

Abstract
Transcription initiation by RNA polymerase II is a complex, multistep process that minimally involves transcription complex assembly, open complex formation, and promoter clearance. Hydrolysis of the β–γ phosphoanhydride bond of ATP has previously been shown to be required for open complex formation, as well as for the phosphorylation of the carboxy-terminal domain of the largest subunit of RNA polymerase II. The observation that ATP-dependent activation of transcription complexes can be blocked by ATP analogues that contain nonhydrolyzable β–γ phosphoanhydride bonds (such as ATPγS) was exploited to develop a functional kinetic assay for ATP-activated transcription complexes. Activated complexes on the promoter present in the long terminal repeat of the proviral DNA of mouse mammary tumor virus were defined as those that could productively initiate transcription in the presence of excess ATPγS. Activation is dependent on treatment of assembled preinitiation complexes with ATP (or dATP) prior to addition of ATPγS, At least 15–35% of the total number of preinitiation complexes present become activated within 2 min in the presence of (d)ATP, and activation appears to be rapidly reversible. The time course of formation of activated complexes in the presence of dATP is characterized by two kinetic phases: a rapid formation followed by a relatively slow decay. Activated complexes were estimated to form with a half-time of less than 1 min.
Keywords: ATP-mediated activation, RNA polymerase II, Mouse mammary tumor virus, Open transcription complexes, Transcription initiation
TRANSCRIPTION initiation by RNA polymerases can involve multiple steps, including preinitiation complex formation, promoter opening, formation of an initial transcription bubble, abortive initiation, productive initiation, and promoter clearance (20,21, 34,41). RNA polymerase II (pol II) requires a complex array of proteins for specific transcription initiation from even the simplest pol II promoter [for a review, see (42)]. These proteins have been termed general transcription factors, and include transcription factors (TF) IIA, IIB, IID, HE, IIF, and IIH. Assembly of the preinitiation complex can be achieved in an ordered stepwise process (2,5), or, alternatively, a preassembled pol II-general transcription factor-coactivator complex (holoenzyme), which contains most or all of the general transcription factors necessary for initiation, can be recruited onto the promoter (25,26,32).
Transcription initiation at pol II promoters requires the hydrolysis of the β—γ phosphoanhydride bond of ATP (4,12,20,21,35,38,41). Open complex formation, a step that involves isomerization of the closed complex to generate a region of melted template near the transcription initiation site, is thought to be mediated by the ATP-dependent helicase activity of TFIIH (9,10,12,17,36). Several lines of evidence indicate that the hydrolysis of ATP is also required in at least one additional step thought to correspond to phosphorylation of the carboxy-terminal domain (CTD) of the largest subunit of pol II by a protein kinase activity of TFIIH (13,21,27,28,37) or another protein kinase such as P-TEFb (30). Phosphorylation of the CTD (or even the presence of the CTD) is not necessarily required for productive transcription initiation in vitro (22); however, phosphorylation appears to limit promoter-proximal pausing of pol II (6,21,30,33,39). Consistent with the (d)ATP requirement for open complex formation, pol II lacking a CTD retains a requirement for (d)ATP hydrolysis for transcription in vitro (27).
Using HeLa cell nuclear extracts, Gralla and co-workers have demonstrated that open complexes can be directly detected by potassium permanganate foot-printing, that the detection of open complexes requires the hydrolysis of ATP, and that, for the promoters that have been examined, opening occurs rapidly (within 2 min) after addition of ATP to preformed (closed) transcription complexes (19–21,41). Although this assay can be used to directly detect open complexes, it is not well suited for kinetic studies because the time required for permanganate modification of single-stranded DNA in the open region is significant when compared to the time required for the open complex to form. Analogous to prokaryotic systems (31), it seems plausible that the transition from a closed to an open complex is a regulated event on at least some pol II promoters. Indeed, transcriptional regulators have been shown to increase the number of open transcription complexes that can be detected in vitro (41).
In this report, we have developed a kinetic assay that allows the rate of ATP-dependent activation of pol II transcription complexes to be assessed. We have used this assay to follow activation on the proviral promoter present in the long terminal repeat of the retrovirus mouse mammary tumor virus (MMTV). The MMTV promoter, unlike many pol II promoters that have been examined in mechanistic detail, is relatively weak in the absence of activation by hormone-activated steroid receptor proteins [for review see (14)]. Our assay provides a means to compare the rates of ATP-dependent activation for pol II promoters that mediate different levels of transcriptional activity.
MATERIALS AND METHODS
Materials
HPLC-purified ribonucleoside 5′-triphosphates were purchased from Sigma. Elution profiles demonstrate that the nucleotides are 98% pure and show no cross-contamination of other nucleotides with ATP. Sarkosyl, tRNA, DTT, dATP, and diethylpyrocarbonate (DEPC) were also from Sigma. ATPγS and proteinase K were from Boehringer Mannheim, [α-32P]CTP (3000 Ci/mmol) was obtained from New England Nuclear. Autoradiographic film and intensifying screens were from DuPont.
Transcription Template
Plasmid pMBPT3 (24) consists of wild-type MMTV promoter sequences from −109 to +14, relative to the transcription start site at +1, inserted upstream of a 151 base pair T-free cassette. Transcription of this template generates a 172 nucleotide U-free transcript in the absence of added UTP.
Template DNA was prepared from large-scale plasmid preparations (1 1) by alkaline extraction (1) and purified by CsCl density gradient centrifugation followed by gel filtration chromatography as described (C. M. Bral, J. W. Steinke, C. K. Kang, and D. O. Peterson, submitted).
In Vitro Transcription Reactions
HeLa cell nuclear extracts were prepared according to the method of Dignam (8) with slight modifications (C. M. Bral, J. W. Steinke, C. K. Kang, and D. O. Peterson, submitted). All solutions except nuclear extract were prepared using DEPC-treated distilled water. Transcription reactions were performed as previously described [(23,24); C. M. Bral, J. W. Steinke, C. K. Kang, and D. O. Peterson, submitted] with modifications stated below. Nuclear extract (60 μg of protein) was incubated on ice for 15 min in TM0.1 buffer [50 mM Tris-HCl (pH 7.9 at room temperature), 1 mM EDTA, 12.5 mM MgCl2, 20% glycerol, 100 mM KC1, and 4 mM DTT]. This treatment with DTT has been shown to prevent loss of activity of nuclear extract during storage at −70°C (data not shown). Preinitiation complexes were assembled on the promoter-containing DNA template by incubating the pretreated nuclear extract with 500 ng of template DNA in a final volume of 25 μl. Assembly was carried out at 30°C for 120 min. RNA synthesis was initiated by the addition of a solution containing nucleotides and Sarkosyl such that the final volume of the reaction was 35 μl and the final concentration of reagents was 150 μM ATP, 150 μM GTP, 50 μM (10 μCi) [α-32P]CTP, and 0.02% Sarkosyl. The reaction was allowed to proceed at 30°C for 10 min, a time determined to be sufficient for full-length RNA synthesis. Reactions were terminated by the addition of stop solution (350 μl) containing 50 mM Tris-HCl (pH 7.5 at room temperature), 1% SDS, 5 mM EDTA, 25 μg/ml tRNA, 100 μg/ml proteinase K, and 3000–7000 cpm of a 32P-labeled recovery control RNA and incubated at 37°C for 15 min. Mixtures were then extracted with an equal volume of a 1:1 mixture of phenolxhloroform saturated with a solution containing 100 mM Tris-HCl (pH 7.5 at room temperature), 10 mM sodium acetate, 100 mM NaCl, and 1 mM EDTA. Precipitated RNA was fractionated by electrophoresis on 8% polyacrylamide gels in the presence of 8 M urea. Dried gels were exposed 15–48 h to Reflection film with an intensifying screen at −70°C. Gels were quantitated using a Fujix BAS 2000 Phosphorimager. Recovery control RNA was synthesized with SP6 RNA polymerase (C. M. Bral, J. W. Steinke, C. K. Kang, and D. O. Peterson, submitted) and used to normalize for MMTV RNA recovery among separate reactions. Data from replicate experiments were normalized to allow for direct comparison of derived values among experiments. All experiments were repeated using multiple nuclear extract preparations.
The correction applied to the number of activated transcription complexes in Fig. 4 was based on the first-order, (d)ATP-dependent decrease in total transcriptional activity observed in Fig. 5. The first-order rate constant (k) derived from a fit to the data in Fig. 5 using the computer program KaleidaGraph (Abel-beck Software) was used to calculate a corrected number of activated complexes at time t (A cor) according to the equation
FIG. 4.
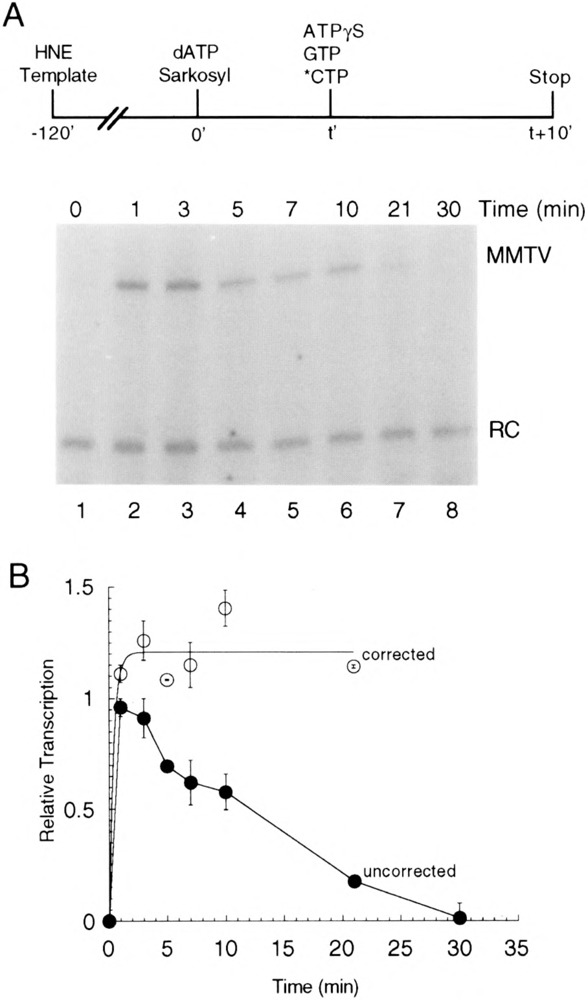
Time course of activated complex formation. (A) Preinitiation complexes were assembled on pMBPT3 template in HeLa nuclear extract (HNE) and then activated at time 0 by the addition of dATP (100 μM). Sarkosyl (0.02%) was added along with the dATP. At time I, activated complexes were allowed to transcribe by adding [α-32P]CTP, GTP, and 10-fold molar excess ATPγS (1 mM) and incubating for 10 min. (B) Quantitation of transcriptional activity from activated complexes (closed circles) and correction for (d)ATP-dependent decay (open circles) (see text for details of the correction). Quantitation was made relative to a recovery control (RC) RNA added with the stop solution. Points represent the average of at least three independent experiments, and error bars represent SEM.
FIG. 5.
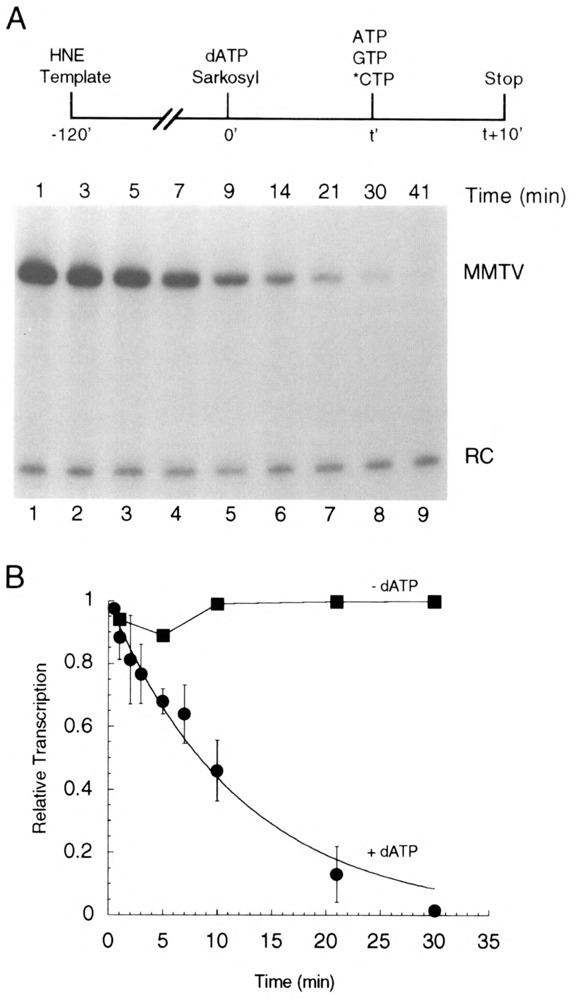
Stability of total transcription complexes. (A) Preinitiation complexes were assembled on pMBPT3 template in HeLa nuclear extract (HNE) and then activated at time 0 by the addition of dATP (20 μM). Sarkosyl (0.02%) was added along with the dATP. At time t, ATP, [α-32P]CTP, and GTP were added and the reactions incubated an additional 10 min, thus allowing all functional transcription complexes (both activated and unactivated) to generate RNA products. (B) Quantitation of transcriptional activity from total complexes treated with dATP as described in (A) (closed circles) or from complexes that were not treated with dATP (closed squares). Quantitation was made relative to a recovery control (RC) RNA added with the stop solution. Points represent the average of at least three independent experiments, and error bars represent SEM.
where A obs is the observed number of activated complexes from Fig. 4.
RESULTS
Experimental Strategy
A requirement for ATP hydrolysis in transcription initiation by pol II has been clearly established (35,41). ATP may be required in two discrete steps: melting of template DNA to form an open complex and CTD phosphorylation. Evidence suggests that these steps are mediated by the ATP-dependent helicase and protein kinase activities of TFIIH, respectively (12,17,21), but other CTD kinases, such as P-TEFb (30), may also be involved. Our strategy to develop an assay that would be useful for assessing the rate of ATP-dependent activation is based on the observation that ATP analogues that do not contain hydrolyzable β—γ phosphoanhydride bonds, such as ATPγS and AMP-PNP, inhibit ATP-dependent activation (4,11,12,19) but are capable of being used as elongation substrates by pol II (4,12,21). The basis of our assay is to assess the time course of the ATP-dependent transition from preinitiation complexes that are sensitive to inhibition by such analogues to activated complexes that have completed steps in initiation that require ATP hydrolysis and are thus resistant to analogue inhibition (Fig. 1A). At the time of addition of the nonhydrolyzable ATP analogue, other NTPs necessary for synthesis of a full-length RNA are also added, and the amount of transcript produced serves as a reporter for the number of activated complexes present at the time of analogue addition. It should be noted that after ATP-dependent activation is blocked, activated complexes have more than one potential fate. For example, in addition to productively initiating transcription, they could revert to closed preinitiation complexes (Fig. 1A). However, as long as a constant fraction of the activated complexes present at the time of nucleotide addition synthesizes a full-length transcript, the number of such transcripts will be proportional to (but not necessarily identical to) the number of activated complexes. Stated another way, our assay assumes that complexes that are equivalently activated have an equal probability of generating a full-length transcript.
FIG. 1.
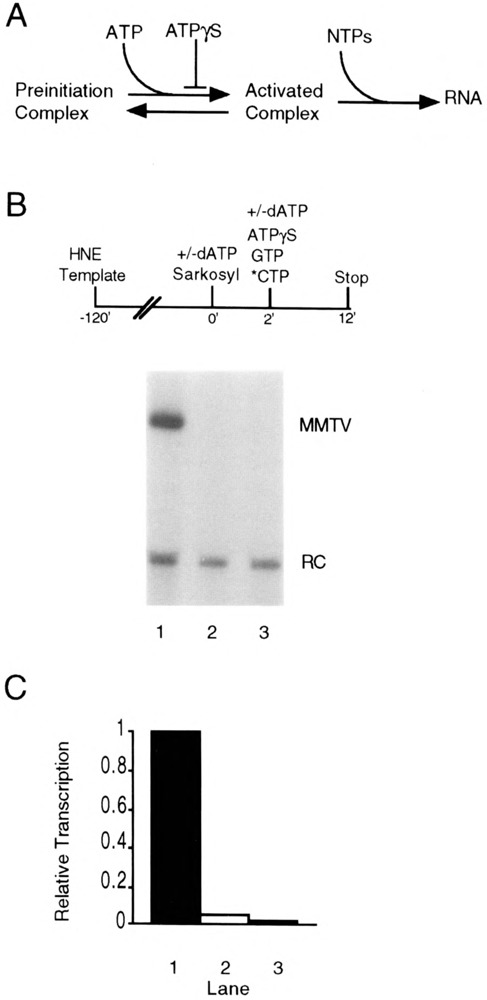
Detection of activated pol II transcription complexes in vitro. (A) ATP-dependent activation of pol II preinitiation complexes (see text for details). (B) Requirement for dATP pretreatment to generate activated complexes. Transcription complexes were assembled on the pMBPT3 template in HeLa nuclear extract (HNE) followed by the addition of dATP (20 μM) and Sarkosyl (0.02%) at time 0. The dATP allows activation of preassembled complexes, whereas Sarkosyl prevents the assembly of any additional complexes and limits transcription to a single round. After a 2-min dATP pretreatment, [α-32P]CTP, GTP, and ATPγS (200 μM) were added, and the incubations allowed to continue for an additional 10 min (lane 1). Control reactions received no dATP (lane 3) or received dATP at the same time as the other nucleotides (lane 2). (C) Quantitation of transcription activity from complexes that received no dATP (stippled bar), dATP at the same time as the other nucleotides (open bar), or a 2-min dATP pretreatment (closed bar). Quantitation was made relative to a recovery control (RC) RNA added with the stop solution.
Detection of Activated Complexes
In order to determine whether we could detect activated complexes by this strategy, we first tested whether an adenine nucleoside triphosphate containing a hydrolyzable β—γ phosphoanhydride bond was required for transcription from the MMTV promoter. Transcription complexes were allowed to assemble on an MMTV promoter-containing template in a reaction in which HeLa cell nuclear extract served as the source of transcription factors (Fig. 1B, top). In this template a portion of the MMTV long terminal repeat (−109 to +14 relative to the transcription initiation site) is linked to a synthetic T-free cassette, and a U-free RNA transcript of 172 nucleotides is synthesized in the absence of added UTP. At time 0, dATP (20 pM) and a low concentration of the anionic detergent Sarkosyl (0.02%) were added. We have shown that this concentration of Sarkosyl inhibits transcription complex assembly and prevents reinitiation in our system with the MMTV promoter [(23); and data not shown], similar to previous observations with other promoters (15,16,23,24). After 2 min in the presence of dATP, which has been shown in other studies to be a time sufficient for transition to an open complex (41), GTP, [α-32P]CTP, and a 10-fold molar excess of ATPyS (200 μM) were added. It has been demonstrated that ATPγS can function efficiently in chain elongation but cannot activate preinitiation complexes in a purified pol II transcription system prepared from rat liver (4,12,21). RNA synthesis was then allowed to proceed for 10 min, and full-length U-free transcripts were detected after fractionation of the transcription products by polyacrylamide gel electrophoresis (Fig. 1B, lane 1). When dATP was omitted (lane 3), or added at the same time as the elongation substrates (lane 2), essentially no detectable transcript was generated. Treatment of preinitiation complexes with dATP prior to addition of the NTPs required for RNA synthesis resulted in at least a 20-to 50-fold increase in transcription (Fig. 1C). Thus, as has been shown for other promoters (4,12,20,21,35,38,41), there is a nearly absolute requirement for (d)ATP-dependent activation in order to detect significant transcription from the MMTV promoter under these conditions.
We performed several additional controls to further validate this assay (Fig. 2). Transcription complexes were assembled on the MMTV promoter-containing template in HeLa nuclear extract. At time 0 Sarkosyl (0.02%) and various NTPs were added. Sarkosyl prevented further transcription complex assembly and limited transcription to a single round. In some cases (lanes 5, 6, 9, and 10) dATP was added at time 0 followed 2 min later by additional NTPs. After allowing 10 min for RNA synthesis, transcripts were fractionated by polyacrylamide gel electrophoresis. When [α-32P]CTP, GTP, and ATP were added at time 0 (lane 2), transcription proceeded efficiently; RNA synthesis under these conditions reflects the maximum signal expected in this series of assays. However, when ATP was omitted (lane 1) or when dATP substituted for ATP at either 10 |iM (lane 3) or 100 μM (lane 7), essentially no transcripts were detected. Similar to the result described in Fig. 1, a significant level of transcription was observed with [α-32P]CTP, GTP, dATP and ATPγS (present in 10-fold excess over dATP) only if the dATP was added 2 min prior to the other nucleotides (compare lanes 4 and 5 and lanes 8 and 9).
FIG. 2.
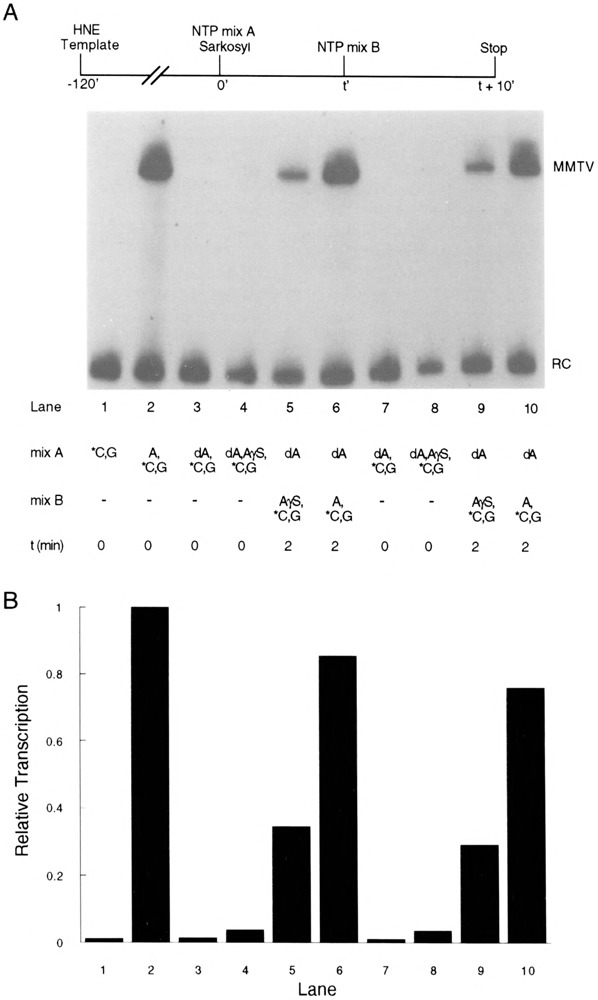
Effect of ATPγS on transcription. (A) Transcription complexes were assembled on pMBPT3 template DNA in HeLa nuclear extract (HNE). At time 0 NTPs (mix A) and Sarkosyl (0.02%) were added. In some experiments (lanes 5, 6, 9, and 10), a second NTP solution (mix B) was added after 2 min. RNA synthesis was terminated 10 min after the final addition of NTPs. All experiments contained 150 μM GTP and 50 μM (10 μCi) [α-32P]CTP added as indicated below and in the figure. Mix A contained the following nucleotides (final concentrations in the assay are indicated): CTP and GTP (lane 1); ATP (150 μM), CTP, and GTP (lane 2); dATP (10 μM), CTP, and GTP (lane 3); dATP (10 μM), ATPγS (100 μM), CTP, and GTP (lane 4); dATP (10 μM) (lanes 5 and 6); dATP (100 μM), CTP, and GTP (lane 7); dATP (100 μM), ATPγS (1 mM), CTP, and GTP (lane 8); dATP (100 μM) (lanes 9 and 10). Mix B contained the following nucleotides (final concentrations in the assay are indicated): ATPγS (100 μM), CTP, and GTP (lane 5); ATP (150 μM), CTP, and GTP (lane 6); ATPγS (1 mM), CTP, and GTP (lane 9); ATP (150 μM), CTP, and GTP (lane 10). (B) Quantitation of transcription shown in (A). Quantitation was made relative to a recovery control (RC) RNA added with the stop solution.
In sum, the results in Figs. 1 and 2 demonstrate that i) ATP (or dATP) is required for transcription from the MMTV promoter, ii) (d)ATP is required prior to the initiation of RNA synthesis, iii) a 10-fold excess of ATPγS can block (d)ATP-dependent step(s) in initiation, and iv) ATPγS can serve as an elongation substrate for pol II when it is the only adenine ribonucleoside triphosphate present. All of these results are completely consistent with the idea that transcription complexes activated by (d)ATP can be detected and quantitated by allowing them to productively initiate RNA synthesis in the presence of excess ATPγS.
Extent of Activated Complex Formation
We have estimated the apparent extent of activated complex formation. Transcription complexes were activated in the presence of dATP at 10 μM (Fig. 2, lanes 5 and 6) or 100 μM (lanes 9 and 10). After 2 min, [α-32P]CTP, GTP, and either excess ATPγS (lanes 5 and 9) or ATP (lanes 6 and 10) was added to initiate RNA synthesis. As described above, transcripts produced in the presence of ATPγS resulted from transcription complexes that were activated at the time of analogue addition, whereas reactions that contained ATP allowed RNA synthesis from all preinitiation complexes, independent of whether they had been activated during the 2-min treatment with dATP. Based on this experiment, the apparent number of activated complexes was roughly 30% of the total (Fig. 2B). Over a number of similar experiments, the extent of formation of activated complexes varied between 15% and 35% of the total number of complexes present (data not shown). As described in the Discussion section, our measurement provides a minimum estimate, and it is likely that the actual number of activated complexes is significantly higher than the number we detect directly.
Stability of Activated Complexes
With a highly purified, reconstituted-transcription system, it has been shown that activated complexes on the adenovirus major late (AdML) promoter rapidly disappear in the presence of excess ATPγS, with a half-time on the order of 1 min (12). However, the fate of these activated complexes was not investigated; loss of transcriptional activity could have been due to reversion to unactivated preinitiation complexes or to dissociation or some other irreversible process. We sought to determine the stability and reversibility of activated MMTV transcription complexes in our experimental system. In this experiment (Fig. 3A, top), preinitiation complexes were assembled, dATP (20 μM) was added, and activation was allowed to proceed for 45 s. As shown below (Fig. 4), activation for 45 s results in near maximal detection of ATPγS-resistant MMTV transcription complexes. Sarkosyl (0.02%) was added with the dATP to prevent further transcription complex assembly and to limit transcription to a single round. After the short period of activation, a 10-fold molar excess of ATPγS (200 μM) was added. Activated complexes were monitored over time by addition of GTP and [α-32P]CTP to aliquots removed at time t and allowing RNA synthesis to proceed for 10 min (Fig. 3A, lanes 1–5). Under these conditions activated complexes disappeared very rapidly with a half-time of about 1 min (Fig. 3B, open circles), similar to results obtained with the AdML promoter (12). The total number of preinitiation complexes, both activated and unactivated, was determined by taking advantage of preliminary experiments in which we varied the ratio of ATPγS to dATP and demonstrated that inhibition of activation by ATPγS can be overcome by dATP at a concentration equal to or greater than that of ATPγS (data not shown). Therefore, transcripts synthesized after addition of dATP (200 μM), GTP, and [α-32P]CTP at time t (Fig. 3A, lanes 6–9) reflect the sum of the activity of activated and unactivated complexes, and were much more stable than activated complexes in the presence of ATPγS [Fig. 3B, compare stability of total (closed circles) and activated (open circles) complexes].
FIG. 3.
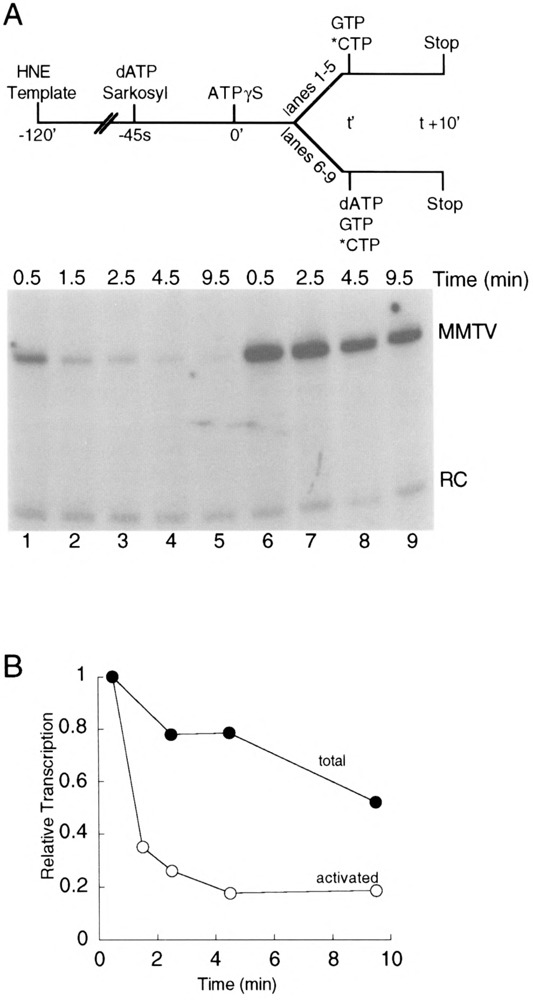
Stability of activated complexes. (A) Transcription complexes were assembled on pMBPT3 template in HeLa nuclear extract (HNE). Sarkosyl (0.02%) and dATP (20 μM) were added, and activated complexes were allowed to form for 45 s. Further activation was blocked by the addition of 10-fold molar excess ATPγS (200 μM) at time 0. Activated complexes (lanes 1–5) were observed by adding [α-32P]CTP and GTP at time t, and incubating an additional 10 min. To observe total complexes (lanes 6–9), the activation block was reversed at time t by the addition of dATP (200 μM), along with [α-32P]CTP and GTP and incubating an additional 10 min. (B) Quantitation of transcriptional activity from activated complexes (open circles) or total complexes (closed circles). Quantitation was made relative to a recovery control (RC) RNA added with the stop solution.
With regard to the reversibility of ATP-dependent transcription complex activation, this experiment was inconclusive. Because the activated complexes we detect constitute only about 15–35% of the total number of complexes present (Fig. 2B), we can account for much of the essentially complete loss of activated complexes in the first 4–5 min after addition of ATPγS by the 20% decrease in the total number of complexes observed over the same time period (Fig. 3B). However, as described in the Discussion section, we believe it is much more likely that most of the decrease in the number of activated complexes we observed in the experiment in Fig. 3 is due to reversion to unactivated complexes.
Time Course of Activated Complex Formation
A major advantage of our assay is that it allows the kinetics of transcription complex activation to be assessed. Transcription complexes were assembled on the MMTV promoter and activated by addition of dATP (100 μM) at time 0; addition of Sarkosyl (0.02%) at time 0 assured that no additional complexes would assemble and that any transcription would be limited to a single round. At time t, GTP, [α-32P]CTP, and ATPγS (1 mM) were added (Fig. 4A, top), and RNA synthesis was allowed to proceed for 10 min. Because ATPγS prevents the formation of additional activated complexes, the number of transcripts observed provides a measure of the number of activated complexes present at time t. The number of activated complexes increased rapidly during the first 1–3 min after addition of dATP and then slowly decreased (Fig. 4A and B, closed circles); after about 30 min, activated complexes were no longer detectable.
To further explore the slow decay in the number of ATPγS-resistant, activated complexes observed in Fig. 4, we performed an experiment in which we examined the effect of dATP on the total number of transcriptionally competent complexes, both activated and unactivated (Fig. 5). MMTV transcription complexes were assembled in HeLa nuclear extract. At time 0, dATP (20 μM) and Sarkosyl (0.02%) were added, followed at time t by addition of ATP, GTP, and [α-32P]CTP. RNA synthesis was allowed to proceed for 10 min. As no ATPγS is present in this assay, there is no inhibition of activation, and therefore the amount of RNA generated reflects the total number of transcription complexes present at time t. In the presence of dATP, but in the absence of initiating nucleotides, total transcription complexes decayed with apparent first-order kinetics (half-time of about 8 min) (Fig. 5B, closed circles). The decrease was completely dependent on the presence of dATP [Fig. 5B, compare presence of dATP (closed circles) to absence of dATP (closed squares)] or ATP but was not observed in the presence of ATPγS (data not shown). Thus, it appears that the β—γ phosphoanhydride bond of ATP is required for the observed decrease. Similar observations were first made by Luse and coworkers (3).
There are several possible explanations for these results. As described in the Discussion section, it is most likely that the observed decrease in (d)ATP-activated, ATPγS-resistant complexes seen in Fig. 4 (after the initial, rapid increase) reflects either an inherent property of activated complexes or a nonspecific, (d)ATP-dependent activity. In either case, a correction (see Materials and Methods for details) can be made based on the measured first-order rate of decay in total transcriptional potential derived from the experiment in Fig. 5. The correction allows calculation of the number of activated complexes that would be present if the (d)ATP-dependent decrease in total transcriptional activity observed in Fig. 5 did not occur. By applying this calculated correction, the formation of ATPγS-resistant activated complexes was found to plateau within 3 min and have a half-time of formation of less than 1 min (Fig. 4B, open circles).
DISCUSSION
Detection of Activated Transcription Complexes
We have exploited the published observation that ATP-dependent activation of pol II transcription complexes can be reversibly blocked by an excess of an ATP analogue containing a nonhydrolyzable β—γ phosphoanhydride bond (4,11,12,19) to assay for the presence of activated complexes by following ATP-dependent transition to analogue resistance. Our experiments confirm the requirement for hydrolyzable ATP for transcription from the MMTV promoter, the ability of ATPγS to block activation of MMTV transcription complexes, and the presence of a (d)ATP-dependent activation step that leads to formation of complexes that are resistant to ATPγS. Furthermore, our results are consistent with other reports that ATPγS can serve as an elongation substrate for pol II in the absence of other adenine ribonucleoside triphosphates.
Extent of Formation of Activated Complexes
The measured extent of activation (approximately 15–35%) provided by the experiment in Fig. 2 and similar experiments should be viewed as only a minimum estimate. Several observations are consistent with the idea that a significant fraction of activated complexes present at the time of addition of ATPγS do not result in full-length transcripts in our assay. In the presence of ATPγS, activated MMTV promoter complexes disappear with a half-time of about 1 min (Fig. 3). This is comparable to the time required to form a stable MMTV transcription elongation complex defined by resistance to a high concentration of Sarkosyl (0.2%) (J. W. Steinke and D. O. Peterson, unpublished observation), which, at least for the AdMF promoter, requires formation of the first few phosphodiester bonds of the transcript (3,15,16). Thus, some activated complexes present at the time of addition of ATPγS may not synthesize a detectable RNA due to partitioning between alternative pathways, only one of which leads to productive initiation in the presence of excess ATPγS. It should be emphasized that within a given experiment, as long as a constant fraction of activated complexes synthesizes an RNA, the number of transcripts produced is proportional to (but not necessarily equal to) the number of activated complexes present at the time of addition of ATPγS. Potential differences in the partitioning of activated complexes between alternative pathways must be considered in interpreting apparent differences in the extent of activated complex formation under different experimental conditions or among different promoters.
Reversibility of Activated Complex Formation
It is interesting to consider the fate of the activated complexes as they decay in the presence of ATPγS (Fig. 3). There are two plausible possibilities: either the complexes revert to unactivated complexes that are capable of reactivation, or the decay reflects irreversible loss of activity due to the inherent instability of activated complexes. Because we detect only a relatively small fraction of the transcription complexes as activated in the presence of dATP, it is difficult to directly determine whether the complexes that decay can be reactivated. However, a rapid and irreversible loss of activated complexes in the absence of the nucleotides necessary for transcription initiation is not consistent with other experiments. That is, if activated complexes rapidly and irreversibly lose their potential to productively initiate, we would expect to see a rapid decay of total transcriptional potential in the presence of dATP alone. Although we do observe a decay (Fig. 5), it is much too slow to result from the same pathway by which activated complexes are lost in Fig. 3. Thus, we believe that an equilibrium between activated and unactivated complexes is rapidly established in the presence of (d)ATP (in the absence of other nucleotides), and that the subsequent loss of activity seen in Fig. 4 occurs by a process distinct from the much more rapid decrease in activated complexes observed in the presence of ATPγS (Fig. 3).
The (d)ATP-dependent loss in transcriptional competence for MMTV preinitiation complexes observed in Fig. 5 is interesting; similar observations were first made by Luse and coworkers with the AdML promoter (3). This loss appears not to be the result of unanticipated initiation due to (unlabeled) nucleotide contamination in our nuclear extract that would allow the synthesis of unlabeled transcripts prior to addition of labeled CTP. A pulse-chase experiment in which incubation (5 min) in the presence of dATP (as in Fig. 5) was accompanied by a low concentration (0.1 μM) of high specific activity (3000 Ci/mmol) [α-32P]CTP and then followed by a chase of ATP, GTP, and excess unlabeled CTP did not result in any detectable full-length transcripts. We calculate that we should have been able to detect a signal if only 1 or 2 of the 64 C residues in the 172-nt transcript were incorporated during the pulse. Thus, we believe it is very unlikely that premature initiation with unlabeled NTPs is responsible for the dATP-dependent decrease in signal we observe. We have also considered that the loss in transcriptional activity might reflect the lack of stability of MMTV transcription complexes at 30°C. However, unactivated MMTV transcription complexes can be incubated at 30°C in the absence of (d)ATP (in the presence of 0.02% Sarkosyl) for over 60 min with little or no loss in ability to generate a full-length transcript when supplied with the appropriate nucleotides (Fig. 5B, closed squares, and data not shown). Alternatively, the activated, but not yet initiated, complexes may represent an inherently unstable intermediate between stable unactivated complexes and elongation complexes, which are also quite stable (3,29). Finally, it is possible that some nonspecific (d)ATP-dependent activity, such as a protease [for a review see (7)], could be present in the nuclear extract and degrade or otherwise destroy the ability of the complexes to productively initiate transcription. This seems particularly relevant in light of the demonstrated susceptibility of preinitiation complexes to low levels of protease activity (3).
A Kinetic Assay for Formation of Activated Transcription Complexes
The time-dependent formation of activated transcription complexes resistant to ATPγS provides a useful kinetic assay for (d)ATP-dependent activation. The time course of such assays is characterized by a rapid formation of activated complexes followed by a slow decay (Fig. 4). It is likely that this decay is the result of the same process that leads to the (d)ATP-dependent decrease in transcriptional activity seen in Fig. 5. Consequently, whether the decrease reflects an inherent property of activated complexes or is an artifact of the nuclear extract transcription system (see above), the experiment in Fig. 5 provides a means of calculating a correction factor (see Materials and Methods) so that the rate of activation can be determined independent of the decay. Applying this correction leads to a plateau in the activation time course with an estimated half-time of less than 1 min (Fig. 4B, open circles).
We have demonstrated that activation is relatively rapid for complexes on the MMTV promoter (Fig. 4), but the rate of activation need not be the same for all promoters or under all experimental conditions. Related to our observations, open pol II transcription complexes on the adenovirus E4 promoter could be detected within 2 min after addition of dATP (10 μM) in a nuclear extract transcription system derived from HeLa cells (41). It will be interesting to directly compare rates of activation for strong and weak pol II promoters and to assess potential roles of transcriptional regulatory proteins in the activation process. To facilitate such comparisons, it may be necessary to decrease the rate of activation by, for example, decreasing the temperature at which the assays are performed.
The kinetic assay we have described does not resolve the two steps in transcription initiation in which a requirement for a hydrolyzable ATP has been reported: formation of an open complex and CTD phosphorylation. To focus on these steps individually, it may be possible to adapt our general strategy of detecting activated complexes by their resistance to nonhydrolyzable ATP analogues. For example, the kinase inhibitor H8 [N-(2-(methylamino)ethyl)-5-is-quinolinesulfonamide] has been shown to inhibit CTD phosphorylation without preventing open complex formation (18,21). Resistance to nonhydrolyzable ATP analogues in the presence of H8 could provide a direct assay for open complex formation, although the absence of CTD phosphorylation could decrease the efficiency of synthesis of full-length transcripts (11,21,30,39). An alternative approach could exploit transcription assays reconstituted with pol II that lacks a CTD (22,27); RNA synthesis under these conditions is dependent on (d)ATP hydrolysis (27), presumably for open complex formation. Furthermore, open transcription complexes constructed by assembling factors onto template DNA that contains mismatched bases in the appropriate region can initiate transcription in the absence of hydrolyzable ATP (17,40). Such templates may allow the effect of CTD phosphorylation on the kinetics and efficiency of initiation to be determined independent of the requirement for ATP in open complex formation.
As the components of the pol II transcription apparatus continue to be defined, it will be increasingly important to develop kinetic assays for discrete steps in transcription to fully explore the biochemical roles of each factor.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant R01 CA32695 from the National Cancer Institute. The support of the Texas Agricultural Experiment Station is also gratefully acknowledged. We thank Gary Kunkel, John Steinke, and Lisa Dandridge for helpful discussions and Katherine Beifuss for technical assistance.
REFERENCES
- 1. Birnboim H. C.; Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513–1523; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buratowski S.; Hahn S.; Guarente L.; Sharp P. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 56:549–561; 1989. [DOI] [PubMed] [Google Scholar]
- 3. Cai H.; Luse D. S. Transcription initiation by RNA polymerase II in vitro. Properties of preinitiation, initiation, and elongation complexes. J. Biol. Chem. 262:298–304; 1987. [PubMed] [Google Scholar]
- 4. Conaway R. C.; Conaway J. W. ATP activates transcription initiation from promoters by RNA polymerase II in a reversible step prior to RNA synthesis. J. Biol. Chem. 263:2962–2968; 1988. [PubMed] [Google Scholar]
- 5. Conaway R. C.; Conaway J. W. General initiation factors for RNA polymerase II. Annu. Rev. Biochem. 62:161–190; 1993. [DOI] [PubMed] [Google Scholar]
- 6. Conaway R. C.; Reines D.; Garrett K. P.; Powell W.; Conaway J. W. Purification of RNA polymerase II general transcription factors from rat liver. Methods Enzymol. 273:194–207; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coux O.; Tanaka K.; Goldberg A. L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65:801–847; 1996. [DOI] [PubMed] [Google Scholar]
- 8. Dignam J. D.; Martin P. L.; Shastry B. S.; Roeder R. G. Eukaryotic gene transcription with purified components. Methods Enzymol. 101:582–598; 1983. [DOI] [PubMed] [Google Scholar]
- 9. Drapkin R.; Reardon J. T.; Ansari A.; Huang J.-C.; Zawel L.; Ahn K.; Sancar A.; Reinberg D. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature 368:769–772; 1994. [DOI] [PubMed] [Google Scholar]
- 10. Drapkin R.; Reinberg D. The multifunctional TFIIH complex and transcriptional control. Trends Biochem. Sci. 19:504–508; 1994. [DOI] [PubMed] [Google Scholar]
- 11. Dvir A.; Conaway R. C.; Conaway J. W. Promoter escape by RNA polymerase II—a role for an ATP co-factor in suppression of arrest by polymerase at promoter-proximal sites. J. Biol. Chem. 271:23352–23356; 1996. [DOI] [PubMed] [Google Scholar]
- 12. Dvir A.; Garrett K. P.; Chalut C.; Egly J. M.; Conaway J. W.; Conaway R. C. A role for ATP and TFIIH in activation of the RNA polymerase II preinitiation complex prior to transcription initiation. J. Biol. Chem. 271:7245–7248; 1996. [DOI] [PubMed] [Google Scholar]
- 13. Feaver W. J.; Gileadi O.; Li Y.; Kornberg R. D. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell 67:1223–1230; 1991. [DOI] [PubMed] [Google Scholar]
- 14. Gynzburg W. H.; Salmons B. Factors controlling the expression of mouse mammary tumour virus. Biochem. J. 283:625–632; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hawley D. K.; Roeder R. G. Separation and partial characterization of three functional steps in transcription initiation by human RNA polymerase II. J. Biol. Chem. 260:8163–8172; 1985. [PubMed] [Google Scholar]
- 16. Hawley D. K.; Roeder R. G. Functional steps in transcription initiation and reinitiation from the major late promoter in a HeLa nuclear extract. J. Biol. Chem. 262:3452–3461; 1987. [PubMed] [Google Scholar]
- 17. Holstege F. C. P.; van der Vliet P. C.; Timmers H. T. M. Opening of an RNA polymerase II promoter occurs in two distinct steps and requires the basal transcription factors IIE and IIH. EMBO J. 15:1666–1677; 1996. [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Y.; Gralla J. D. RNA polymerase II phosphorylation: Uncoupling from GAL4-VP16 directed open complex formation and transcription in a reconstituted system. Nucleic Acids Res. 22:4958–4962; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang Y.; Gralla J. D. Nucleotide requirements for activated RNA polymerase II open complex formation in vitro. J. Biol. Chem. 270:1277–1281; 1995. [DOI] [PubMed] [Google Scholar]
- 20. Jiang Y.; Smale S. T.; Gralla J. D. A common ATP requirement for open complex formation and transcription at promoters containing initiator or TATA elements. J. Biol. Chem. 268:6535–6540; 1993. [PubMed] [Google Scholar]
- 21. Jiang Y.; Yan M.; Gralla J. D. A three-step pathway of transcription initiation leading to promoter clearance at an activated RNA polymerase II promoter. Mol. Cell. Biol. 16:1614–1621; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim W.-Y.; Dahmus M. E. The major late promoter of adenovirus-2 is accurately transcribed by RNA polymerases IIO, IIA, and IIB. J. Biol. Chem. 264:3169–3176; 1989. [PubMed] [Google Scholar]
- 23. Kim M. H.; Peterson D. O. Oct-1 protein promotes functional transcription complex assembly on the mouse mammary tumor virus promoter. J. Biol. Chem. 270:27823–27828; 1995. [PubMed] [Google Scholar]
- 24. Kim M. H.; Peterson D. O. Stimulation of basal transcription from the mouse mammary tumor virus promoter by Oct proteins. J. Virol. 69:4717–4726; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koleske A. J.; Young R. A. An RNA polymerase II holoenzyme responsive to activators. Nature 368:466–469; 1994. [DOI] [PubMed] [Google Scholar]
- 26. Koleske A. J.; Young R. A. The RNA polymerase II holoenzyme and its implications for gene regulation. Trends Biochem. Sci. 20:113–116; 1995. [DOI] [PubMed] [Google Scholar]
- 27. Laybourn P. J.; Dahmus M. Phosphorylation of RNA polymerase IIA occurs subsequent to interaction with the promoter and before the initiation of transcription. J. Biol. Chem. 265:1365–13173; 1990. [PubMed] [Google Scholar]
- 28. Lu H.; Zawel L.; Fisher L.; Egly J.-M.; Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 358:641–645; 1992. [DOI] [PubMed] [Google Scholar]
- 29. Luse D. S.; Kochel T.; Kuempel E. D.; Coppola J. A.; Cai H. Transcription initiation by RNA polymerase II in vitro. At least two nucleotides must be added to form a stable ternary complex. J. Biol. Chem. 262:289–297; 1987. [PubMed] [Google Scholar]
- 30. Marshall N. F.; Peng J. M.; Xie Z.; Price D. H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 271:27176–27183; 1996. [DOI] [PubMed] [Google Scholar]
- 31. McClure W. R. Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem. 54:171–204; 1985. [DOI] [PubMed] [Google Scholar]
- 32. Ossipow V.; Tassan J. P.; Nigg E. A.; Schibler U. A mammalian RNA polymerase II holoenzyme containing all components required for promoter-specific transcription initiation. Cell 83:137–146; 1995. [DOI] [PubMed] [Google Scholar]
- 33. Parada C. A.; Roeder R. G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 384:375–378; 1996. [DOI] [PubMed] [Google Scholar]
- 34. Roeder R. G. The complexities of eukaryotic transcription initiation: Regulation of preinitiation complex assembly. Trends Biochem. Sci. 16(11):402–408; 1991. [DOI] [PubMed] [Google Scholar]
- 35. Sawadogo M.; Roeder R. G. Energy requirement for specific transcription initiation by the human RNA polymerase II system. J. Biol. Chem. 259:5321–5326; 1984. [PubMed] [Google Scholar]
- 36. Schaeffer L.; Roy R.; Humbert S.; Moncollin V.; Vermeulen W.; Hoeijmakers J. H. J.; Chambon P.; Egly J.-M. DNA repair helicase: A component of BTF2 (TFIIH) basic transcription factor. Science 260:58–63; 1993. [DOI] [PubMed] [Google Scholar]
- 37. Serizawa H.; Conaway R. C.; Conaway J. W. A carboxyl-terminal-domain kinase associated with RNA polymerase II transcription factor 5 from rat liver. Proc. Natl. Acad. Sci. USA 89:7476–7480; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serizawa H.; Conaway R. C.; Conaway J. W. Multifunctional RNA polymerase II initiation factor 5 from rat liver. Relationship between carboxyl-terminal domain kinase, ATPase, and DNA helicase activities. J. Biol. Chem. 268:17300–17308; 1993. [PubMed] [Google Scholar]
- 39. Song C. Z. Requirement for phosphorylation of RNA polymerase II C-terminal domain in transcription is both transcript length and promoter dependent. Biochem. Biophys. Res. Commun. 229:810–816; 1996. [DOI] [PubMed] [Google Scholar]
- 40. Tantin D.; Carey M. A heteroduplex template circumvents the energetic requirement for ATP during activated transcription by RNA polymerase II. J. Biol. Chem. 269:17397–17400; 1994. [PubMed] [Google Scholar]
- 41. Wang W.; Carey M.; Gralla J. D. Polymerase II promoter activation: Closed complex formation and ATP-driven start site opening. Science 255:450–453; 1992. [DOI] [PubMed] [Google Scholar]
- 42. Zawel L.; Reinberg D. Advances in RNA polymerase II transcription. Curr. Opin. Cell Biol. 4:488–495; 1992. [DOI] [PubMed] [Google Scholar]