

Abstract
Cancer and stromal cells, which include (cancer‐associated) fibroblasts, adipocytes, and immune cells, constitute a mixed cellular ecosystem that dynamically influences the behavior of each component, creating conditions that ultimately favor the emergence of malignant clones. Ovarian cancer cells release cytokines that recruit and activate stromal fibroblasts and immune cells, so perpetuating a state of inflammation in the stroma that hampers the immune response and facilitates cancer survival and propagation. Further, the stroma vasculature impacts the metabolism of the cells by providing or limiting the availability of oxygen and nutrients. Autophagy, a lysosomal catabolic process with homeostatic and prosurvival functions, influences the behavior of cancer cells, affecting a variety of processes such as the survival in metabolic harsh conditions, the invasive growth, the development of immune and chemo resistance, the maintenance of stem‐like properties, and dormancy. Further, autophagy is involved in the secretion and the signaling of promigratory cytokines. Cancer‐associated fibroblasts can influence the actual level of autophagy in ovarian cancer cells through the secretion of pro‐inflammatory cytokines and the release of autophagy‐derived metabolites and substrates. Interrupting the metabolic cross‐talk between cancer cells and cancer‐associated fibroblasts could be an effective therapeutic strategy to arrest the progression and prevent the relapse of ovarian cancer.
Keywords: autophagy, cancer‐associated fibroblast, cytokines, ovarian cancer
Abbreviations
- ASMA
alpha‐Smooth Muscle Actin
- CAF
cancer‐associated fibroblast
- ECM
Extracellular matrix
- EOC
Epithelial Ovarian Cancer
- FAP
fibroblast activating protein
- LPA
lysophosphatidic acid
- MSC
mesenchymal stem cell
1. INTRODUCTION
Ovarian cancer is the seventh most common cancer in women, with the incidence of around 9.4 and 5.0 age‐standardized rate per 100,000 in developed and developing countries, respectively.1, 2 Despite the progress in drug discovery and improvement in the management, ovarian cancer remains the leading cause of death from gynecological cancer.2, 3 Worldwide, as many as 240,000 women are diagnosed with ovarian cancer, and approximately half of them die each year.2 Early diagnosis could give a high probability of recovery, yet it is difficult because of the unspecificity of the symptoms in the early phases of the disease. On the other hand, late stage ovarian cancers are aggressive, featuring rapid growth, dissemination, chemo/radio resistance, and recurrence.4 Ovarian cancer progression denotes not only the emergence of more aggressive clones, but also reflects the dynamic changes in the microenvironment, in its cellular and molecular composition, as well as in the heterotypic interactions between tumor and stromal cells.5 Alike other solid tumors, the ovarian cancer tissue is composed of epithelial‐derived cancer cells embedded in a tumor stroma, which consists of heterogeneous cell types and a mixture of amorphous components.6 The latter forms the extracellular matrix (ECM) and includes structural and specialized proteins and proteoglycans. Various cell types are found in the stroma of ovarian cancer, including immune and inflammatory cells (such as lymphocytes, macrophages, and mastocytes), endothelial cells, adipocytes, and the “cancer‐associated fibroblasts” (CAFs).7, 8 The latter definitely represent the major cell component in the activated stroma.
For long time the potential role in carcinogenesis of stromal cells has been neglected, as they were regarded just as part of an inflammatory reaction induced by necrotic cancer cells. It is now recognized that the stroma composition and architecture, in terms of vascularization, type of cells, and of their secretion, play a role in the establishment and progression of cancer cells. This fact was appreciated since the “seed and soil” paradigm of metastasis formation formulated by Stephen Paget, in which the stroma (the soil) is a determinant factor in allowing the cancer cell (the seed) to take root.9, 10 It is now well established that the stroma contributes to ovarian tumorigenesis and progression.11, 12
An important notion is that the stroma itself, in terms of cell and molecular composition and architecture, is subject to dynamic changes that parallel the growth and progression of the tumor, which suggests that the two compartments coevolve and influence each other during cancer progression.13
The bidirectional communication between stromal and ovarian cancer cells has an impact on the metabolism, and consequently on the behavior, of the various actors.14 Autophagy is one such metabolic process that occurs in both cancer and stromal cells and that is influenced by and in turn influences the microenvironment.15 Autophagy is a lysosome‐driven process of macromolecules and organelles degradation that plays a fundamental role in cell and tissue homeostasis.16 In cancer cells, autophagy is clearly deregulated and contributes to abnormal growth and development of metastasis and of immune, radio, and chemo resistance.17, 18 Autophagy plays a role in cancer stemness19, 20 and in cell reprogramming,21 as well as in tumor cell dormancy22 and in tumor invasion.23 In the last decade, it has become evident that autophagy in stromal cells, particularly in CAFs, adipocytes, and immune cells also has a role in the carcinogenesis process, since it is involved in the secretion of cytokines and of other soluble factors that impinge on the metabolism of epithelial cancer cells. Recent studies support the view that a “metabolic symbiosis” exists between CAFs and cancer cells. In this model, cancer cells induce a rise of autophagy in CAFs, which in turn provide the cancer cells with energetic metabolic substrates and so reducing their autophagy needs.24, 25 Thus, the metabolic cross‐talk between stromal and cancer cells reciprocally affects autophagy regulation,26 which might reflect in behavioral changes driving cancer relapse and metastasis as we will see later.
In this review, we present the “state of art” of the current knowledge on the role of stromal cells (particularly CAFs) and of their cytokines in the development and progression of ovarian cancer, with an emphasis on the role played by autophagy in the cross‐talk between epithelial cancer cells and CAFs. This latter aspect has not been studied extensively in ovarian cancer, yet the data available support the view that, as demonstrated in other cancer models, autophagy is differentially regulated in ovarian cancer and in CAFs. Understanding how CAFs and ovarian cancer cells reprogram each other's metabolism and, in particular, how autophagy is modulated in both these cells might shed lights on novel signaling pathways that could be targeted for the treatment of ovarian cancer.
2. THE PROGNOSTIC IMPACT OF STROMA IN OVARIAN CANCER
During progression, ovarian cancer cells do not completely dedifferentiate and still preserve some morphological characteristics reminiscent of the anatomical region of origin. On this ground, the pathologists classify ovarian cancer in four main histotypes (serous, clear cell, endometrioid, and mucinous), featuring a diverse grade of the stroma component. Recently, a classification model of ovarian carcinomas in type I and type II, based on morphological, genetic, and clinical characteristics has been proposed.27, 28, 29 Type I tumors are clinically indolent, and comprise three subtypes: (1) low‐grade serous carcinomas; (2) endometriosis‐related tumors that include low‐grade endometrioid, clear cell, and seromucinous carcinomas; and (3) mucinous carcinomas and malignant Brenner tumors. These tumors apparently originate as benign hyperproliferative lesions in extra‐ovary tissues, and later implant on the ovary where eventually undergo malignant transformation. Type II tumors are clinically very aggressive, and include high‐grade serous carcinomas (accounting by far for the majority of ovarian cancers), high‐grade endometrioid carcinomas, undifferentiated carcinomas, and carcinosarcomas. High‐ and low‐grade serous carcinomas originate in the fimbriated end of the fallopian tube and, subsequently, involve the ovary. However, these tumors show a very different genetic landscape and behavior: high‐grade serous ovarian carcinomas generally bear TP53 mutations and BRCA1/2 epi‐mutations and are chemosensitive, while low‐grade serous ovarian carcinomas generally bear Ki‐RAS and B‐RAF mutations and are chemoresistant.28, 29, 30
Epithelial ovarian cancer cells cohabit with a variety of stromal cells embedded in the ECM to form an organoid‐like structure.6 In general, low‐grade serous and mucinous histotypes present with a higher content of the stroma component compared to high‐grade serous and endometrioid carcinomas.31 Characteristically, the clear cell histotype presents with an intense expansion of the ECM with a low infiltration of the stromal cell component.31
Depending on its composition, the stroma can either impede the neoplastic growth or create the conditions for cell growth and cell migration of cancer cells. Although differing from that in normal ovary tissue, the stroma in ovarian cancer may vary in the expression of genes and production of proteins that ultimately affect tumor growth and invasion.32 Transcriptome profiling of genes encoding signaling molecules and cognate receptors in ovarian cancer cells and matched stromal cells isolated from patients revealed the existence of two distinct stromal compartments, one permissive and one less prone to support cancer growth.32 Noteworthy, the former was associated with grade 3 and the latter with grade 2 ovarian serous adenocarcinomas.32 Consistent with the above findings, when coinjected with ovarian cancer cells in nude mice, stromal cells isolated from normal ovary were shown to restrict the tumor growth and stromal cells from cancer tissues instead promoted ovarian cancer progression.33
A few studies correlate the level of reactive stroma with ovarian cancer progression. It has been reported that patients bearing an ovarian carcinoma with a high content of stroma present with a high pathologic stage at diagnosis,34 and display a reduced overall survival and poor prognosis independently from the histotype.35 Along these lines, the extent of CAFs infiltration in the ovarian cancer stroma was significantly correlated with lymph node and omentum metastases and increased number of lymphatic and blood vessels, typical signs of cancer progression.36 Consistently, an increased content of certain ECM components produced by CAFs, such as the glycosaminoglycan hyaluronic acid and its partner glycoprotein versican, was associated with increased microvessel density, platinum resistance, and poorer overall and progression‐free survival in ovarian cancer patients.37, 38, 39
3. THE ROLE OF INFLAMED STROMA IN OVARIAN TUMORIGENESIS
The impressive and intriguing similarity in the cellular and molecular composition of the stroma formed during the healing of a wound and of the stroma surrounding epithelial cancer has been known for a long time. The wound repair by secondary intention is accompanied by a sequence of events in the stroma that include clotting, neovascularization, recruitment of macrophages and lymphocytes, activation of myofibroblasts, and release of pro‐inflammatory cytokines (e.g., TNF‐α, TGF‐β, IL‐6, and IL‐1β). This sequence of events causes an intense remodeling of the ECM, with dynamic alternation of degradation and synthesis of fibrous proteins, eventually leading to the formation of a scar. Strikingly, a similar scenario occurs during the evolution of infiltrative epithelial tumors, with the exception that cancer cell proliferation and necrosis maintain a pro‐inflammatory environment that eventually results in a desmoplastic stroma.40 This analogy has suggested to Dvorak the paradigm that “cancer is a wound that never heals.”40, 41 This paradigm perfectly fits with the inflammatory process that associates with ovarian cancer development and progression.42, 43, 44 At each ovulation, the follicle wall ruptures to release the ovum and is thereafter repaired through controlled inflammatory events much alike the wound healing.45 During this process, macrophages and fibroblasts are recruited to the wounded epithelial surface, and an enormous amount of cytokines/chemokines and matrix‐remodeling enzymes (including prostaglandins, bioactive eicosanoids, plasminogen activators, collagenases, interleukins,33 tumor necrosis factor α [TNF‐α], and various growth factors) are released on site.46, 47, 48 Therefore, the ovarian surface epithelial cells adjacent to the site of ovulation are exposed to an inflammatory and oxidative environment that enhances the risk of malignant transformation.49, 50 In addition, the chemokines and cytokines released in this context attract and promote the adhesion of extra‐ovarian malignant cells to the ovary.51 Thus, the enhanced risk of ovarian carcinogenesis associated with repetitive ovulation would arise from the continuous creation of an inflammatory microenvironment that favors either the local malignant transformation or the homing of extra‐ovarian malignant cells and, thereafter, cancer progression. Conceivably, factors other than inflammation, for instance the balance between estrogens and progesterons, are also involved in ovarian tumorigenesis.52, 53 Yet, it is a fact that inflammation in the peritoneal cavity determines a stromal environment permissive for ovarian cancer progression.54, 55, 56, 57
4. OVARIAN CANCER CELLS PROMOTE THE RECRUITMENT AND ACTIVATION OF STROMAL FIBROBLASTS
Active CAFs are marked by the characteristic expression of alpha‐smooth muscle actin (ASMA) and fibroblast‐activated protein (FAP).58 Studies conducted in a multitude of carcinomas have shown that CAFs may originate from different sources.59 CAFs have been reported to derive from: (1) the conversion of fibroblasts locally present in the ECM,60 (2) the differentiation of bone marrow derived precursor cells,61 (3) the trans‐differentiation of malignant epithelial cells (through EMT, epithelial‐mesenchymal transition)62 or of endothelial cells (through EndMT, endothelial‐mesenchymal transition),63 and (4) the differentiation of mesenchymal stem cells (MSCs).64, 65
Ovarian cancer cells release chemotactic cytokines and growth factors that recruit and activate the cells in the stroma (Fig. 1). The conditioned medium of ovarian (SKOV3) cancer cells, as well as TGF‐β1, could induce the trans‐differentiation of stromal fibroblasts into ASMA‐expressing myofibroblasts.66 The conversion of adipose‐derived and bone marrow derived MSCs and of peritoneal fibroblasts into CAFs was observed in a xenograft transplant of human ovarian cancer SKOV3 cells transgenically expressing the homeobox gene HOXA9.67 This effect was mediated by the release into the peritoneum of TGF‐β2 driven by HOXA9 from the ovarian cancer cells.67 Interestingly, the concomitant transplantation of ovarian cancer cells expressing transgenic green fluorescent protein could exclude the conversion of these cells into CAFs, which suggests that generation of CAFs through EMT of epithelial ovarian cancer cells is a rare event.67 The conversion of adipose‐derived MSC into CAFs within ovarian cancer stroma can be induced also by lysophosphatidic acid (LPA) released by the ovarian cancer cells.64 An important issue is whether the characters acquired by CAFs are stably maintained also in the absence of malignant cells, or whether these phenotypic changes are reversible. Both genetic and epigenetic factors could be involved in stabilizing the phenotype of CAFs. However, loss of heterozygosity and alterations of chromosomal copy number were found extremely rare in CAFs from ovarian cancer cells.68 It is therefore likely that the activated phenotype of CAFs is maintained through the epigenetic regulation of gene expression. In support of this hypothesis, a distinct pattern of DNA methylation has been reported in CAFs isolated from breast and prostate cancers.69, 70 Such a study has yet to be done in CAFs isolated from ovarian cancer. For sure, micro‐RNAs play a big role in modulating the phenotypic characters of fibroblasts in the ovarian cancer microenvironment.71 Differences in the expression of a subset of 11 micro‐RNAs have been reported between normal omental fibroblasts and CAFs from omental tumors, with miR‐31 and miR‐214 being the most downregulated and miR‐155 being the most upregulated in the latter.72 Interestingly, playing with the transfection of specific miRNAs and anti‐miRNAs to mimic this deregulation, it was possible to induce a functional conversion of normal fibroblasts into CAFs and vice versa.72
Figure 1.
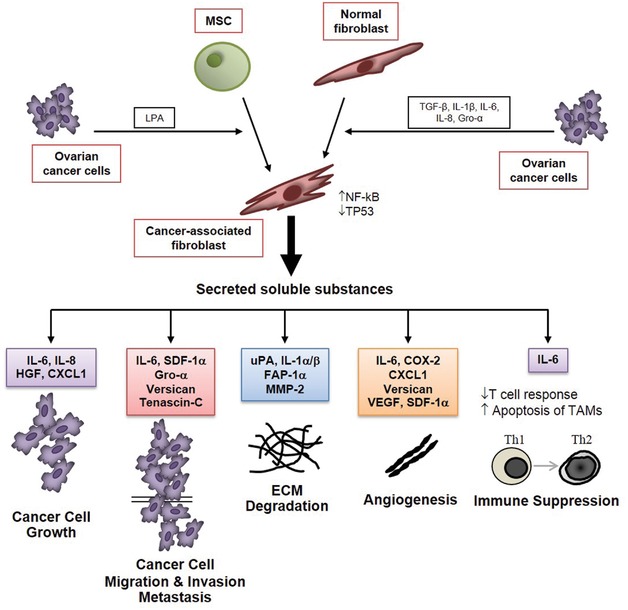
Origin and tumorigenic roles of ovarian cancer associated fibroblasts
Note: Ovarian cancer cells secrete cytokines and soluble factors that induce the recruitment, differentiation, and activation of cancer‐associated fibroblasts (CAFs). The latter secrete a set of soluble factors, including extracellular matrix components and cytokines, that affect the behavior and the fate of ovarian cancer cells (for abbreviations, see the text). The cytokines released in the cancer stroma elicit both paracrine and autocrine stimulations on cancer cells, fibroblasts, immune cells, and endothelial cells, thus amplifying and perpetuating the effects.
5. CANCER‐ASSOCIATED FIBROBLASTS SUPPORT OVARIAN CANCER PROGRESSION
The contribution of CAFs in cancer progression has been widely discussed in many different cancer models,73, 74, 75, 76 including ovarian cancer.5, 8, 31, 77 Here, we will briefly review the principal ways through which CAFs promote ovarian cancer progression (see also Fig. 1).
Ovarian CAFs express and secrete, among others, high levels of IL‐6, cyclo‐oxygenase 2 (COX‐2), and of chemokine (CXC motif) ligand CXCL‐1.31, 78 The latter promotes cell proliferation by binding to its receptor CXCR2, which is highly expressed on ovarian cancer cells.79 Ovarian CAFs could facilitate cancer cell spreading and invasion by releasing the hepatocyte growth factor (HGF),80 and collagenolytic proteases such as the Matrix Metalloprotease MMP‐281, 82 and the urokinase‐like activator of the plasminogen (uPA).83 The numerous cytokines and soluble factors released by CAFs reported to promote ovarian cancer progression are listed in Table 1.
Table 1.
Cytokines and soluble factors secreted by CAFs and proposed activity in ovarian cancer
| Name | Effect | Mechanism/pathway | Reference |
|---|---|---|---|
| CCL5 | Induces resistance to cisplatin | STAT3 and PI3K/AKT | 164 |
| TGF‐β |
|
TGF‐β/SMAD | 12, 165 |
| VCAN | Promotes cell migration and invasion | NF‐κB | 84 |
| MFAP5 | Enhances cell motility and invasion | FAK/CREB/troponin C | 166 |
| NPPB | A novel biomarker for ovarian cancer | NPR1‐dependent pathway (in lung cancer), not determined in ovarian cancer | 167 |
| FAP‐1α | Induces cell proliferation and invasion | Integrin, α3β1, uPAR, and pERK | 168, 169 |
| SDF‐1 (CXCL‐12) |
|
|
170, 171, 172, 173 |
| CXCL‐11 | Mediates cell proliferation and migration | CXCR3‐dependent pathway | 174 |
| CXCL‐1 | Induces cancer‐promoting inflammation | CXCR2‐dependent pathway | 78, 173 |
| IL‐6 | Mediates cancer‐promoting inflammation | IL‐6R/JAK2/STAT3 | 78, 173 |
| HGF | Induces cell migration and invasion | c‐met‐dependent pathway | 175 |
| LPA | Promotes cell proliferation, invasion, and chemoresistance | LPA2‐dependent pathway | 12 |
|
High level in advanced EOC correlates with poor disease‐specific survival | Degrade ECM | 175, 176 |
| MMP‐1 | Activates the production of CXCL1 and CXCL8 from cancer cells | PAR1 activation | 177 |
| VEGF |
|
|
171, 178, 179, 180 |
| TNF‐α | TNF network (TNF, CXCL12, IL6) inducing angiogenesis, inflammation, and leukocyte infiltration | TNFR1‐dependent pathway and NOTCH signaling | 173, 181, 182, 183 |
Note: CCL5, cisplatin‐induced chemokine (C‐C motif) ligand 5; TGF‐β, transforming growth factor β; VCAN, versican; MFAPA5, microfibrillar‐associated protein; NPPB, natriuretic peptide B; NPR1, NPPB receptor; FAP, fibroblast activation protein 1α, SDF‐1, stromal‐derived factor‐1 or CXCL‐12, CXCL‐11, CXCL‐1; IL‐6, interleukin 6; EZH2, enhancer of zeste homologue 2; LPA, lysophosphatidic acid; ECM, extracellular matrix.
The secretion of versican in the ECM is another means through which CAFs may promote ovarian cancer motility, spreading, and invasion. Under TGF‐β stimulation, CAFs release high amount of versican, along with MMP‐9, resulting in increased aggressiveness of ovarian cancer.84
CAFs are found abundantly in the proximity of the neo‐formed blood vessels. It has been demonstrated that under LPA stimulation, the active fibroblasts express and release the CXC chemokine ligand 12/stromal cell derived factor 1α (SDF‐1α), the Vascular Endothelial Growth Factor (VEGF‐A), and IL‐6, which promote the recruitment of endothelial cells and the angiogenic sprouting in the tumor context.85 These events clearly favor the growth and metastasization of ovarian cancers.86 Finally, CAFs contribute to cancer progression also by suppressing the immune response. It has been reported that a high ratio of tumor infiltrating T‐cytotoxic versus T‐regulatory (Treg) lymphocytes associates with a better prognosis in ovarian cancer patients.87, 88 Conversely, the presence of Treg lymphocytes in the tumor microenvironment of ovarian cancer tissues creates an immunosuppressive condition that negatively affects the patient's survival.89, 90 Besides ovarian cancer cells, also the CAFs release immune‐modulatory cytokines that eventually suppress or limit the immune response8, 57 (Fig. 2).
Figure 2.
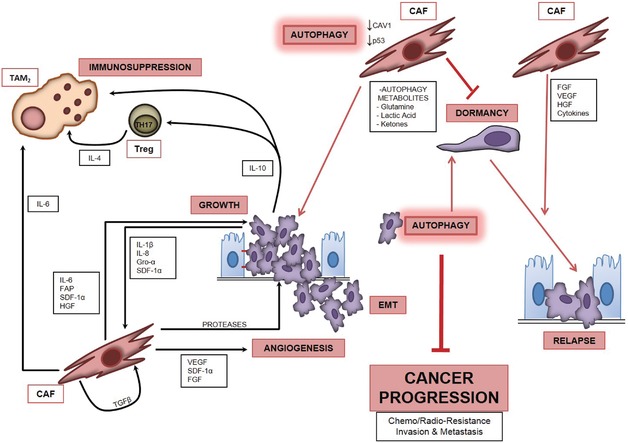
The cross‐talk between ovarian cancer cells and cancer‐associated fibroblasts in the regulation of autophagy and its role in cancer progression
Note: Autophagy in cancer cells opposes malignant progression and promotes dormancy. Cytokines and other factors secreted by ovarian cancer cells activate autophagy in CAFs, which then secrete a vast array of soluble factors including proteases, cytokines, growth factors, and metabolites that ultimately create a microenvironment favorable to cancer growth, metastasization, and onset of chemoresistance. The inflammatory stroma could also create the conditions for awakening the dormant cancer cells, thus favoring cancer relapse (for abbreviations and a detailed description, refer to the text).
6. AUTOPHAGY IN CANCER AND ITS MODULATION BY MICROENVIRONMENTAL FACTORS
Autophagy (literally, self‐eating) is a catabolic process that accomplishes the degradation within lysosomes of aged, redundant, or abnormal molecules and organelles.91 In this process, the autophagy material to be degraded is sequestered within the double‐membrane vesicles called autophagosomes, which eventually fuse with endosomes and lysosomes. For a more in‐depth description of the morphological and biochemical aspects of autophagy, the readers may refer to some excellent reviews.92, 93
In normal quiescent cells, autophagy runs at low basal level and allows the turnover of macromolecules and organelles/membranes without net increases in cell mass. Growth factors and nutrients (including amino acids and glucose) stimulate cell growth and proliferation and concomitantly downregulate autophagy. The progrowth signals activated by these factors converge on and positively activate mTOR.94 In turn, mTOR inhibits the ULK1 and BECLIN‐1–PI3kC3 complexes, thus preventing the formation of autophagosomes.95, 96 In metabolite‐ and energy‐restricted situations, the autophagy increases above the baseline in order to provide the needed substrates through the degradation of redundant constituents.97 In these situations, the lack of oxygen or of energy suppliers activates the AMPk pathway that switches off mTOR and activates ULK1, which then promotes the formation of the BECLIN‐1–PI3kC3 autophagy.97 Activation of the latter leads to the production of phosphatydilinositol‐3‐phosphate (PI3P), the biochemical signal for the recruitment of autophagosomal membranes.95, 96
Autophagy cooperates with the DNA repair systems to prevent chromosomal instability and failure in the upregulation of autophagy under conditions of genotoxic stress may lead to cell transformation.98 As such, autophagy acts as the guardian of both the genome and the proteome, thus accomplishing an anticancer preventive function. Yet, this same function may serve to repair the DNA damage induced by anticancer drugs, thus conferring chemo resistance to cancer cells.99 In a similar fashion, the upregulation of autophagy may turn of advantage for cancer cells by conferring an abnormal resistance to nutrient depletion.100, 101 It is known that the majority of cancer cells in the most inner part of fast growing and insufficiently vascularized tumors die by necrosis because of the lack of energetic nutrients and of oxygen.102 However, despite the lack of external nutrients those cancer clones may survive in a dormant state103 by raising the level of basal autophagy.22
Autophagy is deregulated in cancer cells,104, 105 because of mutations or epimutations of autophagy genes (e.g., BECLIN‐1) or autophagy‐regulating genes (e.g., PTEN, TP53). As a matter of facts, a large number of oncogenes and of oncosuppressor proteins regulate directly, or indirectly, the autophagy process.106 Interestingly, many autophagy genes (e.g., BECLIN‐1) act as oncosuppressors. The expression of autophagy genes and of autophagy‐regulating genes is modulated also at epigenetic level through histone deacetylation and promoter methylation events, as well as by certain micro‐RNAs.107, 108, 109, 110 This fact opens the possibility that environmental factors could affect the level of autophagy in a cell lineage in a chronic “stable” manner. As better illustrated below, cytokines and other soluble factors present in the stroma are capable of modulating the signaling pathways as well as the expression of autophagy‐related proteins that control autophagy. In addition, the vascularization of the stroma and the composition of the ECM (in terms of protein density) determine the availability and the diffusion of the nutrients, thus affecting the metabolic response in cancer cells as well as in stromal cells.111 The availability of oxygen, glucose, and certain amino acids (e.g., glutamine, arginine, and others) influences essentially three energetic metabolic pathways, that is, the glycolysis, the mitochondrial respiration, and autophagy. The mTORC1 complex, which positively regulates protein synthesis and cell growth, functions also as the master negative regulator of autophagy in response to metabolic stresses. In well vascularized area, the presence of growth factors and nutrients activates the PI3kC1‐AKT‐mTORC1 pathway, and maintains autophagy at very low basal level.95 Vice versa, the lack of growth factors or of amino acids reliefs the inhibitory action of the mTORC1 complex and elicits the raise of basal autophagy.112, 113 Further, the drop in the production of ATP that follows the lack of glucose or of oxygen activates the LKB‐AMPK pathway, which then inactivates the mTORC1 complex and directly activates the ULK1 complex, thus triggering autophagy.114
The fact that autophagy plays a pivotal role in the integrated response to all metabolic stresses highlights its role in the development of cancer cells,101 and points to the contribution of the tumor microenvironment in cancer progression through the regulation of this process.15, 115
7. AUTOPHAGY REGULATES OVARIAN CANCER CELL MIGRATION AND DORMANCY: ROLE OF STROMAL FIBROBLASTS
The first evidence that defective autophagy plays a role in ovarian tumorigenesis arose from the observation that transgenic mice hemizygous knock‐out for Beclin‐1, a main regulator of autophagy, spontaneously developed ovarian cancer, among others.116 A parallel study demonstrated that Beclin‐1 acts as a haploinsufficient oncosuppressor.117 Interestingly, monoallelic deletion of BECLIN‐1 had previously been reported in up to 75% of human epithelial ovarian cancers.118 Remarkably, the expression of the autophagy‐active BECLIN‐1 protein has been proposed as a prognostic marker in human ovarian cancer.119, 120 However, these studies did not consider the role of BECLIN‐1‐dependent autophagy in the CAFs surrounding the ovarian cancer cells. In fact, the genetic monoallelic deletion of BECLIN‐1 clearly involves the whole cell populations in the body and therefore the metabolism of cells other than parenchymal ones is likely to be also affected. Besides autophagy, BECLIN‐1 is involved also in the control of receptor endocytosis and associated growth factor signaling,121 and its dysfunctional expression may have great impact on both the epithelial and stromal cells response to extracellular signals as well as on their reciprocal interaction.
Dysfunctional regulation of autophagy in ovarian cancer cells has been recently reviewed.108, 122, 123 Here, we provide an overview of the evidence supporting the involvement of CAFs and of the soluble factors present in the stroma in the regulation of autophagy and of autophagy‐related phenomena in ovarian cancer (Fig. 2).
A number of inflammatory‐related proteins abnormally present in the tumor context or in the ascitic fluid, and associated with ovarian cancer progression, could directly or indirectly affect autophagy.
Perhaps the most abundant cytokine accumulating in the plasma and ascitic fluid of ovarian cancer patients is IL‐6,124 a pro‐inflammatory cytokine secreted in large amount by CAFs and ovarian cancer cells. This cytokine has been shown to induce the anchorage‐independent growth and the migration and invasion of epithelial ovarian carcinoma cells.23, 125, 126 Very recently, we demonstrated that IL‐6 inhibits basal autophagy in ovarian cancer cells.23 More in detail, IL‐6 downregulates the expression of the GTPase Ras homolog ARH‐I/DIRAS3, which acts as a promoter of BECLIN‐1‐dependent autophagy and as an inhibitor of cell locomotion.23 The bioactive phospholipid LPA is another molecule highly secreted by ovarian cancer cells and found in the plasma and serum of the patients. LPA acts in an autocrine manner on ovarian cancer cells as well as in a paracrine manner on CAFs stimulating the secretion of VEGF, of cytokines (including IL‐6 and IL‐8), and of proinvasive soluble factors.85, 127, 128 LPA stimulates the EMT and ovarian cancer cell migration through activation of the Hedgehog pathway.129, 130 LPA was shown to inhibit starvation‐induced autophagy in prostate cancer cells.131 Very recently, we have tested the effects of LPA in ovarian cancer cell lines and found that it inhibits autophagy through induction of the Hedgehog pathway (Ferraresi et al., unpublished). Thus, the presence of LPA in the stroma can limit the autophagy compliance in ovarian cancer cell through a direct autocrine action or via indirect stimulation of IL‐6 by CAFs.
CAFs mediated regulation of autophagy impinges on another phenomenon linked to ovarian cancer progression and relapse, namely cancer cell dormancy. Cell dormancy refers to a low energetic metabolic state of the cell associated with cell quiescence. Dormant cancer cells are radio‐ and chemoresistant, and if rescued from dormancy, these cells restart to grow. Cell dormancy depends on microenvironmental conditions and is under epigenetic control.104 Multicellular spheroids of ovarian epithelial cancer were xenografted subcutaneously in nude mice and could remain in a state of dormancy for nearly 2 months.132 Dormancy was associated with scarce and imperfect neovasculature and no infiltration of stromal cells.133 Regrowth of dormant ovarian cancer cells was obtained upon gonadotropin stimulation, and was associated with angiogenesis and recruitment of ASMA‐positive stromal cells.134 Thus, exit from dormancy and tumor regrowth were marked by infiltration of myofibroblasts, which positively stabilized neoangiogenesis.104, 134 Worthy of note, dormancy of ovarian cancer cells was strictly dependent on the actual level of autophagy in the cancer cells. The group of Robert Bast found that ARH‐I (or DIRAS3) plays a pivotal role in the regulation of autophagy and dormancy in human ovarian cancer cells.22 ARH‐I is a maternally imprinted oncosuppressor downregulated in 60% of ovarian cancers. These authors demonstrated that reexpression of ARH‐I restores autophagy at high level in ovarian cancer cells. However, while the reexpression of ARH‐I caused cell death in cultured ovarian cancer cells, it enabled the autophagy‐dependent survival in a dormant state when these cells were xenografted in mice. This is consistent with the fact that autophagic cell death is a phenomenon mostly observed in vitro and hardly (if at all) in vivo. Of note, in cultured cells, overexpressing the transgenic ARH‐I autophagic cell death was reduced in the presence of growth factors (IGF‐1, M‐CSF), angiogenic factors (VEGF, IL‐8), and matrix proteins found in the xenografts. From these data, the authors concluded that ARH‐I can drive cancer cell dormancy in the presence of factors that promote survival in the cancer microenvironment through modulation of autophagy.22 From a clinical point of view, these findings suggest that relapse of ovarian cancer may result from the breakdown of dormancy induced by changes in the extent of CAFs infiltration in the tumor stroma.
8. AUTOPHAGY IN IMMUNE CELLS DRIVES THE SECRETION OF CYTOKINES
The secretion of proinflammatory cytokines by the immune cells in the tumor environment follows an unconventional route that exploits the vesicular traffic associated with autophagy. Deretic and colleagues showed that stimulation of autophagy enhances the secretion of IL‐1β by macrophages primed with pro‐inflammatory triggers.135 However, in another study the induction of autophagy in macrophages was shown to promote the autophagy‐mediated degradation of pro‐IL‐1β, thus limiting the secretion of mature IL‐1β upon stimulation with a pro‐inflammatory trigger.136 These contradictory results probably relied on the diverse activation of mitophagy, which dampens the mitochondrial release of reactive oxygen species. Yet, another study showed that starvation‐ or interferon‐γ‐induced autophagy in macrophages and T lymphocytes could promote the secretion of TNF‐α.137 Interestingly, IL‐2, a cytokine released by T lymphocytes, promotes the survival and proliferation of stromal fibroblasts through induction of autophagy.138
9. AUTOPHAGY MEDIATES THE CYTOKINE‐INDUCED DIFFERENTIATION OF FIBROBLASTS INTO MYOFIBROBLASTS
A few studies link the activity of cytokines with the autophagy process in the CAFs. Starvation‐induced differentiation of fibroblasts in myofibroblasts, as marked by the de novo synthesis of ASMA and increased expression of stress fibers, strictly depended on induction of autophagy.139 Similarly, one can hypothesize that the differentiation of stromal fibroblasts into myofibroblasts induced by TGF‐β1 and other cytokines secreted by ovarian cancer cells66 occurs through the upregulation of autophagy. In this same line, it is interesting observation that IL‐1β secreted by ovarian cancer cells attenuates the expression of p53 in neighboring CAFs,31 given that cytoplasmic p53 is a known downregulator of autophagy.140 As said before, sustained autophagy (as induced by IL‐2) is necessary for myofibroblasts survival in the stroma.138
10. METABOLIC INTERPLAY BETWEEN CAFS AND OVARIAN CANCER CELLS: AUTOPHAGY AS A DRIVING FORCE TO CANCER PROGRESSION
Autophagy is greatly influenced by an array of factors in the tumor microenvironment, such as hypoxia, pH, oxidative stress, ammonia, glucose and amino acid availability, cytokines, hormones, and growth factors.141, 142, 143, 144, 145, 146, 147 The physical interaction of tumor cell with surrounding cells (inflammatory cells, fibroblasts) in the matrix also influences the autophagy compliance, and consequently also the survival or death, of the tumor cell. In addition, the cytokines released by both CAFs and ovarian cancer cells have an impact on the composition of the stroma by recruiting other cells, and thus contribute to create a microenvironment that ultimately affects the regulation of autophagy in ovarian cancer cells.
It has been proposed that the functional cross‐talk between tumor cells and stromal cells through the exchange of soluble factors finally results in the reprogramming of the latter toward a metabolic state that is permissive for the growth and metastasization of the cancer cells.148 In this respect, autophagy may represent the target and the pivotal driver at the same time of such metabolic reprogramming.
Lisanti and colleagues have recently proposed a new paradigm of the mutual interaction between CAFs and cancer cells, in which the former supply energetic metabolites to the latter.26, 149 In such situation, autophagy in cancer cells would be maintained at low basal level, while autophagy in CAFs would be upregulated. A number of inflammatory cytokines present in the stroma and the hydrogen peroxide released by ovarian cancer cells could induce autophagy in CAFs.150, 151 As a result, CAFs would fuel the metabolism of neighboring cancer cells with autophagy metabolites such as amino acids, fatty acids, ketones, and lactate,26, 149, 152, 153 thus supporting the growth and propagation of cancer cells while keeping autophagy at a minimal level. In this model, the so‐called “Warburg effect,” that is, the aerobic glycolysis, occurs in CAFs rather than in cancer cells, which suggested the term “reverse Warburg effect.”154, 155 In a similar fashion, in pancreatic cancer the tumor‐induced autophagy in CAFs leads to the secretion of alanine, which outcompetes glutamine, in turn, to fuel the cancer cells in low‐glucose microenvironment.25 This same scenario is likely to occur also in the case of ovarian cancer.156 Thus, an integrated model of the interplay between CAFs and ovarian cancer cells cannot disregard the modulation of autophagy in both the cell types (see Fig. 3). Our unpublished data support the view that high infiltration of CAFs associates with a low level of Beclin‐1‐dependent autophagy in ovarian cancer cells. We have previously reported that the patients bearing an ovarian cancer highly expressing autophagy‐active Beclin‐1 could experience a better prognosis as compared to those bearing an autophagy‐defective cancer.120 Also, we have shown that ovarian cancer cell migration induced by IL‐6 occurs via downregulation of autophagy.23 Taken together, it is tempting to speculate that poor prognosis in ovarian cancer patients is due to the downregulation of autophagy in cancer cells operated by CAFs, as predicted in the model proposed by Lisanti and colleagues. If this interpretation is correct, to be effective the therapeutic strategies targeting autophagy should consider the different modulation of autophagy in CAFs and in cancer cells as influenced by the microenvironmental conditions.24
Figure 3.
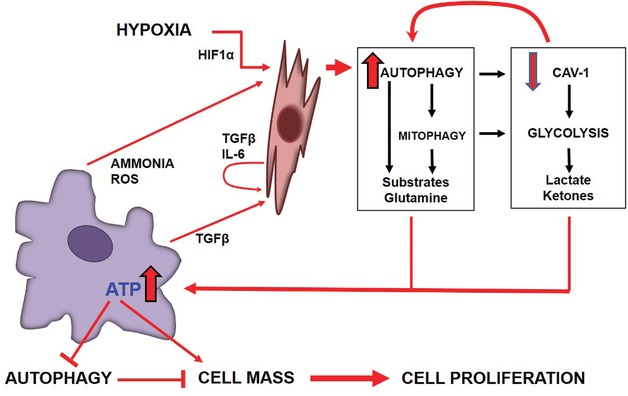
The reciprocal regulation of autophagy between cancer cells and cancer‐associated fibroblasts
Note: The cytokine‐mediated cross‐talk and the exchange of metabolites between cancer cells and CAFs reciprocally regulate the actual level of autophagy in the cells. Cytokines, reactive oxygen species (ROS), and ammonia released by cancer cells as well as hypoxia (which triggers the HIF1α pathway) and lack of nutrients in the tumor microenvironment induce autophagy in CAFs, which leads to loss of mitochondria (mitophagy) and of Caveolin 1. The latter events favor glycolysis. Because of increased autophagy and glycolysis in CAFs, metabolites (including glutamine), lactic acid, and ketones are supplied to cancer cells. As a result, in these cells autophagy is downregulated, while anabolism is stimulated, with consequent growth of cancer.
11. CONCLUDING REMARKS AND PERSPECTIVES
The tumor stroma is characterized by intense infiltration of reactive immune and inflammatory cells, neoformation of a nonfunctional network of blood vessels, recruitment of fibroblasts, and neodeposition of collagen and fibrin, besides other ECM proteins. In such a reactive stroma (also referred to as desmoplastic), fibroblasts, either locally resident or recruited from other anatomical sites, undergo a pronounced alterations in the phenotype and expression profile of cellular and secreted proteins. A complex stromal–epithelial interaction reciprocally influences the dynamic changes in the structure and composition of ECM and of tumor stroma, and alterations in this interaction play an important role in the development of cancer.157
In vivo, the actual level of autophagy in cancer cells is different in the different areas of the tumor considered, depending on the local level of nutrients, oxygen, and on the mixture of soluble factors, as provided by the vasculature and stromal composition. Given the pathophysiological role of autophagy in the regulation of cell survival and cell death in response to metabolic and genotoxic stresses, it appears clear that, by influencing the level of autophagy in the cancer cells, the stroma composition indirectly contributes to the onset of chemo resistance and dormancy in ovarian cancer cells, two conditions that negatively impact on prognosis. CAFs could release exosome in the stromal compartment to deliver metabolic substrates and micro‐RNAs to cancer cells, thus reprogramming the energetic metabolism of the latter.158
The reciprocal stimulation through physical contact and soluble factors between CAFs and cancer cells not only reprograms the expression of secretory factors, but also modifies in a dynamic way the metabolism in both cell types. In brief, we may assume that the stromal microenvironment acts as an epigenetic modifier of autophagy, with a fallout on the behavior of malignant cells and, consequently, the prognosis of cancer's patients.
Presently, autophagy is a target process in the therapy of cancer159 and of ovarian cancer in particular.160 With increasing recognition of the role of CAFs in carcinogenesis and progression, it is believed that targeting the metabolic cross‐talk between epithelial cancer cells and CAFs could open novel therapeutic strategies to fight ovarian cancer.161 Potential therapeutic strategies presently under investigation include, among others, inhibitors of the signaling pathways triggered by TGF‐β and HIF‐1α, two known inducers of autophagy in CAFs (Fig. 3).162, 163 Thus, learning how the actual level of autophagy in cancer cells and in stromal cells modulated by the tumor microenvironment may help to predict whether inhibitors or inducers of autophagy will achieve a long‐term therapeutic benefit.
ACKNOWLEDGMENTS
The authors thank Associazione per la Ricerca Medica Ippocrate‐Rhazi (Novara, Italy) for a “Maria Di Lauro‐Isidoro” fellowship to R.T., and Comoli, Ferrari & SpA (Novara, Italy) for a fellowship to A.F. They also thank the Siriraj Cancer Foundation, Faculty of Medicine Siriraj Hospital (Bangkok, Thailand). Authors are thankful to Dario Michelone for artwork. The authors have declared no conflict of interest.
Biographies
Chanitra Thuwajit is now Associate Professor in Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Thailand. She received her Ph.D. in Biochemistry in 1998 and her M.D. degree in 2001 from Mahidol University. Her current research focuses on: (1) tumor microenvironment in cancer progression; (2) role of IL‐33 in cholangiocarcinoma; (3) role of periostin in colorectal cancer; (4) role of autophagy in ovarian cancer. She has published 17 articles in SCI journals so far.
Alessandra Ferraresi is Ph.D. student in Sciences and Medical Biotechnology at Università del Piemonte Orientale (Novara, Italy), working under the mentorship of Prof. Ciro Isidoro in the laboratory of Molecular Pathology and Nanobioimaging. Her current research focuses on the role of autophagy in ovarian cancer cell stemness and migration. She has co‐authored three articles published in peer reviewed journals, so far.
Rossella Titone received her PhD degree in Biotechnology for Human Health at the Università del Piemonte Orientale in 2015. In 2007, Dr. Titone received her Bachelor of Science degree in Medical Biotechnologies at the University of Palermo. In 2009, she completed her Master's degree in Biomedical Biotechnologies with honors at the University of Siena. Thereafter, she performed her PhD studies under the mentorship of Professor Ciro Isidoro. She also spent one year as PhD visiting student in the Department of Internal Medicine of UT Southwestern Medical Center in Dr. Beth Levine's laboratory. During her PhD training she has studied the involvement and mechanisms of regulation of autophagy in ovarian and in breast cancers. Currently, she is postdoctoral researcher in the Department of Ophthalmology at UT Southwestern Medical Center, where she works in Dr. Danielle Robertson's laboratory studying corneal epithelial cell biology.
Peti Thuwajit is now Assistant Professor in Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Thailand. He received his Ph.D. in Biochemistry from Mahidol University in 1999. His current research focuses on: (1) hormonal effects in cholangiocarcinoma; (2) lipidomics in plasma of cancer patients; (3) development of nanomaterials for detection of cancer cells. He has published 23 articles in SCI journals so far.
Ciro Isidoro (http://www.isidorolab.com) received his doctoral degree (summa cum laude) in Biological Sciences from the University of Torino (Italy) and his doctoral degree (summa cum laude) in Medicine and Surgery from the University of Piemonte Orientale (Novara, Italy). Currently, he is Professor of Pathology and of Experimental Oncology at the School of Medicine of Università del Piemonte Orientale (Novara, Italy). He is also Visiting Professor at the Faculty of Medicine Siriraj Hospital of Mahidol University (Bangkok, Thailand) and at the Department of Cell Biology of the University of Oklahoma College of Medicine (Oklahoma City, United States). He is Professeur Honoraire at the Faculté de Medecine et de Pharmacie de l.Université de Franche‐Comté (Besancon, France). He is member of the scientific board of the « Center for Integrative Cancer Research » of the Georgia Tech Institute, University of Georgia (Atlanta, US). He is Co‐Editor in Chief of the J. of Traditional and Complementary Medicine, and Associate Editor of Autophagy, BMC Cancer, Molecular Carcinogenesis, J. of Ovarian Research, Genes and Cancer, J. of Molecular Signaling, and other journals. His fields of expertise include: lysosome biogenesis and function, autophagy regulation in cancer and in neurodegenerative diseases, cell death pathways, mechanisms of action of natural products, epigenetic regulation of autophagy and of cell death.
Thuwajit C, Ferraresi A, Titone R, et al. The metabolic cross‐talk between epithelial cancer cells and stromal fibroblasts in ovarian cancer progression: autophagy plays a role. Med Res Rev. 2018;38:1235–1254. https://doi.org/10.1002/med.21473
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. Cancer J Clin. 2016;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14(1):9–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. Cancer J Clin. 2016;66(4):271–289. [DOI] [PubMed] [Google Scholar]
- 4. Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced‐stage epithelial ovarian cancer. Nat Rev Clin Oncol. 2013;10(4):211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naora H. Heterotypic cellular interactions in the ovarian tumor microenvironment: biological significance and therapeutic implications. Front Oncol. 2014;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davidson B, Trope CG, Reich R. The role of the tumor stroma in ovarian cancer. Front Oncol. 2014;4:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slaughter KN, Thai T, Penaroza S, et al. Measurements of adiposity as clinical biomarkers for first‐line bevacizumab‐based chemotherapy in epithelial ovarian cancer. Gynecol Oncol. 2014;133(1):11–15. [DOI] [PubMed] [Google Scholar]
- 8. Worzfeld T, Pogge von Strandmann E, Huber M, et al. The unique molecular and cellular microenvironment of ovarian cancer. Front Oncol. 2017;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133(3421):571–573. [PubMed] [Google Scholar]
- 10. Langley RR, Fidler IJ. The seed and soil hypothesis revisited—the role of tumor‐stroma interactions in metastasis to different organs. Int J Cancer. 2011;128(11):2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenaway J, Moorehead R, Shaw P, Petrik J. Epithelial‐stromal interaction increases cell proliferation, survival and tumorigenicity in a mouse model of human epithelial ovarian cancer. Gynecol Oncol. 2008;108(2):385–394. [DOI] [PubMed] [Google Scholar]
- 12. Thibault B, Castells M, Delord JP, Couderc B. Ovarian cancer microenvironment: implications for cancer dissemination and chemoresistance acquisition. Cancer Metast Rev. 2014;33(1):17–39. [DOI] [PubMed] [Google Scholar]
- 13. Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: an easy way out. Cancer Lett. 2013;341(1):80–96. [DOI] [PubMed] [Google Scholar]
- 14. Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co‐evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol. 2014;92(1):43–54. [DOI] [PubMed] [Google Scholar]
- 15. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19(7):428–446. [DOI] [PubMed] [Google Scholar]
- 16. Okamoto K. Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol. 2014;205(4):435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo JY, White E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRAS(G12D)‐driven lung tumors. Autophagy. 2013;9(10):1636–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yue W, Hamai A, Tonelli G, et al. Inhibition of the autophagic flux by salinomycin in breast cancer stem‐like/progenitor cells interferes with their maintenance. Autophagy. 2013;9(5):714–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13(4):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang S, Xia P, Rehm M, Fan Z. Autophagy and cell reprogramming. Cell Mol Life Sci. 2015;72(9):1699–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu Z, Luo RZ, Lu Y, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118(12):3917–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferraresi A, Phadngam S, Morani F, et al. Resveratrol inhibits IL‐6‐induced ovarian cancer cell migration through epigenetic up‐regulation of autophagy. Mol Carcinog. 2017;56(3):1164–1181. [DOI] [PubMed] [Google Scholar]
- 24. Lisanti MP, Martinez‐Outschoorn UE, Sotgia F. Oncogenes induce the cancer‐associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle. 2013;12(17):2723–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinez‐Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer‐associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60. [DOI] [PubMed] [Google Scholar]
- 27. Romero I, Bast RC, Jr. Minireview: human ovarian cancer—biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153(4):1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramalingam P. Morphologic, immunophenotypic, and molecular features of epithelial ovarian cancer. Oncology. 2016;30(2):166–176. [PubMed] [Google Scholar]
- 29. Rojas V, Hirshfield KM, Ganesan S, Rodriguez‐Rodriguez L. Molecular characterization of epithelial ovarian cancer: implications for diagnosis and treatment. Int J Mol Sci. 2016;17(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. BioMed Res Int. 2014;2014:934261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schauer IG, Sood AK, Mok S, Liu J. Cancer‐associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13(5):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lili LN, Matyunina LV, Walker LD, Benigno BB, McDonald JF. Molecular profiling predicts the existence of two functionally distinct classes of ovarian cancer stroma. BioMed Res Int. 2013;2013:846387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parrott JA, Nilsson E, Mosher R, et al. Stromal‐epithelial interactions in the progression of ovarian cancer: influence and source of tumor stromal cells. Mol Cell Endocrinol. 2001;175(1–2):29–39. [DOI] [PubMed] [Google Scholar]
- 34. Athavale R, Thomakos N, Godfrey K, et al. The effect of epithelial and stromal tumor components on FIGO stages III and IV ovarian carcinosarcomas treated with primary surgery and chemotherapy. Int J Gynecol Cancer. 2007;17(5):1025–1030. [DOI] [PubMed] [Google Scholar]
- 35. Labiche A, Heutte N, Herlin P, Chasle J, Gauduchon P, Elie N. Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int J Gynecol Cancer. 2010;20(1):28–33. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y, Tang H, Cai J, et al. Ovarian cancer‐associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011;303(1):47–55. [DOI] [PubMed] [Google Scholar]
- 37. Anttila MA, Tammi RH, Tammi MI, Syrjanen KJ, Saarikoski SV, Kosma VM. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000;60(1):150–155. [PubMed] [Google Scholar]
- 38. Voutilainen K, Anttila M, Sillanpaa S, et al. Versican in epithelial ovarian cancer: relation to hyaluronan, clinicopathologic factors and prognosis. Int J Cancer. 2003;107(3):359–364. [DOI] [PubMed] [Google Scholar]
- 39. Ghosh S, Albitar L, LeBaron R, et al. Up‐regulation of stromal versican expression in advanced stage serous ovarian cancer. Gynecol Oncol. 2010;119(1):114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dvorak HF. Tumors: wounds that do not heal‐redux. Cancer Immunol Res. 2015;3(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. [DOI] [PubMed] [Google Scholar]
- 42. Kisielewski R, Tolwinska A, Mazurek A, Laudanski P. Inflammation and ovarian cancer—current views. Ginekol Pol. 2013;84(4):293–297. [DOI] [PubMed] [Google Scholar]
- 43. Fleming JS, Beaugie CR, Haviv I, Chenevix‐Trench G, Tan OL. Incessant ovulation, inflammation and epithelial ovarian carcinogenesis: revisiting old hypotheses. Mol Cell Endocrinol. 2006;247(1–2):4–21. [DOI] [PubMed] [Google Scholar]
- 44. Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91(17):1459–1467. [DOI] [PubMed] [Google Scholar]
- 45. Wang X, Wang E, Kavanagh JJ, Freedman RS. Ovarian cancer, the coagulation pathway, and inflammation. J Transl Med. 2005;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rainczuk A, Rao J, Gathercole J, Stephens AN. The emerging role of CXC chemokines in epithelial ovarian cancer. Reproduction. 2012;144(3):303–317. [DOI] [PubMed] [Google Scholar]
- 47. Papacleovoulou G, Critchley HO, Hillier SG, Mason JI. IL1alpha and IL4 signalling in human ovarian surface epithelial cells. J Endocrinol. 2011;211(3):273–283. [DOI] [PubMed] [Google Scholar]
- 48. Buscher U, Chen FC, Kentenich H, Schmiady H. Cytokines in the follicular fluid of stimulated and non‐stimulated human ovaries; is ovulation a suppressed inflammatory reaction? Hum Reprod. 1999;14(1):162–166. [DOI] [PubMed] [Google Scholar]
- 49. Bahar‐Shany K, Brand H, Sapoznik S, et al. Exposure of fallopian tube epithelium to follicular fluid mimics carcinogenic changes in precursor lesions of serous papillary carcinoma. Gynecol Oncol. 2014;132(2):322–327. [DOI] [PubMed] [Google Scholar]
- 50. Tone AA, Salvador S, Finlayson SJ, et al. The role of the fallopian tube in ovarian cancer. Clin Adv Hematol Oncol. 2012;10(5):296–306. [PubMed] [Google Scholar]
- 51. Yang‐Hartwich Y, Gurrea‐Soteras M, Sumi N, et al. Ovulation and extra‐ovarian origin of ovarian cancer. Sci Rep. 2014;4:6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wissing ML, Kristensen SG, Andersen CY, et al. Identification of new ovulation‐related genes in humans by comparing the transcriptome of granulosa cells before and after ovulation triggering in the same controlled ovarian stimulation cycle. Hum Reprod. 2014;29(5):997–1010. [DOI] [PubMed] [Google Scholar]
- 53. Chen X, Aravindakshan J, Yang Y, Sairam MR. Early alterations in ovarian surface epithelial cells and induction of ovarian epithelial tumors triggered by loss of FSH receptor. Neoplasia. 2007;9(6):521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Freedman RS, Deavers M, Liu J, Wang E. Peritoneal inflammation—a microenvironment for epithelial ovarian cancer (EOC). J Transl Med. 2004;2(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nowak M, Glowacka E, Szpakowski M, et al. Proinflammatory and immunosuppressive serum, ascites and cyst fluid cytokines in patients with early and advanced ovarian cancer and benign ovarian tumors. Neuro Endocrinol Lett. 2010;31(3):375–383. [PubMed] [Google Scholar]
- 56. Clendenen TV, Lundin E, Zeleniuch‐Jacquotte A, et al. Circulating inflammation markers and risk of epithelial ovarian cancer. Cancer Epidemiol Biomark Prevent. 2011;20(5):799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maccio A, Madeddu C. Inflammation and ovarian cancer. Cytokine. 2012;58(2):133–147. [DOI] [PubMed] [Google Scholar]
- 58. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. [DOI] [PubMed] [Google Scholar]
- 59. Xouri G, Christian S. Origin and function of tumor stroma fibroblasts. Semin Cell Dev Biol. 2010;21(1):40–46. [DOI] [PubMed] [Google Scholar]
- 60. Mueller L, Goumas FA, Affeldt M, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol. 2007;171(5):1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Direkze NC, Hodivala‐Dilke K, Jeffery R, et al. Bone marrow contribution to tumor‐associated myofibroblasts and fibroblasts. Cancer Res. 2004;64(23):8492–8495. [DOI] [PubMed] [Google Scholar]
- 62. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101(4):830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma‐associated fibroblasts. Cancer Res. 2007;67(21):10123–10128. [DOI] [PubMed] [Google Scholar]
- 64. Jeon ES, Moon HJ, Lee MJ, et al. Cancer‐derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast‐like cells. Stem Cell. 2008;26(3):789–797. [DOI] [PubMed] [Google Scholar]
- 65. Spaeth EL, Labaff AM, Toole BP, Klopp A, Andreeff M, Marini FC. Mesenchymal CD44 expression contributes to the acquisition of an activated fibroblast phenotype via TWIST activation in the tumor microenvironment. Cancer Res. 2013;73(17):5347–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao Q, Qu X, Yang Q, Wei M, Kong B. CLIC4 mediates TGF‐beta1‐induced fibroblast‐to‐myofibroblast transdifferentiation in ovarian cancer. Oncol Rep. 2009;22(3):541–548. [DOI] [PubMed] [Google Scholar]
- 67. Ko SY, Barengo N, Ladanyi A, et al. HOXA9 promotes ovarian cancer growth by stimulating cancer‐associated fibroblasts. J Clin Invest. 2012;122(10):3603–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qiu W, Hu M, Sridhar A, et al. No evidence of clonal somatic genetic alterations in cancer‐associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40(5):650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905. [DOI] [PubMed] [Google Scholar]
- 70. Hanson JA, Gillespie JW, Grover A, et al. Gene promoter methylation in prostate tumor‐associated stromal cells. J Natl Cancer Instit. 2006;98(4):255–261. [DOI] [PubMed] [Google Scholar]
- 71. Chou J, Werb Z. MicroRNAs play a big role in regulating ovarian cancer‐associated fibroblasts and the tumor microenvironment. Cancer Discov. 2012;2(12):1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mitra AK, Zillhardt M, Hua Y, et al. MicroRNAs reprogram normal fibroblasts into cancer‐associated fibroblasts in ovarian cancer. Cancer Discov. 2012;2(12):1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cirri P, Chiarugi P. Cancer‐associated‐fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metast Rev. 2012;31(1–2):195–208. [DOI] [PubMed] [Google Scholar]
- 74. Madar S, Goldstein I, Rotter V. “Cancer associated fibroblasts”—more than meets the eye. Trends Mol Med. 2013;19(8):447–453. [DOI] [PubMed] [Google Scholar]
- 75. Raffaghello L, Vacca A, Pistoia V, Ribatti D. Cancer associated fibroblasts in hematological malignancies. Oncotarget. 2015;6(5):2589–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer‐associated fibroblasts in pancreatic cancer. J Hematol Oncol. 2017;10(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fu S, Dong L, Sun W, Xu Y, Gao L, Miao Y. Stromal‐epithelial crosstalk provides a suitable microenvironment for the progression of ovarian cancer cells in vitro. Cancer Invest. 2013;31(9):616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro‐inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013;437(3):397–402. [DOI] [PubMed] [Google Scholar]
- 79. Yang G, Rosen DG, Liu G, et al. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin Cancer Res. 2010;16(15):3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. Effective inhibition of c‐MET‐mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 2015;34(2):144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Westerlund A, Hujanen E, Puistola U, Turpeenniemi‐Hujanen T. Fibroblasts stimulate human ovarian cancer cell invasion and expression of 72‐kDa gelatinase A (MMP‐2). Gynecol Oncol. 1997;67(1):76–82. [DOI] [PubMed] [Google Scholar]
- 82. Furuya M, Ishikura H, Nemori R, Shibata M, Fujimoto S, Yoshiki T. Clarification of the active gelatinolytic sites in human ovarian neoplasms using in situ zymography. Hum Pathol. 2001;32(2):163–168. [DOI] [PubMed] [Google Scholar]
- 83. Noskova V, Ahmadi S, Asander E, Casslen B. Ovarian cancer cells stimulate uPA gene expression in fibroblastic stromal cells via multiple paracrine and autocrine mechanisms. Gynecol Oncol. 2009;115(1):121–126. [DOI] [PubMed] [Google Scholar]
- 84. Yeung TL, Leung CS, Wong KK, et al. TGF‐beta modulates ovarian cancer invasion by upregulating CAF‐derived versican in the tumor microenvironment. Cancer Res. 2013;73(16):5016–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jeon ES, Heo SC, Lee IH, et al. Ovarian cancer‐derived lysophosphatidic acid stimulates secretion of VEGF and stromal cell‐derived factor‐1 alpha from human mesenchymal stem cells. Exp Mol Med. 2010;42(4):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gavalas NG, Liontos M, Trachana SP, et al. Angiogenesis‐related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci. 2013;14(8):15885–15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Leffers N, Gooden MJ, de Jong RA, et al. Prognostic significance of tumor‐infiltrating T‐lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother. 2009;58(3):449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Knutson KL, Maurer MJ, Preston CC, et al. Regulatory T cells, inherited variation, and clinical outcome in epithelial ovarian cancer. Cancer Immunol Immunother. 2015;64(12):1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. [DOI] [PubMed] [Google Scholar]
- 90. Wertel I, Surowka J, Polak G, et al. Macrophage‐derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumour Biol. 2015;36(6):4811–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015;25(7):376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. [DOI] [PubMed] [Google Scholar]
- 95. Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–1435. [DOI] [PubMed] [Google Scholar]
- 98. Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21(11):1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Huang Z, Zhou L, Chen Z, Nice EC, Huang C. Stress management by autophagy: Implications for chemoresistance. Int J Cancer. 2016;139(1):23–32. [DOI] [PubMed] [Google Scholar]
- 100. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Horsman MR, Vaupel P. Pathophysiological basis for the formation of the tumor microenvironment. Front Oncol. 2016;6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Linde N, Fluegen G, Aguirre‐Ghiso JA. The relationship between dormant cancer cells and their microenvironment. Adv Cancer Res. 2016;132:45–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lyu T, Jia N, Wang J, et al. Expression and epigenetic regulation of angiogenesis‐related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics. 2013;8(12):1330–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Guo JY, Xia B, White E. Autophagy‐mediated tumor promotion. Cell. 2013;155(6):1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Maiuri MC, Tasdemir E, Criollo A, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16(1):87–93. [DOI] [PubMed] [Google Scholar]
- 107. Morani F, Titone R, Pagano L, et al. Autophagy and thyroid carcinogenesis: genetic and epigenetic links. Endocr Relat Cancer. 2014;21(1):R13–R29. [DOI] [PubMed] [Google Scholar]
- 108. Titone R, Morani F, Follo C, Vidoni C, Mezzanzanica D, Isidoro C. Epigenetic control of autophagy by microRNAs in ovarian cancer. BioMed Res Int. 2014;2014:343542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15(1):65‐74. [DOI] [PubMed] [Google Scholar]
- 110. Gozuacik D, Akkoc Y, Ozturk DG, Kocak M. Autophagy‐regulating microRNAs and cancer. Front Oncol. 2017;7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Icard P, Kafara P, Steyaert JM, Schwartz L, Lincet H. The metabolic cooperation between cells in solid cancer tumors. Biochim Biophys Acta. 2014;1846(1):216–225. [DOI] [PubMed] [Google Scholar]
- 112. Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12(3):547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wong PM, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen R, Zou Y, Mao D, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014;206(2):173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhao X, He Y, Chen H. Autophagic tumor stroma: mechanisms and roles in tumor growth and progression. Int J Cancer. 2013;132(1):1–8. [DOI] [PubMed] [Google Scholar]
- 116. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100(25):15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Eccles DM, Russell SE, Haites NE, et al. Early loss of heterozygosity on 17q in ovarian cancer. The Abe Ovarian Cancer Genetics Group. Oncogene. 1992;7(10):2069–2072. [PubMed] [Google Scholar]
- 119. Lin HX, Qiu HJ, Zeng F, et al. Decreased expression of Beclin 1 correlates closely with Bcl‐xL expression and poor prognosis of ovarian carcinoma. PLoS One. 2013;8(4):e60516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Valente G, Morani F, Nicotra G, et al. Expression and clinical significance of the autophagy proteins BECLIN 1 and LC3 in ovarian cancer. BioMed Res Int. 2014;2014:462658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rohatgi RA, Janusis J, Leonard D, et al. Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene. 2015;34(42):5352–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Peracchio C, Alabiso O, Valente G, Isidoro C. Involvement of autophagy in ovarian cancer: a working hypothesis. J Ovarian Res. 2012;5(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Orfanelli T, Jeong JM, Doulaveris G, Holcomb K, Witkin SS. Involvement of autophagy in cervical, endometrial and ovarian cancer. Int J Cancer. 2014;135(3):519–528. [DOI] [PubMed] [Google Scholar]
- 124. Plante M, Rubin SC, Wong GY, Federici MG, Finstad CL, Gastl GA. Interleukin‐6 level in serum and ascites as a prognostic factor in patients with epithelial ovarian cancer. Cancer. 1994;73(7):1882–1888. [DOI] [PubMed] [Google Scholar]
- 125. Obata NH, Tamakoshi K, Shibata K, Kikkawa F, Tomoda Y. Effects of interleukin‐6 on in vitro cell attachment, migration and invasion of human ovarian carcinoma. Anticancer Res. 1997;17(1a):337–342. [PubMed] [Google Scholar]
- 126. Wang Y, Li L, Guo X, et al. Interleukin‐6 signaling regulates anchorage‐independent growth, proliferation, adhesion and invasion in human ovarian cancer cells. Cytokine. 2012;59(2):228–236. [DOI] [PubMed] [Google Scholar]
- 127. Fang X, Yu S, Bast RC, et al. Mechanisms for lysophosphatidic acid‐induced cytokine production in ovarian cancer cells. J Biol Chem. 2004;279(10):9653–9661. [DOI] [PubMed] [Google Scholar]
- 128. Jesionowska A, Cecerska‐Heryc E, Matoszka N, Dolegowska B. Lysophosphatidic acid signaling in ovarian cancer. J Recept Signal Transduct Res. 2015;35(6):578–584. [DOI] [PubMed] [Google Scholar]
- 129. Ward JD, Ha JH, Jayaraman M, Dhanasekaran DN. LPA‐mediated migration of ovarian cancer cells involves translocalization of Galphai2 to invadopodia and association with Src and beta‐pix. Cancer Lett. 2015;356(2 Pt B):382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ha JH, Ward JD, Radhakrishnan R, Jayaraman M, Song YS, Dhanasekaran DN. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Galphai2, Src, and HIF1alpha signaling nexus. Oncotarget. 2016;7(25):37664–37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Chang CL, Liao JJ, Huang WP, Lee H. Lysophosphatidic acid inhibits serum deprivation‐induced autophagy in human prostate cancer PC‐3 cells. Autophagy. 2007;3(3):268–270. [DOI] [PubMed] [Google Scholar]
- 132. Gilead A, Neeman M. Dynamic remodeling of the vascular bed precedes tumor growth: MLS ovarian carcinoma spheroids implanted in nude mice. Neoplasia. 1999;1(3):226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gilead A, Meir G, Neeman M. The role of angiogenesis, vascular maturation, regression and stroma infiltration in dormancy and growth of implanted MLS ovarian carcinoma spheroids. Int J Cancer. 2004;108(4):524–531. [DOI] [PubMed] [Google Scholar]
- 134. Granot D, Addadi Y, Kalchenko V, Harmelin A, Kunz‐Schughart LA, Neeman M. In vivo imaging of the systemic recruitment of fibroblasts to the angiogenic rim of ovarian carcinoma tumors. Cancer Res. 2007;67(19):9180–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy‐based unconventional secretory pathway for extracellular delivery of IL‐1beta. EMBO J. 2011;30(23):4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Harris J, Hartman M, Roche C, et al. Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J Biol Chem. 2011;286(11):9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kleinnijenhuis J, Oosting M, Plantinga TS, et al. Autophagy modulates the Mycobacterium tuberculosis‐induced cytokine response. Immunology. 2011;134(3):341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kang R, Tang D, Lotze MT, Zeh HJ, 3rd . AGER/RAGE‐mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6‐pSTAT3 pathway. Autophagy. 2012;8(6):989–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bernard M, Dieude M, Yang B, Hamelin K, Underwood K, Hebert MJ. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy. 2014;10(12):2193–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22(2):181–185. [DOI] [PubMed] [Google Scholar]
- 141. Song J, Guo X, Xie X, et al. Autophagy in hypoxia protects cancer cells against apoptosis induced by nutrient deprivation through a Beclin1‐dependent way in hepatocellular carcinoma. J Cell Biochem. 2011;112(11):3406–3420. [DOI] [PubMed] [Google Scholar]
- 142. Wojtkowiak JW, Rothberg JM, Kumar V, et al. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012;72(16):3938–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Xu T, Su H, Ganapathy S, Yuan ZM. Modulation of autophagic activity by extracellular pH. Autophagy. 2011;7(11):1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III‐dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci. 2010;117(1):152–162. [DOI] [PubMed] [Google Scholar]
- 145. Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3(119):ra31. [DOI] [PubMed] [Google Scholar]
- 146. Morani F, Phadngam S, Follo C, et al. PTEN deficiency and mutant p53 confer glucose‐addiction to thyroid cancer cells: impact of glucose depletion on cell proliferation, cell survival, autophagy and cell migration. Genes Cancer. 2014;5(7–8):226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Shuvayeva G, Bobak Y, Igumentseva N, et al. Single amino acid arginine deprivation triggers prosurvival autophagic response in ovarian carcinoma SKOV3. BioMed Res Int. 2014;2014:505041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bhowmick NA, Moses HL. Tumor‐stroma interactions. Curr Opin Genet Dev. 2005;15(1):97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Martinez‐Outschoorn UE, Pavlides S, Howell A, et al. Stromal‐epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43(7):1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Martinez‐Outschoorn UE, Whitaker‐Menezes D, Lin Z, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin‐1 as a key regulator. Cell Cycle. 2011;10(11):1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Martinez‐Outschoorn UE, Balliet RM, Lin Z, et al. Hereditary ovarian cancer and two‐compartment tumor metabolism: epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFkappaB activation in the tumor stroma. Cell Cycle. 2012;11(22):4152–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Capparelli C, Guido C, Whitaker‐Menezes D, et al. Autophagy and senescence in cancer‐associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11(12):2285–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Guido C, Whitaker‐Menezes D, Capparelli C, et al. Metabolic reprogramming of cancer‐associated fibroblasts by TGF‐beta drives tumor growth: connecting TGF‐beta signaling with “Warburg‐like” cancer metabolism and L‐lactate production. Cell Cycle. 2012;11(16):3019–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Pavlides S, Vera I, Gandara R, et al. Warburg meets autophagy: cancer‐associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16(11):1264–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Carito V, Bonuccelli G, Martinez‐Outschoorn UE, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11(18):3403–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Suh YA, Jo SY, Lee HY, Lee C. Inhibition of IL‐6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int J Oncol. 2015;46(3):1405–1411. [DOI] [PubMed] [Google Scholar]
- 157. Weaver VM, Gilbert P. Watch thy neighbor: cancer is a communal affair. J Cell Sci. 2004;117(Pt 8):1287–1290. [DOI] [PubMed] [Google Scholar]
- 158. Zhao H, Yang L, Baddour J, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife. 2016;5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhan L, Zhang Y, Wang W, et al. Autophagy as an emerging therapy target for ovarian carcinoma. Oncotarget. 2016;7(50):83476–83487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ko SY, Naora H. Therapeutic strategies for targeting the ovarian tumor stroma. World J Clin Cases. 2014;2(6):194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Deep G, Agarwal R. Targeting tumor microenvironment with silibinin: promise and potential for a translational cancer chemopreventive strategy. Curr Cancer Drug Targets. 2013;13(5):486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhang J, Liu J. Tumor stroma as targets for cancer therapy. Pharmacol Ther. 2013;137(2):200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yan H, Guo BY, Zhang S. Cancer‐associated fibroblasts attenuate Cisplatin‐induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem Biophy Res Commun. 2016;470(4):947–954. [DOI] [PubMed] [Google Scholar]
- 165. Xu LN, Xu BN, Cai J, Yang JB, Lin N. Tumor‐associated fibroblast‐conditioned medium promotes tumor cell proliferation and angiogenesis. Genet Mol Res. 2013;12(4):5863–5871. [DOI] [PubMed] [Google Scholar]
- 166. Leung CS, Yeung TL, Yip KP, et al. Calcium‐dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun. 2014;5:5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Lawrenson K, Grun B, Lee N, et al. NPPB is a novel candidate biomarker expressed by cancer‐associated fibroblasts in epithelial ovarian cancer. Int J Cancer. 2015;136(6):1390–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Chen H, Yang WW, Wen QT, Xu L, Chen M. TGF‐beta induces fibroblast activation protein expression; fibroblast activation protein expression increases the proliferation, adhesion, and migration of HO‐8910PM [corrected]. Exp Mol Pathol. 2009;87(3):189–194. [DOI] [PubMed] [Google Scholar]
- 169. Yang W, Han W, Ye S, et al. Fibroblast activation protein‐alpha promotes ovarian cancer cell proliferation and invasion via extracellular and intracellular signaling mechanisms. Exp Mol Pathol. 2013;95(1):105–110. [DOI] [PubMed] [Google Scholar]
- 170. Scotton CJ, Wilson JL, Scott K, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62(20):5930–5938. [PubMed] [Google Scholar]
- 171. Kryczek I, Lange A, Mottram P, et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005;65(2):465–472. [PubMed] [Google Scholar]
- 172. Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F. Involvement of SDF‐1alpha/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. International journal of cancer. 2008;122(1):91–99. [DOI] [PubMed] [Google Scholar]
- 173. Kulbe H, Chakravarty P, Leinster DA, et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res. 2012;72(1):66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lau TS, Chung TK, Cheung TH, et al. Cancer cell‐derived lymphotoxin mediates reciprocal tumour‐stromal interactions in human ovarian cancer by inducing CXCL11 in fibroblasts. J Pathol. 2014;232(1):43–56. [DOI] [PubMed] [Google Scholar]
- 175. Cai J, Tang H, Xu L, et al. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis. 2012;33(1):20–29. [DOI] [PubMed] [Google Scholar]
- 176. Kamat AA, Fletcher M, Gruman LM, et al. The clinical relevance of stromal matrix metalloproteinase expression in ovarian cancer. Clin Cancer Res. 2006;12(6):1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Agarwal A, Tressel SL, Kaimal R, et al. Identification of a metalloprotease‐chemokine signaling system in the ovarian cancer microenvironment: implications for antiangiogenic therapy. Cancer Res. 2010;70(14):5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Belotti D, Paganoni P, Manenti L, et al. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 2003;63(17):5224–5229. [PubMed] [Google Scholar]
- 179. Furuya M, Yonemitsu Y, Aoki I. III. Angiogenesis: complexity of tumor vasculature and microenvironment. Curr Pharm Des. 2009;15(16):1854–1867. [DOI] [PubMed] [Google Scholar]
- 180. Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18(2):185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR. Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression. J Clin Invest. 1993;91(5):2194–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kulbe H, Thompson R, Wilson JL, et al. The inflammatory cytokine tumor necrosis factor‐alpha generates an autocrine tumor‐promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67(2):585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Charles KA, Kulbe H, Soper R, et al. The tumor‐promoting actions of TNF‐alpha involve TNFR1 and IL‐17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119(10):3011–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]