

Abstract
Autophagy is an evolutionarily conserved cellular process that facilitates the continuous recycling of intracellular components (organelles and proteins) and provides an alternative source of energy when nutrients are scarce. Recent studies have implicated autophagy in many disorders, including pulmonary diseases. However, the role of autophagy in endothelial cell (EC) barrier dysfunction and its relevance in the context of acute lung injury (ALI) remain uncertain. Here, we provide evidence that autophagy is a critical component of EC barrier disruption in ALI. Using an aerosolized bacterial lipopolysaccharide (LPS) inhalation mouse model of ALI, we found that administration of the autophagy inhibitor 3-methyladenine (3-MA), either prophylactically or therapeutically, markedly reduced lung vascular leakage and tissue edema. 3-MA was also effective in reducing the levels of proinflammatory mediators and lung neutrophil sequestration induced by LPS. To test the possibility that autophagy in EC could contribute to lung vascular injury, we addressed its role in the mechanism of EC barrier disruption. Knockdown of ATG5, an essential regulator of autophagy, attenuated thrombin-induced EC barrier disruption, confirming the involvement of autophagy in the response. Similarly, exposure of cells to 3-MA, either before or after thrombin, protected against EC barrier dysfunction by inhibiting the cleavage and loss of vascular endothelial cadherin at adherens junctions, as well as formation of actin stress fibers. 3-MA also reversed LPS-induced EC barrier disruption. Together, these data imply a role of autophagy in lung vascular injury and reveal the protective and therapeutic utility of 3-MA against ALI.
Keywords: adherens junctions, autophagy, endothelial cells, lung vascular injury
INTRODUCTION
The disruption of vascular endothelial cell (EC) adherens junctions (AJs) is a major determinant of protein-rich edema formation and inflammatory cell infiltration that underlie diseases such as acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (1, 24, 27). AJs, formed by tethering of vascular endothelial (VE)-cadherin between two contiguous EC, serve to maintain EC barrier integrity and, thus, allow minimal filtration of fluids and selective passage of ions and molecules, such as electrolytes and proteins (14, 29). Disassembly of AJs is primarily regulated by loss of VE-cadherin from the cell surface, which is further aided by the contractile forces generated by actin-myosin interaction (actin stress fiber formation) (8, 14, 27, 46, 47). Loss of cell surface VE-cadherin is, in part, regulated by proteolytic cleavage/degradation or endocytosis of VE-cadherin (42). Thus VE-cadherin disassembly and actin-myosin interactions are major mechanisms of AJ disruption and increased EC permeability caused by proinflammatory mediators (8, 14, 27, 46, 47).
Autophagy is an evolutionarily conserved cellular process that facilitates the continuous recycling of intracellular components (organelles and proteins) and provides an alternative source of energy when nutrients are scarce (33, 53). During autophagy, cells sequester cytoplasmic contents in a double-membrane vesicle, the autophagosome, which delivers them to the lysosome for degradation (33, 53). Autophagy is facilitated by a large number (>30) of autophagy-related proteins (Atg genes) and is accomplished in several sequential stages (initiation, nucleation, elongation, and maturation) (33, 53). Autophagy is initiated by uncoordinated 51-like kinase [ULK1, the mammalian ortholog of the yeast autophagy-related (ATG1)] complex in which ATG13 and FAK family-interacting protein of 200 kDa (FIP200) are substrates of the ULK1 Ser/Thr kinase (53). ATG5-ATG12 conjugate and beclin 1 (ATG6) are essential for autophagosome formation (19, 53). Another protein essential for autophagosome formation is microtubule-associated protein-1 light chain-3 [LC3 (ATG8)] (33, 53). LC3 is cleaved to release a COOH-terminal glycine required for its conjugation to phospholipids mediated by ATG5–ATG12 (33, 53).
Recent studies have revealed novel roles of autophagy in embryogenesis, development, cell death, immunity, and inflammation and provided evidence of associations between autophagic dysfunction and disease (33, 43, 53). Aberrant regulation of autophagy is associated with aging and human diseases, including cancer, neurodegeneration, inflammatory bowel disease, and pulmonary diseases (5, 6, 18, 41, 43, 44, 55). However, the role of autophagy in EC barrier function and its relevance in the context of ALI are unclear. Here, we provide evidence that autophagy is a critical component of EC barrier dysfunction and that targeting autophagy via 3-methyladenine (3-MA) may be a viable therapeutic approach to control ALI.
MATERIALS AND METHODS
Reagents.
Lipopolysaccharide (LPS) of Escherichia coli origin, the autophagy inhibitor 3-MA, and the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 were obtained from Sigma-Aldrich (St. Louis, MO); human thrombin from Enzyme Research Laboratories (South Bend, IN); antibodies to VE-cadherin from Abcam (Cambridge, MA) and BD Biosciences (San Jose, CA); and antibodies to ATG5 and β-actin from Cell Signaling Technology (Beverly, MA) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. All other materials were obtained from Thermo Fisher Scientific (Waltham, MA).
Murine model of ALI.
Male 8- to 10-wk-old wild-type C57BL/6 mice (Jackson, Bar Harbor, ME) were exposed to an aerosol of saline alone or saline containing Escherichia coli LPS (0.5–0.75 mg/ml, 12 ml) for 30 min, as described elsewhere (2, 39). All animal care and treatment procedures were approved by the University of Rochester Committee on Animal Resources and performed in accordance with National Institutes of Health guidelines.
Evaluation of lung inflammation and injury.
After the experiment, bronchoalveolar lavage (BAL) fluids and lungs were collected as described elsewhere (2, 40). BAL fluids were analyzed for total protein levels using the bicinchoninic acid kit (Pierce, Rockford, IL) and for albumin levels using ELISA kits (R & D Systems, Minneapolis, MN). Lung tissues were homogenized in radioimmunoprecipitation (RIPA) buffer [50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 0.25 mM EDTA, pH 8.0, 1% deoxycholic acid, 1% Triton X, 5 mM NaF, and 1 mM sodium orthovanadate supplemented with protease inhibitor cocktail (Sigma-Aldrich)] or in hexadecyltrimethylammonium bromide buffer (pH 6.0), as described elsewhere (2, 10). The levels of proinflammatory mediators (vascular cell adhesion molecule-1, monocyte chemoattractant protein-1, and IL-1β) in mouse lung homogenates were determined using ELISA kits (R & D Systems), as described elsewhere (2). Myeloperoxidase (MPO) activity in the lung tissues was monitored to determine sequestration of polymorphonuclear leukocytes in the lung, as described elsewhere (2, 3, 10).
Wet-to-dry lung weight ratio and Evans blue-conjugated albumin (EBA) were measured in mice that were not subjected to BAL. At 1 h before they were euthanized, these mice were injected retroorbitally with EBA (30 mg/kg) to monitor vascular leak, as described elsewhere (31, 45). Blood was removed from the lungs by gentle infusion of 10 ml of phosphate-buffered saline containing 5 mM EDTA through the right ventricle. The lungs were excised en bloc and blotted dry, and the right lungs were snap-frozen in liquid nitrogen. The left lungs were weighed before and after being dried at 60°C for 24 h to calculate lung wet-to-dry weight ratio. Dried lungs were incubated with formamide at 60°C for 24 h and centrifuged, and supernatant absorbance at 620 and 740 nm was recorded. Tissue heme pigment contamination was corrected using readings of absorbance at 740 nm. The extravasated EBA concentration in lung homogenate was calculated against a standard curve and expressed as micrograms of EBA per milliliter.
Lung histopathology and immunohistochemistry.
After the experiment, lungs were harvested from mice that did not undergo BAL. The lungs were fixed in 10% formalin, embedded in paraffin, cut into 5-μm sections, mounted onto slides, and stained with hematoxylin-eosin for histological analysis (10). Sections were imaged with a Nikon Eclipse E400 microscope, and the representative images were taken at ×400.
Endothelial cells.
Human pulmonary artery EC (HPAEC) were obtained from Lonza (Walkersville, MD) and cultured as described elsewhere (30) in gelatin-coated flasks with use of endothelial basal medium 2 (EBM2) with BulletKit additives (BioWhittaker, Walkersville, MD). Experiments were performed in HPAEC below passage 6.
RNAi knockdown.
Predesigned short-interfering RNA (siRNA) specific for human ATG5 (si-ATG5) and a nontargeting siRNA control (si-Con) were purchased from Dharmacon (Lafayette, CO). HPAEC were transfected with si-ATG5 or si-Con using DharmaFect1 siRNA transfection reagent (Dharmacon) essentially as described elsewhere (2). Briefly, 50–100 nM siRNA and DharmaFect1 were mixed together and then added to cells that were 50–60% confluent. At 24–36 h after transfection, cells were used for experiments in which transendothelial electrical resistance (TER) was measured or cells were lysed to monitor ATG5 levels.
Immunoblot analysis.
Immunoblotting was performed as described previously (9). Briefly, equal amounts of protein from cell lysates or lung homogenates prepared in RIPA buffer were subjected to SDS-PAGE and then transferred onto nitrocellulose membranes for Western blotting, as described elsewhere (9). Representative blots were from the same membrane, which may have more samples in various groups.
Immunofluorescence.
Confluent HPAEC monolayers grown on gelatin-coated coverslips were subjected to immunofluorescence staining, as described elsewhere (2, 11). Alexa Fluor 488-phalloidin (Fisher Scientific, Pittsburg, PA) and VE-cadherin antibody (BD Biosciences, San Jose, CA) were used to visualize F-actin filaments and AJs, respectively. DNA staining with Hoechst dye was used to visualize nuclei. Images were obtained using a fluorescence microscope (Nikon Instech, Tokyo, Japan).
Measurement of endothelial permeability by TER.
The integrity of the endothelial barrier was determined using an electrical cell-substrate impedance-sensing system (Applied Biophysics, Troy, NY), as described elsewhere (2, 36). Briefly, confluent HPAE monolayers grown on gelatin-coated gold microelectrodes were treated with thrombin or LPS in the presence or absence of 3-MA (2 mM) and/or LY294002 (30 μM) as indicated. TER was measured over time and normalized to baseline resistance.
Statistical analysis.
Values are means ± SE. Data were analyzed by standard one-way ANOVA. Significance between the groups was determined using Tukey’s test (Prism 5.0, GraphPad Software, San Diego, CA). In some cases (Figs. 1, 5, 6, and 7), Student’s t-test was performed for comparisons between experimental groups. P < 0.05 was considered statistically significant.
Fig. 1.
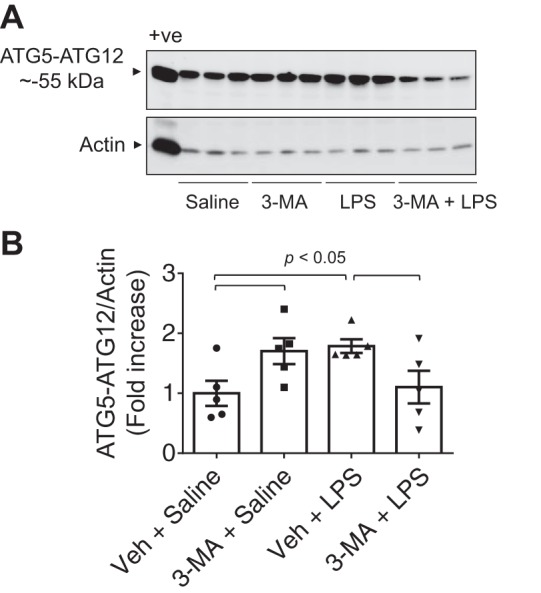
Effect of 3-methyladenine (3-MA) on lipopolysaccharide (LPS)-induced autophagy in the lung. Mice were treated with 3-MA (35 mg/kg ip) at 7 and 1 h before and 7 h after LPS challenge as illustrated in Fig. 2A. After 18 h of LPS inhalation, an antibody that detects ATG5 conjugated with ATG12 (ATG5-ATG12) was used to analyze ATG5 levels in lung homogenates. A and B: immunoblots and quantitative analysis of ATG5-ATG12 (~55 kDa) abundance in lungs from mice exposed to LPS in the presence of 3-MA or saline control. +ve, positive control (HEK 293 cell lysate); Veh, vehicle. Values are means ± SE (n = 5 for each condition) and expressed as fold increase over saline control.
Fig. 5.
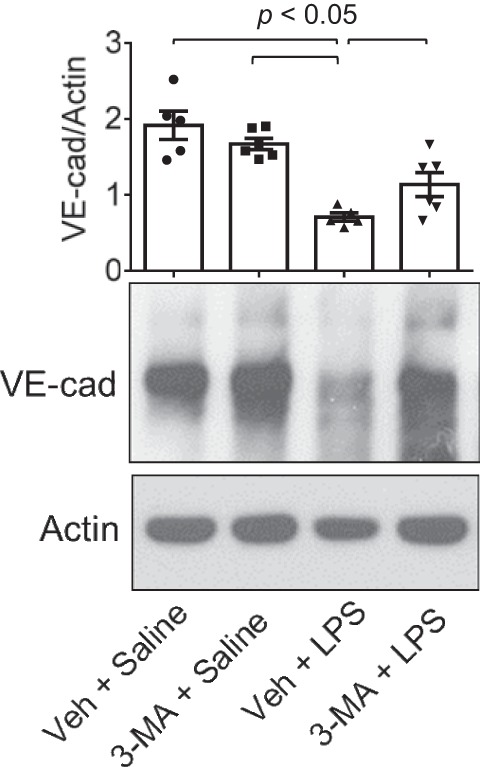
Protective effect of 3-MA on LPS-induced decrease in VE-cadherin (VE-Cad) levels. Mice were treated with 3-MA and LPS as shown in Fig. 2A. At 18 h after LPS challenge, VE-cadherin levels in lung homogenates were analyzed by immunoblotting. Actin levels were used to monitor loading. Effect of 3-MA on LPS-induced decrease in VE-cadherin levels was normalized to actin levels. Values are means ± SE (n = 5–6 for each condition).
Fig. 6.

Effect of 3-MA on endothelial barrier function. A: confluent human pulmonary artery endothelial cells (HPAEC) grown on gold electrode plates were treated with 3-MA (2 mM) for ~35 min and then challenged with thrombin (2.5 U/ml). Changes in transendothelial electrical resistance (TER) were measured to monitor endothelial barrier function. Values are means ± SE (n = 4 for each condition). *P < 0.05, thrombin vs. 3-MA + thrombin (control). B: confluent HPAEC grown on gold electrode plates were treated with 3-MA (2 mM) or left untreated (control), and changes in TER were measured to monitor endothelial barrier integrity. Values are means ± SE (n = 3–4 for each condition). *P < 0.05, 3-MA vs. untreated. C and D: confluent HPAEC grown on coverslips were treated with 3-MA for 5 min (C) or 15 min (D) after thrombin challenge, and changes in TER were measured to monitor endothelial barrier function. Values are means ± SE (n = 3–5 for each condition). *P < 0.05, thrombin-treated vs. thrombin + 3-MA (control). E: confluent HPAEC grown on coverslips were treated with 3-MA at ~0.5 h after LPS challenge, and changes in TER were measured to monitor endothelial barrier function. Values are means ± SE (n = 4–8 for each condition). *P < 0.05, LPS vs. LPS + 3-MA (control).
Fig. 7.

Effect of the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 on endothelial barrier function. A: confluent HPAEC grown on gold electrode plates were pretreated with LY294002 (LY, 30 μM) for ~30 min and then challenged with thrombin (2.5 U/ml). Changes in TER were measured to monitor endothelial barrier function. Values are means ± SE (n = 3 for LY alone; n = 6 for both thrombin and LY + thrombin). *P < 0.05, thrombin vs. LY + thrombin (control). B: confluent HPAEC grown on gold electrode plates were pretreated with LY294002 (LY, 30 μM) and 3-MA (2 mM) for ~55 and 15 min, respectively, and then challenged with thrombin (2.5 U/ml). Changes in TER were measured to monitor endothelial barrier function. Values are means ± SE (n = 5 for each condition). *P < 0.05, LY + thrombin vs. LY + 3-MA + thrombin-(control).
RESULTS
Protective and therapeutic utility of the autophagy inhibitor 3-MA prevents and treats LPS-induced lung vascular inflammation and injury.
We tested the possibility that autophagy plays an important role in lung vascular inflammation and injury. We first determined if autophagy is induced in an aerosolized bacterial LPS inhalation mouse model of ALI (2, 39). A marked increase in autophagy, as evidenced by an increase in the levels of ATG5 [conjugated with ATG12 (ATG5-ATG12), ~55 kDa], an essential regulator of autophagy (33, 35, 53), was noted in the lungs of mice exposed to LPS (Fig. 1). Intriguingly, lungs from mice exposed to the autophagy inhibitor 3-MA alone also showed an increase in ATG5 levels; however, lungs from mice exposed to 3-MA before LPS (3-MA + LPS) showed a significant decrease in ATG5 levels compared with those exposed to LPS alone. These data suggest that while 3-MA increases the constitutive (basal) autophagy, it protects against LPS (stress)-induced autophagy. We next determined if the induction of autophagy contributes to lung inflammatory injury evoked by LPS. We performed a “protection study (3-MA+LPS),” in which mice were exposed to the autophagy inhibitor 3-MA both before and after LPS challenge (Fig. 2A; hereafter referred to as “before”). We also evaluated the therapeutic utility of autophagy inhibition against ALI by performing a “treatment study (LPS + 3-MA),” in which mice were first challenged with LPS to induce lung injury and then treated with 3-MA (Fig. 2B; hereafter referred to as “after”). Histological examination of the lung showed increased interstitial thickening and neutrophil infiltration in mice exposed to LPS (Fig. 2, Ca vs. Cc). By contrast, in mice treated with 3-MA before or after LPS challenge, these markers of lung inflammatory injury were substantially reduced (Fig. 2, Cc vs. Cd and Ce). Consistent with this finding, LPS induced an increase in the levels of proinflammatory mediators (vascular cell adhesion molecule-1, monocyte chemoattractant protein-1, and IL-1β) and neutrophil infiltration in the lung, but these responses were significantly inhibited in mice treated with 3-MA before or after LPS challenge (Fig. 3). Analysis of BAL fluid showed that LPS induced a nearly fourfold increase in BAL protein and albumin levels; however, this response was inhibited in mice treated with 3-MA before or after LPS challenge (Fig. 4, A and B). We also determined the effect of 3-MA on LPS-induced EBA extravasation and increase in lung wet-to-dry weight ratio, important indexes of lung vascular leak and tissue edema. We found a significant decrease in LPS-induced lung vascular leak and tissue edema in mice treated with 3-MA before or after LPS challenge (Fig. 4, C and D). Because the levels of VE-cadherin, a major regulator of endothelial barrier integrity (14, 50, 51), are reduced in inflamed lungs (13, 16), we tested the possibility that autophagy regulates lung vascular leak by decreasing VE-cadherin levels. Indeed, we observed a significant decrease in VE-cadherin levels in the lungs of mice challenged with LPS (Fig. 5); however, this response was inhibited in mice treated with 3-MA (Fig. 5). Collectively, these data identify a critical role of autophagy in the mechanism of lung vascular inflammation and injury. These data also underscore the importance of targeting autophagy via 3-MA as a viable therapeutic approach to control lung injury.
Fig. 2.

Timeline of protective (Prot) and therapeutic (Ther) administration of 3-MA and its effects on LPS-induced lung pathology. A and B: mice were treated with 3-MA (35 mg/kg ip) at 7 and 1 h before and 7 h after LPS challenge (A) or 1 and 7 h after LPS challenge (B). C: at 18 h after LPS inhalation, lung sections were prepared, and alterations in lung histology were detected by hematoxylin-eosin staining. Images are representative of similar images from 4–5 mice exposed to LPS in the presence or absence of 3-MA.
Fig. 3.

Protective and therapeutic effect of 3-MA on LPS-induced increase in proinflammatory mediators and neutrophil sequestration in the lung. Mice were treated with 3-MA and LPS as shown in Fig. 2, A and B. At 18 h after LPS challenge, lungs were analyzed for levels of proinflammatory mediators [vascular cell adhesion molecule-1 (VCAM-1), monocyte chemoattractant protein-1 (MCP-1), and interleukin (IL)-1β] by ELISA (A–C) and neutrophil sequestration by tissue myeloperoxidase (MPO) activity (D). A460, absorbance at 460 nm. Values are means ± SE [n = 4–5 (saline-treated groups) and 4–6 (LPS-treated groups)].
Fig. 4.

Protective and therapeutic effect of 3-MA on LPS-induced lung vascular leak and tissue edema. Mice were treated with 3-MA and LPS as shown in Fig. 2, A and B. At 18 h after LPS challenge, lungs were analyzed for bronchoalveolar lavage (BAL) protein (A), BAL albumin (B), Evans blue albumin (EBA) extravasation (C), and wet-to-dry weight ratio (D). Values are means ± SE [n = 4–6 (saline-treated groups) and 5–7 (LPS-treated groups)].
Inhibition of autophagy protects against EC barrier dysfunction by suppressing VE-cadherin disassembly and actin stress fiber formation.
We investigated the possibility that 3-MA exerts its protective effect on lung vascular injury by its ability to prevent EC barrier disruption caused by thrombin, an edemagenic and proinflammatory agonist, the concentration of which is elevated in plasma and lavage fluids of patients with ALI/acute respiratory distress syndrome (12, 20). We determined the effect of 3-MA on thrombin-induced change in TER as measured by electric cell-substrate impedance sensing. A characteristic time-dependent decrease in TER with maximal change at ~0.5 h was observed upon thrombin challenge of confluent EC monolayers (Fig. 6A) (2, 28). Treatment of cells with 3-MA significantly increased the baseline TER (Fig. 6B) and was also effective in reducing and reversing the thrombin-induced decrease in TER (Fig. 6A). We also evaluated if 3-MA can restore EC barrier integrity after it is disrupted. Indeed, 3-MA was effective in restoring EC barrier function when added after thrombin or LPS challenge (Fig. 6, C–E). To exclude the possibility that the protective effect of 3-MA is mediated by a PI3K-inihibitable pathway other than autophagy, we assessed the effect of the PI3K inhibitor LY294002 on thrombin-induced EC barrier disruption. Interestingly, we found that, unlike 3-MA, LY294002 not only failed to protect the barrier from disruption but also inhibited recovery of barrier function after thrombin challenge (Fig. 7A). These data are consistent with an earlier report showing a role of PI3K in barrier enhancement (21). To further reinforce the conclusion that the effect of 3-MA on EC barrier disruption is uncoupled from its ability to inhibit PI3K, we determined if 3-MA can exert its protective effect in the presence of LY294002. Indeed, 3-MA was effective in protecting against thrombin-induced barrier disruption, even in the presence of LY294002 (Fig. 7B). To definitively establish the role of autophagy in EC barrier disruption, we determined the effect of siRNA-mediated knockdown of ATG5, an essential regulator of autophagy (17, 34, 35), on EC barrier disruption. Depletion of ATG5 by this approach (Fig. 8, A and B) also attenuated thrombin-induced EC barrier disruption (Fig. 8C). These data indicate an important role of autophagy in EC barrier disruption and the utility of 3-MA in restoring the barrier integrity of EC.
Fig. 8.

Effect of ATG5 knockdown on thrombin-induced endothelial barrier disruption. HPAEC were transfected with siRNA-control (si-Con) or siRNA-ATG5 (si-ATG5). A: after 24–36 h, total cell lysates were immunoblotted with an anti-ATG5 antibody. Total protein levels (bottom blot) were used to monitor loading. M, markers. B: quantitative analysis of ATG5 abundance in endothelial cells transfected with si-Con or si-ATG5. Values are means ± SE (n = 4 for each condition). C: after 24–36 h, cells were reseeded on gold electrode plates and allowed to reach confluency. Confluent monolayers were treated with thrombin, and changes in TER were measured to determine endothelial barrier function. Value are means ± SE (n = 4–6 for each condition). *P < 0.05, si-Con + thrombin vs. si-ATG5 + thrombin (control).
Because formation of actin stress fibers and loss of VE-cadherin at AJs are the key determinants of EC barrier dysfunction (4, 7, 8, 27, 29), we examined if 3-MA protects EC barrier integrity by preventing these events (actin stress fiber formation and VE-cadherin loss at AJs). Visualization of actin stress fibers showed that thrombin challenge increased stress fiber formation and that pretreatment of cells with 3-MA attenuated this this response (Fig. 9A). Similarly, we observed that thrombin caused a marked decrease in immunostaining of VE-cadherin at AJs and that this response was inhibited in 3-MA-treated cells (Fig. 9B). Notably, 3-MA also increased the basal immunostaining of VE-cadherin at AJs (Fig. 9B), consistent with the 3-MA-mediated increase in baseline TER (Fig. 6B). Studies have shown that thrombin and other stimuli induce cleavage of VE-cadherin, which also contributes, at least in part, to disassembly of AJs (7, 15, 42). Therefore, we determined the possibility that inhibition of autophagy promotes AJ integrity by preventing cleavage of VE-cadherin. Thrombin induced cleavage of VE-cadherin in a time-dependent manner (Fig. 9C). Importantly, the time course of VE-cadherin cleavage was consistent with changes in TER caused by thrombin (Fig. 9C, Fig. 6, A, C, and D, and Fig. 7, A and B). We found that thrombin-induced VE-cadherin cleavage was significantly inhibited in 3-MA-treated cells (Fig. 9D). Together, these data show a role of autophagy in regulating EC barrier dysfunction by virtue of promoting actin cytoskeleton rearrangement and AJ disruption via VE-cadherin cleavage.
Fig. 9.

Effect of 3-MA on thrombin-induced actin stress fiber formation and loss of cell surface VE-cadherin. A: confluent HPAEC grown on coverslips were treated with 3-MA (2 mM) for 1 h and then challenged with thrombin (2.5 U/ml) for 15 min. Alexa 488-labeled phalloidin was used to visualize actin stress fibers by fluorescence microscopy. Images are representative of 3 experiments. B: confluent HPAEC monolayers grown on coverslips were treated with 3-MA (2 mM) for 1 h and then challenged with thrombin (2.5 U/ml) for 15 min. Immunofluorescence was performed using VE-cadherin antibody to visualize adherens junctions. Arrows indicate disruption of VE-cadherin staining. Images are representative of 3 experiments. C: confluent HPAEC monolayers were treated with thrombin (2.5 U/ml) for the indicated time periods. Total cell lysates were analyzed by immunoblotting to monitor cleavage of VE-cadherin (cVE-Cad, ~90 kDa). Actin levels were used to monitor loading. Top: quantitative analysis of thrombin-induced time-dependent generation of cVE-Cad. Values are means ± SE (n = 7 for each condition). Bottom: immunoblots. D: confluent HPAEC monolayers were treated with 3-MA (2 mM) for 1 h and then challenged with thrombin (2.5 U/ml) for 15 min. Total cell lysates were analyzed by immunoblotting to monitor cleavage of VE-cadherin (cVE-Cad, ~90 kDa). Actin levels were used to monitor loading. Top: quantitative analysis of thrombin-induced generation of cVE-Cad in the presence of 3-MA. Values are means ± SE (n = 3 for each condition). Bottom: immunoblots.
DISCUSSION
The major finding of this study is the identification of a novel role of autophagy in endothelial barrier dysfunction and its relevance in the pathogenesis of ALI. Using an aerosolized bacterial LPS inhalation mouse model of ALI, we found that the autophagy inhibitor 3-MA produced beneficial effects against lung vascular injury and neutrophil sequestration both prophylactically and therapeutically. The protective effect of 3-MA on lung inflammatory injury correlates with its ability to protect against the decrease in VE-cadherin levels and increase in inflammatory mediators and neutrophil sequestration in the lungs of mice exposed to LPS. Consistent with this finding, our in vitro studies using cultured EC showed that inhibition of autophagy attenuated barrier dysfunction by preventing disassembly of VE-cadherin and formation of stress fibers caused by thrombin. Thus the contribution of autophagy in ALI relies, at least in part, on its ability to cause EC barrier dysfunction in the inflamed lung.
The vascular EC that form the inner lining of blood vessels serve to separate the blood contents from extravascular space and provide the first barrier for inflammatory cells migrating to the interstitial and alveolar spaces (7, 27, 38, 51). Consequently, loss of endothelial barrier integrity following exposure to noxious (chemical, cellular, or mechanical) agents that are either inhaled or delivered to the lung through the pulmonary circulation results in protein-rich pulmonary edema formation and inflammatory cell infiltration (1, 25, 27, 40, 48). To understand the role of autophagy in the mechanism of lung vascular injury caused by LPS inhalation, we first determined the ability of LPS to induce autophagy in the lung. We provide evidence that autophagy is induced in the lung following exposure of mice to LPS. It is intriguing that autophagy is also induced in the lungs of mice exposed to 3-MA alone. However, mice exposed to 3-MA before LPS showed a decrease in lung autophagy, suggesting a protective effect of 3-MA on LPS-induced autophagy. Thus, while 3-MA can increase the constitutive autophagy, it reduces stress (LPS)-induced autophagy. The differential effects of 3-MA are consistent with an earlier study showing that 3-MA has a dual role in modulation of autophagy: whereas 3-MA suppresses starvation-induced autophagy, prolonged treatment with 3-MA in nutrient-rich conditions increases constitutive autophagy (52). In contrast to a clear prosurvival role for constitutive autophagy (17, 22, 23), stress-induced autophagy has been associated with both salutary and deleterious effects in many disorders, including lung diseases (32, 41, 43). In the heart, ischemia induces autophagy via an AMP-activated protein kinase (AMPK)-dependent mechanism, whereas ischemia-reperfusion increases autophagy via a beclin 1-dependent, but AMPK-independent, mechanism (26). However, autophagy induced by these mechanisms plays distinct roles: while it serves a protective function in ischemia, it is detrimental during reperfusion (26). Similarly, while constitutive autophagy in the heart is a homeostatic mechanism for maintaining cardiomyocyte size and global cardiac structure and function, pressure overload-induced cardiac autophagy is a maladaptive response that contributes to heart failure progression (35, 56). Consistent with these findings, our results show that an increase in constitutive autophagy by 3-MA itself has no detrimental effects on the lung and is associated with increased baseline TER and cell surface VE-cadherin staining in EC (Figs. 6B and 9B). In contrast, exposure of mice to 3-MA before or after LPS challenge showed a marked protection against lung vascular leak and tissue edema. Moreover, inhibition of autophagy was effective in protecting against the loss of VE-cadherin in the lungs of mice exposed to LPS. Similarly, inhibition of autophagy also attenuated LPS-induced lung neutrophil sequestration. These data identify an important role of autophagy in LPS-induced ALI and show the preventive and therapeutic potential of 3-MA against ALI.
Our data are consistent with earlier reports (44, 55) showing a protective effect of inhibiting autophagy in other mouse models of ALI. Sun et al. (44) showed that inhibition of autophagy by 3-MA ameliorated ALI and mortality caused by avian influenza A H5N1 infection. The protective effect of autophagy inhibition in this case was attributed, in major part, to its ability to inhibit autophagic death of alveolar epithelial cells (44). Similarly, Zhang et al. (55) demonstrated that autophagy plays a critical role in the pathogenesis of ventilator-induced lung injury via activation of nucleotide-binding domain- and leucine-rich repeat-containing protein (NLRP3) inflammasome signaling in pulmonary macrophages. Xu et al. (54) reported that inhibition of autophagy by 3-MA substantially reduced pulmonary tissue edema associated with hepatopulmonary syndrome. The present study identifies autophagy as an important determinant of lung vascular injury caused by LPS inhalation and provides evidence that autophagy is mechanistically linked to ALI via its ability to induce, at least in part, endothelial barrier dysfunction. For example, administration of 3-MA showed a marked protection against LPS-induced decrease in the levels of VE-cadherin, an EC-specific protein, in the lungs of mice. Moreover, EC exposed to 3-MA showed a marked protection against thrombin-induced cleavage and loss of VE-cadherin from AJs and contractile forces generated by actin-myosin interaction (actin stress fiber formation) and, thereby, barrier disruption caused by thrombin. Consistent with this, exposure of cells to 3-MA before and after thrombin challenge was effective in reducing and reversing the barrier disruption. Moreover, the effect of the PI3K inhibitor LY294002 in augmenting thrombin-induced EC barrier dysfunction and the ability of 3-MA to exert its protective effect, even in the presence of LY294002, show that the effect of 3-MA on EC barrier disruption is uncoupled from its ability to inhibit PI3K. Furthermore, knockdown of ATG5, an essential regulator of autophagy (17, 34, 35), also protected against thrombin-induced EC barrier disruption. Together, these data identify a previously unrecognized role of autophagy in regulating endothelial barrier dysfunction in ALI. However, these data do not exclude the involvement of autophagy in alveolar epithelial cells and pulmonary macrophages in this model of ALI, particularly in view of their reported role in ALI associated with H5N1 infection (44) and mechanical ventilation (55), respectively. Therefore, additional comprehensive studies using mice with cell-specific deletion of essential autophagy genes such as ATG5 or ATG7 are required to address the precise contribution of autophagy in endothelial vs. epithelial cells or macrophages in ALI (37, 49).
In summary, this study shows a critical role of autophagy in regulating EC barrier dysfunction associated with ALI and supports the concept that inhibiting autophagy by 3-MA could prove a viable therapeutic strategy to control ALI.
GRANTS
This work was supported by National Institutes of Health Grants HL-116632, HL-130870, and ES-01247.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A.S., A.L., and V.G. performed experiments; S.A.S. and A.L. analyzed data; S.A.S., F.F., and A.R. interpreted results of experiments; S.A.S. and A.R. prepared figures; S.A.S., A.L., V.G., F.F., and A.R. approved final version of manuscript; F.F. and A.R. conceived and designed research; F.F. and A.R. edited and revised manuscript; A.R. drafted manuscript.
REFERENCES
- 1.Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol 75: 593–615, 2013. doi: 10.1146/annurev-physiol-030212-183756. [DOI] [PubMed] [Google Scholar]
- 2.Bijli KM, Fazal F, Slavin SA, Leonard A, Grose V, Alexander WB, Smrcka AV, Rahman A. Phospholipase C-ε signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury. Am J Physiol Lung Cell Mol Physiol 311: L517–L524, 2016. doi: 10.1152/ajplung.00069.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bijli KM, Kanter BG, Minhajuddin M, Leonard A, Xu L, Fazal F, Rahman A. Regulation of endothelial cell inflammation and lung polymorphonuclear lymphocyte infiltration by transglutaminase 2. Shock 42: 562–569, 2014. doi: 10.1097/SHK.0000000000000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birukova AA, Shah AS, Tian Y, Moldobaeva N, Birukov KG. Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell Signal 28: 541–551, 2016. doi: 10.1016/j.cellsig.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brest P, Corcelle EA, Cesaro A, Chargui A, Belaïd A, Klionsky DJ, Vouret-Craviari V, Hebuterne X, Hofman P, Mograbi B. Autophagy and Crohn’s disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med 10: 486–502, 2010. doi: 10.2174/156652410791608252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 107: 18880–18885, 2010. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dejana E, Vestweber D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci 116: 119–144, 2013. doi: 10.1016/B978-0-12-394311-8.00006-6. [DOI] [PubMed] [Google Scholar]
- 8.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 91: 1487–1500, 2001. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 9.Fazal F, Bijli KM, Minhajuddin M, Rein T, Finkelstein JN, Rahman A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 21047–21056, 2009. doi: 10.1074/jbc.M109.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fazal F, Bijli KM, Murrill M, Leonard A, Minhajuddin M, Anwar KN, Finkelstein JN, Watterson DM, Rahman A. Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS One 8: e59965, 2013. doi: 10.1371/journal.pone.0059965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fazal F, Minhajuddin M, Bijli KM, McGrath JL, Rahman A. Evidence for actin cytoskeleton-dependent and -independent pathways for RelA/p65 nuclear translocation in endothelial cells. J Biol Chem 282: 3940–3950, 2007. doi: 10.1074/jbc.M608074200. [DOI] [PubMed] [Google Scholar]
- 12.Gando S, Nanzaki S, Morimoto Y, Kobayashi S, Kemmotsu O. Systemic activation of tissue-factor dependent coagulation pathway in evolving acute respiratory distress syndrome in patients with trauma and sepsis. J Trauma 47: 719–723, 1999. doi: 10.1097/00005373-199910000-00017. [DOI] [PubMed] [Google Scholar]
- 13.Gao R, Ma Z, Hu Y, Chen J, Shetty S, Fu J. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am J Physiol Lung Cell Mol Physiol 308: L847–L853, 2015. doi: 10.1152/ajplung.00274.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 26: 441–454, 2013. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 15.Golovkine G, Faudry E, Bouillot S, Voulhoux R, Attrée I, Huber P. VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog 10: e1003939, 2014. doi: 10.1371/journal.ppat.1003939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, Zhao YY, Komarova YA, Vogel SM, Malik AB, Komorova YA, Vogel SM, Malik AB. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125: 652–664, 2015. doi: 10.1172/JCI77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889, 2006. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 18.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8: 108–117, 2011. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 19.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 22: 140–149, 2010. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idell S, Gonzalez K, Bradford H, MacArthur CK, Fein AM, Maunder RJ, Garcia JG, Griffith DE, Weiland J, Martin TR, McLarty J, Fair DS, Walsh PN, Colman RW. Procoagulant activity in bronchoalveolar lavage in the adult respiratory distress syndrome. Contribution of tissue factor associated with factor VII. Am Rev Respir Dis 136: 1466–1474, 1987. doi: 10.1164/ajrccm/136.6.1466. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi K, Sato K, Kida T, Omori K, Hori M, Ozaki H, Murata T. Stromal cell-derived factor-1α/C-X-C chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and Rac1 activation. Arterioscler Thromb Vasc Biol 34: 1716–1722, 2014. doi: 10.1161/ATVBAHA.114.303890. [DOI] [PubMed] [Google Scholar]
- 22.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884, 2006. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 23.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 24.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77: 1763–1772, 2009. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol 49: 119–133, 2008. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922, 2007. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 27.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 28.Mehta D, Rahman A, Malik AB. Protein kinase C-α signals ρ-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 276: 22614–22620, 2001. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 29.Mehta D, Ravindran K, Kuebler WM. Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol 307: L924–L935, 2014. doi: 10.1152/ajplung.00318.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minhajuddin M, Bijli KM, Fazal F, Sassano A, Nakayama KI, Hay N, Platanias LC, Rahman A. Protein kinase C-δ and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-κB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 4052–4061, 2009. doi: 10.1074/jbc.M805032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittal M, Tiruppathi C, Nepal S, Zhao YY, Grzych D, Soni D, Prockop DJ, Malik AB. TNFα-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc Natl Acad Sci USA 113: E8151–E8158, 2016. doi: 10.1073/pnas.1614935113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizumura K, Cloonan S, Choi ME, Hashimoto S, Nakahira K, Ryter SW, Choi AM. Autophagy: friend or foe in lung disease? Ann Am Thorac Soc 13 Suppl 1: S40–S47, 2016. doi: 10.1513/AnnalsATS.201507-450MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N. Autophagy: process and function. Genes Dev 21: 2861–2873, 2007. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 34.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152: 657–668, 2001. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624, 2007. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 36.Paria BC, Vogel SM, Ahmmed GU, Alamgir S, Shroff J, Malik AB, Tiruppathi C. Tumor necrosis factor-α-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol 287: L1303–L1313, 2004. doi: 10.1152/ajplung.00240.2004. [DOI] [PubMed] [Google Scholar]
- 37.Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei Y, Lan L, Deng X, Liang H, Ma F, Wu M. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol 198: 3205–3213, 2017. doi: 10.4049/jimmunol.1601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahman A, Fazal F. Blocking NF-κB: an inflammatory issue. Proc Am Thorac Soc 8: 497–503, 2011. doi: 10.1513/pats.201101-009MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reutershan J, Basit A, Galkina EV, Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 289: L807–L815, 2005. doi: 10.1152/ajplung.00477.2004. [DOI] [PubMed] [Google Scholar]
- 40.Rizzo AN, Sammani S, Esquinca AE, Jacobson JR, Garcia JG, Letsiou E, Dudek SM. Imatinib attenuates inflammation and vascular leak in a clinically relevant two-hit model of acute lung injury. Am J Physiol Lung Cell Mol Physiol 309: L1294–L1304, 2015. doi: 10.1152/ajplung.00031.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryter SW, Nakahira K, Haspel JA, Choi AM. Autophagy in pulmonary diseases. Annu Rev Physiol 74: 377–401, 2012. doi: 10.1146/annurev-physiol-020911-153348. [DOI] [PubMed] [Google Scholar]
- 42.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res 102: 1192–1201, 2008. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 306: 990–995, 2004. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, Zhao Y, Yan Y, Tang J, Zhao C, Yang P, Liu K, Wang S, Lu H, Li X, Tan L, Gao R, Song J, Gao X, Tian X, Qin Y, Xu KF, Li D, Jin N, Jiang C. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 5: ra16, 2012. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 45.Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, Mehta D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res 103: 1164–1172, 2008. doi: 10.1161/01.RES.0000338501.84810.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian Y, Gawlak G, O’Donnell JJ 3rd, Birukova AA, Birukov KG. Activation of vascular endothelial growth factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J Biol Chem 291: 10032–10045, 2016. doi: 10.1074/jbc.M115.690487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708, 2006. doi: 10.1080/10739680600930347. [DOI] [PubMed] [Google Scholar]
- 48.Tiruppathi C, Shimizu J, Miyawaki-Shimizu K, Vogel SM, Bair AM, Minshall RD, Predescu D, Malik AB. Role of NF-κB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. J Biol Chem 283: 4210–4218, 2008. doi: 10.1074/jbc.M703153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, Connelly PS, Daniels MP, Komatsu M, Cao L, Finkel T. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 19: 1281–1287, 2013. doi: 10.1038/nm.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trani M, Dejana E. New insights in the control of vascular permeability: vascular endothelial-cadherin and other players. Curr Opin Hematol 22: 267–272, 2015. doi: 10.1097/MOH.0000000000000137. [DOI] [PubMed] [Google Scholar]
- 51.Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol 28: 223–232, 2008. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 52.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285: 10850–10861, 2010. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9: 1102–1109, 2007. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 54.Xu D, Chen B, Gu J, Chen L, Belguise K, Wang X, Yi B, Lu K. Inhibition of autophagy ameliorates pulmonary microvascular dilation and PMVECs excessive proliferation in rat experimental hepatopulmonary syndrome. Sci Rep 6: 30833, 2016. doi: 10.1038/srep30833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 307: L173–L185, 2014. doi: 10.1152/ajplung.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117: 1782–1793, 2007. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]