

Broadly neutralizing antibodies (bNAbs) prevent HIV infection in monkey challenge models and suppress HIV replication in infected humans. Combinations of bNAbs are more effective at suppression, and antibody-like molecules engineered to have two or three bNAb combining sites also inhibit HIV replication in monkeys and other animal models. To expand the available array of multispecific HIV inhibitors, we designed single-component molecules that incorporate two (bispecific) or three (trispecific) bNAb binding sites that recognize the HIV envelope glycoprotein (Env) or the HIV coreceptor (CCR5) or that cross-target both Env and CCR5. Several of the bi- and trispecific molecules neutralized most viruses in a diverse cross-clade panel, with greater breadth and potency than those of the individual parental bNAbs. The molecules described here provide additional options for preventing or suppressing HIV infection.
KEYWORDS: CrossMAb, HIV envelope, HIV-1, anti-CCR5, bispecific antibody, trispecific antibody
ABSTRACT
Protection against acquiring human immunodeficiency virus (HIV) infection may not require a vaccine in the conventional sense, because broadly neutralizing antibodies (bNAbs) alone prevent HIV infection in relevant animal challenge models. Additionally, bNAbs as therapeutics can effectively suppress HIV replication in infected humans and in animal models. Combinations of bNAbs are generally even more effective, and bNAb-derived multivalent antibody-like molecules also inhibit HIV replication both in vitro and in vivo. To expand the available array of multispecific HIV inhibitors, we designed single-component molecules that incorporate two (bispecific) or three (trispecific) bNAbs that recognize HIV Env exclusively, a bispecific CrossMAb targeting two epitopes on the major HIV coreceptor, CCR5, and bi- and trispecifics that cross-target both Env and CCR5. These newly designed molecules displayed exceptional breadth, neutralizing 98 to 100% of a 109-virus panel, as well as additivity and potency compared to those of the individual parental control IgGs. The bispecific molecules, designed as tandem single-chain variable fragments (scFvs) (10E8fv-N6fv and m36.4-PRO 140fv), displayed median 50% inhibitory concentration (IC50s) of 0.0685 and 0.0131 μg/ml, respectively. A trispecific containing 10E8-PGT121-PGDM1400 Env-specific binding sites was equally potent (median IC50 of 0.0135 μg/ml), while a trispecific molecule targeting Env and CCR5 simultaneously (10E8Fab-PGDM1400fv-PRO 140fv) demonstrated even greater potency, with a median IC50 of 0.007 μg/ml. By design, some of these molecules lacked Fc-mediated effector function; therefore, we also constructed a trispecific prototype possessing reconstituted CH2-CH3 domains to restore Fc receptor binding capacity. The molecules developed here, along with those described previously, possess promise as prophylactic and therapeutic agents against HIV.
IMPORTANCE Broadly neutralizing antibodies (bNAbs) prevent HIV infection in monkey challenge models and suppress HIV replication in infected humans. Combinations of bNAbs are more effective at suppression, and antibody-like molecules engineered to have two or three bNAb combining sites also inhibit HIV replication in monkeys and other animal models. To expand the available array of multispecific HIV inhibitors, we designed single-component molecules that incorporate two (bispecific) or three (trispecific) bNAb binding sites that recognize the HIV envelope glycoprotein (Env) or the HIV coreceptor (CCR5) or that cross-target both Env and CCR5. Several of the bi- and trispecific molecules neutralized most viruses in a diverse cross-clade panel, with greater breadth and potency than those of the individual parental bNAbs. The molecules described here provide additional options for preventing or suppressing HIV infection.
INTRODUCTION
Natural human immunodeficiency virus type 1 (HIV-1) infection elicits neutralizing antibodies; however, most individuals do not generate broadly neutralizing antibodies (bNAbs). For the minority that do so, breadth often takes several years to evolve, and in the rarer, elite neutralizers, often longer (1). Immunization with HIV-1 envelope glycoprotein (Env) trimers fails to generate bNAbs in most animal models, although breadth has been achieved in cows (2) and in some transgenic mice following Env immunization (3). The development of a broad neutralizing response directed toward the external HIV Env protein is limited by quaternary packing, conformational masking, and extensive N-glycan shielding. All of these attributes evolved via strong host antibody selection to occlude exposure of conserved regions to the human immune system (4). Efficient screening of HIV-1-infected patients now yields dozens of bNAbs (5–11), along with structural determination of the sites of Env vulnerability for many of these bNAbs. General features of bNAbs include increased somatic hypermutation (SHM) (12), insertions/deletions (indels) (13), and relatively long HCDR3s (14). In general, bNAbs target epitopes involving conserved amino acids and, often, proximal N-glycans that are common between multiple viral strains, thereby accomplishing neutralization breadth. The use of bNAbs isolated from elite neutralizers, both alone and in combination, has demonstrated potent antiviral activity in macaque challenge studies (15–19) and humanized mice (20, 21) and has shown a therapeutic impact in infected humans (22, 23). In relation to the efficacy of the latter, bNAbs are being evaluated in HIV-infected patients to control virus replication, to reduce the latent viral reservoir, or, more recently, to prevent acquisition of infection (24). For example, the CD4 binding site (CD4bs)-specific bNAbs VRC01 (25) and 3BNC117 (22) both reduce viremia when infused therapeutically into human subjects.
Selected bNAbs demonstrate incomplete neutralization (26), while combinations of bNAbs are often more effective (27, 28) than using a single bNAb to cover natural resistance (29). Combinations of the more potent, more recently isolated bNAbs generate an additive effect that enhances in vitro neutralization (30), consistent with earlier studies using a small set of less potent bNAbs (31–33). Several groups have exploited bNAb recognition of distinct conserved determinants to create multiple formats of bi- and trispecific antibody-like molecules. Other investigators have created “oligo-MAbs” that target different neutralizing determinants on HIV-1 Env, exploiting the breadth of antibodies such as PG9 and PG16 as tandem single-chain variable fragments (scFvs) or as Ig fusions (34), as well as 3BNC117 and 10-1074 or PGT135 as CrossMAbs (cMAbs) (35, 36). A very recent design (37) is a modified IgG with bNAb binding sites arrayed in tandem, with one on top of the other, a variation of the Morrison multispecific antibody design (38).
HIV-1 preferentially uses CCR5 as its coreceptor in vivo (39, 40). To exploit this in vivo bias, Brinkman et al. generated scFvs specific for both the CCR5 N terminus (Nt) and second extracellular loop (ECL2) appended at the N or C terminus of an IgG to potentially create dual specificity (31). Alternatively, bispecific CrossMAbs target HIV Env and the primary receptor, CD4, or Env and CCR5 simultaneously (34). Another Ig-like design contains two N-terminal domains of CD4 fused to an Ig Fc, followed by C-terminal CCR5 Nt residues (to interact with the coreceptor binding site of gp120 [41]). However, inclusion of CD4 brings about the possibility of enhancement of infection, as detected when low levels of soluble CD4 enhance HIV entry in an Env-dependent manner (42, 43).
Generally, large bispecific molecules exhibit more desirable pharmacokinetic properties than those of lower-molecular-weight molecules. However, larger mass may come at the expense of tissue penetration, for which smaller molecules are more efficient (44). Molecules that have an intermediate size, larger than a Fab but smaller than a full IgG, may possess both desired properties of a longer in vivo half-life and better tissue penetration, resulting in more efficacious therapeutics.
In this study, we sought to determine if a diverse array of bi- and trispecific molecules would display increased coverage or potency in a single molecular entity. Accordingly, we designed a set of inhibitors based on three distinct strategies: first, targeting HIV Env alone; second, targeting two domains of CCR5 alone; and third, targeting both Env and CCR5 simultaneously. The bispecifics were in the format of tandem scFvs (45), similar to bispecific T-cell engagers (BiTEs) that target T cells to tumor cells (46). The trispecific design was in the Fab(scFv)2 format, which exploits the Fab molecule as a scaffold to which two scFvs are appended through connecting linkers at the respective N and C termini (47, 48). Here we describe the in vitro broad neutralization properties of the bi- and trispecific designs, adding to the tools available to the field for therapeutic or preventative applications.
RESULTS
Design of bi- and trispecific molecules targeting distinct HIV Env neutralizing determinants.
We selected four major neutralizing epitopes located at different regions of the HIV Env trimer for proof-of-principle design of bi- and trispecific antibody-based inhibitors in combinations or formats not fully tested previously. The well-described cross-conserved neutralizing sites themselves, from the base of the trimer to the V-region top cap, included the membrane-proximal external region (MPER), the CD4 binding site (CD4bs), the N332 glycan “supersite” (base of V3), and the V2 trimer apex (Fig. 1). We used the prototypic bNAb sequences derived from the equally well-described bNAbs 10E8 (MPER), N6 (CD4bs), PGT121 (N332 supersite), and PGDM1400 (V2 apex).
FIG 1.
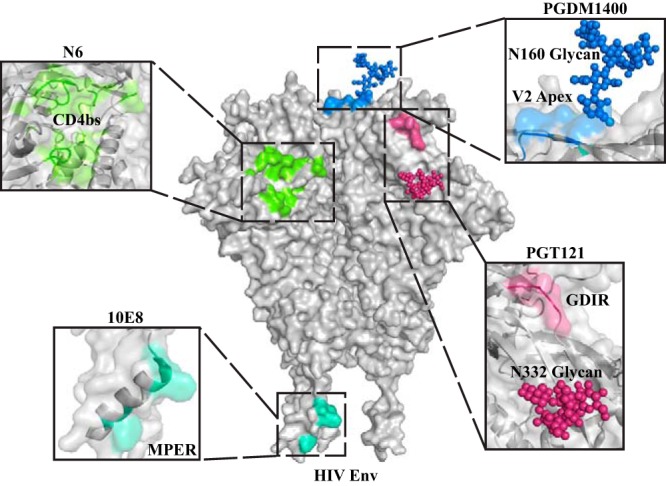
Epitopes on HIV Env targeted by selected bNAbs. The Env trimer is shown as a space-filling model, and the epitopes targeted are highlighted clockwise from bottom left, as follows: the MPER epitope (10E8; sea green), the CD4bs (N6; light green), the V2 apex (PGDM1400; blue, with the N160 glycan highlighted), and the N332 glycan, with the V3 GDIR residues shown in dark pink (PGT121). The binding sites are enlarged in the flanking black boxes. Approximate distances between these epitopes are as follows: 10E8 to N6, 94 Å; N6 to PGDM1400, 33 Å; and PGDM1400 to PGT121, 54 Å.
To begin, we designed a bispecific molecule to simultaneously target the CD4bs and the MPER on Env. We engineered sequences encoding the heavy (H) and light (L) chain variable regions of the CD4bs-directed bNAb N6 and the MPER-directed bNAb 10E8 in the form of scFvs, each containing a short H-to-L linker. Each scFv was connected by a second linker sequence, (G4S)3, to generate a bispecific antibody-like molecule (Fig. 2, top panel). We selected these two bNAbs because of their outstanding neutralization breadth as IgGs (5, 6), anticipating broad coverage. Initial screening of the 10E8fv-N6fv bispecific against a 12-virus global indicator panel, which contained several highly resistant tier 3 isolates, resulted in neutralization of 10 of the 12 viruses tested (data not shown). Based upon this neutralization profile, we assessed the 10E8fv-N6fv bispecific molecule against a modified Seaman panel (49) which included 109 viruses, and it neutralized 100% of the viruses tested with reasonable potency (Fig. 2, bottom panel).
FIG 2.

Design and neutralization profile of the bispecific molecule 10E8fv-N6fv. (Top) The design of the tandem scFv molecule is shown as a linear box schematic along with a cartoon depiction of the bispecific. (Bottom) Neutralization potencies (IC50s, in micrograms per milliliter) in the TZM-bl cell in vitro neutralization assay. In this and subsequent figures, “>40” indicates that the IC50 was not detectable at the highest concentration used, 40 μg/ml. ND, not determined.
Bolstered by the neutralization breadth of this initial bispecific design, we shifted to a trispecific design to potentially increase potency by using a configuration more amenable to restoration of Fc-mediated effector function. Accordingly, we generated trispecific molecules designed as two chains, designated A and B, which interacted via their respective CH1 and CL domains, linking two scFvs on each respective A and B component. The scFvs were again connected with (G4S)3 linkers (Fig. 3A). The initial design combined the antigen binding sites derived from the H and L chains of three bNAbs, namely, 10E8, PGT121, and PGDM1400. Again, these bNAbs were selected to potentially neutralize most known HIV isolates, based upon their individual neutralization profiles (6, 9, 50). We first designed 10E8 as the Fab, PGT121 as the scFv A arm, and PGDM1400 as the scFv B arm. To determine empirically if other configurations would enhance cross-neutralizing capacity in a head-to-head manner, other, “shuffled” combinations of the individual V region gene segments were designed, with the goal of maintaining trispecific recognition. Therefore, we did not generate combinations that would result in repetition of one of the three bNAbs in the three potential binding sites (Fab, scFv A arm, or scFv B arm). This process resulted in a total of eight different 10E8Fab-PGT121fv-PGDM1400fv trispecific molecules with the three bNAb-based specificities in different orientations, generating versions V1 to V8 (Fig. 3A, bottom section). We first confirmed the functional binding of the trispecifics to well-ordered native flexibly linked (NFL) Env trimers by enzyme-linked immunosorbent assay (ELISA). Shown in Fig. 3B is a representative example with 10E8Fab-PGT121fv-PGDM1400fv.V8 compared to the parental IgGs. The monovalent trispecific molecule bound to the trimer in a manner similar that of PGDM1400 IgG, which binds one quaternary epitope per trimer. Increased binding by PGT121 IgG, which has three binding sites per trimer, was observed as expected.
FIG 3.

Design and neutralization profile of trispecific 10E8Fab-PGT121fv-PGDM1400fv variants. (A) Schematic representation of the trispecific design. Chains A and B (top) were combined to form the trispecific molecule (middle). scFv, single-chain variable fragment; VH, heavy chain variable region; VL, light chain variable region; IL-2 SS, interleukin-2 signal sequence; L, linker. The various orientations of the scFvs are shown at the bottom (H1 to H4 and L1 to L4), with different combinations used to make the eight variants tested (e.g., H4 + L1 was used to form the V8 trispecific). (B) Binding of 10E8Fab-PGT121fv-PGDM1400fv.V8 to NFL trimers compared to that of the parental IgGs by ELISA. (C) Neutralization profiles of the eight 10E8Fab-PGT121fv-PGDM1400fv variants, the individual parental IgGs (10E8, PGT121, and PGDM1400), and the three parental IgGs in combination against a 28-virus panel initially selected based on susceptibility or resistance to the parental IgGs. The three parental IgGs were combined at a concentration of 0.33 μg/ml each (total concentration, 1 μg/ml). IC50 values (in micrograms per milliliter) are shown.
To better assess the functionality of these eight related 10E8Fab-PGT121fv-PGDM1400fv trispecifics, a panel of 28 relatively resistant clinical isolates was selected for analysis, along with the parental IgG controls. The viruses that we selected generally displayed resistance or susceptibility to each of the three bNAbs in assessments of independent neutralization capacity. For example, since the viral strain CAAN5342.A3 is highly sensitive to PGT121 but relatively resistant to 10E8 and PGDM1400, it can be used as an indicator of the PGT121fv functionality/specificity of the trispecific molecule. Similarly, 6540.v4.c1 can serve as an indicator of PGDM1400 activity, and BJOX010000.06.2 can be an indicator of 10E8 activity. Given the high potency of the parental bNAbs, we used a relatively low starting concentration (1 μg/ml) for this initial neutralization panel to better evaluate independent neutralization capacity and to rank order the different trispecific molecules. As shown in Fig. 3C, the parental bNAb neutralization patterns were generally consistent with previously reported specificities and potencies (9, 51). With this panel, we observed that if a bNAb potently neutralized a given virus (i.e., at concentrations of <0.009 μg/ml), then most 10E8Fab-PGT121fv-PGDM1400fv trispecific variants also neutralized such viruses. For example, T250-4 is neutralized potently by both PGT121 and PGDM1400, and we anticipated that most of the trispecifics would also neutralize this virus similarly, which was borne out by the data. Occasionally, a lower potency than that of the parental bNAbs was detected, especially if the parental bNAb(s) also neutralized the virus with low potency (e.g., ZM109) or if the neutralization was mostly driven by PGT121. Since the trispecific design now renders the binding monovalent, this was not entirely unexpected.
While the trispecifics were less potent than the parental IgGs in some cases, there was a marked improvement in breadth. The individual parental antibodies 10E8, PGT121, and PGDM1400 neutralized 43%, 61%, and 50% of the viruses of the 28-virus panel, respectively, in this assay format. When the three bNAbs were combined to a total IgG concentration of 1 μg/ml, 100% of the viruses tested were neutralized (Fig. 3C, last column), indicating that this combination of bNAbs in the proper trispecific configuration should display broad neutralization capacity in a single inhibitory molecule. Exhibiting additivity relative to the potencies of individual IgGs, the trispecific 10E8Fab-PGT121fv-PGDM1400fv versions V3 and V5 each neutralized 24 of 28 (86%) viruses in this resistant panel. Neutralization breadth was slightly diminished if both of the scFv VHs were at the C termini (V2 and V4). Version V8 neutralized 23 of 28 (82%) viruses tested, with a lower median 50% inhibitory concentration (IC50) (0.081 μg/ml) than those of the other trispecific variants. We therefore selected 10E8Fab-PGT121fv-PGDM1400fv.V8 for screening against the larger, 109-virus panel of diverse clinical isolates. To better determine the breadth against the larger panel of viruses, we used the less stringent starting concentration of 40 μg/ml. The 10E8Fab-PGT121fv-PGDM1400fv.V8 trispecific was 99% effective against this panel, displaying markedly increased breadth and potency (IC50 = 0.0135 μg/ml) compared to those of the parental bNAbs (Fig. 4).
FIG 4.

Neutralization profile of trispecific variant 10E8Fab-PGT121fv-PGDM1400fv.V8 against a 109-virus panel. Neutralization potencies of 10E8Fab-PGT121fv-PGDM1400fv.V8 (IC50s, in micrograms per milliliter) in the TZM-bl cell neutralization assay are given. ND, not determined. Parental IgGs that formed the basis for the trispecific were also tested, and their IC50s are shown.
Modified Env-targeting trispecific designs to restore Fc elements and to increase solubility.
Next, we evaluated whether we could further improve the potency of the 10E8Fab-PGT121fv-PGDM1400fv.V8 trispecific design. We constructed three variants with distinct modifications. First, we focused on restoring the Fc region for potential Fc receptor (FcR) interaction. Since the Fc region has been shown to enhance protective efficacy in vivo (52) and Fc-mediated interaction with the neonatal (n) FcRn contributes to a longer in vivo half-life (53), reconstitution of these effector elements will be valuable for future in vivo testing. Accordingly, we incorporated the CH2-CH3 domains of the IgG H chain between the Fab and the scFvs to reconstitute FcR interaction. The 10E8Fab-PGT121fv-PGDM1400fv.V8Fc trispecific retained a comparable neutralizing capacity and, in some cases, demonstrated enhanced neutralization potency against isolates not readily neutralized by the original V8 variant (Fig. 5A). For the second modification, we utilized a longer linker (LL), (G4S)6 instead of (G4S)3, between the 10E8 Fab component and the two scFvs. The trispecific molecule, 10E8Fab-PGT121fv-PGDM1400fv.V8LL, demonstrated equivalent or occasionally enhanced neutralization compared to that with the original V8 design against a 12-virus panel (Fig. 5B). Third, we incorporated structure-based substitutions that improve 10E8 IgG solubility (54), and potentially function, generating the 10E8Fab-PGT121fv-PGDM1400fv.V8.4DS trispecific. The 4DS variant showed improved neutralization potency compared to that with the original V8 design against a small virus test panel (Fig. 5C). Consistent with the neutralization data, both the V8LL and V8.4DS variants bound to well-ordered NFL trimers comparably as assessed by ELISA (Fig. 5D).
FIG 5.

Design and neutralization profiles of trispecific V8 variants. Neutralization profiles of the following trispecific 10E8Fab-PGT121fv-PGDM1400fv.V8 variants are given, with cartoon schematics shown above: V8Fc (Fc region incorporated for binding to FcRn) (A), V8LL (longer linker between the Fab and scFvs) (B), and V8.4DS (improved solubility) (C). IC50 values (in micrograms per milliliter) are indicated. ND, not determined. (D) Binding of the V8LL and V8.4DS variants to NFL trimers compared to that of the parental trispecific 10E8Fab-PGT121fv-PGDM1400fv.V8 and PGDM1400 IgG by ELISA.
Design of a cMAb to target two sites on CCR5 with potentially enhanced avidity.
Besides directly targeting the virus, we reasoned that it might be valuable to improve the potency of a CCR5-directed antibody, PRO 140 (55), to potentially increase the avidity for the predominant HIV coreceptor. To do so, besides targeting the extracellular loop 2 (ECL2)-directed epitope recognized by PRO 140, we generated a bispecific IgG by combining the PRO 140 specificity with that of RoAb13, which recognizes the CCR5 Nt region (56) (Fig. 6A). The bispecific RoAb13-PRO 140 antibody was constructed based on the CrossMAb (cMAb) design. In this design, arms of the IgG were encoded using either RoAb13- or PRO 140-derived nucleotide sequences. We used the published “knob-in-hole” (KIH) approach, in which the antibody is engineered to possess mutated residues in the CH3 domain, allowing association of only the desired heavy and light chains via a complementary “knob” and “hole.” As described previously, a domain exchange of the CH1 and CL regions was introduced to ensure proper association of a given light chain with the correct heavy chain (57) (Fig. 6B; see Materials and Methods). Expression (and formation) of the cMAb was confirmed by SDS-PAGE analysis and compared to that of the parental IgGs (Fig. 6B, right panel).
FIG 6.

cMAb design and binding characteristics. (A) Crystal structure of CCR5 (PDB entry 4MBS), with the extracellular loop 2 (ECL2) region shown in red and the N-terminal (Nt) region (PDB entry 2L87) superimposed in blue. The orange lines represent the top and bottom layers of the plasma membrane, with the transmembrane region in between. The top view is shown on the right. (B) Cartoon representation of the cMAb, including the T366W change in the RoAb13 CH3 domain to give the knob (red) and the modification of three residues (T366S/L368A/Y407V) to create the hole (blue) in the CH3 domain of the other corresponding heavy chain on PRO 140. The key or knob-in-hole (KIH) design was used to ensure that the two different heavy chains would pair up. The constant domain of the light chain (CL) (gray) and constant domain 1 of the heavy chain (CH1) (black) on the PRO 140 side of the IgG molecule were swapped to ensure correct association of the light chains. The KIH design and the CL/CH1 swap on one of the IgG arms ensure that the IgG molecules are in the bispecific cMAb format, eliminating the undesirable monospecific IgG formats. On the right is an SDS-PAGE gel showing PRO 140 IgG (lanes 1), RoAb13 (IgG) (lanes 2), and the cMAb (lanes 3), run under nonreducing (NR) and reducing (R) conditions. (C) Human (PRO 140, RoAb13, and cMAb) and mouse (CTC8 and 45523) anti-CCR5 antibodies are represented as cartoons. PRO 140 and 45523 are ECL2 specific, while RoAb13 and CTC8 are Nt-specific antibodies. The cMAb is dually specific, recognizing both Nt and ECL2 epitopes. (D) Table summarizing the cross-competition results between the human antibodies (PRO 140, RoAb13, and cMAb) and the mouse antibodies (CTC8 and 45523) to confirm the binding specificity of the cMAb. MFI, mean fluorescence intensity; APC, allophycocyanin; FITC, fluorescein isothiocyanate.
To confirm independent binding of the humanized RoAb13 component to the CCR5 N terminus and of humanized PRO 140 to ECL2 independently, we performed a cross-competition assay using murine antibodies specific for these sites. Control RoAb13 (Nt) and PRO 140 were expressed as human IgG1 antibodies. The competing mouse antibodies specific to the same epitopes, 45523 (ECL2) and CTC8 (Nt), were obtained commercially (Fig. 6C). To establish a cross-competition assay, samples containing equivalent numbers of cells were first incubated with a human IgG (e.g., PRO 140 IgG), followed by incubation with the competing mouse antibody for the same site (e.g., 45523). A noncompeting mouse antibody was used as a negative control. Fluor-labeled secondary antibodies were used to detect binding of the primary antibodies as assessed by fluorescence-activated cell sorter (FACS) analysis. When we performed this assay using the cMAb, greatly diminished signals from the competing mouse antibodies were detected (Fig. 6D), indicating that the cMAb could bind to both epitopes effectively. Since we also detected comparable binding of both PRO 140 and CTC8 (or RoAb13 and 45523) in the same sample, these data suggest that the Nt and ECL2 epitopes are sufficiently far apart to not interfere with their respective ligands. Individually, PRO 140 and RoAb13 are quite broad, as they neutralized 97% and 92% of the viruses, with mean values of 0.323 μg/ml and 0.177 μg/ml, respectively. When we tested the PRO 140-RoAb13 cMAb, all viruses in the global panel were neutralized (data not shown). When neutralization was assessed against the larger virus panel, the cMAb neutralized 99% of the viruses, with a mean potency of 0.272 μg/ml (Fig. 7). In the rare case where both PRO 140 and RoAb13 could not neutralize a given virus, such as WEAU, the cMAb was also ineffective. As expected, if either one of the antibodies could effectively neutralize a given virus, then the cMAb could as well.
FIG 7.

Neutralization breadth of the RoAb13-PRO 140 cMAb. Cartoons of RoAb13, PRO 140, and the RoAb13-PRO 140 cMAb are shown at the top, with their neutralization potencies (IC50s, in micrograms per milliliter) given below.
Bispecifics and trispecifics cross-targeting both HIV Env and the CCR5 coreceptor.
In our third overall approach, epitopes on HIV Env and CCR5 were simultaneously targeted using antibody binding regions designed as either bispecific or trispecific antibody-like molecules. We initially designed two bispecific molecules, with the rationale that their respective epitopes are likely to be proximal during receptor-coreceptor interaction during the initial stages of the HIV entry process. Accordingly, a bispecific molecule using PGDM1400 (V2 apex) and PRO 140 (CCR5) was constructed as tandem scFvs (PGDM1400fv-PRO 140fv) (Fig. 8). This bispecific displayed considerable breadth of neutralization and was more effective against clade C viruses. Next, we sought to incorporate a CD4-induced (CD4i) specificity to assess the impact on neutralization capacity. To do so, we selected, m36.4, a small, single-VH-domain inhibitor that was originally found by screening a phage library to identify small domains with capacities similar to that of the CD4i MAb X5 (58). By itself, m36.4 neutralized only 3 of 12 viruses in the global panel (data not shown), but we reasoned that colocalization of this CD4i-directed variable domain with the CCR5 coreceptor might be effective, since they are proximal during viral entry. Accordingly, we paired the m36.4 VH domain with the PRO 140 scFv to generate a bispecific molecule cross-targeting Env CD4i residues and the ECL2 region of CCR5. The m36.4-PRO 140fv bispecific neutralized 100% of the viruses, with an impressive median IC50 of 0.0131 μg/ml (Fig. 8). Among the bispecific designs, m36.4-PRO 140fv was the most potent inhibitor, even after adjustment on a molar basis for the slightly lower molecular mass.
FIG 8.


Neutralization by Env-CCR5 cross-targeting molecules. Neutralization profiles are given for the Env-CCR5-targeting molecules without (−) or with (+) TPST-1 coexpression for designs incorporating PGDM1400. IC50 values (in micrograms per milliliter) are provided. ND, not determined.
We also generated three trispecifics in this Env-CCR5 cotargeting class, revealing that the PRO 140Fab-PGDM1400fv-PGT121fv molecule neutralized 97% of viruses tested (Fig. 8). Since several of the Env apex-specific bNAbs contain sulfated tyrosines, we wanted to ensure maximal posttranslational sulfation by coexpression of the tyrosyl protein sulfotransferase enzyme (TPST-1). Sulfated Ys can enhance bNAb neutralizing potency (e.g., for PG9, PGT145, PGDM1400, and CAP256 [9, 53, 54, 59]), whereas the lack of Y sulfation has been reported to decrease potency (60). Cotransfection of a TPST-1-expressing plasmid did not greatly alter the neutralization capacity (Fig. 8), suggesting that there was adequate posttranslational modification of these trispecifics when they were expressed from the 293 producer cells. Another trispecific molecule in this cross-targeting series, 10E8Fab-PGDM1400fv-PRO 140fv, was the most potent trispecific, neutralizing 98% of the viruses with a relatively high potency (0.011 μg/ml) (Fig. 8). TPST-1 coexpression had a minimal but slightly enhanced effect on the potency (0.0071 μg/ml) of this trispecific. In addition, the trispecific RoAb13Fab-PGDM1400fv-PGT121fv combination neutralized 94% of the viruses tested, with moderate potencies (Fig. 8). Other trispecifics, using RoAb13Fab in combination with PRO 140fv-PGDM400fv or with PGT121fv-PRO 140fv, neutralized most of the viruses in the smaller global panel but were not exceptionally potent (data not shown).
DISCUSSION
Here we describe multiple bi- and trispecific HIV inhibitory molecules designed to incorporate broad HIV neutralization in a single-component protein. We first showed that a bispecific molecule incorporating 10E8 and N6, two of the broadest bNAbs against Env described to date, neutralized 100% of the viruses tested in a large panel, with reasonable potency. Next, we developed trispecific molecules by using different orientations of 10E8, PGT121, and PGDM1400 to increase potency. The 10E8Fab-PGT121fv-PGDM1400fv.V8 variant neutralized 99% of the viruses, but with increased potency compared to that of the 10E8fv-N6fv bispecific for the same virus panel. Additional modifications of this trispecific, including changes in linker length, Fc reconstitution, and changes in 10E8 solubility, enhanced potency and coverage against difficult-to-neutralize viruses. Next, we generated a bispecific cMAb that targeted two epitopes on CCR5, and it demonstrated increased breadth and potency compared to those of the parental IgGs, neutralizing 99% of viruses tested. In addition, we developed m36.4-PRO 140fv, a unique combination that targets Env and CCR5 simultaneously. This bispecific neutralized 100% of the viruses tested, with remarkable potency. Finally, by leveraging the results from the previous designs, we engineered, among others, the 10E8Fab-PGDM1400fv-PRO 140fv trispecific, which is among the most potent and broad molecules, displaying excellent coverage with a high degree of potency. We note that all the multivalent inhibitors were made with a His tag for ease of purification, which would be eliminated for in vivo assessment.
The 10E8fv-N6fv bispecific neutralized 100% of the viruses tested, perhaps because the design allows coengagement of the CD4bs and the MPER simultaneously. We tested a tandem VH-VL orientation and showed that this variant neutralized 100% of the viruses tested. Most other molecules targeting HIV Env were trispecific molecules constructed in selected orientations. Generally, these molecules also displayed relatively “modest to good” potencies against the selected virus panel. The orientations of the VH and VL regions of the scFvs in the trispecifics can influence the potency of such molecules, as we detected subtle differences in the different ordering of the gene segment subunits derived from the same three bNAb building blocks when the trispecifics were analyzed in a head-to-head manner. The trispecifics invariably neutralized more viruses than the parental IgGs did, with median IC50s that ranged from 0.081 to 0.178 μg/ml against the panel (Fig. 3C). This range in potency indicated that differences in the orientation of the variable regions imparted differences in how the molecules interacted with their respective binding sites to mediate neutralization. Other related molecules targeting Env, namely, the version with enhanced 10E8 solubility, the Fc-containing version, and the version with a longer linker, all displayed moderate gains in potency, with each offering its own advantages over the original version 8 trispecific. For example, the addition of the Fc CH2 and CH3 domains to the trispecific design was done to reintroduce the normal IgG effector function, which is likely necessary for in vivo viral clearance. The Fc-restored trispecific molecule was substantially larger and also more potent than the original trispecific lacking the Fc region. This might be due to its larger mass physically interfering with virus entry. The improved potency of the trispecific containing the more soluble 10E8 variant (V8.4DS) may be due to enhanced activity in solution.
The CCR5-targeting bispecific cMAb, though more potent than the individual parental IgGs, displayed a moderate gain in potency. Coverage was excellent, as the cMAb neutralized 99% of viruses in the large panel tested. Since targeting either Env or CCR5 individually generated breadth but gave moderate potency, we reasoned that cross-engaging these regions concurrently might improve both aspects of neutralization, as seen previously for certain Env-receptor combinations (34, 61, 62). Given the paucity of spikes reported to be present on each virus particle, it is possible that two Env trimer spikes will never be in proximity to be engaged by one trispecific molecule at the same time. However, with the right combining sites in a given trispecific molecule, cross-engagement with more than one epitope on Env might be more effective via avidity for either the same spike or another spike. Many of the trispecifics designed here to engage Env were not necessarily designed to bind multiple epitopes of a given Env simultaneously but rather were designed to have an additive effect on coverage, endowed by the overlapping breadth of the three bNAbs. In any case, multiple bNAb Env specificities make it more difficult for the virus to escape neutralization than it would be with a single targeting specificity, as indicated in previous studies (28).
The most potent trispecifics that we characterize here are designed to engage HIV Env and either bind to or interfere with CCR5 simultaneously, creating an effective impediment to virus entry, consistent with previous reports (41, 42). For example, some bNAb combinations that we selected as trispecifics were used in earlier studies (63) or in the context of bispecific cMAbs (42). In the latter study, two combinations, 10E8-PRO 140 (MPER-CCR5) and 10E8-iMAb (MPER-CD4), displayed good potency against a 118-virus panel. These molecules have been described as the most potent therapeutic agents, to date (64), compared to those of other designs that are under development. Our best candidate, the 10E8FAb-PGDM1400fv-PRO 140fv trispecific, is equally potent, with a median IC50 of 0.007 μg/ml, with the added advantage that it is smaller than the cMAb IgG molecule and hence may possess better tissue penetration. Our other candidate molecule, m36.4-PRO 140fv, with a median IC50 of 0.013 μg/ml, should offer excellent tissue penetration due to its smaller mass. Other recent studies used bNAbs alone or in combination and showed slightly lower potencies. For example, in a study by Kong et al., four bNAbs, targeting the CD4bs, V3 glycans, the trimer apex, and MPER in combination, achieved 100% coverage, with a median IC50 of 0.01 μg/ml (30), whereas our single molecules achieved higher potency while also maintaining breadth. Since in vitro neutralization potency is directly associated with protection in simian-human immunodeficiency virus (SHIV) challenge studies (16, 17, 65), it is likely that the single-component molecules we developed here, targeting Env or Env and CCR5 in tandem, have potential for further testing as anti-HIV therapeutics or prophylactics.
MATERIALS AND METHODS
IgG and bi- and trispecific construct cloning.
10E8, PGT121, PGDM1400, N6, PRO 140, and RoAb13 were cloned as human IgG1 antibodies in cytomegalovirus (CMV)-driven expression plasmids (66). Bispecific constructs were essentially two scFvs connected by a G4S linker sequence of two or three repeats. The initial eight trispecific design variations of 10E8Fab-PGT121fv-PGDM1400fv were generated by Creative Biolabs, and they all contained C-terminal His tags for ease of purification. Subsequently, we cloned all the constructs into the pCMV-1 expression vector, between the HindIII and EcoRI restriction sites. Trispecific molecules were of a Fab design. For both the N and C termini, the sequences for two scFvs were cloned in frame with sequences encoding connecting G4S linkers. Four “heavy” and four “light” constructs in multiple orientations were generated to express eight different trispecifics by transfecting one heavy and one light construct into 293F cells at a 1:1 plasmid DNA ratio. In the trispecific coding sequences, within the portion for the linker connecting the Fab part and the scFv, is a BamHI site which allows other bNAb scFvs to be exchanged. Variations of the original trispecific version 8 (V8) design included using the variable regions of the more soluble version of 10E8, named V8.4DS (54); using a longer linker (V8LL), with (G4S)6 between the Fab and the scFv regions; and incorporating the Fc region of the IgG (V8Fc) between the Fab and scFv regions. Designs including the PGDM1400 scFv were also expressed with and without the addition of a plasmid encoding the enzyme TPST-1 to enhance posttranslational sulfation of tyrosines.
Expression and purification of bi- and trispecific molecules and IgGs.
The bi- and trispecific molecules were produced by transient transfection of expression plasmids into FreeStyle 293F suspension cells (Invitrogen) and purified from cell supernatants by use of a His tag purification resin (Roche), using chelation chemistry directed toward the C-terminal His tags. Following chelation on a nickel affinity column, the molecules were eluted and buffer exchanged into phosphate-buffered saline (PBS), pH 7.4, using either 30-kDa-cutoff Amicon Ultra-4 centrifugal filter units (Millipore) or Slide-A-Lyzer G2 dialysis cassettes with 20-kDa-cutoff membranes (Thermo Fisher Scientific). Parental IgGs were purified by passing supernatants collected after transient transfection through 1-ml affinity columns of protein A Sepharose beads (GE Healthcare). Each column was washed with PBS, and the column-bound IgG was eluted with 100 mM glycine-HCl buffer, pH 2.7, neutralized by addition of 2 M Tris-HCl, pH 8.5, and dialyzed extensively against PBS, pH 7.4.
cMAb design and cloning.
The anti-CCR5 bispecific cMAb utilizing the knob-in-hole (KIH) and light chain domain crossover formats was constructed as previously reported (57). Briefly, four plasmids were required to express the cMAb. The first-component plasmid encoded the PRO 140 heavy chain with the VH region followed by the light chain constant region, the hinge region, and CH2-CH3 (Fc portion). The CH3 domain on the PRO 140 side had the “hole” (T366S/L368A/Y407V) engineered into it. The second-component plasmid encoded the PRO 140 light chain with the VL region followed by the CH1 domain. The third component was the RoAb13 heavy chain, in which the “knob” (T366W) was engineered into its CH3 domain. The fourth plasmid encoded the RoAb13 light chain. The plasmids were transiently transfected in equal amounts into 293F cells, and the cMAb was purified from the supernatant by use of a protein A Sepharose affinity column as described above. Individual mutations required to generate cMAb component plasmids were created using a QuikChange II site-directed mutagenesis kit (Agilent). Plasmids that required domain crossovers were constructed by gene synthesis of fragments (GeneArt) and seamless cloning (Invitrogen) into expression vectors according to the manufacturer's instructions. Note that the PRO 140 antibody used clinically is an IgG4 to limit activation of complement and cell lysis, which is an additional modification that may be used for in vivo testing of the cMAb in preclinical models or the clinic.
Flow cytometric cross-competition assay.
Cf2Th cells overexpressing CCR5, obtained from the NIH AIDS repository, were used to show that both arms of the cMAb are active by binding to both the Nt and ECL2 epitopes on CCR5. The human antibodies RoAb13 (Nt) and PRO 140 (ECL2) and the cMAb (Nt and ECL2) were incubated on ice for 15 min, followed by fixation with 1% paraformaldehyde (PFA) and washing, and then the corresponding competing antibody or the noncompeting mouse antibody was added to different tubes to evaluate if the mouse antibody could bind the epitope potentially occupied by the corresponding human antibody. The human and mouse antibodies were detected with fluor-labeled secondary antibodies as assessed by FACS analysis. The mouse antibodies 45523 (ECL2) and CTC8 (Nt) were purchased from R&D Systems.
ELISA.
ELISA plates were coated with the trispecific molecules or IgGs in PBS, pH 7.4, at 4°C overnight. Plates were blocked with 5% nonfat dry milk in PBS-T (PBS with 0.2% Tween 20) at room temperature (RT) for 2 h. Wells were then incubated with serial dilutions of biotinylated JRFL NFL TD CC+ trimers at RT for 90 min, followed by washing in PBS-T and subsequent incubation with streptavidin-horseradish peroxidase (HRP) (Sigma) to detect binding of the trimer. Plates were washed in PBS-T and developed using 3,3′,5,5′-tetramethylbenzidine chromogenic substrate solution (Life Technologies).
Virus neutralization assay.
Virus neutralization was assessed with a single-cycle assay using huCD4+ CCR5+ TZM-bl target cells and HIV-1 pseudoviruses as described previously (49). To determine the antibody concentration that resulted in a 50% reduction in entry (IC50; determined in relative luciferase units), which reflects neutralization of HIV, we performed serial dilutions of the parental antibodies or the bi- and trispecific molecules and fitted the neutralization dose-response curves by nonlinear regression. The sources of the Env-encoding plasmids derived from the HIV-1 isolates used in the neutralization assays were described previously (67). We used a modified Seaman panel containing 109 of the 117 viruses, as our clones for THRO4156.18 (virus 8 in the panel), Ce1086_B2 (virus 38), Ce0682_E4 (virus 42), Q259.d2.17 (virus 65), Q842.d12 (virus 66), 9004SS_A3_4 (virus 71), 231966.c02 (virus 105), and 191821_E6_1 (virus 106) did not express.
ACKNOWLEDGMENTS
We thank Elise Landais for providing molecular clones of select pseudovirus constructs for initial neutralization screening and Raiees Andrabi for excellent discussions and technical advice.
This work was funded by NIH P01 grant AI104722 (HIVRAD), Scripps CHAVI-ID grant AI100663, and the International AIDS Vaccine Initiative (IAVI) and its generous donors. The full list of IAVI donors is available at www.iavi.org. This study was made possible by the generous support of the Bill & Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery and the American people, through USAID.
The contents are the responsibility of IAVI and do not necessarily reflect the views of USAID or the U.S. government.
REFERENCES
- 1.McCoy LE, Burton DR. 2017. Identification and specificity of broadly neutralizing antibodies against HIV. Immunol Rev 275:11–20. doi: 10.1111/imr.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sok D, Le KM, Vadnais M, Saye-Francisco KL, Jardine JG, Torres JL, Berndsen ZT, Kong L, Stanfield R, Ruiz J, Ramos A, Liang CH, Chen PL, Criscitiello MF, Mwangi W, Wilson IA, Ward AB, Smider VV, Burton DR. 2017. Rapid elicitation of broadly neutralizing antibodies to HIV by immunization in cows. Nature 548:108–111. doi: 10.1038/nature23301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee K, Klasse PJ, Sanders RW, Pereyra F, Michael E, Lu M, Walker BD, Moore JP. 2010. IgG subclass profiles in infected HIV type 1 controllers and chronic progressors and in uninfected recipients of Env vaccines. AIDS Res Hum Retroviruses 26:445–458. doi: 10.1089/aid.2009.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlsson Hedestam GB, Guenaga J, Corcoran M, Wyatt RT. 2017. Evolution of B cell analysis and Env trimer redesign. Immunol Rev 275:183–202. doi: 10.1111/imr.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, O'Dell S, Chuang GY, Druz A, Georgiev IS, Schramm CA, Zheng A, Joyce MG, Asokan M, Ransier A, Darko S, Migueles SA, Bailer RT, Louder MK, Alam SM, Parks R, Kelsoe G, Von Holle T, Haynes BF, Douek DC, Hirsch V, Seaman MS, Shapiro L, Mascola JR, Kwong PD, Connors M. 2016. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity 45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouquet H, Scharf L, Euler Z, Liu Y, Eden C, Scheid JF, Halper-Stromberg A, Gnanapragasam PN, Spencer DI, Seaman MS, Schuitemaker H, Feizi T, Nussenzweig MC, Bjorkman PJ. 2012. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci U S A 109:E3268–E3277. doi: 10.1073/pnas.1217207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong CH, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA. 2011. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sok D, van Gils MJ, Pauthner M, Julien JP, Saye-Francisco KL, Hsueh J, Briney B, Lee JH, Le KM, Lee PS, Hua Y, Seaman MS, Moore JP, Ward AB, Wilson IA, Sanders RW, Burton DR. 2014. Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc Natl Acad Sci U S A 111:17624–17629. doi: 10.1073/pnas.1415789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. 2010. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, Gnanapragasam PN, Oliveira TY, Seaman MS, Kwong PD, Bjorkman PJ, Nussenzweig MC. 2013. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153:126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kepler TB, Liao HX, Alam SM, Bhaskarabhatla R, Zhang R, Yandava C, Stewart S, Anasti K, Kelsoe G, Parks R, Lloyd KE, Stolarchuk C, Pritchett J, Solomon E, Friberg E, Morris L, Karim SS, Cohen MS, Walter E, Moody MA, Wu X, Altae-Tran HR, Georgiev IS, Kwong PD, Boyd SD, Fire AZ, Mascola JR, Haynes BF. 2014. Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe 16:304–313. doi: 10.1016/j.chom.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, O'Dell S, Patel N, Shahzad-ul Hussan S, Yang Y, Zhang B, Zhou T, Zhu J, Boyington JC, Chuang GY, Diwanji D, Georgiev I, Kwon YD, Lee D, Louder MK, Moquin S, Schmidt SD, Yang ZY, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Burton DR, Koff WC, Walker LM, Phogat S, Wyatt R, Orwenyo J, Wang LX, Arthos J, Bewley CA, Mascola JR, Nabel GJ, Schief WR, Ward AB, Wilson IA, Kwong PD. 2011. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baba TW, Liska V, Hofmann-Lehmann R, Vlasak J, Xu W, Ayehunie S, Cavacini LA, Posner MR, Katinger H, Stiegler G, Bernacky BJ, Rizvi TA, Schmidt R, Hill LR, Keeling ME, Lu Y, Wright JE, Chou TC, Ruprecht RM. 2000. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med 6:200–206. doi: 10.1038/72309. [DOI] [PubMed] [Google Scholar]
- 16.Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, Parren PW, Marx PA, Burton DR. 2009. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med 15:951–954. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. 2009. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog 5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann-Lehmann R, Vlasak J, Rasmussen RA, Jiang S, Li PL, Baba TW, Montefiori DC, Bernacky BJ, Rizvi TA, Schmidt R, Hill LR, Keeling ME, Katinger H, Stiegler G, Cavacini LA, Posner MR, Ruprecht RM. 2002. Postnatal pre- and postexposure passive immunization strategies: protection of neonatal macaques against oral simian-human immunodeficiency virus challenge. J Med Primatol 31:109–119. doi: 10.1034/j.1600-0684.2002.01014.x. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann-Lehmann R, Vlasak J, Rasmussen RA, Smith BA, Baba TW, Liska V, Ferrantelli F, Montefiori DC, McClure HM, Anderson DC, Bernacky BJ, Rizvi TA, Schmidt R, Hill LR, Keeling ME, Katinger H, Stiegler G, Cavacini LA, Posner MR, Chou TC, Andersen J, Ruprecht RM. 2001. Postnatal passive immunization of neonatal macaques with a triple combination of human monoclonal antibodies against oral simian-human immunodeficiency virus challenge. J Virol 75:7470–7480. doi: 10.1128/JVI.75.16.7470-7480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deruaz M, Moldt B, Le KM, Power KA, Vrbanac VD, Tanno S, Ghebremichael MS, Allen TM, Tager AM, Burton DR, Luster AD. 2016. Protection of humanized mice from repeated intravaginal HIV challenge by passive immunization: a model for studying the efficacy of neutralizing antibodies in vivo. J Infect Dis 214:612–616. doi: 10.1093/infdis/jiw203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horwitz JA, Halper-Stromberg A, Mouquet H, Gitlin AD, Tretiakova A, Eisenreich TR, Malbec M, Gravemann S, Billerbeck E, Dorner M, Buning H, Schwartz O, Knops E, Kaiser R, Seaman MS, Wilson JM, Rice CM, Ploss A, Bjorkman PJ, Klein F, Nussenzweig MC. 2013. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A 110:16538–16543. doi: 10.1073/pnas.1315295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP Jr, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, Horwitz JA, Shimeliovich I, Ben-Avraham S, Witmer-Pack M, Platten M, Lehmann C, Burke LA, Hawthorne T, Gorelick RJ, Walker BD, Keler T, Gulick RM, Fatkenheuer G, Schlesinger SJ, Nussenzweig MC. 2015. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522:487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehandru S, Vcelar B, Wrin T, Stiegler G, Joos B, Mohri H, Boden D, Galovich J, Tenner-Racz K, Racz P, Carrington M, Petropoulos C, Katinger H, Markowitz M. 2007. Adjunctive passive immunotherapy in human immunodeficiency virus type 1-infected individuals treated with antiviral therapy during acute and early infection. J Virol 81:11016–11031. doi: 10.1128/JVI.01340-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, Manrique A, Huber M, Rehr M, Oxenius A, Weber R, Stiegler G, Vcelar B, Katinger H, Aceto L, Gunthard HF. 2005. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med 11:615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 25.Ledgerwood JE, Coates EE, Yamshchikov G, Saunders JG, Holman L, Enama ME, DeZure A, Lynch RM, Gordon I, Plummer S, Hendel CS, Pegu A, Conan-Cibotti M, Sitar S, Bailer RT, Narpala S, McDermott A, Louder M, O'Dell S, Mohan S, Pandey JP, Schwartz RM, Hu Z, Koup RA, Capparelli E, Mascola JR, Graham BS, VRC 602 Study Team. 2015. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin Exp Immunol 182:289–301. doi: 10.1111/cei.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy LE, Falkowska E, Doores KJ, Le K, Sok D, van Gils MJ, Euler Z, Burger JA, Seaman MS, Sanders RW, Schuitemaker H, Poignard P, Wrin T, Burton DR. 2015. Incomplete neutralization and deviation from sigmoidal neutralization curves for HIV broadly neutralizing monoclonal antibodies. PLoS Pathog 11:e1005110. doi: 10.1371/journal.ppat.1005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, Stephenson KE, Chang HW, Shekhar K, Gupta S, Nkolola JP, Seaman MS, Smith KM, Borducchi EN, Cabral C, Smith JY, Blackmore S, Sanisetty S, Perry JR, Beck M, Lewis MG, Rinaldi W, Chakraborty AK, Poignard P, Nussenzweig MC, Burton DR. 2013. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 503:224–228. doi: 10.1038/nature12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shingai M, Nishimura Y, Klein F, Mouquet H, Donau OK, Plishka R, Buckler-White A, Seaman M, Piatak M Jr, Lifson JD, Dimitrov DS, Nussenzweig MC, Martin MA. 2013. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature 503:277–280. doi: 10.1038/nature12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, Zang T, Dorner M, Billerbeck E, Labitt RN, Gaebler C, Marcovecchio P, Incesu RB, Eisenreich TR, Bieniasz PD, Seaman MS, Bjorkman PJ, Ravetch JV, Ploss A, Nussenzweig MC. 2012. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 492:118–122. doi: 10.1038/nature11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong R, Louder MK, Wagh K, Bailer RT, deCamp A, Greene K, Gao H, Taft JD, Gazumyan A, Liu C, Nussenzweig MC, Korber B, Montefiori DC, Mascola JR. 2015. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J Virol 89:2659–2671. doi: 10.1128/JVI.03136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkmann U, Kontermann RE. 2017. The making of bispecific antibodies. MAbs 9:182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mascola JR, Louder MK, VanCott TC, Sapan CV, Lambert JS, Muenz LR, Bunow B, Birx DL, Robb ML. 1997. Potent and synergistic neutralization of human immunodeficiency virus (HIV) type 1 primary isolates by hyperimmune anti-HIV immunoglobulin combined with monoclonal antibodies 2F5 and 2G12. J Virol 71:7198–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, Parren PW. 2001. Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J Virol 75:12198–12208. doi: 10.1128/JVI.75.24.12198-12208.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pace CS, Song R, Ochsenbauer C, Andrews CD, Franco D, Yu J, Oren DA, Seaman MS, Ho DD. 2013. Bispecific antibodies directed to CD4 domain 2 and HIV envelope exhibit exceptional breadth and picomolar potency against HIV-1. Proc Natl Acad Sci U S A 110:13540–13545. doi: 10.1073/pnas.1304985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asokan M, Rudicell RS, Louder M, McKee K, O'Dell S, Stewart-Jones G, Wang K, Xu L, Chen X, Choe M, Chuang G, Georgiev IS, Joyce MG, Kirys T, Ko S, Pegu A, Shi W, Todd JP, Yang Z, Bailer RT, Rao S, Kwong PD, Nabel GJ, Mascola JR. 2015. Bispecific antibodies targeting different epitopes on the HIV-1 envelope exhibit broad and potent neutralization. J Virol 89:12501–12512. doi: 10.1128/JVI.02097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bournazos S, Gazumyan A, Seaman MS, Nussenzweig MC, Ravetch JV. 2016. Bispecific anti-HIV-1 antibodies with enhanced breadth and potency. Cell 165:1609–1620. doi: 10.1016/j.cell.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, Lord DM, Wei RR, Deng G, Louder M, Schmidt SD, Mankoff Z, Wu L, Asokan M, Beil C, Lange C, Leuschner WD, Kruip J, Sendak R, Do Kwon Y, Zhou T, Chen X, Bailer RT, Wang K, Choe M, Tartaglia LJ, Barouch DH, O'Dell S, Todd JP, Burton DR, Roederer M, Connors M, Koup RA, Kwong PD, Yang ZY, Mascola JR, Nabel GJ. 2017. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 358:85–90. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coloma MJ, Morrison SL. 1997. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol 15:159–163. doi: 10.1038/nbt0297-159. [DOI] [PubMed] [Google Scholar]
- 39.Kozak SL, Platt EJ, Madani N, Ferro FE Jr, Peden K, Kabat D. 1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J Virol 71:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, Ho DD. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 41.Gardner MR, Kattenhorn LM, Kondur HR, von Schaewen M, Dorfman T, Chiang JJ, Haworth KG, Decker JM, Alpert MD, Bailey CC, Neale ES Jr, Fellinger CH, Joshi VR, Fuchs SP, Martinez-Navio JM, Quinlan BD, Yao AY, Mouquet H, Gorman J, Zhang B, Poignard P, Nussenzweig MC, Burton DR, Kwong PD, Piatak M Jr, Lifson JD, Gao G, Desrosiers RC, Evans DT, Hahn BH, Ploss A, Cannon PM, Seaman MS, Farzan M. 2015. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature 519:87–91. doi: 10.1038/nature14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y, Yu J, Lanzi A, Yao X, Andrews CD, Tsai L, Gajjar MR, Sun M, Seaman MS, Padte NN, Ho DD. 2016. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell 165:1621–1631. doi: 10.1016/j.cell.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan N, Sun Y, Binley J, Lee J, Barbas CF III, Parren PW, Burton DR, Sodroski J. 1998. Determinants of human immunodeficiency virus type 1 envelope glycoprotein activation by soluble CD4 and monoclonal antibodies. J Virol 72:6332–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L. 2010. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol 28:355–362. doi: 10.1016/j.tibtech.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. 1988. Single-chain antigen-binding proteins. Science 242:423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 46.Huehls AM, Coupet TA, Sentman CL. 2015. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol 93:290–296. doi: 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller KM, Arndt KM, Strittmatter W, Pluckthun A. 1998. The first constant domain (C(H)1 and C(L)) of an antibody used as heterodimerization domain for bispecific miniantibodies. FEBS Lett 422:259–264. doi: 10.1016/S0014-5793(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 48.Schoonjans R, Willems A, Schoonooghe S, Fiers W, Grooten J, Mertens N. 2000. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J Immunol 165:7050–7057. doi: 10.4049/jimmunol.165.12.7050. [DOI] [PubMed] [Google Scholar]
- 49.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Georgiev IS, Doria-Rose NA, Zhou T, Kwon YD, Staupe RP, Moquin S, Chuang GY, Louder MK, Schmidt SD, Altae-Tran HR, Bailer RT, McKee K, Nason M, O'Dell S, Ofek G, Pancera M, Srivatsan S, Shapiro L, Connors M, Migueles SA, Morris L, Nishimura Y, Martin MA, Mascola JR, Kwong PD. 2013. Delineating antibody recognition in polyclonal sera from patterns of HIV-1 isolate neutralization. Science 340:751–756. doi: 10.1126/science.1233989. [DOI] [PubMed] [Google Scholar]
- 52.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. 2014. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158:1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ko SY, Pegu A, Rudicell RS, Yang ZY, Joyce MG, Chen X, Wang K, Bao S, Kraemer TD, Rath T, Zeng M, Schmidt SD, Todd JP, Penzak SR, Saunders KO, Nason MC, Haase AT, Rao SS, Blumberg RS, Mascola JR, Nabel GJ. 2014. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature 514:642–645. doi: 10.1038/nature13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon YD, Georgiev IS, Ofek G, Zhang B, Asokan M, Bailer RT, Bao A, Caruso W, Chen X, Choe M, Druz A, Ko SY, Louder MK, McKee K, O'Dell S, Pegu A, Rudicell RS, Shi W, Wang K, Yang Y, Alger M, Bender MF, Carlton K, Cooper JW, Blinn J, Eudailey J, Lloyd K, Parks R, Alam SM, Haynes BF, Padte NN, Yu J, Ho DD, Huang J, Connors M, Schwartz RM, Mascola JR, Kwong PD. 2016. Optimization of the solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure-based design. J Virol 90:5899–5914. doi: 10.1128/JVI.03246-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olson WC, Rabut GE, Nagashima KA, Tran DN, Anselma DJ, Monard SP, Segal JP, Thompson DA, Kajumo F, Guo Y, Moore JP, Maddon PJ, Dragic T. 1999. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J Virol 73:4145–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ji C, Brandt M, Dioszegi M, Jekle A, Schwoerer S, Challand S, Zhang J, Chen Y, Zautke L, Achhammer G, Baehner M, Kroetz S, Heilek-Snyder G, Schumacher R, Cammack N, Sankuratri S. 2007. Novel CCR5 monoclonal antibodies with potent and broad-spectrum anti-HIV activities. Antiviral Res 74:125–137. doi: 10.1016/j.antiviral.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, Schwaiger M, Stubenrauch KG, Sustmann C, Thomas M, Scheuer W, Klein C. 2011. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 108:11187–11192. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen W, Xiao X, Wang Y, Zhu Z, Dimitrov DS. 2010. Bifunctional fusion proteins of the human engineered antibody domain m36 with human soluble CD4 are potent inhibitors of diverse HIV-1 isolates. Antiviral Res 88:107–115. doi: 10.1016/j.antiviral.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andrabi R, Su CY, Liang CH, Shivatare SS, Briney B, Voss JE, Nawazi SK, Wu CY, Wong CH, Burton DR. 2017. Glycans function as anchors for antibodies and help drive HIV broadly neutralizing antibody development. Immunity 47:524.e3–537.e3. doi: 10.1016/j.immuni.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choe H, Li W, Wright PL, Vasilieva N, Venturi M, Huang CC, Grundner C, Dorfman T, Zwick MB, Wang L, Rosenberg ES, Kwong PD, Burton DR, Robinson JE, Sodroski JG, Farzan M. 2003. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell 114:161–170. doi: 10.1016/S0092-8674(03)00508-7. [DOI] [PubMed] [Google Scholar]
- 61.Pace CS, Fordyce MW, Franco D, Kao CY, Seaman MS, Ho DD. 2013. Anti-CD4 monoclonal antibody ibalizumab exhibits breadth and potency against HIV-1, with natural resistance mediated by the loss of a V5 glycan in envelope. J Acquir Immune Defic Syndr 62:1–9. doi: 10.1097/QAI.0b013e3182732746. [DOI] [PubMed] [Google Scholar]
- 62.Sun M, Pace CS, Yao X, Yu F, Padte NN, Huang Y, Seaman MS, Li Q, Ho DD. 2014. Rational design and characterization of the novel, broad and potent bispecific HIV-1 neutralizing antibody iMabm36. J Acquir Immune Defic Syndr 66:473–483. doi: 10.1097/QAI.0000000000000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sievers SA, Scharf L, West AP Jr, Bjorkman PJ. 2015. Antibody engineering for increased potency, breadth and half-life. Curr Opin HIV AIDS 10:151–159. doi: 10.1097/COH.0000000000000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagh K, Seaman MS, Zingg M, Fitzsimons T, Barouch DH, Burton DR, Connors M, Ho DD, Mascola JR, Nussenzweig MC, Ravetch J, Gautam R, Martin MA, Montefiori DC, Korber B. 2018. Potential of conventional & bispecific broadly neutralizing antibodies for prevention of HIV-1 subtype A, C & D infections. PLoS Pathog 14:e1006860. doi: 10.1371/journal.ppat.1006860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moldt B, Rakasz EG, Schultz N, Chan-Hui PY, Swiderek K, Weisgrau KL, Piaskowski SM, Bergman Z, Watkins DI, Poignard P, Burton DR. 2012. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci U S A 109:18921–18925. doi: 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. 2008. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods 329:112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sundling C, Forsell MN, O'Dell S, Feng Y, Chakrabarti B, Rao SS, Lore K, Mascola JR, Wyatt RT, Douagi I, Karlsson Hedestam GB. 2010. Soluble HIV-1 Env trimers in adjuvant elicit potent and diverse functional B cell responses in primates. J Exp Med 207:2003–2017. doi: 10.1084/jem.20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]