

Abstract
The discovery that heterozygous and homozygous mutations in the gene encoding progranulin are causally linked to frontotemporal dementia and lysosomal storage disease, respectively, reveals previously unrecognized roles of the progranulin protein in regulating lysosome biogenesis and function. Given the importance of lysosomes in cellular homeostasis, it is not surprising that progranulin deficiency has pleiotropic effects on neural circuit development and maintenance, stress response, innate immunity and ageing. This Progress article reviews recent advances in progranulin biology emphasizing its roles in lysosomal function and brain innate immunity, and outlines future avenues of investigation that may lead to new therapeutic approaches for neurodegeneration.
Progranulin was originally described as a growth factor that regulates wound healing, vasculogenesis and tumour growth1. However, two landmark studies in 2006 revealed that mutations in GRN, the gene that encodes progranulin, are causally linked to a familial form of frontotemporal lobar degeneration (FTLD) with distinct neuropathological features consisting of ubiquitin-positive protein aggregates in the nucleus and cytoplasm of cortical neurons2,3. These aggregates were later found to be highly enriched in TAR DNA-binding protein 43 (TDP43)4,5. The recognition of distinct proteinopathies for FTLD provided an important conceptual framework to investigate disease mechanisms caused by several major genes linked to familial frontotemporal dementia (FTD), including GRN and the hexanucleotide repeat expansion in C9ORF72 (REFS 6,7).
The identification of autosomal dominant mutations in the GRN gene as a common cause for FTLD has sparked renewed interest in this biologically relevant molecule2,8. In the ensuing decade, several potential functions have been proposed for progranulin in the CNS, ranging from trophic factor support for neurons to suppression of microglial activation9, but the exact mechanism by which GRN haploinsufficiency results in neurodegeneration remains poorly understood. The discovery that patients with homozygous GRN mutations developed neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disease, has drawn further attention to progranulin position in the lysosome and its potential role in neuronal proteostasis10,11.
This Progress article focuses on the fundamental biology of progranulin by highlighting its evolutionarily conserved function (or functions) in the lysosome with multisystem impacts on growth and/or survival, immunity and proteostasis. We further describe how progranulin deficiency in microglia and neurons contributes to the pathogenesis and circuit-specific neurodegeneration in FTLD and potentially Alzheimer disease (AD). Together, these recent discoveries provide new insights into how dysregulated levels of progranulin and granulin result in such diverse human pathologies. They also establish a foundation on which future investigations can connect progranulin biology with key neurodegenerative disease phenotypes, including TDP43 proteinopathy and other failures of protein homeostasis.
Evolution of progranulin
Progranulin and granulins
Progranulin is a phylogenetically ancient, cysteine-rich, secreted protein with several interesting features. First, progranulin serves as a precursor protein (or holoprotein) that can be cleaved into granulin. Granulins seem to oppose the activity of the holoprotein, at least in wound healing, inflammation and neuroprotection12–14. Each granulin consists of an approximately 60 amino-acid motif repeated in a tandem manner throughout the protein. Most commonly, a granulin domain consists of 12 cysteines (two individual cysteines at the amino and carboxyl ends bookending four sets of vicinal pairs) that form six disulfide bonds. The disulfide bonds formed by cysteines confer granulins with a stable, structurally compact, stacked β-sheet configuration that is potentially protease resistant15. However, granulin variants are found with fewer cysteines. For example, the human petite granulin has 6 cysteines, and granulin G has 10 cysteines. The structural and functional differences conferred by differential cysteine number are currently not known.
The number of granulin repeats are highly variable through phylogeny: whereas Dictyostelium discoideum and flowering plants have a single granulin, Caenorhabditis elegans has 3 granulin repeats, humans have 7 granulin repeats (apart from petite granulin), and exon duplications in zebrafish have expanded the number of granulin repeats to at least 11, possibly 12 (REF. 16) (BOX 1; FIG. 1). This diversity in granulin repeats suggests that they have an active biological role, with different granulins taking on distinct specificities. Structurally, granulins belong to a larger family of cysteine-rich mini-proteins, which includes oxytocin, epidermal growth factor and defensins17. These small cysteine-rich proteins function as hormones, growth factors, ion channel modulators and enzyme inhibitors. Exploring potential analogies to other cysteine-rich mini-proteins could shed light on the function of both the progranulin holoprotein and the cleaved granulins.
Box 1. Evolutionary dynamics of granulin domains and progranulin genes.
Progranulins are evolutionary ancient proteins that emerged about 1.5 billion years ago1. Granulin-domain-containing proteins are found in unicellular eukaryotes, plants, metazoan animals and vertebrates, and they are among the first extracellular regulatory proteins still used by multicellular animals16. Across species, the granulin domains share conserved cysteine residues. However, the number of granulin domains per progranulin is highly variable from species to species. A single granulin domain is found in basal eukaryotes such as Dictyostelium discoideum, whereas Caenorhabditis elegans has 3 granulin repeats, Xenopus tropicalis has 14 and the purple sea urchin has 27 granulin domains90. This domain diversity is achieved by the duplication and/or loss of the combination of exons encoding granulin peptides16. In some lineages, granulins are also associated with other domains, such as a cysteine protease domain in many plants including Arabidopsis thaliana. No progranulin gene has been identified in Drosophila species.
In addition to duplication of its granulin domains, the progranulin gene family has expanded in many vertebrate lineages, and this evolutionary history may provide hints about progranulin functions. Whereas most vertebrates have a single copy of GRN (which encodes progranulin), fish genomes contain multiple grn paralogues, often comprising long-form paralogues with multiple granulin domains and short-form paralogues with only one or two granulin domains91. For example, killifish and zebrafish have two long-form grn and one or two short-form grn, respectively. These two sets of paralogues have distinct origins. Short-form progranulins originated in vertebrates, were lost in tetrapods but were preserved in some teleost for more than 500 million years16. Fish long-form progranulins were created by whole-genome duplication about 300 million years ago, and the duplicated paralogues were preserved, a pattern consistent with vertebrate genes that show greater association with disease92. The evolutionary dynamics of GRN reflect its crucial regulatory function and may be instrumental to understanding how a primitive regulatory protein is employed in complex tissue functions during metazoan evolution.
Figure 1. Evolutionary dynamics of granulin domains and progranulin genes.

A comparison of progranulin protein across selected species and model organisms is shown. Species cladogram based on established species phylogeny is shown on the left, and the corresponding progranulin proteins with complete granulin domains are shown on the right. Protein sequences corresponding to the longest progranulin isoform in each species as identified by Basic Local Alignment Search Tool (BLAST) were obtained from National Center for Biotechnology Information (NCBI) Protein database. Granulin domains were identified using the NCBI Conserved Domain Database and were plotted on the protein sequence using custom scripts.
Although progranulin can be cleaved into individual granulin domains, one must be mindful not to conflate progranulin levels with that of its cleavage products. In other words, levels of progranulin and granulin may be independently regulated, tethered in the same direction (progranulin↑, granulin ↑) or in opposite directions (progranulin ↓, granulin ↑). Progranulin also undergoes regulated glycosylation18,19, protecting it from lysosomal enzyme-mediated degradation and adding a layer of complexity to its localization and half-life.
Progranulin expression patterns
Progranulin is broadly expressed by many cell types, although the expression is low in muscles or resting endothelium20,21. Within the CNS, GRN mRNA is produced by a wide range of cell types including neurons, microglia, endothelial cells and astrocytes22. Although its putative role is in the lysosome, an amino-terminal secretion signal directs progranulin to secretory vesicles in which it undergoes regulated exocytosis23. This confers both potential autocrine and endocrine roles to progranulin. A case in point is progranulin in microglia, in which basal progranulin levels and secretion are low; upon activation, microglial progranulin expression increases significantly and is likely to have an impact on neuronal function and synaptic density24. Thus, when examining progranulin levels, it is important to consider the source of progranulin and whether it is derived from an intracellular or an extracellular source, or some combination of both. Indeed, results from a large-scale study show poor correlations of progranulin levels in plasma and cerebrospinal fluid (CSF)25.
Progranulin as a neurotrophic factor
In addition to its potential roles as a growth factor, structural similarities between progranulin and other secreted proteins have led to the hypothesis that progranulin, and perhaps the cleaved granulins, may function as neurotrophic factors to promote neuronal survival and differentiation. In support of this idea, progranulin levels are reduced in the CSF of patients with FTD with GRN mutations25. Further, some reports found that human recombinant full-length progranulin and granulin E peptide (amino acids 494–594) can seemingly promote survival and neurite outgrowth in rat spinal motor neurons, cortical neurons and hippocampal neurons in vitro26–29, whereas exogenous progranulin rescues the neurite outgrowth phenotypes in Grn−/− neurons. By contrast, FTD-associated pathogenic mutations in progranulin interfere with the neurite outgrowth promoting activity27–29. Although these results suggest that progranulin functions as an exogenous trophic factor to promote neurite outgrowth and neuronal survival, it is interesting to note that knocking down progranulin in hippocampal neurons and SH-SY5Y cells also compromises their survival, neurite outgrowth and synapse formation28,30, further suggesting that progranulin functions via autocrine mechanisms. Indeed, progranulin has been shown to be co-transported with brain-derived neurotrophic factor (BDNF) in dendrites and axons of cultured hippocampal neurons in both anterograde and retrograde manners, and the secretion of progranulin seems to be modulated by neuronal activity23.
Despite these observations, one major challenge in validating the neurotrophic property of progranulin is the lack of evidence for a definitive receptor that interacts with progranulin and initiates signal transduction pathways to promote survival and neurite outgrowth in neurons. Although tumour necrosis factor (TNF) receptors and sortilin have been implicated as progranulin receptors31,32, evidence supporting these proteins as the bona fide receptors that transduce progranulin signals is still elusive. Sortilin traffics extracellular progranulin to the lysosome rather than serving as a signalling hub. Further, the reported interactions between progranulin and TNF receptor were not reproduced by co-immunoprecipitation or surface plasmon resonance33. Thus, it is unclear how progranulin could regulate neuronal survival and neurite outgrowth via the TNF-mediated mechanism. Curiously, recombinant progranulin continues to promote neurite outgrowth in hippocampal neurons lacking sortilin in vitro29, and inhibition of the granulin E–sortilin interaction does not diminish the ability of granulin E to promote neurite outgrowth34. Interestingly, a recent study showed that progranulin binds to ephrin type-A receptor 2 (EPHA2) on the cell surface and that such interaction activates the tyrosine kinase activity of EPHA2 and the downstream kinase AKT35. The interaction between progranulin and EPHA2 promotes capillary morphogenesis and the autoregulation of GRN mRNA, which does not require sortilin. Although these new results provide novel insights into the role of progranulin as a growth factor that promotes angiogenesis, the role of EPHA2 in neurite outgrowth and in neurodegeneration remains to be determined.
Progranulin dosage and disease
Several lines of evidence indicate that progranulin has gene-dosage-dependent effects on neurodegeneration. As previously indicated, GRN haploinsufficiency causes familial forms of FTD2,3. GRN mutations that are associated with neurodegenerative disease (for a list of mutations, see http://www.molgen.ua.ac.be/FTDmutations/) largely result in premature stop codons and nonsense mediated decay, although a few familial mutations impair secretion or produce full-length protein with loss of a key cysteine residue27,36,37. GRN-mutation carriers have serum levels of progranulin that are more than 50% lower than the levels in non-carriers38, and pathologically these patients develop FTLD with neuronal cytoplasmic protein aggregates enriched in hyperphosphorylated, cleaved TDP43 (REF. 39) (BOX 2).
Box 2. Frontotemporal dementia and frontotemporal lobar degeneration.
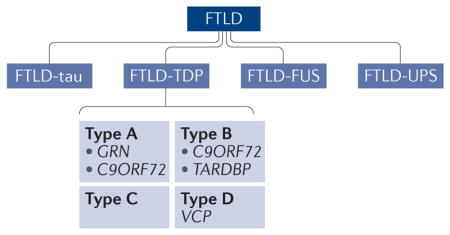
Frontotemporal dementia (FTD) describes a clinical condition of progressive neurodegenerative syndrome causing cognitive changes in the frontal executive, language and/or behavioural domains93. The neuropathology in frontotemporal lobar degeneration (FTLD) ranges from diffuse symmetric atrophy in bilateral frontal and temporal lobes to lobar atrophy that asymmetrically affects one cerebral hemisphere and to focal lesions that selectively affect the language areas. Microscopically, FTLD can be further classified based on the accumulation of misfolded proteins in neurons or glia, including tau (FTLD-tau), TAR DNA-binding protein 43(TDP43; which is encoded by TARDBP) (FTLD-TDP) and FUS (FTLD-FUS)94,95. A small number of patients with FTLD show no definitive proteinopathy but have clear evidence of abnormalities in the ubiquitin-proteasome system (FTLD-UPS).
Following the discovery of mutations in GRN (which encodes progranulin), it was reported that TDP43 is an abundant polyubiquitylated protein in the brain tissues of patients with FTLD-UPS4,5. Depending on the distribution and subcellular localization of TDP43 aggregates, FTLD-TDP can be further divided into four subtypes: type A (mutations in GRN and C9ORF72), type B (mutations in C9ORF72 and TARDBP), type C, and type D (mutation in valosin-containing protein (VCP)).
Individuals with GRN mutations typically present with behavioural variant FTD (characterized by apathy, irritability, social withdrawal, loss of empathy and compulsive, impulsive or inappropriate behaviour) or primary progressive aphasia (decreased fluency and word-finding difficulty) subtypes, although symptoms can be accompanied by parkinsonism and/or aspects of corticobasal degeneration96. The propensity for FTD associated with GRN mutations to cause clinically heterogeneous disease within families suggests additional genetic, epigenetic or environmental contributions to phenotype.
Individuals who carry two mutant GRN alleles, and thus completely lack progranulin and its cleavage products (functional null), develop a lysosomal storage disease that is now known as NCL (or ceroid neuronal lipofuscinosis-11 (CLN11))10,11,40. These patients have undetectable levels of progranulin in the plasma and develop neurological deficits, such as vision loss, myoclonic seizures and cerebellar ataxia between 20 and 30 years of age. Skin biopsy reveals lipofuscin accumulation in a vacuole-like organelle, confirming the NCL diagnosis. Interestingly, several studies show that cortical neurons and lymphoblasts from patients with FTLD with GRN mutations also contain a marked increase in lysosomal storage materials41,42. Together, these results further support the gene-dosage effects of progranulin deficiency in neurodegeneration.
The dosage-dependent effects of progranulin deficiency can also be detected in mouse models. For instance, heterozygous Grn+/− mice develop age-dependent social and behavioural deficits but show no evidence of gliosis or neurodegeneration43. By contrast, homozygous Grn−/− mice display premature death and age-dependent gliosis and lipofuscin deposits, recapitulating aspects of lysosomal storage disease24,44–47. Although the function of progranulin in the lysosomes is still unclear, these results have pivoted the field towards this organelle to understand how progranulin exerts its pleiotropic activities.
Trafficking of progranulin
How might progranulin deficiency cause lysosomal defects? During synthesis, progranulin is translated directly into the endoplasmic reticulum lumen where it associates with protein disulfide isomerases48, presumably for co-translational folding and, in humans, formation of its up to 44 disulfide bonds. From there, it is likely to be trafficked through the Golgi via transport vesicles, although the characteristics of these vesicles and the circumstances surrounding their secretion are incompletely understood. Once in the extracellular space, potential fates for progranulin include cleavage into granulins by an extracellular proteases12,14,49–51 or uptake into target cells via binding of its C terminus to sortilin31. Sortilin ultimately delivers progranulin via trafficking from endosomes to the lysosome, where it may be cleaved and/or degraded in a manner that regulates intracellular levels31,52 (FIG. 2).
Figure 2. Intracellular trafficking of progranulin and its role in lysosome biogenesis and function.

Progranulin has been described as a secreted protein that undergoes endocytosis and intracellular trafficking to reach the lysosomes in microglia, neurons and tumour cells. a | In wild-type cells, progranulin is present in the Golgi apparatus and is secreted probably via the secretory pathway. Another structurally related protein, prosaposin, heterodimerizes with progranulin, and regulates the transport and secretion of progranulin. Once released in the extracellular space, progranulin undergoes sortilin-mediated endocytosis to reach the lysosome via a mechanism that is poorly understood. Another alternative and highly plausible mechanism for progranulin to reach the lysosome is through the recycling endosome, multivesicular body (MVB), late endosome and eventually to early lysosome. The transcription of GRN (which encodes progranulin) and of many lysosomal genes is under the control of the transcription factor TFEB. b | In the absence of progranulin, mouse Grn−/−microglia show an expansion in the size of the late endosome and early lysosome but no defect in the expression of sortilin. Consistent with these findings, transcriptomic profiling in different regions of Grn−/−mouse brain, including the cerebral cortex, hippocampus and cerebellum, shows age-dependent increases in the expression of genes related to lysosomal functions and to the innate immune system, including a marked increase the production of complement proteins, C1qa and C3 products, by microglia. For reasons that are not entirely clear, loss of progranulin increases the transcriptional activity of TFEB, which promotes a marked upregulation of lysosomal genes. As a consequence, Grn−/−microglia and neurons exhibit features of lysosomal storage disease. Interestingly, over-expression of granulins can also lead to similar lysosomal defects. These results support the idea that a delicate balance of progranulin and granulin is crucial for the proper formation and function of lysosomes. ER, endoplasmic reticulum.
Recently, an alternative route to the lysosome involving another potentially secreted glycoprotein, prosaposin, has been described. Prosaposin and progranulin exhibit striking parallels. Like progranulin, prosaposin cleavage results in a set of cysteine-rich peptides, saposins53, which function in the lysosome to promote sphingolipid hydrolysis54 and utilize sortilin as a trafficking receptor55–57. A homozygous loss-of-function mutation in the gene encoding prosaposin leads to a lysosomal storage disease known as sphingolipidoses, which is similar to NCL in that it results from failed lysosomal macromolecular degradation54,58. Thus, in some ways, it is not surprising that progranulin and prosaposin form heterodimers59,60 and that prosaposin can regulate both intracellular and secreted progranulin levels61 (FIG. 2). In addition, prosaposin provides a direct route for progranulin to reach the lysosome, serving as a molecular bridge between progranulin and mannose-6-phosphate receptor (M6PR) or low-density lipoprotein receptor-related protein 1 (LRP1)59. Although much remains to be learned about the relationship between progranulin and prosaposin, their similarities and intertwined fates suggest a cellular coordination that is important in lysosome function and regulation9.
Lysosome and neurodegeneration
Despite the subcellular localization of progranulin, the exact functions of intracellular progranulin remain obscure. Evidence supporting a role for progranulin in the lysosome includes its delivery to the endolysosomal system by sortilin, its colocalization with lysosome-associated membrane protein 1 (LAMP1), as well as the finding that Grn−/− mice demonstrate increased reactivity for LAMP1 in certain brain regions31,62,63. These results support a role for progranulin in regulating the formation and function of lysosomes.
As a major site of macromolecular breakdown and recycling, lysosomes occupy an integral role in cellular proteostasis. Lysosome and autophagy dysfunction have been key players in neurodegenerative disease pathogenesis64. For example, mutations in the gene GBA (which encodes β-glucocerebrosidase) increase the risk of Parkinson disease and Lewy body disease65. Homozygous loss of GBA has been associated with a form of Gaucher disease, a juvenile onset lysosomal storage disease. Other variants of NCL-associated genes, including cathepsin D (CTSD) and probable cation-transporting ATPase 13A2 (ATP13A2; also known as PARK9) are also linked to AD and Parkinson disease, respectively66,67. Recently, attention has focused on the lysosome as a nutrient sensor and signal integration centre through the activity of mechanistic target of rapamycin (mTOR)68. Progranulin also has links to the master regulator of lysosome biogenesis, the transcription factor TFEB69,70. Upon translocation into the nucleus, TFEB activates its target genes to induce lysosomal biogenesis, which increases the degradative capacity of the cell (FIG. 2). Both progranulin and prosaposin are targets of TFEB69,71. In fact, the promoter of GRN contains TFEB binding sites, and progranulin expression is upregulated by TFEB overexpression69,72. As such, progranulin is highly co-regulated with other lysosomal genes72.
A recent study provided evidence that the cleaved granulins may play a part in lysosome function by impairing degradation of TDP43. In C. elegans, when granulins are expressed in the absence of the full-length protein, they increase levels of neuronal TDP43 and exacerbate its toxicity13. This first demonstration of a neurotoxic role for granulins supports a role for full-length progranulin and processed granulins converging in the lysosome. Based on these results, it is tempting to propose that a delicate balance of progranulin and granulins in the lysosome is crucial to regulate the biogenesis and function of this important intracellular organelle. Thus, both a complete absence of progranulin and excessive levels of granulins seem to negatively modulate lysosome function. Regardless, further studies exploring in more detail the relationship, regulation and potential biological interactions between the holoprotein and the granulins are warranted.
Progranulin and brain innate immunity
Given the prominent functions of lysosomes in phagocytic cells, including macrophages and microglia, it is not surprising that progranulin has been implicated in the function of the innate immune system. Progranulin has been shown to be a microglial chemoattractant73, whereas lower-molecular-mass granulins bind to, and potentiate signalling from, macrophage toll-like receptor 9 (TLR9), which is involved in innate immunity74. Several studies indicate that complete loss of progranulin causes an aberrant increase in phagocytosis and elevated production of pro-inflammatory cytokines in microglia and macrophages45,46,75. For instance, acute exposure to the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induces more robust microglial activation and neurodegeneration in Grn−/− mice and microglia-specific conditional Grn-knockout (CD11b–Cre;Grnfl/fl) mice45. In this model, many Grn−/− and CD11b–Cre;Grnfl/fl microglial cells exhibit prominent reactive features, as if they are actively engulfing injured neurons. These results support the idea that progranulin is normally required to suppress excessive microglial activation in an acute neural injury paradigm. Interestingly, progranulin seems to have similar roles in suppressing microglial activation during ageing. Transcriptomic profiling of several brain regions from a large cohort of ageing Grn+/+, Grn+/− and Grn−/− mice shows a prominent age-dependent upregulation of genes that are related to lysosomal functions and innate immunity in Grn−/− mouse brain24. Most genes that are dysregulated in the absence of progranulin belong to microglia, suggesting an essential role of progranulin in suppressing microglial activation in the ageing brain. Interestingly, in wild-type microglia, progranulin is localized to late endosomes, recycling endosomes and early lysosomes24, suggesting that progranulin regulates the formation and/or trafficking of these compartments (FIG. 2). By contrast, Grn−/− microglia show a marked increase in the size and number of lysosomes24. Consistent with a role for progranulin in regulating intracellular trafficking from endosomes to lysosomes, Grn−/− microglia exhibit increased synaptic pruning activity and preferentially remove inhibitory synapses in the ventral thalamus24.
How might progranulin deficiency promote microglial activation in Grn−/− mice during ageing? Previous studies showed that loss of progranulin leads to the upregulation of pro-inflammatory cytokines, including TNF and IL-6, in microglia45,46. In addition, some complement genes, including C1qa, C1qb, C1qc and C3, were upregulated in Grn−/− mice before the onset of neurodegenerative features. System-level analysis of these dysregulated genes shows extensive functional interactions between the innate immunity genes and lysosomal genes. As the classical complement pathway is a well-recognized arm of the innate immunity surveillance system76, these results suggest that aberrant activation of the complement pathway has key mechanistic contributions to the excessive synaptic pruning observed in Grn−/− mice (FIG. 3). Indeed, genetic deletion of C1qa reduces synaptic pruning activity in Grn−/− microglia and protects against the excessive synaptic loss that disrupts sensorimotor integration in the thalamocortical neural circuit. Taken together, these results suggest that neurodegeneration due to progranulin deficiency may be partially caused by altered lysosome function and subsequent aberrant microglial activation. This increased activity then results in overly aggressive synapse removal, thus suggesting a crucial role for microglia as potential drivers of neurodegeneration (FIG. 3).
Figure 3. Progranulin deficiency disrupts glia–neuron homeostasis and promotes neurodegeneration during ageing.

Emerging evidence indicates that, under physiological conditions, both microglia and astrocytes are required to maintain homeostasis in the brain. Results from transcriptomic and functional analyses of progranulin-deficient (Grn−/−) mouse brains support the idea that progranulin functions as a ‘brake’ to suppress excessive microglial activation. In the ageing process, Grn−/− microglia show age-dependent activation, characterized by enlarged cytoplasm and retracted processes, and a robust activation of the innate immunity system by increased production of complement proteins (such as C1qa and iC3b, the inactive product of cleaved C3b) and pro- inflammatory cytokines, which promotes synaptic pruning and causes cytotoxicity in neurons, respectively. Furthermore, microglial activation in Grn−/−brains is accompanied by a marked increase in Grn−/−astrocytes. These results suggest that loss of progranulin in microglia and astrocytes disrupts the homeostatic balance between these two cell types to promote neurodegeneration. Finally, loss of progranulin in neurons has also been shown to increase cellular stress and lysosomal defects that can further promote TAR DNA-binding protein 43 (TDP43) proteinopathy and cytotoxicity.
One prominent and unexpected neurodegenerative feature in Grn−/− mice is the preferential loss of inhibitory synapses in the thalamocortical circuit. Although both excitatory and inhibitory synapses are tagged with complements, only inhibitory synapses show an age-dependent loss24. As a consequence, the thalamocortical circuit in Grn−/− mice exhibits hyperexcitability that contributes to the excessive grooming behaviour in these mice. Consistent with the prerequisite role of complement activation in synaptic pruning, removal of C1qa protects the loss of inhibitory synapses, mitigates the hyperexcitability in the thalamocortical circuit, reduces excessive grooming and prolongs the survival of Grn−/−C1qa−/− mice. The microglial dysfunction and hyper-excitability in the thalamocortical circuit seen in Grn−/− mice during ageing bring important insights into the pathogenesis of human disease. Indeed, patients with FTLD with GRN mutations show a progressive increase in levels of complements C1qa and C3 in the CSF, as well as intense microglial infiltration in the frontal cortex. Future studies should determine whether GRN carriers also exhibit evidence of hyperexcitability in the thalamocortical circuit.
Therapeutics discovery
In humans and model organisms, age has been observed to increase progranulin levels25,75. In addition, stressful stimuli, such as hypoxia, acidosis, hyperosmolarity and inhibition of lysosomal function by artificial alkalinization52, seem to increase progranulin production and secretion72,77,78, alter its glycosylation18 and possibly promote its cleavage12,14,50,51. Low circulating levels of progranulin in GRN carriers38 suggest that progranulin expression from the wild-type allele might be a potential therapeutic target. Consistent with this, Cenik and colleagues79 designed a luciferase-based screen and identified the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) as an enhancer of progranulin expression. However, dose–response relationship suggests that SAHA reaches a ‘ceiling’ effect in upregulating progranulin mRNA and protein levels. These results imply that progranulin expression is tightly regulated by epigenetic modifications of the GRN locus and by additional post-translational mechanisms.
Indeed, it has been suggested that prosaposin forms heterodimers with progranulin and regulates the intracellular and secreted levels of progranulin59–61 (FIG. 2). In addition, secretory leukocyte protease inhibitor (SLPI) seems to directly bind to progranulin to prevent its cleavage14. Similarly, lysosome alkalinization increases expression of progranulin, presumably by altering its transport, degradation or cleavage into granulins52. Finally, several microRNAs, including miR-659, miR-29b and miR-107 (REFS 80–83), have been shown to regulate progranulin protein level by altering translation and/or mRNA stability. Individuals who are TT homozygotes at the rs5848 single-nucleotide polymorphism (SNP) locus show tighter miR-659 binding, which leads to a decrease in progranulin levels and to a 3.2-fold increase in risk of FTLD with TDP43 accumulation80. Additional understanding of how progranulin expression and cleavage are regulated may aid in future development of therapeutic strategies.
Future perspectives
The discovery that progranulin may affect formation and function of intracellular organelles and activate microglial innate immunity in the CNS provides new and exciting insights into disease mechanisms and potential therapeutic targets for FTLD. First, the profound microglial activation phenotype in the brains of Grn−/− mice and patients with FTLD with GRN mutations raises the intriguing possibility that progranulin-deficient microglia may disrupt the homeostatic balance of glia–neuron interactions and promote neurodegeneration in the ageing brain (FIG. 3). Although this can be achieved by activating innate immunity pathways, including pathways related to the complement system and pro-inflammatory cytokines in progranulin-deficient microglia, it is possible that progranulin-deficient microglia could recruit and activate astrocytes that may have additional cytotoxic effects on neurons. In support of this idea, a recent study shows that in a lipopolysaccharide-induced neuroinflammation model, microglia can release IL-1α, TNF and C1qa to activate astrocytes84. In future studies, it will be important to determine whether Grn−/− microglia use similar or entirely different mechanism to activate Grn−/− astrocytes. Alternatively, loss of progranulin may convert astrocytes into a more aggressive phenotype such that Grn−/− microglia and Grn−/− astrocytes cooperatively promote neurodegeneration (FIG. 3).
Second, another important question regarding the fundamental role of progranulin in lysosomes is how this function affects cells beyond microglia and macrophages. Given the presence of progranulin in many cell types, it is likely that progranulin may have broader roles in regulating lysosomes in other cell types, including neurons, with consequences on proteostasis and stress response. In support of this idea, neurons in Grn−/− mice show pathological features similar to those seen in NCL44–47. Similar lysosomal phenotypes have also been detected in patients with FTLD with GRN mutations41,42. Future studies are required to determine whether simultaneous losses of progranulin in microglia, astrocytes and neurons have synergistic effects to promote neurodegeneration (FIG. 3).
Third, the highly evolutionarily conserved granulin repeat motif and the diverse downstream effects of progranulin loss, deficiency and excess suggest that progranulin and granulin have broader roles in ageing and proteostasis outside of the CNS. Indeed, a recent study in African turquoise killifish shows that a variant in the killifish grn gene is in a region that is genetically linked to lifespan regulation85. Although multiple other potential lifespan genes are also present in that region, the grn gene is a strong candidate to underlie lifespan differences not only because it is in the region that is the most clearly associated with differences in lifespan but also because it harbours coding variants in the granulin repeat domain analogous to FTD variants. Clearly, precise maintenance of progranulin levels is important, as demonstrated by both its disease associations and the multiple layers of regulation around gene expression (promoter and microRNAs), modification (disulfide bonding and glycosylation), trafficking (sortilin, prosaposin and M6PR), processing (into granulins) and degradation. Evidence even exists supporting a role for progranulin in phagocytosis and removal of amyloid plaques86.
Last, progranulin is certainly not alone as a neurodegenerative disease gene implicated in microglial functions. In fact, abnormal activation of innate immunity is a common feature of several neurodegenerative diseases. Although several clinical studies have identified rare GRN-mutation carriers with clinical, imaging and neuropathological evidence of AD87–89, it remains unclear how patients with those mutations or other GRN variants have a higher propensity to develop AD pathology. Given the complex neurodegenerative features in patients with AD, future studies using a larger series of GRN-mutation carriers and quantitative measurements will be required to provide more mechanistic insights into how reduced progranulin levels may affect the development of amyloid and tau pathology. In addition to the potential role of progranulin in AD pathology, it is possible that reduced progranulin level may promote proteostasis defects via related processes such as stress response and protein clearance.
Conclusion
Although regulation of progranulin and its cleavage products affords great therapeutic potential in multiple disease modalities, clarity regarding how they modulate growth, innate immunity and proteostasis is needed. New model systems to explore these questions, as well as the role of progranulin and granulin in ageing and potentially other yet-to-be identified disorders, would also benefit the field. Efforts to boost progranulin levels have somewhat outpaced our knowledge as to its precise responsibilities in the lysosome in phagocytic and other cell types. Further, little is known regarding how cleaved granulins are produced and regulated, and about their function. Such knowledge could shift efforts, for example, away from enhancing progranulin levels in microglia towards increasing neuronal progranulin. Alternatively, an approach such as inhibiting progranulin cleavage into granulins could be explored. Until a more cogent understanding of progranulin and granulin function is gained, efforts to manipulate their levels for clinical application should proceed cautiously. Nonetheless, progranulin and granulin remain complex yet integral factors in human health with pleiotropic downstream effects that aptly illustrate how the precise regulation of a protein and its cleavage products can contribute to cellular homeostasis and prevention of disease.
Acknowledgments
The authors thank W. W. Seeley, E. H. Bigio and I. R. Mackenzie for sharing the neuropathology findings in patients with frontotemporal lobar degeneration with mutations in the gene encoding progranulin. This work has been supported by US Public Health Service grants NS095257 (A.W.K.) and NS098516 (E.J.H.), the Tau Consortium (A.W.K.), the Consortium for Frontotemporal Dementia Research (E.J.H.), VA Merit Award BX002978 (E.J.H.), and the Glenn Foundation for Medical Research (A.M., P.P.S. and A.B.)
Glossary
- Deficiency
A nonspecific term used to describe both null and heterozygous loss-of-function alleles.
- Frontotemporal dementia (FTD)
A clinical term describing a group of disorders caused by progressive neuron loss in the frontal and/or temporal lobes of the brain. Symptoms typically manifest as personality, behaviour and language changes and can be accompanied by motor features (for additional details, see BOX 2).
- Frontotemporal lobar degeneration (FTLD)
Umbrella term for a group of neurodegenerative conditions that affect primarily, or first, the frontal and/or temporal lobes of the brain, and that are characterized by a diverse array of neuronal inclusions comprising tau, TAR DNA-binding protein 43 or FUS.
- Granulin
An approximately 60 amino-acid motif characterized by highly conserved cysteines that are arranged singly or in pairs, which form six disulfide bonds.
- Haploinsufficiency
A loss-of-function mutation in one gene allele.
- Innate immunity
A relatively nonspecific part of the immune response, consists of physical barriers (such as skin and mucosa), phagocytic cells (microglia, macrophages, dendritic cells, and so on) and circulating factors (tumour necrosis factor, interferon and complement) that co-ordinately provide protection from invading pathogens, and participate in repair and maintenance of cells and organ systems.
- Lysosomal storage disease
A group of approximately 50 metabolic disorders that result from defective lysosomal degradation of cellular constituents, primarily affecting terminally differentiated neurons. Symptoms include seizure, blindness and developmental delay.
- Lysosome
A subcellular organelle found in eukaryotes containing cathepsins and other acid hydrolases that are responsible for degrading and recycling cellular constituents.
- Microglia
A type of non-neuronal support cells found in the CNS. Considered the resident macrophages of the brain and spinal cord, microglia are part of the innate immune system and originate from yolk sac progenitors.
- Neuronal ceroid lipofuscinosis (NCL)
A relatively rare subset of lysosomal storage diseases in which protein–lipid adducts known as lipofuscin accumulate in various tissues. Symptoms include blindness, epilepsy and cognitive decline.
- Nonsense mediated decay
A process that induces degradation of mRNAs that contain premature translation-termination codons, and constitutes an mRNA-surveillance mechanism that prevents the synthesis of truncated, potentially toxic, proteins.
- Null
A situation in which both alleles of a gene are mutated, leading to complete loss of gene expression.
- Overexpression
Excessive expression of a gene beyond the normal biologically defined levels. It can be found in pathological conditions such as cancer or it can be induced as part of experimental manipulation of a gene.
- Progranulin
Also known as proepithelin, granulin-epithelin precursor, acrogranin, PC cell-derived growth factor and epithelial transforming growth factor, this precursor protein has pleotropic effects in neurons owing to its effects on lysosomal function.
- Proteostasis
A linguistic blend of ‘protein’ and ‘homeostasis’ that refers to the cellular processes regulating the production, folding, trafficking and degradation of proteins.
- TAR DNA-binding protein 43 (TDP43)
The protein encoded by the TARDBP gene. TDP43 acts as a transcriptional regulator that shuttles between the nucleus and the cytoplasm. In certain forms of frontotemporal lobar degeneration, including those due to mutations in the genes encoding progranulin and C9ORF72, it forms intra-cytoplasmic aggregates of hyperphosphorylated, cleaved proteins.
Footnotes
Competing interests statement
The authors declare no competing interests.
Contributor Information
Aimee W. Kao, Department of Neurology, University of California San Francisco, 675 Nelson Rising Lane, San Francisco, California 94158, USA
Andrew McKay, Department of Genetics, Stanford University, 300 Pasteur Drive, Stanford, California 94305, USA.
Param Priya Singh, Department of Genetics, Stanford University, 300 Pasteur Drive, Stanford, California 94305, USA.
Anne Brunet, Department of Genetics, Stanford University, 300 Pasteur Drive, Stanford, California 94305, USA.
Eric J. Huang, Department of Pathology, University of California San Francisco and Pathology Service 113B, San Francisco VA Medical Center, 513 Parnassus Avenue, San Francisco, California 94143, USA
References
- 1.Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 2.Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 3.Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 4.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 5.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 6.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruts M, Kumar-Singh S, Van Broeckhoven C. Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr Alzheimer Res. 2006;3:485–491. doi: 10.2174/156720506779025251. [DOI] [PubMed] [Google Scholar]
- 9.Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem. 2012;287:32298–32306. doi: 10.1074/jbc.R112.399170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith KR, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almeida MR, et al. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. 2016;41:200.e1–201.e5. doi: 10.1016/j.neurobiolaging.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Kessenbrock K, et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salazar N, et al. The progranulin cleavage products, granulins, exacerbate TDP-43 toxicity and increase TDP-43 levels. J Neurosci. 2015;35:9315–9328. doi: 10.1523/JNEUROSCI.4808-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 15.Tolkatchev D, et al. Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 2008;17:711–724. doi: 10.1110/ps.073295308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palfree RG, Bennett HP, Bateman A. The evolution of the secreted regulatory protein progranulin. PLoS ONE. 2015;10:e0133749. doi: 10.1371/journal.pone.0133749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavergne V, Taft RJ, Alewood PF. Cysteine-rich mini-proteins in human biology. Curr Top Med Chem. 2012;12:1514–1533. doi: 10.2174/156802612802652411. [DOI] [PubMed] [Google Scholar]
- 18.Kanazawa M, et al. Multiple therapeutic effects of progranulin on experimental acute ischaemic stroke. Brain. 2015;138:1932–1948. doi: 10.1093/brain/awv079. [DOI] [PubMed] [Google Scholar]
- 19.Songsrirote K, Li Z, Ashford D, Bateman A, Thomas-Oates J. Development and application of mass spectrometric methods for the analysis of progranulin N-glycosylation. J Proteomics. 2010;73:1479–1490. doi: 10.1016/j.jprot.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/ granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–599. doi: 10.1002/dvdy.10341. [DOI] [PubMed] [Google Scholar]
- 21.Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petoukhov E, et al. Activity-dependent secretion of progranulin from synapses. J Cell Sci. 2013;126:5412–5421. doi: 10.1242/jcs.132076. [DOI] [PubMed] [Google Scholar]
- 24.Lui H, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165:921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson AM, et al. Progranulin protein levels are differently regulated in plasma and CSF. Neurology. 2014;82:1871–1878. doi: 10.1212/WNL.0000000000000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Damme P, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, et al. Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem. 2010;112:1305–1315. doi: 10.1111/j.1471-4159.2009.06546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao X, et al. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3β. Protein Cell. 2010;1:552–562. doi: 10.1007/s13238-010-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gass J, et al. Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener. 2012;7:33. doi: 10.1186/1750-1326-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tapia L, et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci. 2011;31:11126–11132. doi: 10.1523/JNEUROSCI.6244-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu F, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang W, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332:478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, et al. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J Neurosci. 2013;33:9202–9213. doi: 10.1523/JNEUROSCI.5336-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Muynck L, et al. The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol Aging. 2013;34:2541–2547. doi: 10.1016/j.neurobiolaging.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 35.Neill T, et al. EphA2 is a functional receptor for the growth factor progranulin. J Cell Biol. 2016;215:687–703. doi: 10.1083/jcb.201603079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gass J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 37.Shankaran SS, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- 38.Finch N, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64:1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 40.Carcel-Trullols J, Kovacs AD, Pearce DA. Cell biology of the NCL proteins: what they do and don’t do. Biochim Biophys Acta. 2015;1852:2242–2255. doi: 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 41.Ward ME, et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aah5642. [DOI] [PMC free article] [PubMed]
- 42.Gotzl JK, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014;127:845–860. doi: 10.1007/s00401-014-1262-6. [DOI] [PubMed] [Google Scholar]
- 43.Filiano AJ, et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33:5352–5361. doi: 10.1523/JNEUROSCI.6103-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed Z, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggests a role for progranulin in successful aging. Am J Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martens LH, et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–3959. doi: 10.1172/JCI63113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin F, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin F, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010;24:4639–4647. doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Almeida S, Zhou L, Gao FB. Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS ONE. 2011;6:e26454. doi: 10.1371/journal.pone.0026454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butler GS, Dean RA, Tam EM, Overall CM. Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of matrix metalloproteinase-14 (MT1-MMP) mediated membrane protein shedding. Mol Cell Biol. 2008;28:4896–4914. doi: 10.1128/MCB.01775-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bai XH, et al. ADAMTS-7, a direct target of PTHrP, adversely regulates endochondral bone growth by associating with and inactivating GEP growth factor. Mol Cell Biol. 2009;29:4201–4219. doi: 10.1128/MCB.00056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suh HS, Choi N, Tarassishin L, Lee SC. Regulation of progranulin expression in human microglia and proteolysis of progranulin by matrix metalloproteinase-12 (MMP-12) PLoS ONE. 2012;7:e35115. doi: 10.1371/journal.pone.0035115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Capell A, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kishimoto Y, Hiraiwa M, O’Brien JS. Saposins: structure, function, distribution, and molecular genetics. J Lipid Res. 1992;33:1255–1267. [PubMed] [Google Scholar]
- 54.Meyer RC, Giddens MM, Coleman BM, Hall RA. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014;1585:1–12. doi: 10.1016/j.brainres.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lefrancois S, Zeng J, Hassan AJ, Canuel M, Morales CR. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 2003;22:6430–6437. doi: 10.1093/emboj/cdg629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hassan AJ, Zeng J, Ni X, Morales CR. The trafficking of prosaposin (SGP-1) and GM2AP to the lysosomes of TM4 Sertoli cells is mediated by sortilin and monomeric adaptor proteins. Mol Reprod Dev. 2004;68:476–483. doi: 10.1002/mrd.20096. [DOI] [PubMed] [Google Scholar]
- 57.Zeng J, Racicott J, Morales CR. The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp Cell Res. 2009;315:3112–3124. doi: 10.1016/j.yexcr.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 58.Hulkova H, et al. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum Mol Genet. 2001;10:927–940. doi: 10.1093/hmg/10.9.927. [DOI] [PubMed] [Google Scholar]
- 59.Zhou X, et al. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol. 2015;210:991–1002. doi: 10.1083/jcb.201502029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicholson AM, et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat Commun. 2016;7:11992. doi: 10.1038/ncomms11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiesberger T, et al. Cellular uptake of saposin (SAP) precursor and lysosomal delivery by the low density lipoprotein receptor-related protein (LRP) EMBO J. 1998;17:4617–4625. doi: 10.1093/emboj/17.16.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gowrishankar S, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci USA. 2015;112:E3699–E3708. doi: 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun. 2014;2:78. doi: 10.1186/s40478-014-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 65.Aharon-Peretz J, Badarny S, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype–genotype correlation. Neurology. 2005;65:1460–1461. doi: 10.1212/01.wnl.0000176987.47875.28. [DOI] [PubMed] [Google Scholar]
- 66.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 67.Schuur M, et al. Cathepsin D gene and the risk of Alzheimer’s disease: a population-based study and meta-analysis. Neurobiol Aging. 2011;32:1607–1614. doi: 10.1016/j.neurobiolaging.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 68.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 70.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience. 2013;250:8–19. doi: 10.1016/j.neuroscience.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 72.Belcastro V, et al. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 2011;39:8677–8688. doi: 10.1093/nar/gkr593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pickford F, et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol. 2011;178:284–295. doi: 10.1016/j.ajpath.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park B, et al. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity. 2011;34:505–513. doi: 10.1016/j.immuni.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 75.Kao AW, et al. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci USA. 2011;108:4441–4446. doi: 10.1073/pnas.1100650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 77.Guerra RR, Kriazhev L, Hernandez-Blazquez FJ, Bateman A. Progranulin is a stress-response factor in fibroblasts subjected to hypoxia and acidosis. Growth Factors. 2007;25:280–285. doi: 10.1080/08977190701781222. [DOI] [PubMed] [Google Scholar]
- 78.Holler CJ, et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 2016;11:46. doi: 10.1186/s13024-016-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cenik R, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. 2011;286:16101–16108. doi: 10.1074/jbc.M110.193433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rademakers R, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsiung GY, Fok A, Feldman HH, Rademakers R, Mackenzie IR. rs5848 polymorphism and serum progranulin level. J Neurol Sci. 2011;300:28–32. doi: 10.1016/j.jns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiao J, Herl LD, Farese RV, Gao FB. Micro RNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE. 2010;5:e10551. doi: 10.1371/journal.pone.0010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang WX, et al. miR-107 regulates granulin/ progranulin with implications for traumatic brain injury and neurodegenerative disease. Am J Pathol. 2010;177:334–345. doi: 10.2353/ajpath.2010.091202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valenzano DR, et al. The African turquoise killifish genome provides insights into evolution and genetic architecture of lifespan. Cell. 2015;163:1539–1554. doi: 10.1016/j.cell.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Minami SS, et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat Med. 2014;20:1157–1164. doi: 10.1038/nm.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kelley BJ, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging. 2007;30:739–751. doi: 10.1016/j.neurobiolaging.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perry DC, et al. Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol. 2013;70:774–778. doi: 10.1001/2013.jamaneurol.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rademakers R, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C→T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 90.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Herrero J, et al. Ensembl comparative genomics resources. Database (Oxford) 2016;2016:bav096. doi: 10.1093/database/bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Singh PP, et al. On the expansion of “dangerous” gene repertoires by whole-genome duplications in early vertebrates. Cell Rep. 2012;2:1387–1398. doi: 10.1016/j.celrep.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 93.Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci. 2011;45:330–335. doi: 10.1007/s12031-011-9538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mackenzie IR, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mackenzie IR, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;7:965–974. doi: 10.1016/S1474-4422(08)70194-7. [DOI] [PubMed] [Google Scholar]