

Abstract
The hormone relaxin has been long recognized for its involvement in maternal adaptation during pregnancy. However, discoveries during the last two decades on the mechanism of action of relaxin, its family of receptors and newly described roles in attenuating ischemia/reperfusion injury, inflammation and arrhythmias have prompted vast interest in exploring its therapeutic potential in cardiovascular disease. These observations have inspired clinical trials in patients with acute heart failure that were concluded recently. This review will discuss our current understanding of the protective signaling pathways elicited by relaxin in the heart and highlight important new breakthroughs about relaxin signaling that may pave the way to more carefully-designed future trials.
Keywords: Relaxin, RXFP1, Myocardial Infarction, Heart Failure, Fibrosis
Scope and Relevance
The peptide hormone relaxin has traditionally been linked to the maternal adaptation of the cardiovascular system during the first trimester of pregnancy[1]. By activating several molecular signaling events, relaxin has been proposed as a pleiotropic and cardioprotective hormone. In fact, pre-clinical studies were able to demonstrate that relaxin promotes vasodilatation and angiogenesis, ameliorates ischemia/reperfusion (I/R) injury, regulates extracellular matrix turnover and remodeling following acute myocardial infarction (AMI) (see glossary), suppresses arrhythmias post MI and reverses fibrosis[2–5]. In the RELAX-AHF phase 3 clinical trial, intravenous administration of serelaxin (recombinant human relaxin-2) in the first 48 hours following admission for acute heart failure (AHF) was shown to be safe and was associated with remarkable survival benefit at 180 days[6]; however, the recently completed RELAX-AHF-2 trial did not yield significant clinical benefits, despite significant reduction in biomarkers of injury and worsening of HF through day 5. A recent article called into question the design of several HF trials and highlighted that acute HF is not a “48-hour illness”[7]. Due to its safety profile in patients and based on compelling preclinical reports suggesting a protective role of relaxin against AMI[2–5] and other cardiovascular diseases, more rigorous trials involving relaxin with carefully designed strategies are warranted to provide a fair assessment of its clinical efficacy in patients with cardiovascular disease. In this review article, we present updated information on signaling cascades associated with relaxin and its receptor RXFP1. In subsequent sections, we examine cardiac pathophysiology relevant to MI and adverse remodeling while limiting discussion to pathways specifically countered by relaxin signaling. After reviewing the preclinical data demonstrating the cardioprotective effects of relaxin, we present emerging evidence supporting the prospect of biased signaling at RXFP1 and its potential therapeutic implications.
Relaxin-2 and Its Receptor: Current Knowledge and Significance
The endocrine system specializes in evoking various physiological functions throughout the body in response to environmental or innate stimuli, and the advantage in its design allows for a pleiotropic response, wherein a single hormone, including the 6 kDa peptide relaxin, can orchestrate the dynamic interplay of significant downstream events. Relaxin-2, the active form in humans, is secreted by the corpus luteum in the luteal phase of the human ovarian cycle, and by the placenta[8]. However, mRNA levels were also detected in kidney[9] and heart[8]. Apart from its direct influence on pelvic girdle softening, cervical ripening and uterine contractility in the context of female reproduction [8], relaxin specializes in modulating hemodynamic and cardiovascular function during the latter stages of mammalian pregnancy. Cardiac output (CO) and systemic vascular compliance were increased to mid-term pregnancy levels when relaxin was chronically administered to non-pregnant rats[10].
Relaxin-2 exerts its physiologic effects by primarily acting on its cognate receptor – the Relaxin Family Peptide Receptor 1 (RXFP1), a G-protein Coupled Receptor (GPCR)[11]. Other receptors include RXFP2 (which shares several structural features with RXFP1, and is activated by the insulin like peptide INSL3), RXFP3 (receptor for neuropeptide relaxin-3) and RXFP4 (activated by the gut hormone INSL5)[11]. While its expression in vascular, neuronal, and reproductive tissue is well established[11], RXFP1 has also recently been documented in atrial and ventricular tissues of the heart[12,13], and more specifically, in the ventricular cardiomyocyte[2]. It has been well documented in the Langendorff isolated-perfused rat heart setting that relaxin also induces positive inotropy and chronotropy[14].
RXFP1 Signaling
RXFP1 belongs to a class of GPCRs – Leucine-Rich Repeat (LRR) containing GPCRs (LGRs) – that possess a unique LRR domain on the N-terminal extracellular side of the receptor. RXFP1 attains further specialization via the attachment of an LDL-A domain to the LRR[15]. The resulting three-dimensional structure allows for unique receptor conformations upon ligand activation. The B-chain of relaxin interacts with aspartate and glutamate residues present within the LRR domain, facilitating the interaction between LDL-A and the extracellular loops 1 and 2 (ECL1 and 2) of the transmembrane domain of RXFP1. RXFP1 with truncated LDL-A exhibits no signs of activation despite the presence of relaxin[15]
RXFP1 activates three distinct Gα proteins upon initiation of its canonical cascade by nanomolar levels of relaxin. These Gα proteins – Gαs, GαoB and Gαi3 – exhibit spatiotemporally varied activation profiles evident in the biphasic cAMP response generated at the onset of receptor activation. The initial surge of cAMP generation upon activation of Gαs is curbed by GαoB activity; a more gradual cAMP response is elicited by Gαi3 activation. The latter phase of cAMP generation is in part attributed to Gβγ-induced PI3K activation, as initially implicated in THP-1 cells, where cAMP response was diminished upon pharmacologic inhibition of PI3K[16]. In human atrial tissue, positive inotropy was associated with Gi-PI3K-induced cAMP accumulation[13]. Long-term exposure of serelaxin, recombinant relaxin-2, in human umbilical arterial smooth muscle cells (HUASMCs) and human cardiac fibroblasts (HCFs) also led to increased nNOS-driven NO generation, along with vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs)-2 and 9. Serelaxin induced phosphorylation of ERK1/2 and cGMP signaling in both of these cell lines[17]. Recent evidence also points to PKA-induced activation of p38 MAPK in hepatic stellate cells[18]. Therefore, promiscuous coupling of RXFP1 to its G proteins leads to a varied response contingent upon cell type and further studies are required to elucidate the diversity of pathways evoked.
Pathologic Signaling in Ischemic Heart Disease
The prevalence of ischemic heart disease and the multitude of pathophysiological mechanisms at play necessitate an ever-increasing demand for novel therapies that counter aberrant signaling, minimize loss of functional tissue and improve quality of life for affected patients. MI involves cessation of blood supply to the affected region of the heart, causing necrosis and extravasation of inflammatory cells into infarcted tissue. Ischemia also damages mitochondria and impairs the functionality of the electron transport chain[19]. Although timely reperfusion is essential to salvage myocardium and limit the extent of injury, reintroducing oxygen in the presence of deranged mitochondria leads to the generation of reactive oxygen species (ROS) – which can exacerbate the ischemic insult and associated pathophysiology. In the wake of I/R injury to the myocardium, adverse remodeling coupled with systemic neurohormonal dysregulation and inflammation ultimately lead to clinical presentation of HF[20,21]. Within the broader picture of adverse remodeling, the following subtopics are explored in further detail as they particularly pertain to the cardiac actions of relaxin.
Inflammation
Prolonged myocardial ischemia leads to tissue necrosis and release of intracellular contents upon cell rupture. The resulting cell debris consists of damage-associated molecular patterns (DAMPs), which act on pattern recognition receptors (PRRs) expressed on the surface of resident cells[21]. This initiates a sterile inflammatory cascade resulting in the aggregation of a macromolecular complex – the inflammasome, which consists of the sensor NLRP3 (cryopyrin), the scaffold ASC, and the effector enzyme caspase-1[21]. The consequences of inflammasome activation in the post-infarcted heart vary with the cell type: leukocytes produce a surge in active IL-1β, whereas cardiomyocytes respond via increased pyroptosis and apoptosis[22,23]. Genomic knockdown of NLRP3[24], caspase-1 or ASC[25] in mice led to significant reduction in infarct size upon I/R injury, validating the causality between inflammasome activation and adverse remodeling.
Events leading up to activation of the inflammasome involve priming, and subsequent triggering[21]. The inflammasome is ‘primed’ by DAMPs and other mediators of innate sterile inflammation released upon cardiac cell death. Priming leads to transcription of inflammasome components and post translational modification of cryopyrin[26]. The ‘triggers’ are more specific, involving ATP released upon cell death; the ATP receptor P2X7 is directly implicated. A mouse I/R model employing either pharmacologic or RNA silencing inhibition of the receptor leads to a reduction in infarct size and cardiac enlargement following MI. In addition to ATP, ROS generated from the reperfusion injury also activates the inflammasome in isolated cardiomyocytes subjected to hypoxia-reoxygenation (HR) injury[27]. Both of these pathways (P2X7 signaling and ROS generation) converge to result in mitochondrial dysfunction and oxidization of mitochondrial DNA (mtDNA)[28], which also triggers inflammasome activity[28,29].
Cardiac Fibrosis and Adverse Remodeling
The acute inflammatory phase seen in response to MI eventually subsides as mediators of chronic inflammation direct the course of fibrosis and adverse remodeling. Fibrotic tissue is less compliant than ventricular muscle and leads to elevated diastolic filling pressures in the failing ventricle. Upregulation of renin-angiotensin-aldosterone system (RAAS) due to disruption within the cardiorenal axis mediates the signaling mechanisms observed in fibrosis after MI[30,31]. Elevated levels of Angiotensin II induce expression of TGF-β within cells, which also occurs in the presence of ROS[32,33].
TGF-β is a key modulator of the fibrotic program in the ischemic heart. Activation of its target receptor leads to SMAD-2 and SMAD-3 phosphorylation, which after combining with SMAD-binding element (SBE) increases transcript levels of pro-fibrotic genes[30,34]. SMAD 3 knockout mice had reduced cardiac extracellular collagen deposition and mitigated diastolic dysfunction in comparison with WT mice post MI, despite both having similar infarct size[35]. MMPs and their inhibitors (TIMPs) play a crucial role in scar remodeling, and SMAD3-null cardiac tissue demonstrated a profile tipped in favor of an anti-fibrotic profile of MMPs[35].
Cardiac Arrhythmias
Macrostructural disarray within the cardiac muscle due to fibrotic infiltration and disruptions of ionic currents and conductance at a cellular level predispose the infarcted heart to increased arrhythmic activity[36]. The incidence of atrial fibrillation (AF), one of the most prevalent arrhythmias encountered clinically, is estimated to be between 6–21% after MI[37], and is correlated with a higher mortality and adverse events during and after in-hospital period. Inflammatory mediators such as C-reactive protein (CRP), IL-6, and TNFα are elevated in AF, lending credence to I/R-associated inflammation as a possible etiology[38]. A potential correlation between ROS generation after MI and AF exists, as patients with new-onset AF after cardiac surgery had a significantly higher oxidized protein and serum peroxide levels[39]. Myocardial remodeling orchestrated by RAAS contributes to atrial fibrosis, a risk factor highly associated with AF[36,38].
Rampant electrical remodeling also contributes to arrhythmogenesis in AF. Maintenance of AF is possible due to a shorter atrial refractory period within the myocyte; sustained atrial tachycardia within canine atrial myocytes reduces inward Ca2+ current and consequently action potential duration (APD), which leads to a shorter refractory period[40]. Atrial tachypacing also reduces inward Na+ current, along with reduced channel mRNA levels in canine cells[41]. Reduction in Na+ current can lead to a slower action potential upstroke, reducing conduction velocity (CV) – a mechanism observed in Nav1.5 mutation-induced AF[42,43]. Lowered CV is also observed as a result of reduced connexin expression[43].
The interplay of inflammation, oxidant injury, fibrosis and aberrant electrical activity present after MI necessitates treatment strategies countering multiple cascades of pathologic signaling via pleiotropic activity.
Therapeutic Potential of Relaxin for Cardiovascular Disease
The prospect of relaxin as a cardioprotective agent has been investigated in several preclinical animal models of cardiovascular disease. We present the following evidence in the context of pathophysiology in the order discussed in the previous section.
Inflammation
Relaxin administration prior to ischemia reduced the extent of I/R-associated damage, neutrophil extravasation and activity, mast cell granule release and Ca2+ overload in the rat heart after MI[44]. Relaxin therapy upon reperfusion in a swine model of MI yielded dose-dependent reduction in cardiac troponin release, apoptotic markers (caspase 3, TUNEL), and improved ventricular performance. The protective mechanism was attributed to a decrease in oxidative injury as shown by reduced malondialdehyde (MDA) within treated tissue[45]. Guinea pig hearts subjected to I/R injury also had a significant decrease in MDA production and improved contractility with relaxin administration and the protective effect was blunted upon treatment with L-NMMA, a NOS inhibitor[46]. The benefits of NO signaling in the context of I/R injury have been reported comprehensively in literature[47]. Relaxin has also been shown to reduce human and rabbit platelet aggregation in vitro in a NO dependent manner[48]. A recent study investigated the cardioprotective effect of serelaxin in adult mice at the onset of reperfusion. In this study, serelaxin was shown to reduce mortality and infarct size while preserving LV function. However, these protective effects were abrogated in endothelial (e)NOS knockout mice. Serelaxin treatment also attenuated caspase-1 activity following MI indicating attenuation of NLRP3 inflammasome activation; an effect that was absent in eNOS knockout mice[2]. In vitro studies in macrophages provide evidence to suggest the inhibitory role of NO in attenuating caspase-1 activity[49]. However, the precise role of eNOS-induced NO production on inflammasome activity is yet to be elucidated.
Cardiac Fibrosis and Adverse Remodeling
The anti-fibrotic effects of relaxin have been well studied in the context of cardiac disease. TGF-β-stimulated cardiac fibroblasts increased MMP-2 expression and decreased collagen expression upon treatment with relaxin[50]. Rats grafted with genetically modified C2C12 myoblasts expressing relaxin showed improved histologic outcomes pertaining to cardiac fibrosis after being subjected to permanent ligation of the left anterior descending coronary artery[51]. Anti-fibrotic effects were also observed in mice with permanent left coronary artery ligation receiving exogenous relaxin, illustrated by reduced TGF-β1 expression and myocyte apoptosis, along with improved MMP13 production[52]. Mechanistic studies determined that relaxin-induced inhibition of TGFβ and phosphorylation of Smad-3 were dependent on the Notch-1 pathway, whereby pharmacologic inhibition of Notch-1, or its ligand Jagged-1, significantly abolished the inhibitory effect exerted by relaxin on TGF-β signaling[53]. Treatment of H9C2 cardiomyoblasts with relaxin demonstrated that upregulation of MMP2 and MMP9 and downregulation of TIMP-1 were dependent on activation of sphingosine kinase 1 (Sphk1)[54]. Evidence implicating Erk1/2 in Sphk1 phosphorylation and its subsequent activation corroborates this mechanism[54,55], although the significance of this pathway in adult cardiomyocytes in the context of pathologic fibrosis is unclear. Nevertheless, the consensus from preclinical studies evaluating the effect of relaxin on fibrosis following cardiac injury indicates significant attenuation of pro-fibrotic signaling and cardiac scar formation.
Cardiac Arrhythmias
Relaxin therapy suppresses ventricular and atrial arrhythmias by countering electrical remodeling, in addition to preserving the cardiac substrate by thwarting fibrotic changes. To this end, incidence of ventricular arrhythmias was significantly reduced in a swine model of MI after treatment with relaxin at the onset of reperfusion[56]. Interestingly, the reported cardioprotective effects of relaxin were not exclusive to ischemic heart disease. Spontaneously hypertensive rats (SHR) treated with serelaxin for 2 weeks reversed atrial hypertrophy, increased CV and connexin-43 phosphorylation, and improved restitution kinetics (reduced dependence of APD on diastolic interval)[57]. Relaxin also upregulated Na+ current density in induced pluripotent stem cell-derived cardiomyocytes (iPS-CMs), possibly through Nav1.5 channel overexpression[57]. Similar effects were observed in aged rats, whereby voltage-clamp studies indicated a 46% increase in atrial Na+ current after 2 days of relaxin treatment[3]. Another study in rats subjected to MI evaluated the influence of relaxin on inducibility of tachyarrhythmias and electrophysiological remodeling. Relaxin treatment for 2 weeks improved connexin 43 physiology at the infarct border zone. Treatment also reduced dispersion of APD after MI[58]. In a mouse cryoinfarction model, relaxin therapy reduced mean duration of AF episodes, while improving CV in the atrial tissue[5]. In another cryoinfarction model, chronic low-dose administration of serelaxin following MI reduced inducibility of ventricular tachycardia, but no changes in connexin or ion channel expression were found[59]. Taken together, these studies demonstrate the ability of relaxin to attenuate substrate and electrical remodeling. However, the mechanism behind improved gap junction function and increased Na+ channel expression is yet to be uncovered.
Clinical trials investigating the efficacy of relaxin as a therapeutic agent were motivated by its vasodilatory actions through NO stimulation and functional antagonism of endothelin-1 activity[60]. Currently available vasodilatory agents such as nitrates and nesiritide have limited applicability in the context of AHF due to reservations pertaining to tolerance and long-term safety[61]. The benefits of relaxin treatment were hypothesized to significantly improve outcomes in a subset of AHF patients with normal to high blood pressure. Results from Pre-RELAX-AHF, a randomized, double-blind, placebo-controlled phase IIb study, showed improvement of dyspnea at 24 hours and reduced mortality and readmission rates owing to heart or renal failure at day 60[61]. In the phase III RELAX-AHF trial, 48-hour intravenous infusion of serelaxin improved the primary endpoint of dyspnea measured via visual analogue scale area under the curve (VAS AUC), but did not increase the Likert scale proportion of patients with dyspnea improvement. The additional endpoint of reduced 180 day mortality was satisfied[6]. Serelaxin was subsequently rejected by FDA due to failure to address all the primary endpoints, and insufficiency of single-trial data to justify improvement in symptomatic endpoints[62], although it received “novel breakthrough therapy for HF” designation from the FDA. Post-hoc analyses of the RELAX-AHF are summarized in Table 1. Clinical objectives were reformulated in RELAX-AHF2 trial to include reduced cardiovascular death and worsening HF as primary endpoints, but 48-hour infusion of serelaxin did not lead to beneficial changes in these outcomes[7]. These results, while significant in a clinical sense, fail to underscore the complexity of pathophysiology in AHF, which is not a 48-hour illness, but rather a chronic syndrome with profound ramifications[7]. In this context, recent studies have shown the potential utility of assaying endogenous relaxin-2 levels as a biomarker for predicting disease severity in chronic HF patients[63,64]. In fact, assessing relaxin-2 in addition to BNP had better diagnostic predictability when compared to BNP alone[64].
Table 1.
Summary of post-hoc analyses of RELAX-AHF
| Study, year | Topic of investigation | Results | Notes | Ref. |
|---|---|---|---|---|
| Metra et al, Jan 2013 | Biomarkers of organ damage in patients with acute heart failure | Improvement in cardiac (troponin T), renal (creatinine and cystatin-C) and liver (ALT, AST) biomarkers upon administration of serelaxin, correlating with reduced 180-day mortality | [75] | |
| Metra et al, Oct 2013 | Assessment of dyspnea relief, CV and 180-day mortality in patient sub-groups with AHF upon treatment with serelaxin | Forest plots of subgroup analyses show no subgroup based variations in dyspnea relief upon treatment. CV mortality was reduced in patients ≥ 75 years (P=0.0337), and with no HF hospitalization in previous year (P = 0.0119), no baseline beta blocker use (P=0.0432) and with ≤12% blood lymphocytes (P = 0.0137) and eGFR < 50 ml/min/m2 (P=0.0286). Similar subgroup based trends observed in 180 day all-cause mortality reductions | Pre-defined subgroups: demographic (age, race, sex, region), time from presentation to randomization, eGFR, SBP, past medical history (AF, diabetes, ischemic heart disease, cardiac devices and i.v. adminstration at the time of randomization) | [76] |
| Filippatos et al, Apr 2014 | Response of AHF patients with HFpEF to serelaxin treatment | Serelaxin well tolerated in patients with HFpEF. Dyspnea relief measured via VAS-AUC was similar to observed results in patients with HFrEF; however, Likert scale results for moderate or marked dyspnea improvement at 6,12 and 24 hours show difference in effects, with increased odds ratio for improvement in patients with HFpEF (P=0.03). Survival outcomes were similar in both groups | 26% of recruited patients in RELAX-AHF had HFpEF | [77] |
| Cotter et al, Nov 2015 | Association of growth differentiation factor – 15 (GDF 15) levels with RELAX AHF end points | Large increase in GDF-15 associated with higher risk of 60 and 180-day CV mortality. Randomization to serelaxin treatment correlated with decrease in GDF-15 levels at days 2 and 5. | GDF-15 is a member of the TGF-ß super family, and is associated with poor outcomes in chronic heart failure | [78] |
| Liu et al, Sept 2016 | Effect of treatment with serelaxin on renal function | Renal impairment was defined in patients with an eGFR < 60 ml/min/1.73 m2. Renally impaired patients on serelaxin had further all-cause mortality reduction (HR 0.53, 95% CI 0.34–0.83), compared to patients without impairment (HR 1.30, 95% CI 0.51–3.29) | Renal dysfunction led to an overall increase in CV and all-cause mortality in RELAX AHF | [79] |
| Filippatos et al, Feb 2017 | Assessment of RELAX-AHF endpoints in patients with and without AF | Serelaxin demonstrated similar efficacy and safety profile in patients with and without atrial fibrillation. Although stroke incidence was higher in AF patients in general, a trend of lower incidence was observed in patients treated with serelaxin (OR 0.31, P=0.0759) versus placebo (OR 3.88, P=0.2255; interaction P=0.0518) | [80] |
Biased Signaling at RXFP1: Potential for Novel Drug Development
The ligand-GPCR complex was previously thought to regulate the magnitude of a static signaling response, whether it involved activation in the case of agonism, or cessation of the same through antagonism. However, advances in GPCR physiology have led to the understanding that a diversity of pharmacological profiles can be evoked at the same receptor by different ligands – a phenomenon termed as biased signaling[65]. Biased signaling could lead to variable recruitment of G proteins or other signal transducers, leading to a cellular response that is qualitatively distinct from that produced by the orthosteric, cognate ligand. Such ligand-receptor behavior has been observed in a variety of GPCRs[66], including RXFP1. Concentration bias also exists at RXFP1, whereby the receptor recruits a signalosome, which characterizes its constitutive activity. The signalosome involves an adenylyl cyclase 2 (AC2) and a β-arrestin-2-associated scaffold[67]. Therefore, this apparatus generates cAMP despite the absence of ligand stimulation. Sub-picomolar levels of relaxin amplify this pathway, and at a nanomolar concentration, the signalosome disassembles to lead way to canonical G-protein signaling[67].
Two ligands, namely B7-33 and ML290 have been reported to elicit biased signaling at RXFP1[68,69]. B7-33 is a modified B-chain of relaxin that lacks the A chain present in the native peptide[70]. B7-33 biases the receptor response to prioritize Erk1/2 phosphorylation over cAMP synthesis, as evident by increased MMP2 production in HEK293 cells and reduced fibrosis in an ISO-induced mouse HF model[69]. ML290 is a recently characterized small molecule agonist at human RXFP1. Evidence from site-directed mutagenesis experiments showed that ML290 bound allosterically to RXFP1; residues within transmembrane domain 7 (TM7) and ECL3 of the receptor were found to be crucial to produce a dose-dependent cAMP response[71]. The measured cAMP response generated upon ML290 activity was abolished when the residues G659 and T660 within the ECL3 domain of human RXFP1 were mutated. Interestingly, these residues are not conserved in murine RXFP1, or in human RXFP2[71,72]. The possibility of a biased, cAMP-independent response cannot be ruled out. In fact, a cGMP biased response was observed in human cardiac fibroblasts (HCF) treated with ML290, inhibiting TGF-β-induced SMAD-2 or SMAD-3 phosphorylation. ML290 influenced receptor-G protein interactions, strengthening Gαs, GαoB over Gαi3 recruitment[68]. More studies on cAMP-independent pathways elicited by RXFP1 activation to investigate previously undiscovered protective mechanisms employable through biased signaling are warranted.
Concluding Remarks and Future Perspectives
The preponderance of preclinical evidence and some clinical outcomes outlined in this review are suggestive of a therapeutic role for relaxin in MI and HF. Nevertheless, the requirement for intravenous administration, short terminal half-life (1–4 hours), and cost associated with recombinant protein therapy limit the utility of serelaxin for chronic treatment[73]. Therefore, modulation of the route of administration of relaxin or identification of novel agonists at RXFP1 that can overcome these practical hindrances and offer cardioprotective benefits is a vital area of ongoing research. In fact, the development of gastro-protected porcine oral relaxin that was tested in patients with peripheral arterial disease is a promising approach and provided first clinical evidence of oral relaxin in long-term treatment in patients[74]. While awaiting publication of post-hoc analyses from RELAX-AHF2 in the near future, which will provide valuable details on serelaxin treatment in AHF, it is notable that studies investigating the expression profile of RXFP1 in cardiovascular diseases are lacking. Whether cardiac RXFP1 expression is altered in patients with cardiovascular disease, and depending on the profile change in different etiologies, such knowledge may provide new insight into the potential efficacy of exogenous recombinant relaxin-2 treatment or novel RXFP1 agonists. This will also pave the way for precision cardiovascular medicine by taking advantage of focusing relaxin therapy in patients with preserved RXFP1 expression. Clearly, extensive studies are needed to address this important issue. The new knowledge of biased signaling at RXFP1 also provides a wide range of opportunities to better understand downstream protective signaling and also to develop novel drugs with selective pathway activation, depending on disease etiology. To this end, B7-33 and ML290 have significantly improved our knowledge about RXFP1 signaling and new ML290 analogs are being investigated[74].
Finally, although most preclinical findings suggest a protective role of relaxin against MI and its related inflammation, adverse remodeling, arrhythmias and HF, translating these findings in patients with MI requires careful evaluation of the safety profile of relaxin in the setting of emergent cardiac catheterization. The encouraging outcomes reported in various animal models of cardiovascular disease, however, provide a strong foundation for this promising field of research.
Figure 1.
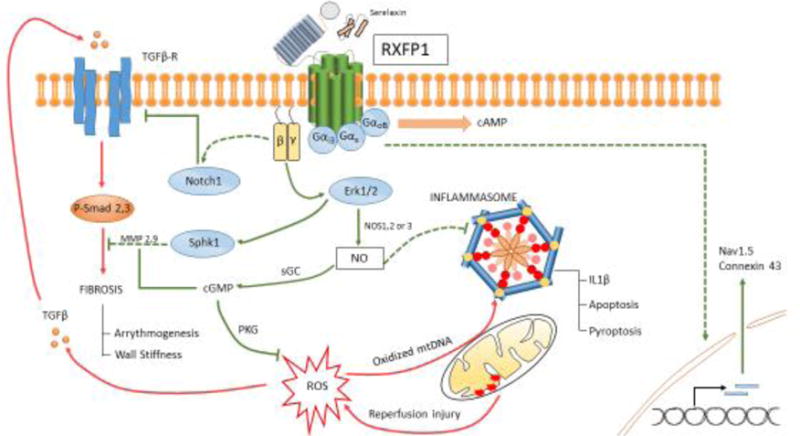
Schematic outlining pathologic signaling after myocardial I/R injury over the course of adverse remodeling (red) and protective mechanisms conferred upon activation of RXFP1 by serelaxin (green) in the mammalian heart. Pathways that require further validation for mechanistic insight are also indicated (dashed). NO produced downstream of βγ subunit activation is central to protection. This involves, but is not limited to, cGMP generation and activation of Protein Kinase G (PKG): targets attributed to mitigating ROS injury and countering fibrosis. NO also counters activation of the inflammasome. RXFP1 signaling can upregulate sodium channel and connexin 43 expression, which lead to increased conduction velocity.
Figure 2.
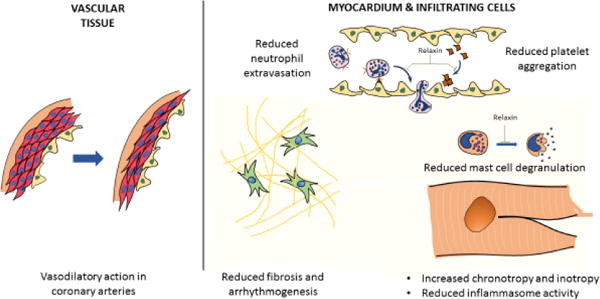
Pleotropic cardioprotective actions of relaxin: The physiological roles of relaxin involve mediating vasodilatation by interacting with smooth muscle and endothelial cells within vascular beds, and increasing cardiac inotropy and chronotropy. In the context of MI, relaxin attenuates inflammation by reducing mast cell degranulation, neutrophil extravasation (infiltrating cells) and inflammasome activity (in resident and infiltrating cells of myocardium). Relaxin also reduces platelet aggregation and counters adverse remodeling by inhibiting fibrosis and arrhythmogenesis in the post-infarcted heart.
Trends.
Relaxin is an endogenous hormone associated with mammalian pregnancy. It acts on relaxin family peptide receptor 1 (RXFP1), a G protein-coupled receptor to generate cell type specific responses.
In the context of myocardial ischemia/reperfusion (IR) injury, preclinical evidence demonstrates the efficacy of relaxin therapy in countering acute inflammation and its consequences.
Relaxin therapy prevents worsening of cardiac function after I/R injury by limiting fibrosis and suppressing arrhythmogenesis. Investigative efforts are focused on uncovering precise mechanisms underlying the protective effects.
Other RXFP1 agonists – including peptide mimetics and small molecules – evoke biased signaling at the receptor and may offer promising future therapeutic alternatives to recombinant relaxin.
Outstanding Questions.
What can be inferred from the outcomes of RELAX-AHF and RELAX-AHF2 when placed in context with preclinical evidence suggesting a protective role for relaxin signaling after acute MI? Clinical investigation of the efficacy of serelaxin in combating adverse remodeling remains unexplored.
How does relaxin signaling associate with decrease in inflammasome activity? Apart from NO mediated signaling and mitoprotection, do other protective pathways play a role?
What specific mechanisms underlie the observed ion channel and gap junction protein expression changes with relaxin therapy?
Can biased signaling at RXFP1 be exploited for therapeutic purposes? Differential recruitment of G proteins and resultant variation in cyclic nucleotide-associated downstream signaling could have meaningful implications in cardiovascular disease.
How is the expression of RXFP1 affected in various cardiac disease states? Could significant differences within populations account for varied responses in observed benefits? Gain/loss-of-function studies involving RXFP1 can fill the gap in knowledge and raise prospects for targeted gene therapy.
Acknowledgments
Dr. Salloum is supported by grants from the National Institutes of Health (HL133167 and AG053654).
Glossary terms
- MI
Myocardial Infarction. Ischemia (loss of blood supply) to the myocardium can lead to ATP depletion, cell swelling due to failure of active transport mechanisms, and increased intracellular Ca2+ concentration. These changes result in irreversible tissue necrosis
- Heart failure
Failure of the heart to pump blood to body organs at a level appropriate for homeostasis. Variety of etiologies exist; ischemic heart failure is characterized by depletion of cardiac muscle from a prior ischemic insult. Heart failure can clinically manifest as either reduced ejection fraction (HFrEF) or impairing ventricular filling despite no overt changes in ejection fraction (HFpEF).
- Acute Heart Failure
Acute exacerbation of heart failure due to decompensation. Clinically manifests as volume overload, dyspnea and exercise intolerance. Potentially life-threatening
- GPCRs
G protein coupled receptors. These cell surface receptors play a crucial role in mediating cell signaling in various organ systems. GPCRs have an extracellular ligand-interacting domain, a transmembrane region within the lipid bilayer, and an intracellular domain that interacts with G proteins or other mediators of signal transduction
- Inotropy
The contractile function of the heart. Mechanisms that increase the strength of cardiac muscle contractions are defined as positive inotropic agents
- Chronotropy
Pertains to heart rate. Positive chronotropic agents increase heart rate
- Reactive oxygen species
A by-product of several biochemical reactions in the cell. Detrimental increases are observed in the context of inflammation and cell damage. Highly unstable due to the presence of an unpaired electron, and can therefore initiate chain reactions
- RAAS
Renin-angiotensin-aldosterone system. The feedback mechanisms exercised by these hormones are critical in maintaining plasma volume and renal function. RAAS disruption in heart failure leads to elevated levels of plasma angiotensin, which mediates pro-fibrotic signaling
- Atrial Fibrillation
Impulse generation at several ectopic foci within the atrial tissue, leading to rapid fibrillations. Can lead to hemostasis in the atrial chamber and subsequent embolus formation, increasing risk for cerebrovascular stroke
- Action potential duration
The total duration of an electrical impulse in a cell – starting from depolarization and ending at resolution to resting membrane potential. Abnormal prolongation or shortening of this interval can be proarrhythmic
- Conduction velocity
Velocity of impulse conduction within cardiac tissue
- Connexins
Components of gap junctions present at the functional syncytium between cardiomyocytes. Critical for maintaining conduction velocity
- β-arrestin
Scaffolding protein that has been associated with GPCR internalization and cessation of activity. However, relevant isoforms are now identified to play a role in transducing biased signaling mechanisms
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Raleigh JMV, et al. Relaxin’ the Heart: A Novel Therapeutic Modality. J Cardiovasc Pharmacol Ther. 2015 doi: 10.1177/1074248415617851. [DOI] [PubMed] [Google Scholar]
- 2.Valle Raleigh J, et al. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovasc Res. 2017;2:cvw246. doi: 10.1093/cvr/cvw246. [DOI] [PubMed] [Google Scholar]
- 3.Henry BL, et al. Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na+ channels. Hear Rhythm. 2016;13:983–991. doi: 10.1016/j.hrthm.2015.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, et al. Relaxin abrogates renal interstitial fibrosis by regulating macrophage polarization via inhibition of Toll-like receptor 4 signaling. Oncotarget. 2017;8:21044–21053. doi: 10.18632/oncotarget.15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beiert T, et al. Relaxin reduces susceptibility to post-infarct atrial fibrillation in mice due to anti-fibrotic and anti-inflammatory properties. Biochem Biophys Res Commun. 2017;490:643–649. doi: 10.1016/j.bbrc.2017.06.091. [DOI] [PubMed] [Google Scholar]
- 6.Teerlink JR, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): A randomised placebo-controlled trial. Lancet. 2013;381:29–39. doi: 10.1016/S0140-6736(12)61855-8. [DOI] [PubMed] [Google Scholar]
- 7.McCullough PA. How Trialists and Pharmaceutical Sponsors Have Failed Us by Thinking That Acute Heart Failure Is a 48-Hour Illness. Am J Cardiol. 2017;120:505–508. doi: 10.1016/j.amjcard.2017.04.056. [DOI] [PubMed] [Google Scholar]
- 8.Bathgate RaD, et al. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–80. doi: 10.1152/physrev.00001.2012. [DOI] [PubMed] [Google Scholar]
- 9.Samuel CS, et al. Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 2004;65:2054–2064. doi: 10.1111/j.1523-1755.2004.00628.x. [DOI] [PubMed] [Google Scholar]
- 10.Conrad KP, et al. Relaxin Modifies Systemic Arterial Resistance and Compliance in Conscious. Nonpregnant Rats. 2004;145:3289–3296. doi: 10.1210/en.2003-1612. [DOI] [PubMed] [Google Scholar]
- 11.Halls ML, et al. International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1-4 the receptors for relaxin family peptides. Pharmacol Rev. 2015;67:389–440. doi: 10.1124/pr.114.009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore XL, et al. Diverse Regulation of Cardiac Expression of Relaxin Receptor by α1- and β1-Adrenoceptors. Cardiovasc Drugs Ther. 2014;28:221–228. doi: 10.1007/s10557-014-6525-x. [DOI] [PubMed] [Google Scholar]
- 13.Dschietzig T, et al. The positive inotropic effect of relaxin-2 in human atrial myocardium is preserved in end-stage heart failure: Role of Gi-phosphoinositide-3 kinase signaling. J Card Fail. 2011;17:158–166. doi: 10.1016/j.cardfail.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Coulson CC, et al. Central hemodynamic effects of recombinant human relaxin in the isolated, perfused rat heart model. Obstet Gynecol. 1996;87:610–2. doi: 10.1016/0029-7844(95)00493-9. [DOI] [PubMed] [Google Scholar]
- 15.Kong RCK, et al. The relaxin receptor (RXFP1) utilizes hydrophobic moieties on a signaling surface of its N-terminal low density lipoprotein class a module to mediate receptor activation. J Biol Chem. 2013;288:28138–28151. doi: 10.1074/jbc.M113.499640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen BT, et al. Phosphoinositide 3-Kinase Activity Is Required for Biphasic Stimulation of Cyclic Adenosine 3′,5′-Monophosphate by Relaxin. Mol Endocrinol. 2003;17:1075–1084. doi: 10.1210/me.2002-0284. [DOI] [PubMed] [Google Scholar]
- 17.Sarwar M, et al. Serelaxin-mediated signal transduction in human vascular cells: Bell-shaped concentration-response curves reflect differential coupling to G proteins. Br J Pharmacol. 2015;172:1005–1019. doi: 10.1111/bph.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh S, et al. Relaxin activates peroxisome proliferator-activated receptor γ (PPAR γ)through a pathway involving PPARγ coactivator 1α (PGC1α) J Biol Chem. 2015;290:950–959. doi: 10.1074/jbc.M114.589325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lesnefsky EJ, et al. Mitochondrial Dysfunction in Cardiac Disease: Ischemia–Reperfusion, Aging, and Heart Failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 20.Bhatt AS, et al. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr Cardiol Rep. 2017;19 doi: 10.1007/s11886-017-0876-4. [DOI] [PubMed] [Google Scholar]
- 21.Toldo S, et al. The Inflammasome in Myocardial Injury and Cardiac Remodeling. Antioxid Redox Signal. 2014;22 doi: 10.1089/ars.2014.5989. 150127063122000. [DOI] [PubMed] [Google Scholar]
- 22.Mezzaroma E, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toldo S, et al. Induction of microrna-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ Cardiovasc Genet. 2014;7:311–320. doi: 10.1161/CIRCGENETICS.113.000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandanger Ø, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–174. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 25.Kawaguchi M, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 26.Juliana C, et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi M, et al. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS One. 2017;12:1–14. doi: 10.1371/journal.pone.0176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada K, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gurung P, et al. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21:193–201. doi: 10.1016/j.molmed.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schirone L, et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. 2017;2017 doi: 10.1155/2017/3920195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zucker IH, et al. The central renin–angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci. 2014;126:695–706. doi: 10.1042/CS20130294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagler MA, et al. TGFβ signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc Res. 2013;99:175–184. doi: 10.1093/cvr/cvt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu RM, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo Y, et al. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J Mol Cell Cardiol. 2017;110:109–120. doi: 10.1016/j.yjmcc.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bujak M, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 36.Francis Stuart SD, et al. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol. 2016;91:114–122. doi: 10.1016/j.yjmcc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vukmirovic M, et al. Predictors and outcomes of new-onset atrial fibrillation in patients with acute myocardial infarction. Vojnosanit Pregl. 2016 doi: 10.2298/VSP150224257V. [DOI] [Google Scholar]
- 38.Zhao Z, et al. Relaxin as novel strategy in the management of atrial fibrillation: Potential roles and future perspectives. Int J Cardiol. 2014;171:e72–e73. doi: 10.1016/j.ijcard.2013.11.103. [DOI] [PubMed] [Google Scholar]
- 39.Ramlawi B, et al. Oxidative stress and atrial fibrillation after cardiac surgery: A case-control study. Ann Thorac Surg. 2007;84:1166–1172. doi: 10.1016/j.athoracsur.2007.04.126. [DOI] [PubMed] [Google Scholar]
- 40.Yue L, et al. Ionic Remodeling Underlying Action Potential Changes in a Canine Model of Atrial Fibrillation. Circ Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- 41.Yagi T, et al. Density and function of inward currents in right atrial cells from chronically fibrillating canine atria. Cardiovasc Res. 2002;54:405–415. doi: 10.1016/s0008-6363(02)00279-1. [DOI] [PubMed] [Google Scholar]
- 42.Olson TM, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. Jama. 2005;293:447–54. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nattel S, et al. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 44.Bani D, et al. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am J Pathol. 1998;152:1367–76. [PMC free article] [PubMed] [Google Scholar]
- 45.Perna AM, et al. Novel drug development opportunity for relaxin in acute myocardial infarction: evidences from a swine model. 2005;19:1525–1527. doi: 10.1096/fj.04-3664fje. [DOI] [PubMed] [Google Scholar]
- 46.Masini E, et al. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: Evidence for an involvement of nitric oxide. Endocrinology. 1997;138 doi: 10.1210/endo.138.11.5520. [DOI] [PubMed] [Google Scholar]
- 47.Totzeck M, et al. Nitrite-Nitric Oxide Signaling and Cardioprotection. Mitochondrial Dynamics in Cardiovascular Medicine. 2017;982:335–346. doi: 10.1007/978-3-319-55330-6_18. [DOI] [PubMed] [Google Scholar]
- 48.Bani D, et al. A novel, simple bioactivity assay for relaxin based on inhibition of platelet aggregation. Regul Pept. 2007;144:10–16. doi: 10.1016/j.regpep.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Kim YM, et al. Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme) J Immunol. 1998;161:4122–8. [PubMed] [Google Scholar]
- 50.Samuel CS, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. doi: 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- 51.Bonacchi M, et al. Functional and histopathological improvement of the post-infarcted rat heart upon myoblast cell grafting and relaxin therapy. J Cell Mol Med. 2009;13:3437–3448. doi: 10.1111/j.1582-4934.2008.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuel CS, et al. Relaxin remodels fibrotic healing following myocardial infarction. Lab Investig. 2011;91:675–690. doi: 10.1038/labinvest.2010.198. [DOI] [PubMed] [Google Scholar]
- 53.Sassoli C, et al. Relaxin Prevents Cardiac Fibroblast-Myofibroblast Transition via Notch-1-Mediated Inhibition of TGF-β/Smad3 Signaling. PLoS One. 2013;8:1–12. doi: 10.1371/journal.pone.0063896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frati A, et al. Role of Sphingosine Kinase/S1P Axis in ECM Remodeling of Cardiac Cells Elicited by Relaxin. Mol Endocrinol. 2015;29:53–67. doi: 10.1210/me.2014-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrano-Sanchez M, et al. Signaling pathways involved in sphingosine kinase activation and sphingosine-1-phosphate release in rat myometrium in late pregnancy: role in the induction of COX2. Endocrinology. 2008;149:en.2007–1756. doi: 10.1210/en.2007-1756. [DOI] [PubMed] [Google Scholar]
- 56.Nistri S, et al. Mast cell inhibition and reduced ventricular arrhythmias in a swine model of acute myocardial infarction upon therapeutic administration of relaxin. Inflamm Res. 2008;57:7–8. doi: 10.1007/s00011-007-0602-6. [DOI] [PubMed] [Google Scholar]
- 57.Parikh A, et al. Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ Res. 2013;113:313–321. doi: 10.1161/CIRCRESAHA.113.301646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang D, et al. Effects of relaxin on cardiac fibrosis, apoptosis, and tachyarrhythmia in rats with myocardial infarction. Biomed Pharmacother. 2016;84:348–355. doi: 10.1016/j.biopha.2016.09.054. [DOI] [PubMed] [Google Scholar]
- 59.Beiert T, et al. Chronic lower-dose relaxin administration protects from arrhythmia in experimental myocardial infarction due to anti-inflammatory and anti-fibrotic properties. Int J Cardiol. 2018;250:21–28. doi: 10.1016/j.ijcard.2017.09.017. [DOI] [PubMed] [Google Scholar]
- 60.Leo CH, et al. Vascular actions of relaxin: nitric oxide and beyond. Br J Pharmacol. 2016 doi: 10.1111/bph.13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teerlink JR, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–1439. doi: 10.1016/S0140-6736(09)60622-X. [DOI] [PubMed] [Google Scholar]
- 62.Center for Drug Evaluation and Research, F. FDA Briefing Document Addendum for the Cardiovascular and Renal Drugs Advisory Committee (CRDAC) 2014 [Google Scholar]
- 63.Xie J, et al. H2 relaxin expression and its effect on clinical outcomes in patients with chronic heart failure. Int J Clin Exp Med. 2015;8:4420–4. [PMC free article] [PubMed] [Google Scholar]
- 64.Han L, et al. Combined Assessment of Relaxin and B-Type Natriuretic Peptide Improves Diagnostic Value in Patients With Congestive Heart Failure. Am J Med Sci. 2017;354:480–485. doi: 10.1016/j.amjms.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Bologna Z, et al. Biased G Protein-Coupled Receptor Signaling : New Player in Modulating Physiology and Pathology. 2017;25:12–25. doi: 10.4062/biomolther.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dewire SM, Violin JD. Biased ligands for better cardiovascular drugs: Dissecting g-protein-coupled receptor pharmacology. Circ Res. 2011;109:205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- 67.Halls ML. Constitutive formation of an RXFP1-signalosome: A novel paradigm in GPCR function and regulation. Br J Pharmacol. 2012;165:1644–1658. doi: 10.1111/j.1476-5381.2011.01470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kocan M, et al. ML290 is a biased allosteric agonist at the relaxin receptor RXFP1. Sci Rep. 2017;7:2968. doi: 10.1038/s41598-017-02916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hossain MA, et al. A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chem Sci. 2016;7:3805–3819. doi: 10.1039/c5sc04754d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hossain MA, Wade JD. Synthetic relaxins. Curr Opin Chem Biol. 2014;22:47–55. doi: 10.1016/j.cbpa.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 71.Hu X, et al. Structural insights into the activation of human relaxin family peptide receptor 1 by small molecule agonists. Biochemistry. 2016 doi: 10.1021/acs.biochem.5b01195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang Z, et al. Activation of relaxin family receptor 1 from different mammalian species by relaxin peptide and small molecule agonist ML290. Front Endocrinol (Lausanne) 2015;6:1–12. doi: 10.3389/fendo.2015.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neverova N, Teerlink JR. Serelaxin : a potential new drug for the treatment of acute heart failure. Expert Opin Investig Drugs. 2014;23:1017–26. doi: 10.1517/13543784.2014.924504. [DOI] [PubMed] [Google Scholar]
- 74.Sonaglia F, et al. Efficacy and safety of oral porcine relaxin (pRLX) in adjunct to physical exercise in the treatment of peripheral arterial disease (PAD) Ital J Anat Embryol. 2013;118:84–91. [PubMed] [Google Scholar]
- 75.Metra M, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the relaxin in acute heart failure (RELAX-AHF) development program: Correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. doi: 10.1016/j.jacc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Metra M, et al. Effects of serelaxin in subgroups of patients with acute heart failure: Results from RELAX-AHF. Eur Heart J. 2013;34:3128–3136. doi: 10.1093/eurheartj/eht371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Filippatos G, et al. Serelaxin in acute heart failure patients with preserved left ventricular ejection fraction: Results from the RELAX-AHF trial. Eur Heart J. 2014;35:1041–1050. doi: 10.1093/eurheartj/eht497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cotter G, et al. Growth differentiation factor 15 (GDF-15) in patients admitted for acute heart failure: Results from the RELAX-AHF study. Eur J Heart Fail. 2015;17:1133–1143. doi: 10.1002/ejhf.331. [DOI] [PubMed] [Google Scholar]
- 79.Liu LCY, et al. Effects of serelaxin in acute heart failure patients with renal impairment: results from RELAX-AHF. Clin Res Cardiol. 2016;105:727–737. doi: 10.1007/s00392-016-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filippatos G, et al. Serelaxin in acute heart failure patients with and without atrial fibrillation: a secondary analysis of the RELAX-AHF trial. Clin Res Cardiol. 2017;106:444–456. doi: 10.1007/s00392-016-1074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]