM1
sequestosome 1
- TFEB
transcription factor EB
- ULK1
unc-51 like autophagy activating kinase 1
- USP44
ubiquitin specific peptidase 44
- WT
wild type
- ZKSCAN3
zinc finger protein with KRAB and SCAN domains 3
Acknowledgments
We thank the Utrecht Sequencing Facility.
Disclosure statement
No potential conflict of interest was reported by the authors.
Conflict of interest
The authors declare no conflict of interest.
Financial disclosure
The authors declare no financial interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Klionsky DJ, Emr SD.. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N. Autophagy in Protein and Organelle Turnover. Cold Spring Harb Symp Quant Biol. 2011;76:397–402. [DOI] [PubMed] [Google Scholar]
- [4].Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2013;15:65–74. [DOI] [PubMed] [Google Scholar]
- [7].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science (80-.). 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van der Vos KE, Gomez-Puerto C, Coffer PJ. Regulation of autophagy by Forkhead box (FOX) O transcription factors. Adv Biol Regul. 2012;52:122–136. [DOI] [PubMed] [Google Scholar]
- [9].Chauhan S, Goodwin JG, Chauhan S, et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell. 2013;50:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shaw J, Yurkova N, Zhang T, et al. Antagonism of E2F-1 regulated Bnip3 transcription by NF- B is essential for basal cell survival. Proc Natl Acad Sci. 2008;105:20734–20739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Copetti T, Bertoli C, Dalla E, et al. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29:2594–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ling J, Kang Y, Zhao R, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tamatani M, Che YH, Matsuzaki H, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. [DOI] [PubMed] [Google Scholar]
- [14].Artal-Martinez de Narvajas A, Gomez TS, Zhang J-S, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol. 2013;33:3983–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wei F-Z, Cao Z, Wang X, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. 2015;11:2309–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shin H-JR, Kim H, Oh S, et al. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Füllgrabe J, Lynch-Day MA, Heldring N, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen S, Jing Y, Kang X, et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res. 2016;45:gkw1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kourmouli N, Jeppesen P, Mahadevhaiah S, et al. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J Cell Sci. 2004;117:2491–2501. [DOI] [PubMed] [Google Scholar]
- [20].Chen H, Fan M, Pfeffer LM, et al. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012;40:6534–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Earle WR, Schilling EL, Stark TH, et al. Production of malignancy in vitro. IV. The mouse fibroblast cultures and changes seen in the living cells. JNCI J Natl Cancer Inst. 1943;4:165–212. [Google Scholar]
- [22].Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann–pick C1. Nature. 2011;477:340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. [DOI] [PubMed] [Google Scholar]
- [25].Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci. 2008;105:9000–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mews P, Zee BM, Liu S, et al. Histone methylation has dynamics distinct from those of histone acetylation in cell cycle reentry from quiescence. Mol Cell Biol. 2014;34:3968–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Biga PR, Latimer MN, Froehlich JM, et al. Distribution of H3K27me3, H3K9me3, and H3K4me3 along autophagy-related genes highly expressed in starved zebrafish myotubes. Biol Open. 2017;6:1720–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Karim MR, Kawanago H, Kadowaki M. A quick signal of starvation induced autophagy: transcription versus post-translational modification of LC3. Anal Biochem. 2014;465:28–34. [DOI] [PubMed] [Google Scholar]
- [30].Milbrandt J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science. 1987;238:797–799. [DOI] [PubMed] [Google Scholar]
- [31].Zhang F, Lin M, Abidi P, et al. Specific interaction of Egr1 and c/EBPβ Leads to the Transcriptional Activation of the human low density lipoprotein receptor gene. J Biol Chem. 2003;278:44246–44254. [DOI] [PubMed] [Google Scholar]
- [32].Peng W, Wan Y, Gong A, et al. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis. 2017;6:e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu J, Grogan L, Nau MM, et al. Physical interaction between p53 and primary response gene Egr-1. Int J Oncol. 2001;18:863–870. [DOI] [PubMed] [Google Scholar]
- [34].Wang S-B, Zhang C, Xu X-C, et al. Early growth response factor 1 is essential for cigarette smoke-induced MUC5AC expression in human bronchial epithelial cells. Biochem Biophys Res Commun. 2017;490:147–154. [DOI] [PubMed] [Google Scholar]
- [35].Chen Z-H, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Hotchin N Editor PLoS One. 2008;3:e3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Reynolds PR, Cosio MG, Hoidal JR. Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am J Respir Cell Mol Biol. 2006;35:314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peng W, Xiong E, Ge L, et al. Egr-1 promotes hypoxia-induced autophagy to enhance chemo-resistance of hepatocellular carcinoma cells. Exp Cell Res. 2016;340:62–70. [DOI] [PubMed] [Google Scholar]
- [38].Lee S-J, Smith A, Guo L, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Di Biase S, Shim HS, Kim KH, et al. Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 2017;15:e2001951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gitenay D, Baron VT. Is EGR1 a potential target for prostate cancer therapy? Future Oncol. 2009;5:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Silverman ES, Collins T. Pathways of Egr-1-mediated gene transcription in vascular biology. Am J Pathol. 1999;154:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao K, Yu M, Zhu Y, et al. EGR-1/ASPP1 inter-regulatory loop promotes apoptosis by inhibiting cyto-protective autophagy. Cell Death Dis. 2017;8:e2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].He J, Yu -J-J, Xu Q, et al. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015;11:373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pagel J-I, Deindl E. Early growth response 1–a transcription factor in the crossfire of signal transduction cascades. Indian J Biochem Biophys. 2011;48:226–235. [PubMed] [Google Scholar]
- [45].Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aggeli IKS, Beis I, Gaitanaki C. ERKs and JNKs mediate hydrogen peroxide-induced Egr-1 expression and nuclear accumulation in H9c2 cells. Physiol Res. 2010;59:443–454. [DOI] [PubMed] [Google Scholar]
- [47].Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. [DOI] [PubMed] [Google Scholar]
- [48].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Berasi SP, Huard C, Li D, et al. Inhibition of gluconeogenesis through transcriptional activation of EGR1 and DUSP4 by AMP-activated kinase. J Biol Chem. 2006;281:27167–27177. [DOI] [PubMed] [Google Scholar]
- [50].Wu Y-F, Wang H-K, Chang H-W, et al. High glucose alters tendon homeostasis through downregulation of the AMPK/Egr1 pathway. Sci Rep. 2017;7:44199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Worringer KA, Rand TA, Hayashi Y, et al. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell. 2014;14:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].E-N N, Kajihara KK, Hsieh I, et al. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Peeters JGC, de Graeff N, Lotz M, et al. Increased autophagy contributes to the inflammatory phenotype of juvenile idiopathic arthritis synovial fluid T cells. Rheumatology. 2017;56:1694–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ji H, Jiang H, Ma W, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McLeay RC, Bailey TL. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics. 2010;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing and ChIP-sequencing data from this publication have been deposited in the NCBI GEO database and together assigned the identifier GSE107603 (RNA-sequencing: GSE107600; ChIP-sequencing: GSE107599).