

Abstract
Purpose
An association between mutational burden and response to immune checkpoint therapy has been documented in several cancer types. The potential for such a mutational burden threshold to predict response to immune checkpoint therapy was evaluated in several clinical datasets, where mutational burden was measured either by whole-exome sequencing or by using commercially available sequencing panels.
Methods
Whole-exome sequencing and RNA sequencing data of 33 solid cancer types from The Cancer Genome Atlas were analyzed to determine whether a robust immune checkpoint–activating mutation (iCAM) burden threshold associated with evidence of immune checkpoint activation exists in these cancers that may serve as a biomarker of response to immune checkpoint blockade therapy.
Results
We found that a robust iCAM threshold, associated with signatures of immune checkpoint activation, exists in eight of 33 solid cancers: melanoma, lung adenocarcinoma, colon adenocarcinoma, endometrial cancer, stomach adenocarcinoma, cervical cancer, estrogen receptor–positive/human epidermal growth factor receptor 2–negative breast cancer, and bladder-urothelial cancer. Tumors with a mutational burden higher than the threshold (iCAM positive) also had clear histologic evidence of lymphocytic infiltration. In published datasets of melanoma, lung adenocarcinoma, and colon cancer, patients with iCAM-positive tumors had significantly better response to immune checkpoint therapy compared with those with iCAM-negative tumors. Receiver operating characteristic analysis using The Cancer Genome Atlas predictions as the gold standard showed that iCAM-positive tumors are accurately identifiable using clinical sequencing assays, such as FoundationOne (Foundation Medicine, Cambridge, MA) or StrandAdvantage (Strand Life Sciences, Bangalore, India). Using the FoundationOne-derived threshold, an analysis of 113 melanoma tumors showed that patients with iCAM-positive disease have significantly better response to immune checkpoint therapy. iCAM-positive and iCAM-negative tumors have distinct mutation patterns and different immune microenvironments.
Conclusion
In eight solid cancers, a mutational burden threshold exists that may predict response to immune checkpoint blockade. This threshold is identifiable using available clinical sequencing assays.
INTRODUCTION
Anti–programmed death-1 (PD-1) and anti–cytotoxic T-lymphocyte antigen-4 (CTLA-4) therapy results in prolonged response in some solid cancers.1-5 Response to CTLA-4 blockade in melanoma1,2 and PD-1 blockade in lung,3 colorectal,4 and urothelial5 cancers is associated with increased mutational burden. This has led to the hypothesis that high mutational–burden tumors are more likely to harbor peptide neoantigens, which may induce a cytotoxic T-cell response that tumors must block to survive, making them vulnerable to immune checkpoint disruption. Although this association between mutational burden and response to immune checkpoint therapy is documented in some cancers,1-5 it is unclear whether this relationship exists in other cancers. Moreover, there is no established method to identify an optimal mutational burden threshold in different cancers. Using RNA sequencing (RNA-seq) and whole-exome sequencing data for all solid cancers in The Cancer Genome Atlas (TCGA), we asked the following three questions: (1) Is there a mutational burden threshold above which there is a signature of immune activation and immune checkpoint pathway upregulation in some cancers? (2) Can this threshold be identified using clinical sequencing assays that interrogate a subset of the exome? (3) Does this threshold identify responders to immune checkpoint therapy?
We found that a robust immune checkpoint–activating mutation (iCAM) threshold satisfying the first two conditions exists in eight classes of solid tumors. In published datasets where condition (3) is testable, patients with iCAM-positive cancers have significantly better clinical response to immune checkpoint therapy than patients with iCAM-negative cancers. Furthermore, an iCAM threshold is accurately identifiable using existing clinical sequencing assays. Analysis of a cohort of patients with melanoma, sequenced using the FoundationOne assay (Foundation Medicine, Cambridge, MA),6 demonstrated that patients with iCAM-positive melanomas had significantly better response to PD-1 blockade than patients with iCAM-negative melanomas. Finally, for these cancer types, distinct patterns of mutations are present in iCAM-positive versus iCAM-negative tumors, suggesting potentially distinct underlying disease mechanisms.
METHODS
Data Collection and Processing
Mutation data were obtained from TCGA (https://wiki.nci.nih.gov/display/TCGA/TCGA+MAF+Files). All Mutation Annotation Format files current as of January 31, 2016 (public and protected) were downloaded, mapped from hg18 to hg19 using liftOver if necessary (https://genome.ucsc.edu/cgi-bin/hgLiftOver), re-annotated using Oncotator,7 merged, and de-duplicated. Clinical data were obtained from the TCGA Data Portal (https://tcga-data.nci.nih.gov/docs/publications/tcga/). The data were subsequently relocated to the Genomic Data Commons Legacy Archive (https://portal.gdc.cancer.gov/legacy-archive/search/f). For each tumor, RNAseqV2-scaled estimates from the Broad Institute Broad Genome Data Analysis Center (http://gdac.broadinstitute.org) and TCGA Data Portal were divided by the median value per sample and used to quantify immune infiltration and leukocyte composition by ESTIMATE8 and CIBERSORT,9 respectively. Only unambiguous (P < .05) CIBERSORT outputs were used. ERBB2 focal copy number data and ESR1 mRNA expression data from the Broad Genome Data Analysis Center were used to classify breast cancer samples into clinical subtypes (estrogen receptor [ER] positive/human epidermal growth factor receptor 2 [HER2] negative, ER negative/HER2 negative, or HER2 positive), and these subtypes were analyzed separately. The luminal-A/B status of ER-positive/HER2-negative tumors was obtained from TCGA.10
Software and Statistical Tests Used
MATLAB R2015a-R2016a (MathWorks, Natick, MA) and R 3.1.0-3.3.2 were used for analysis, unless specified otherwise. In addition to CIBERSORT9 P values, two-sided (P < .05) Fisher’s test and log-rank test were used to compare response and survival rates, respectively. The two-sided (P < .05) Wilcoxon rank sum test was used for all other comparisons. All bracketed ranges throughout this article correspond to 95% confidence intervals.
Determination of iCAM Threshold in TCGA
If nonsynonymous mutational burden is associated with immune checkpoint activation, there should be a mutational burden threshold above which tumors have the following: (1) CD8-positive T-cell response (Data Supplement) and (2) overexpression of immune checkpoint pathways (Data Supplement). In a cancer type cohort with N + 1 unique nonsynonymous mutational burden values ([x0 < x1 < … < xN]), there are N possible values of this threshold ([x1, ..., xN]). For each xi, we tested whether tumors with mutational burden ≥ xi had significantly higher CD8-positive T-cell activation and PD-1/CTLA-4 pathway upregulation compared with tumors with mutational burden < xi (Data Supplement). In eight cancer types, criteria (1) and (2) were simultaneously satisfied for several x-values, confirming an association between mutational burden and immune checkpoint activation in those cancer types. x-values that satisfied a maximum number of subcriteria were selected (Data Supplement) and ranked by P value for each satisfied criterion. The optimal x-value (ie, the iCAM threshold) was identified by minimizing the sum of ranks over these subcriteria (Data Supplement).
Pathology-Based Lymphocyte Infiltration Score
Images for hematoxylin and eosin–stained histologic sections of 15 iCAM-positive and 15 iCAM-negative tumors in each cancer type from the Cancer Digital Slide Archive (http://cancer.digitalslidearchive.net) were pathologically evaluated for tumor-infiltrating lymphocytes in a blinded fashion using a modified Black et al11 system (Data Supplement). The presence of tumor-infiltrating lymphocytes was assessed in ≥ 10 fields at the highest magnification, and tumors were graded for lymphocytic density and distribution, on all slides for each case. The score per slide was the cumulative grade of density and distribution, with the highest score for each case taken as the final score.
Melanoma Samples Used for Prospective Validation of iCAM Threshold
Mutation, treatment, and outcome data in a cohort of 196 de-identified samples were obtained under institutional review board–approved protocols to test the iCAM threshold for the FoundationOne assay.6 One hundred eleven samples from the Rutgers Cancer Institute of New Jersey and 85 from either the Vanderbilt-Ingram Cancer Center or Massachusetts General Hospital had data from either the 315- or 236-gene FoundationOne panels. One hundred thirteen samples from patients treated with single-agent PD-1 therapy (28 from Rutgers Cancer Institute of New Jersey, 85 from Vanderbilt-Ingram Cancer Center and Massachusetts General Hospital) were stratified by iCAM status using the mutational burden threshold of either 14 or nine mutations for the FoundationOne 315- and 236-gene panels, respectively (Data Supplement).
RESULTS
Identification of iCAM Threshold
Using TCGA whole-exome sequencing and RNA-seq data for 33 solid cancers, we looked for an iCAM mutational burden threshold, such that iCAM-positive tumors (ie, tumors with mutational burden above the threshold) had evidence of the following: (1) immune activation: high CD8A mRNA levels, evidence of immune infiltration (measured by ESTIMATE8), and high CD8-positive T-cell fraction in leukocytes (measured by CIBERSORT9); and (2) upregulation of immune checkpoint pathway gene expression (ie, PD-1, CTLA-4, and their ligands; Data Supplement). Although these tumor features have been previously shown to associate with response to immune checkpoint therapy,4,5,12 in this study we used RNA-seq data to indirectly measure these criteria.
Mutational burden has been identified to predict response to immune checkpoint blockade in melanoma,1,2 lung adenocarcinoma,3 colon adenocarcinoma,4 and bladder-urothelial cancer.5 We found that, in addition, a robust iCAM threshold also exists in endometrial cancer, stomach adenocarcinoma, cervical cancer, and ER-positive/HER2-negative breast cancers (Data Supplement). Figure 1A shows the mutational burden densities in each cancer type and the optimal stratification into iCAM-positive and iCAM-negative classes. Figure 1B shows the RNA-seq–based criteria of immune checkpoint activation that are significantly higher in iCAM-positive tumors compared with iCAM-negative tumors for each cancer type. A robust mutational burden threshold could not be identified for triple-negative breast cancer and renal cell cancer—cancer types where immune checkpoint therapy has some clinical activity.13,14
Fig 1.

A robust immune checkpoint–activating mutation (iCAM) threshold associated with evidence of immune checkpoint activation can be identified in eight cancer types in The Cancer Genome Atlas (TCGA). (A) Distributions of nonsynonymous mutational burden in eight cancer types (in log10 scale) separate the samples into iCAM-positive (blue) and iCAM-negative (gold) subsets. (B) Immune checkpoint activation criterion (Data Supplement) significantly higher in iCAM-positive compared with iCAM-negative is shown in gold for the eight cancer types. (C) Results from blinded pathologic quantification of lymphocyte infiltration in high-resolution histologic images from TCGA of 15 iCAM-positive and 15 iCAM-negative tumors in each of the eight cancer types. iCAM-positive samples (blue) and iCAM-negative samples (gold); the shape widths are proportional to number of samples. BLCA, bladder cancer; CESC, cervical cancer; COAD, colon adenocarcinoma; CTLA-4; cytotoxic T-lymphocyte antigen-4; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; LUAD, lung adenocarcinoma; PD-1; programmed death-1; PD-L1, programmed death ligand-1; PD-L2, programmed death ligand-2; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, endometrial cancer.
As an independent test, a blinded pathologic analysis of high-resolution, whole-slide images of hematoxylin and eosin–stained sections from TCGA of 15 iCAM-positive and 15 iCAM-negative tumors for each cancer type showed that iCAM-positive tumors had significantly higher lymphocyte infiltration than iCAM-negative tumors in all eight cancer types (Fig 1C). On a log10 scale, the average iCAM threshold was 2.28 ± 0.21 for these cancers. Note that these thresholds are for the TCGA dataset and specific to our use of the union of mutation calls from TCGA centers. For different definitions of mutational burden or different sequencing depths, the thresholds need to be recomputed.
iCAM Status Predicts Response to PD-1 and CTLA-4 Blockade in Published Data
To test the utility of iCAM, the iCAM thresholds were evaluated in published datasets for patients treated with CTLA-4 blockade in skin melanoma1,2 and PD-1 blockade in lung adenocarcinoma3 and colon adenocarcinoma4 (Fig 2), with samples stratified into iCAM-positive and iCAM-negative classes, as described in the Data Supplement. For consistency with the TCGA dataset, we excluded uveal-melanoma samples from the Snyder et al1 dataset, mucosal-melanoma samples from the Van Allen et al2 dataset, lung-squamous samples from the Rizvi et al3 dataset, and small bowel and ampulla of Vater samples from the Le et al4 dataset.
Fig 2.
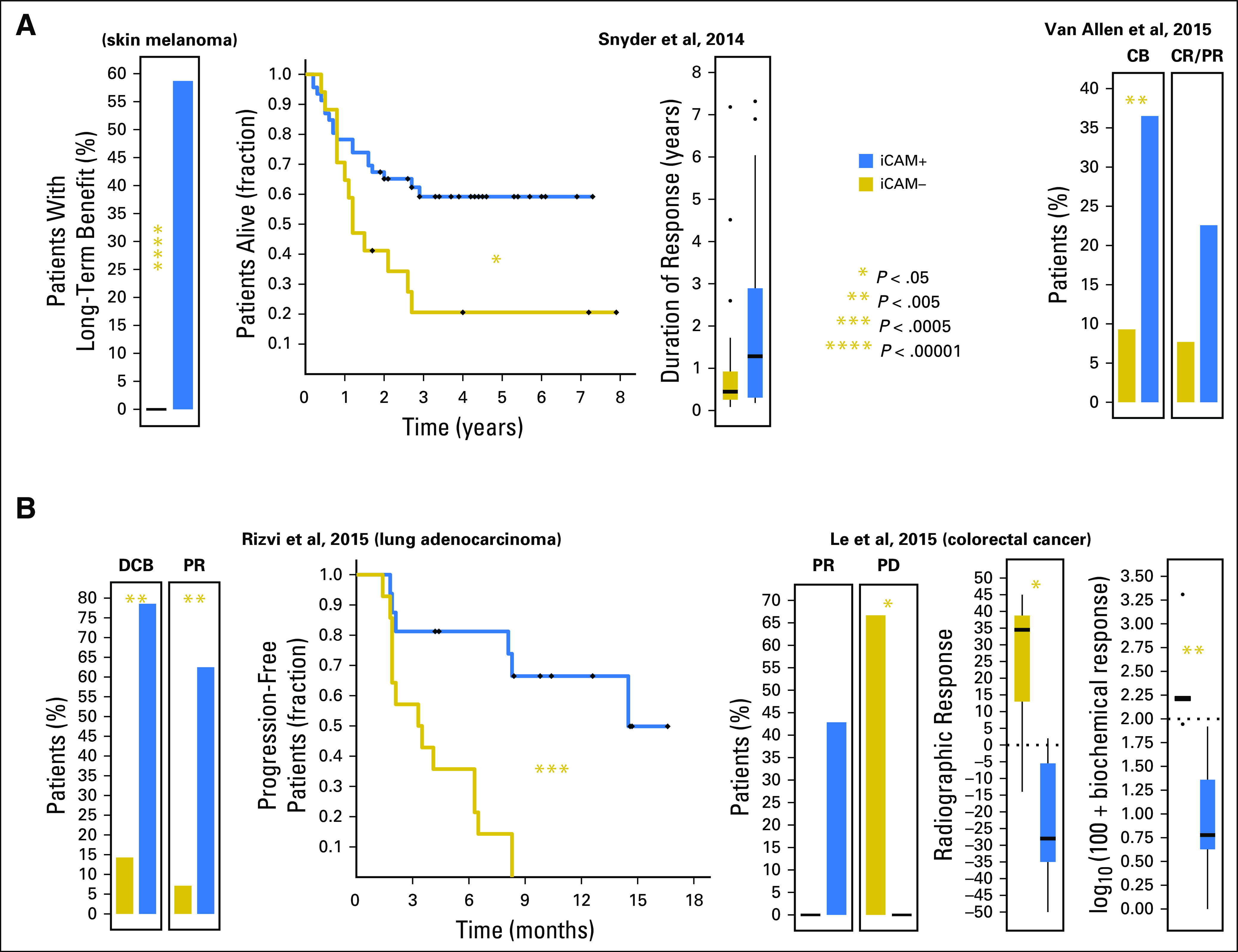
The immune checkpoint–activating mutation (iCAM) threshold is associated with clinical outcomes in patients treated with immune checkpoint blockade in published datasets. (A) Comparison of response in patients with skin melanoma to cytotoxic T-lymphocyte antigen-4 (CTLA-4) targeted therapy by iCAM status in two independent datasets.1,2 (B) Comparison of response in patients with lung adenocarcinoma3 and colorectal cancer4 to programmed death-1 targeted therapy by iCAM status. CB, clinical benefit; CR, complete response; DCB, durable clinical benefit; PD, progressive disease; PR, partial response.
For CTLA-4 blockade in melanoma (Fig 2A), in the Snyder et al1 dataset, 27 (approximately 60%) of 46 patients with iCAM-positive melanomas had long-term benefit compared with zero of 17 with iCAM-negative melanomas. Patients with iCAM-positive melanomas had significantly longer overall survival (hazard ratio [HR], 0.33 [0.14 to 0.78]) and longer duration of response (median, 66 v 22 weeks). In the Van Allen et al2 dataset, 23 (> 35%) of 63 patients with iCAM-positive melanomas had clinical benefit compared with four (< 10%) of 43 patients with iCAM-negative melanomas (odds ratio [OR], 5.6 [1.8 to 17.7]). Objective response rate was higher (OR, 3.5 [0.9 to 13.1]) for patients with iCAM-positive (14 of 62) versus iCAM-negative (three of 39) melanomas. The differences in response rates in these two datasets is probably the result of differences in the patient populations, with the Snyder et al dataset having a higher proportion of patients with long-term benefit compared with the Van Allen et al dataset.
For PD-1 blockade (Fig 2B), in the Rizvi et al3 data set for lung adenocarcinoma, 11 (approximately 80%) of 14 of the iCAM-positive subset had durable clinical benefit compared with 2 (< 15%) of 14 of the iCAM-negative subset (OR, 22.0 [3.1 to 157.3]). Ten (> 60%) of 16 of the iCAM-positive subset had partial response compared with one (< 10%) of 14 of the iCAM-negative subset (OR, 21.7 [2.2 to 210.1]). Progression-free survival was significantly better for patients with iCAM-positive compared with iCAM-negative cancers (HR, 0.13 [0.05 to 0.36]). In the Le et al4 colon adenocarcinoma dataset, objective response rate was higher in iCAM-positive (three of seven) versus iCAM-negative (zero of six) subsets; the fraction of patients with progressive disease was significantly lower in iCAM-positive (zero of seven) versus iCAM-negative (four of six) subsets, and radiographic and biochemical responses were significantly better in iCAM-positive versus iCAM-negative subsets (median, −28 v 34.5 and −94 v 62.5, respectively).
iCAM-Positive Tumors Are Identifiable Using FoundationOne or StrandAdvantage Assays
To test whether assays that interrogate a fraction of the exome can identify iCAM-positive tumors, the mutational burden of TCGA tumors was recomputed by restricting the data to genomic regions assayed by two commercial assays, FoundationOne6 (315 or 236 genes), and StrandAdvantage15 (selected regions of 152 genes). Receiver operating characteristic curves (Fig 3) for iCAM-positive versus iCAM-negative class prediction of tumors from TCGA, using mutations in genomic regions sequenced by these assays (Data Supplement), showed that FoundationOne6 (area under the curve, 0.85 to 0.99) and StrandAdvantage15 (area under the curve, 0.78 to 0.98) can identify iCAM-positive tumors accurately.
Fig 3.

Immune checkpoint–activating mutation (iCAM)–positive tumors can be identified with high accuracy using routine clinical assays. Receiver operating characteristic curves for iCAM status determination of The Cancer Genome Atlas (TCGA) samples using only the exons used in FoundationOne6 (blue) and StrandAdvantage15 (gold) assays. Whole-exome sequencing TCGA classification was used as the gold standard. The area under the receiver operating characteristic curve for these two assays, respectively, was as follows: melanoma (0.98, 0.94), lung (0.94, 0.86), colon (0.99, 0.96), endometrial (0.98, 0.95), stomach (0.99, 0.98), cervical (0.88, 0.78), estrogen receptor (ER)–positive/human epidermal growth factor receptor 2 (HER2)–negative breast (0.85, 0.78), and bladder (0.90, 0.86). BLCA, bladder cancer; CESC, cervical cancer; COAD, colon adenocarcinoma; LUAD, lung adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, endometrial cancer.
Validation of iCAM Threshold in Predicting Response to Immune Checkpoint Blockade in Melanoma Using the FoundationOne Assay
One hundred ninety-six skin melanoma samples were sequenced using the FoundationOne assay6 (Data Supplement) and the number of nonsynonymous mutations was calculated per tumor by summing known or likely mutations and variants of unknown significance. The iCAM threshold projected from TCGA was estimated as nine or 14 nonsynonymous mutations for FoundationOne assays that interrogated 236 or 315 genes, respectively (Data Supplement). Evaluation of response data in 113 patients treated with single-agent PD-1 blockade showed that patients with iCAM-positive tumors had significantly higher objective responses (44 of 76 v six of 37 patients; OR, 7.10 [2.65 to 19.04]) and longer progression-free and overall survival (HR, 0.32 [0.18 to 0.57] and HR, 0.37 [0.20 to 0.69], respectively) compared with patients with iCAM-negative tumors (Data Supplement, Fig 4). These findings are consistent with earlier observations16 that mutation load per megabase from the FoundationOne assay6 can predict response to PD-1 blockade in a subset (n = 65) of this cohort.
Fig 4.
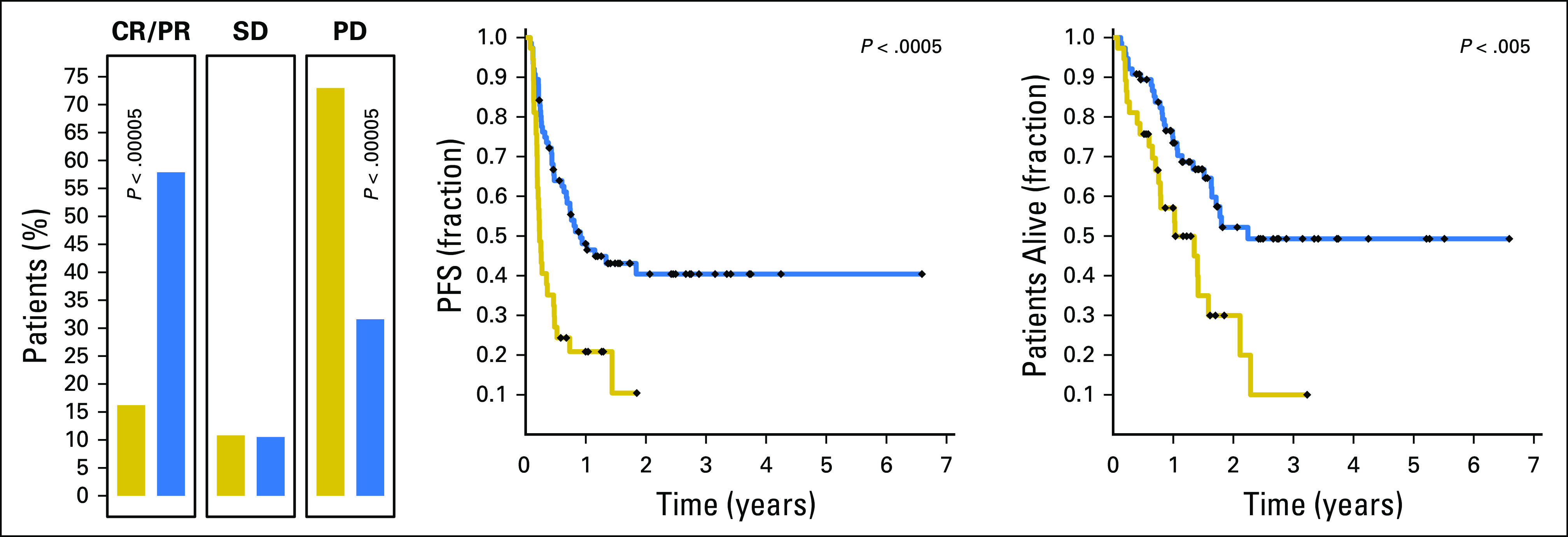
Prospective validation of immune checkpoint–activating mutation (iCAM) threshold for melanoma using FoundationOne6 assay results predicts outcome of immune checkpoint blockade in an independent cohort of patients with melanoma. Shown are comparisons of response rates, progression-free survival (PFS), and overall survival in 113 patients with metastatic melanoma treated with single-agent programmed death-1 (PD-1)–targeted therapy, stratified by iCAM status using the FoundationOne assay.6 CR, objective complete response; PD, progressive disease; PR, objective partial response; SD, stable disease (includes mixed response).
Significantly Mutated Genes in iCAM-Positive Tumors
The MutSigCV17 algorithm was used to identify significantly mutated genes in iCAM-positive and iCAM-negative classes in each cancer type (false discovery rate < 0.1). In lung adenocarcinoma (Fig 5), EGFR was significantly mutated in iCAM-negative tumors but not in iCAM-positive tumors. In colon adenocarcinoma, mutations in APC, KRAS, and TP53 were frequently observed in iCAM-negative tumors, consistent with the Fearon-Vogelstein model of carcinogenesis.18 However, neither APC nor TP53 were significantly mutated in iCAM-positive tumors; instead, iCAM-positive tumors (but not iCAM-negative tumors) had mutations in RNF43, ARID1A, BRAF, BMPR2, ACVR2A, and RPL22. EGFR mutations in lung cancer and APC and TP53 mutations in colon cancer were associated with lower incidences of iCAM-positive tumors, whereas mutations in ARID1A in lung cancer and mutations in RNF43, ARID1A, BRAF, BMPR2, and ACVR2A in colon cancer were associated with higher incidences of iCAM-positive tumors (Data Supplement). Tumor suppressor genes RNF43,19 ACVR2A,20 and RPL2221 were significantly mutated in iCAM-positive colon adenocarcinoma (Fig 5), endometrial cancer, and stomach adenocarcinoma (Data Supplement), but rarely in iCAM-negative tumors. In the other four cancer types, several genes were significantly mutated in iCAM-positive but not iCAM-negative tumors and vice versa (Data Supplement). These findings suggest that iCAM-positive tumors may have distinct underlying driver mutations compared with iCAM-negative tumors.
Fig 5.
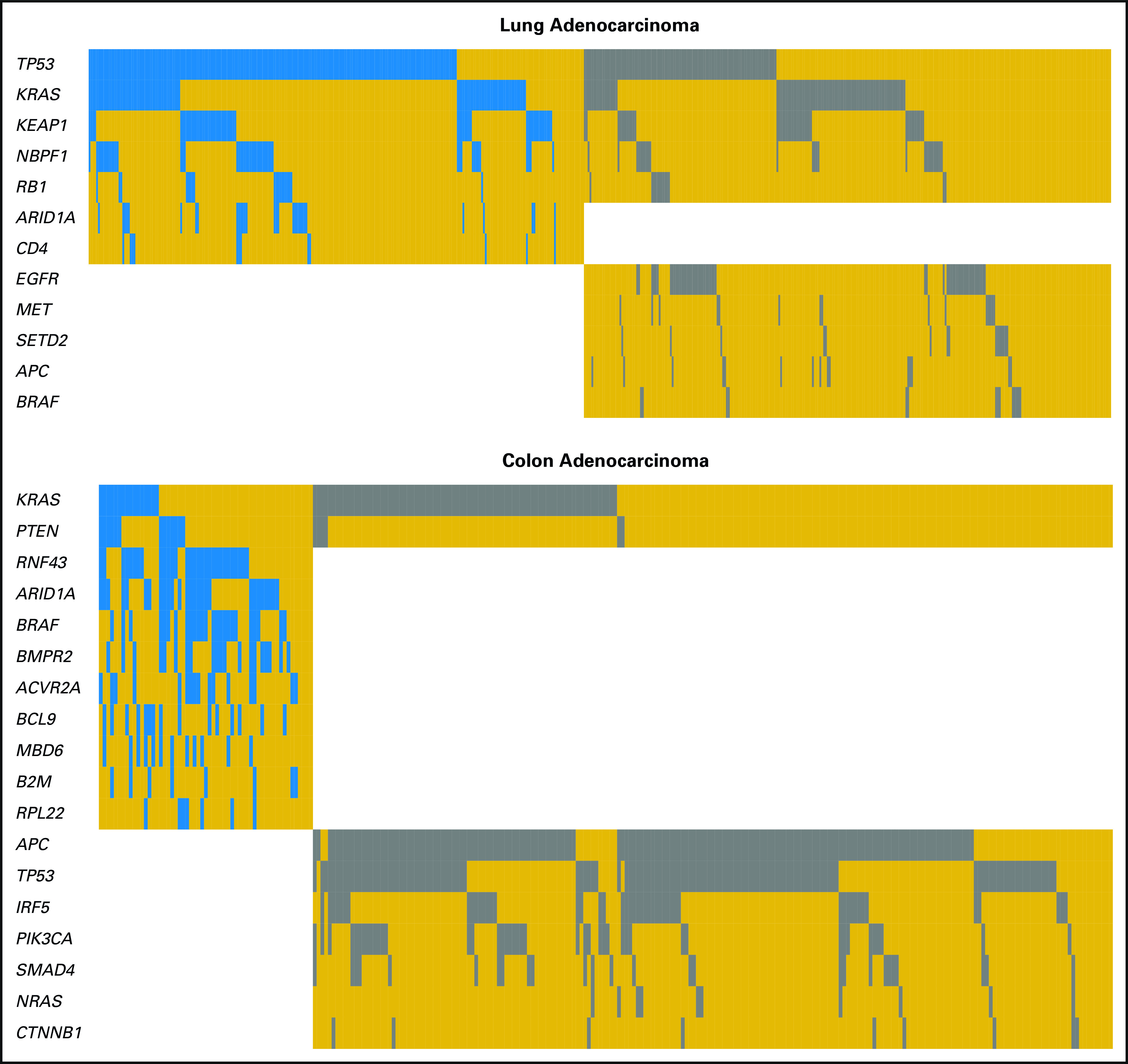
Selected significantly mutated genes (MutSigCV,17 false discovery rate < 0.1) in immune checkpoint–activating mutation (iCAM)–positive and iCAM-negative tumors. Columns are samples and rows are genes (white, gene not significantly mutated in that class; gold, gene significantly mutated in that class but not mutated in that sample; blue and gray, gene significantly mutated in iCAM-positive [blue]/iCAM-negative [gray] class and mutated in that sample).
Mutational Etiology of iCAM-Positive Tumors
For each tumor, we estimated the fractional contribution of 30 mutation signatures (Data Supplement) from the Catalogue of Somatic Mutations in Cancer22,23 (http://cancer.sanger.ac.uk/cosmic/signatures). Compared with iCAM-negative tumors, iCAM-positive tumors had a higher contribution from an ultraviolet signature in melanoma and a smoking signature in lung adenocarcinoma (Fig 6A), consistent with known pathogenesis of melanoma24 and lung25 cancers. Smoking history was also strongly associated with iCAM status in lung adenocarcinoma (Data Supplement).
Fig 6.
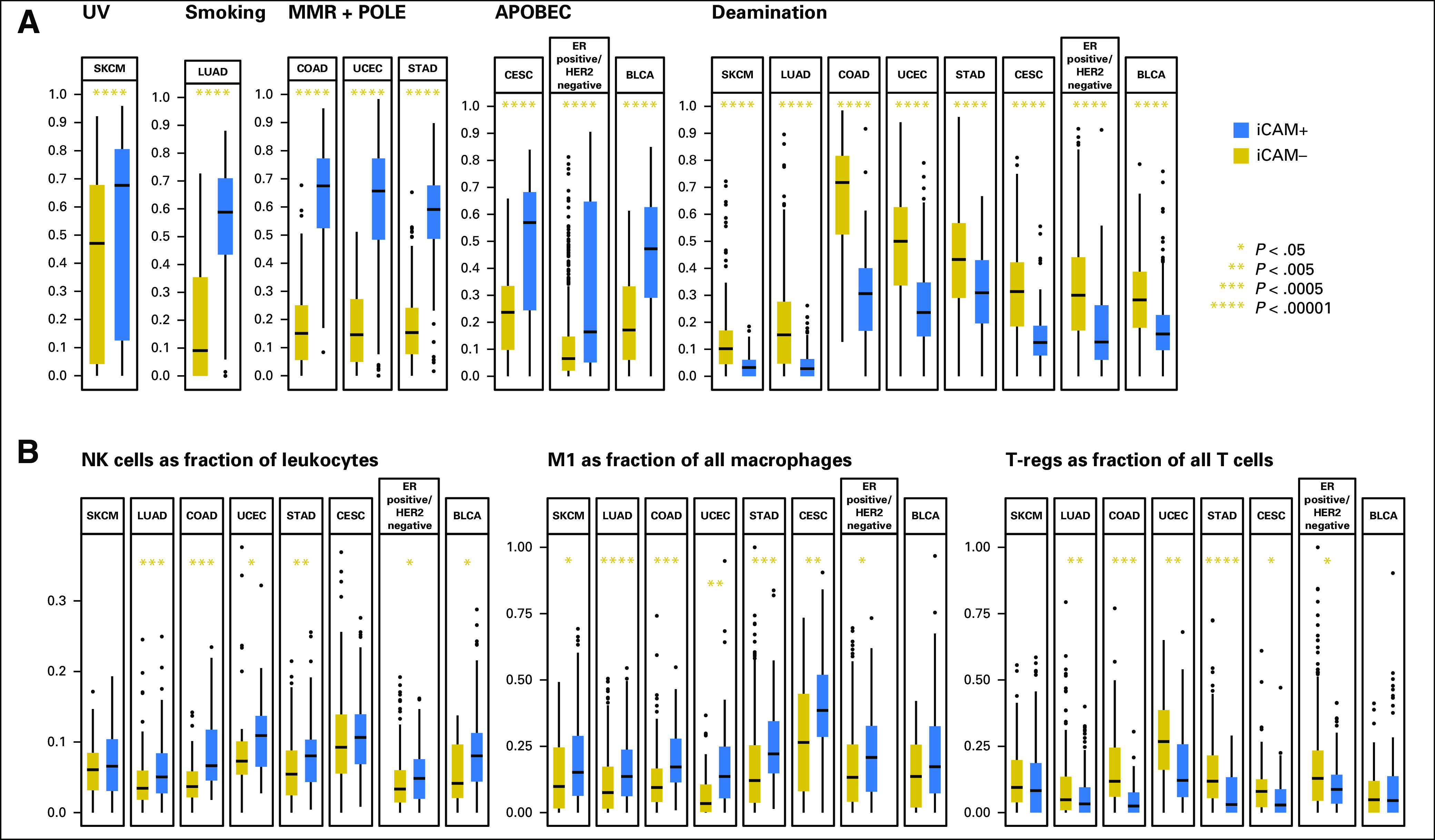
Mutation and immune signatures of immune checkpoint–activating mutation (iCAM)–positive versus iCAM-negative tumors. (A) Comparison of the fractional contribution of mutation signatures from Catalogue of Somatic Mutations in Cancer for etiologies associated with iCAM-positive versus iCAM-negative status in the eight cancer types. (B) Other immune signatures associated with iCAM status (natural killer [NK] fraction in leukocytes, M1 fraction in macrophages, and regulatory T cell (T-regs) fraction in T cells in iCAM-positive and iCAM-negative tumors). The numbers of iCAM-negative and iCAM-positive tumors, respectively, in (B) are as follows: SKCM (71, 211), LUAD (218, 203), COAD (81, 36), UCEC (39, 37), STAD (187, 55), CESC (84, 49), estrogen receptor (ER)–positive/human epidermal growth factor receptor 2 (HER2)–negative (391, 80), and BLCA (34, 134). Only tumors with a CIBERSORT P value < .05 were included in (B). APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; BLCA, bladder cancer; CESC, cervical cancer; COAD, colon adenocarcinoma; Deamination, deamination of 5-methylcytosine; LUAD, lung adenocarcinoma; MMR, mismatch repair defect; POLE, polymerase epsilon proof-reading defect; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, endometrial cancer; UV, ultraviolet.
In colon adenocarcinoma, endometrial cancer, and stomach adenocarcinoma, iCAM-positive tumors were enriched in either mismatch repair or POLE proofreading defects (Fig 6A). Most iCAM-positive tumors in these three cancer types either had microsatellite instability or POLE mutation (Data Supplement). These etiologies are clinically relevant because POLE mutations in endometrial cancer26 and mismatch repair defects in colorectal cancer4 are known to associate with response to PD-1 blockade.
In cervical cancer, ER-positive/HER2-negative breast cancer, and bladder-urothelial cancer, an iCAM-positive status was associated with apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like mutation signatures (Fig 6A), which builds on the previously reported presence of apolipoprotein B mRNA editing enzyme, catalytic polypeptice-like signatures in these cancers.27 In ER-positive/HER2-negative breast cancer, approximately 25% of luminal-B tumors were iCAM positive, whereas < 10% of luminal-A tumors were iCAM positive (Data Supplement). In all eight cancer types, iCAM-negative tumors had a higher fractional contribution from deamination of 5-methylcytosine compared with iCAM-positive tumors (Fig 6A).
Additional Immunologic Properties of iCAM-Positive Tumors
In most of the eight cancer types, iCAM-positive tumors had significantly higher fractions of natural killer cells in leukocytes and M1 cells in macrophages, and significantly lower fractions of regulatory T cells in T cells (Fig 6B), by CIBERSORT9 analysis. Natural killer cells are involved in immune surveillance,28 M1 macrophages suppress angiogenesis and induce apoptosis,29 and regulatory T cells suppress immune response.30 Thus iCAM-positive tumors may have a more favorable immune microenvironment for immune checkpoint therapy. These data are consistent with reports suggesting that specific features of immune microenvironments may associate with response to immune checkpoint therapy.31,32
Sixty-one genes were significantly overexpressed in iCAM-positive tumors compared with iCAM-negative tumors in all eight cancer types (Data Supplement), and approximately 70% of these genes were immune-system related: associated with the interferon-gamma pathway (IFNG, CXCL9, CXCL10, and CXCL11) and cytotoxic T cells (CD8A, PRF1, GZMA, and GZMB). Expression of interferon-pathway genes is associated with recruitment of cytotoxic T cells to the tumor microenvironment.33 The immune checkpoint gene LAG3 was significantly overexpressed in iCAM-positive tumors in all eight cancers, suggesting an additional drug target pathway.
DISCUSSION
In eight of 33 solid cancers in TCGA, we found a mutational burden threshold (iCAM) that identified tumors with signatures of immune checkpoint activation. Analysis of published datasets for melanoma, lung adenocarcinoma, and colon cancer showed that patients with iCAM-positive tumors had better responses to immune checkpoint therapy. The iCAM threshold can be accurately identified using routine clinical sequencing assays. Applied to an independent melanoma data set using the FoundationOne assay,6 iCAM-positive status was able to identify responders to single-agent PD-1 blockade with high accuracy. Although FoundationOne6 and StrandAdvantage15 assays were used in this study, any hybrid capture–based assay that identifies mutations in a substantial portion of the exome could be used. Note that these assays do not have germline controls, so some variant calls may be rare germline polymorphisms. However, these should not affect the presence of an iCAM threshold. The thresholds found in this study could be further refined by removing likely germline variants, increasing sequencing depth, analyzing more samples, and conducting a detailed analysis of tumor purity and mutant allele frequency. Although mutational burden probably associates with response to immune checkpoint blockade in these cancers, other factors (including specific features of the immune microenvironment, germline single-nucleotide polymorphisms, and epigenetic signatures) may also influence response.
In many cancer types, a robust mutational burden threshold associated with evidence of immune checkpoint activation could not be identified. This includes triple-negative breast cancer and renal cell carcinoma, which are cancers with substantial response rates to single-agent immune checkpoint blockade.13,14 In these cancers, features other than tumor mutational load are probably responsible for immune checkpoint activation. For example, in triple-negative breast cancer, the genomic landscape is dominated by genomic rearrangements,34 which are not always captured by exome sequencing. Similarly, other features, such as viral etiology, seen in Merkel cell cancer, may be strongly immunogenic, yet do not associate with a high nonsynonymous mutational burden.35,36
The patterns of mutations present in iCAM-positive tumors reflect the underlying nature of mutagenesis. In the TCGA lung adenocarcinoma cohort (Data Supplement), iCAM status strongly associated with a molecular signature of smoking and smoking history, suggesting that immune checkpoint therapy may work better for heavy smokers or recently reformed smokers, in agreement with a study3 where high transversion rates associated with response to anti–PD-1 therapy. Almost 75% of EGFR-mutant lung cancers were iCAM negative, suggesting that these tumors may be less responsive to immune checkpoint blockade.
In colon adenocarcinoma, endometrial cancer, and stomach adenocarcinoma (Data Supplement), > 85% of iCAM-positive tumors had microsatellite instability-high status and/or a POLE/POLD1 mutation compared with < 5% of iCAM-negative tumors. In ER-positive/HER2-negative breast cancer (Data Supplement), luminal-B tumors were more likely to be iCAM positive (28 of 116 v 19 of 217 tumors; OR, 3.32 [1.76 to 6.25]), which suggests that immune checkpoint therapy may be more effective in these tumors with poor prognosis.37
Not all patients with iCAM-positive cancers responded to PD-1 or CTLA-4 blockade (Fig 2), possibly because immune-evasion mechanisms other than PD-1/CTLA-4 pathway upregulation are operative (eg, abundant myeloid-derived suppressor cells, a known immune suppressor38; mutations in class I HLA genes39). Similarly, some patients with iCAM-negative cancers did respond to PD-1 or CTLA-4 blockade, possibly because of immune responses triggered by mechanisms other than high mutational burden (eg, expression of exogenous viruses,35,36 expression of endogenous retroviruses40). Note that whereas high mutation burden increases the likelihood of immunogenic neoepitopes, some low mutation–burden tumors may also express immunogenic neoepitopes by chance, leading to response. Alternatively, low tumor purity may affect the ability to measure mutational burden accurately. Hence, iCAM status is only one of several potential biomarkers of response to immune checkpoint therapy. Combining iCAM status with methods to quantify the immune microenvironment, such as Immunoscore,41 may be more informative than either approach alone.
In conclusion, quantitative analysis of mutational burden may be a clinically applicable marker of response to immune checkpoint therapy in these cancer types but will need to be rigorously evaluated in prospective trials that meet reporting recommendations for tumor marker prognostic studies criteria.
ACKNOWLEDGMENT
We thank Hossein Khiabanian, PhD, for help with analysis of the Foundation Medicine mutation data for the Rutgers Cancer Institute of New Jersey samples in the prospective cohort.
AUTHOR CONTRIBUTIONS
Conception and design: Anshuman Panda, Gyan Bhanot, Shridar Ganesan
Financial support: Shridar Ganesan
Administrative support: Shridar Ganesan
Provision of study material or patients: Jeffrey S. Ross, Ann Silk, Siraj Ali, Janice M. Mehnert, Christine M. Lovly, Jeffrey Sosman, Douglas B. Johnson, Shridar Ganesan
Collection and assembly of data: Anshuman Panda, Anil Betigeri, Kalyanasundaram Subramanian, Jeffrey S. Ross, Dean C. Pavlick, Siraj Ali, Paul Markowski, Ann Silk, Howard L. Kaufman, Janice M. Mehnert, Ryan Sullivan, Christine M. Lovly, Jeffrey Sosman, Douglas B. Johnson, Gyan Bhanot, Shridar Ganesan
Data analysis and interpretation: Anshuman Panda, Anil Betigeri, Kalyanasundaram Subramanian, Edmund Lattime, Janice M. Mehnert, Douglas B. Johnson, Gyan Bhanot, Shridar Ganesan
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Anshuman Panda
No relationship to disclose
Anil Betigeri
Employment: Strand Life Sciences
Kalyanasundaram Subramanian
Employment: Strand Life Sciences, Syngene International
Patents, Royalties, Other Intellectual Property: Patent to predict drug toxicity
Jeffrey S. Ross
Leadership: Foundation Medicine
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Dean C. Pavlick
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Siraj Ali
Employment: Foundation Medicine
Stock and Other Ownership Interests: Exelixis, Blueprint Medicines, Agios
Patents, Royalties, Other Intellectual Property: Patents via Foundation Medicine, patents via Seres Health on microbiome stuff in non-neoplastic disease (I)
Paul Markowski
No relationship to disclose
Ann Silk
Research Funding: Amgen (Inst), Bristol-Myers Squibb (Inst), Merck (Inst), Paometheus (Inst), Viralytics (Inst)
Howard L. Kaufman
Employment: Compass Therapeutics
Leadership: Compass Therapeutics
Honoraria: Amgen, EMD Serono, Merck, Celldex, Prometheus, Turnstone Bio, Compass Therapeutics
Consulting or Advisory Role: Amgen, Merck, Merck Serono, Paometheus, Celldex, Turnstone Bio, Bristol-Myers Squibb, Compass Therapeutics
Speakers’ Bureau: Merck
Research Funding: Amgen (Inst), Merck (Inst)
Travel, Accommodations, Expenses: EMD Serono, Turnstone Bio
Edmund Lattime
Stock and Other Ownership Interests: Johnson & Johnson (I)
Patents, Royalties, Other Intellectual Property: I am an inventor of an oncolytic virus, for which the patent is held by Thomas Jefferson University, where I worked from 1989 to 1998. The patent is licensed by Silagen. I receive a small annual fee from Thomas Jefferson University.
Janice M. Mehnert
Honoraria: Genentech, EMD Serono
Consulting or Advisory Role: Merck Sharp & Dohme, Amgen
Research Funding: Merck (Inst), Sanofi (Inst), Novartis (Inst), Polynoma (Inst), Immunocore (Inst), Amgen (Inst), AstraZeneca (Inst)
Travel, Accommodations, Expenses: EMD Serono, Merck Sharp & Dohme
Other Relationship: Amgen, EMD Serono, Merck
Ryan Sullivan
Honoraria: Genentech
Consulting or Advisory Role: Novartis, Biodesix, Paometheus, Amgen, Takeda, WorldCare Clinical, ACI Clinical, Merck, BioLineRx
Research Funding: Amgen (Inst), Eli Lilly (Inst), BioMed Valley Discoveries (Inst), Merck (Inst), Deciphera (Inst), Genentech (Inst)
Other Relationship: Boehringer Ingelheim
Christine M. Lovly
Honoraria: Novartis, Sequenom, Qiagen, Pfizer, NCCN, Takeda
Consulting or Advisory Role: ARIAD, Clovis Oncology, Genoptix, Novartis, Foundation Medicine
Research Funding: AstraZeneca, Novartis
Travel, Accommodations, Expenses: Pfizer
Jeffrey Sosman
Honoraria: Amgen, Merck, Array BioPharma, Bristol-Myers Squibb
Consulting or Advisory Role: Amgen, Merck, Array BioPharma, Bristol-Myers Squibb
Douglas B. Johnson
Consulting or Advisory Role: Bristol-Myers Squibb, Genoptix, Merck, Novartis, Incyte
Research Funding: Incyte
Patents, Royalties, Other Intellectual Property: Intellectual property and patents pending surrounding use of MHC-II and response to immune therapy
Gyan Bhanot
No relationship to disclose
Shridar Ganesan
Employment: Merck (I)
Stock and Other Ownership Interests: Ibris, Inspirata, Merck (I)
Consulting or Advisory Role: Inspirata, Novartis
Patents, Royalties, Other Intellectual Property: I hold two patents for digital imaging that may be licensed to Ibris and Inspirata.
Travel, Accommodations, Expenses: Inspirata
REFERENCES
- 1.Snyder A, Makarov V, Merghoub T, et al. : Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189-2199, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Allen EM, Miao D, Schilling B, et al. : Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207-211, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi NA, Hellmann MD, Snyder A, et al. : Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124-128, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le DT, Uram JN, Wang H, et al. : PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509-2520, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg JE, Hoffman-Censits J, Powles T, et al. : Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387:1909-1920, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos AH, Lichtenstein L, Gupta M, et al. : Oncotator: Cancer variant annotation tool. Hum Mutat 36:E2423-E2429, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshihara K, Shahmoradgoli M, Martínez E, et al. : Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman AM, Liu CL, Green MR, et al. : Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453-457, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Network : Comprehensive molecular portraits of human breast tumours. Nature 490:61-70, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Black MM, Speer FD, Opler SR: Structural representations of tumor-host relationships in mammary carcinoma; biologic and prognostic significance. Am J Clin Pathol 26:250-265, 1956 [DOI] [PubMed] [Google Scholar]
- 12.Tumeh PC, Harview CL, Yearley JH, et al. : PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568-571, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nanda R, Chow LQM, Dees EC, et al. : Pembrolizumab in patients with advanced triple-negative breast Cancer: Phase Ib KEYNOTE-012 study. J Clin Oncol 34:2460-2467, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motzer RJ, Escudier B, McDermott DF, et al. : Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373:1803-1813, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sen M, Katragadda S, Ravichandran A, et al. : StrandAdvantage test for early-line and advanced-stage treatment decisions in solid tumors. Cancer Med 6:883-901, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson DB, Frampton GM, Rioth MJ, et al. : Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 4:959-967, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence MS, Stojanov P, Polak P, et al. : Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214-218, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 61:759-767, 1990 [DOI] [PubMed] [Google Scholar]
- 19.Koo B-K, Spit M, Jordens I, et al. : Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488:665-669, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Deacu E, Mori Y, Sato F, et al. : Activin type II receptor restoration in ACVR2-deficient colon cancer cells induces transforming growth factor-beta response pathway genes. Cancer Res 64:7690-7696, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Rao S, Stadanlick JE, Cai KQ, et al. : Loss of Rpl22 promotes tumor progression through regulation of angiogenesis. Cancer Res 75, 2015. (suppl 15; abstr 521) [Google Scholar]
- 22.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB: Understanding the origins of human cancer. Science 350:1175-1177, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Brash DE: UV mutagenic photoproducts in Escherichia coli and human cells: A molecular genetics perspective on human skin cancer. Photochem Photobiol 48:59-66, 1988 [DOI] [PubMed] [Google Scholar]
- 25.Hernandez-Boussard TM, Hainaut P: A specific spectrum of p53 mutations in lung cancer from smokers: Review of mutations compiled in the IARC p53 database. Environ Health Perspect 106:385-391, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehnert JM, Panda A, Zhong H, et al. : Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest 126:2334-2340, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts SA, Lawrence MS, Klimczak LJ, et al. : An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45:970-976, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldhauer I, Steinle A: NK cells and cancer immunosurveillance. Oncogene 27:5932-5943, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Yuan A, Hsiao Y-J, Chen H-Y, et al. : Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci Rep 5:14273, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou W: Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6:295-307, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Huang AC, Postow MA, Orlowski RJ, et al. : T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545:60-65, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guillerey C, Huntington ND, Smyth MJ: Targeting natural killer cells in cancer immunotherapy. Nat Immunol 17:1025-1036, 2016 [DOI] [PubMed] [Google Scholar]
- 33.Corrales L, Matson V, Flood B, et al. : Innate immune signaling and regulation in cancer immunotherapy. Cell Res 27:96-108, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nik-Zainal S, Davies H, Staaf J, et al. : Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47-54, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nghiem PT, Bhatia S, Lipson EJ, et al. : PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med 374:2542-2552, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufman HL, Russell J, Hamid O, et al. : Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 17:1374-1385, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorlie T, Tibshirani R, Parker J, et al. : Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 100:8418-8423, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marvel D, Gabrilovich DI: Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J Clin Invest 125:3356-3364, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shukla SA, Rooney MS, Rajasagi M, et al. : Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 33:1152-1158, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiappinelli KB, Strissel PL, Desrichard A, et al. : Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162:974-986, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galon J, Mlecnik B, Bindea G, et al. : Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 232:199-209, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]