

Abstract
Deep sequencing analysis of an asymptomatic grapevine revealed a virome containing five RNA viruses and a viroid. Of these, Grapevine leafroll-associated virus 7 (GLRaV-7), an unassigned closterovirus, was by far the most prominently represented sequence in the analysis. Graft-inoculation of the infection to another grape variety confirmed the lack of the leafroll disease symptoms, even though GLRaV-7 could be detected in the inoculated indicator plants. A 16,496 nucleotide-long genomic sequence of this virus was determined from the deep sequencing data. Its genome architecture and the sequences encoding its nine predicted proteins were compared with those of other closteroviruses. The comparison revealed that two other viruses, Little cherry virus-1 and Cordyline virus-1 formed a well supported phylogenetic cluster with GLRaV-7.
Keywords: Grapevine viruses, Leafroll disease, Closteroviridae family, GLRaV-7, Deep sequence analysis, LChV-1, CoV-1
1. Introduction
Cultivation of grapevine (Vitis vinifera) and wine production have deep historical roots and major economic significance in the temperate climate zones around the globe (Bisson et al., 2002). Controlling viral, bacterial and fungal diseases of grapevine is an on-going challenge for the grape industry. Virus-caused grapevine leafroll disease is among the most damaging and widespread of the diseases of V. vinifera. Infected vines exhibit down-rolling of leaf margins and inter-veinal reddening or chlorosis (reddish in red-fruit varieties, chlorotic in white-fruit varieties) that increases through the season (Martelli, 1993); the infection reduces grape yields.
Despite decades of research, the relative contributions of the viruses associated with leaftoll disease, and the pathogenicities of each have yet to be resolved. All leafroll disease viruses identified so far belong to the family Closteroviridae. They show long filamentous particles containing positive-sense RNA genomes (Dolja et al., 2006). The list of these viruses includes eleven species (Martelli et al., 2002; Alkowni et al., 2004; Maliogka et al., 2008; Abou Ghanem-Sabanadzovicet al., 2010) not counting the mis-identified Grapevine leafroll associated virus (GLRaV)-8 (Bertsch et al., 2009).
Our objective in this study was to clarify the contextualization of GLRaV-7 among the rest of the leafroll disease causing viruses of grapevine. The rapidly growing list of newly discovered closteroviruses with diverse host ranges, life cycles, and genome architectures (Abou Ghanem-Sabanadzovic et al., 2010; Maliogka et al., 2008; Menzel et al., 2009; Tzanetakis and Martin, 2007; Tzanetakis et al., 2005, 2011) necessitates a phylogenetic and taxonomic update of this large and economically important virus family. The recent co-designation of GLRaVs -4, -5, -6, and -9 as members of the GLRaV-4 subgroup (Abou Ghanem-Sabanadzovic et al., 2011; Thompson et al., 2011) is one step in this reorganization.
The current separation of the Closteroviridae into three genera is in accord with the transmission of each by distinct insect vectors and reflects a general trend in the family evolution (Dolja et al., 2006; Karasev, 2000). However, GLRaV-7 is not classified among these genera, but remains a taxonomically unassigned member of this group. This virus acquired its name on the basis of limited sequence information (Choueiri et al., 1996) although the eponymous disease symptoms have not been definitively attributed to it. Its detection in California was reported by Morales and Monis (2007).
Complete sequences are available for members of the GLRaV-4 subgroup (Abou Ghanem-Sabanadzovic et al., 2011; Thompson et al., 2011), GLRaV-2 (Zhu et al., 1998), and GLRaV-3 (Ling et al., 2004); partial sequence is available for GLRaV-1 (Fazeli and Rezaian, 2000). Subspecies variability has recently been noted for strains of GLRaV-1 (Alabi et al., 2011), GLRaV-3 (Wang et al., 2011), and GLRaV-7 (Morales and Monis, 2007; Al Rwahnih et al., 2011).
We present here a 16,496 nt sequence of the GLRaV-7 genome. The genome encodes a papain-like leader protease, capping enzyme, and helicase; the RNA-dependent RNA polymerase (RdRp); an Hsp70h; a three-gene block encoding the major (CP) and two minor (p61 and CPm) capsid proteins; and two uncharacterized proteins, p25 and p27, at the 3′-terminus. A previously published 590 nt sequence for the GLRaV-7 HSP70h gene (Saldarelli et al., 1998) was found to be 93% identical to its homolog in this sequence; a preliminary GLRaV-7 genomic sequence report (Mikona et al., 2009) was also similar to the GLRaV-7 sequence. Comparative analysis of the GLRaV-7 sequence described here with the sequences of other members of the Closterovirudae suggest that GLRaV-7 could be placed into a fourth genus in that family.
2. Materials and methods
2.1. Virus sources
GLRaV-7 strain Swi was identified in V. vinifera, cv Pinot Noir clone 23 (PN23), a source vine maintained by Foundation Plant Services (FPS) at UC Davis. The infection was assayed by specific RT-PCR testing (Turturo et al., 2000). The PN23 source vine tested positive for GLRaV-7 and negative for all other known grapevine leafroll associated viruses.
2.2. Biological indexing
GLRaV-7-infected PN23 canes were graft inoculated to Cabernet Franc, which is a standard indicator host variety for grapevine leafroll diseases. Nine replicates of 3 indicators each (total 27 plants) were bud chip inoculated from the PN23 source plant. Two bud chips per indicator were grafted, and the grafted plants maintained in the greenhouse for one month for the graft to heal. After two weeks to a month of acclimatization in a shade house, grafted plants were planted in the field. Symptoms were evaluated 1.5 years after inoculation. Two replicates of three plants each of graft inoculated from GLRaV-2 and GLRaV-3 infected vines were grown in parallel as positive controls.
2.3. High throughput sequencing
For sequencing and characterization of the GLRaV-7 Swi isolate double stranded RNA (dsRNA) was extracted from 90 g of cambial scrapings from the PN-23 source vine as described (Routh et al., 1998) without the DNase and RNase enzymatic digestion steps. Complementary DNA (cDNA) libraries were made using the SuperScript® II Reverse Transcriptase kit (Invitrogen, Carlsbad, CA) primed with random hexamers (300 ng/μl, Invitrogen, Carlsbad, CA) and amplified with GenomePlex® complete whole genome amplification kit (Sigma, San Louis, MO) following the manufacturer's instructions. The amplified DNA preparation was cleaned with the PCR cleanup kit (Sigma, St Louis, MO) and DNA quality was checked as described previously (Al Rwahnih et al., 2009). Samples were subjected to 454 Life Sciences (Branford, CT, USA) high-throughput pyrosequencing, using the Genome Sequencer FLX platform.
A second set of cDNA libraries prepared as above were subjected to deep sequencing using the Illumina Genome Analyzer II platform (Illumina, San Diego CA). The High-Speed Sequence Search Suite (HS3) algorithm from GenomeQuest (Westborough, Mass.) was used to sort the sequences (Al Rwahnih et al., 2009). Reads were subjectedtoboth BLASTN and BLASTX analysis (Altschul et al., 1997) (http://www.ncbi.nlm.nih.gov). The contiguous GLRaV-7 genome sequence was assembled using the assembly tools in the SeqMan NGen and SeqMan Pro software suites from DNAstar.
2.4. Multiple sequence alignment and phylogenetic analysis
Comparative analyses of members and tentative members of the viral family Closteroviridae (excluding those for which only partial sequences have been described) produced phylogenetic trees based on amino acid sequences of the heat shock protein 70 homolog (HSP70h) gene, and the coat protein (CP) gene.
Multiple alignments were made with the default options of multiple sequence alignment program Clustal X 1.8 (Thompson et al., 1997). Alternatively, multiple sequence alignments were constructed using the MUSCLE program (Edgar, 2004) and are presented in the ClustalW format. Phylogenetic analyses were conducted using the minimum evolution method as well as Maximum Likelihood and Maximum Parsimony methods from Molecular Evolutionary Genetic Analysis software MEGA version 5 (Tamura et al., 2011). The support for the tree nodes was estimated using 1000 bootstrap replicates with default parameters. Accession numbers of the viruses used in the alignment and in phylogenetic analysis are listed in Table S1, as is that of the unpublished GLRaV-7 sequence of Mikona et al. (2009).
Protein secondary structure predictions for GLRaV-7 p25 and p27 were made using the PSIPRED program; confidence levels in the predictions, on a scale of 0–1, are reflected in the height and the blue color intensity of the bars (McGuffin et al., 2000).
3. Results
3.1. Identification of the asymptomatic GLRaV-7 infection
We analyzed a row of Pinot Noir 23 vines propagated from the PN-23 source vine at Foundation Plants Services (FPS) (http://fps.ucdavis.edu) at the University of California, Davis. All of these clones appeared asymptomatic over the ten years since their planting; all tested positive for GLRaV-7 using the specific RT-PCR assay of Turturo et al. (2000). The asymptomatic GLRaV-7 infection was confirmed in a retest of twenty two of these plants using a second set of RT-PCR primers designed for the specific detection of the viral HSP70h gene (Al Rwahnih et al., 2011). Analysis of the same material with the full panel of tests for all other known leafroll virus species (Klaassen et al., 2011) revealed the presence of no other leafroll viruses.
3.2. Biological indexing analysis
Cane samples from the original PN-23 source vine were graft inoculated to Cabernet franc, which is a standard indicator host for grapevine leafroll diseases (Rowhani et al., 2005). Over the course of the analysis, the inoculated indicator plants were all found to have become positive for GLRaV-7 infection according to the RT-PCR tests. But they showed no leafroll disease symptoms during 2 years of observation. Control inoculations using GLRaV-2 and -3 infected canes induced PCR-detectable and symptomatic infections in the indicator host in both cases.
3.3. Deep sequencing analysis of the virome
We characterized the virome of the GLRaV-7-infected vines using 454 Life Sciences high throughput sequencing of the dsRNA fraction recovered from cane tissue. This total genomic analysis produced 42,226 reads containing 8.7mb of sequence data. BLAST analysis of the high quality reads against the GenBank database (Altschul et al., 1997) revealed 22,910 virus related sequences. The virome was found to include five positive-strand RNA viruses and a viroid (Table 1). The presence of each virus was confirmed by RT-PCR analysis using virus-specific primers (Al Rwahnih et al., 2009). The vast majority of the virus-derived reads, 18,962, were identified as GLRaV-7.
Table 1.
High throughput sequencing reads for grapevine viruses and viroids identified in whole genome analyses of the Pinot Noir 32 vine, using two sequencing protocols, and including qualitative results for virus-specific RT-PCR tests.
| Pathogen | Number of reads | ||
|---|---|---|---|
|
|
|||
| 454 | Illumina | RT-PCR | |
| Grapevine leafroll associated virus-7 | 18,962 | 7,703,496 | + |
| Rupestris stem pitting-associated virus | 2940 | 295,162 | + |
| Grapevine rupestris vein feathering virus | 841 | 16,037 | + |
| Grapevine syrah virus-1 | 139 | 2211 | + |
| Grapevine red globe virus | 25 | 86 | + |
| Hop stunt viroid | 3 | 12 | + |
Although the 454 Life Sciences pyrosequencing run provided nearly complete coverage of the GLRaV-7 genome, a second deep sequencing analysis from the same plant was done using the Illu-mina protocol, to fill in a few short gaps, and to assess positional variations averaged into the consensus sequence. 17,706,338 high quality reads averaging 36nt in length were identified in this secondary analysis. BLAST analysis of these reads identified the same viruses as had been identified in the 454 Life Sciences analysis. The Illumina results supported the 454 identification of GLRaV-7 as the sole leafroll virus in the infection (Table 1).
3.4. Genomic analysis of GLRaV-7 Swi
The total of 7,722,458 GLRaV-7 reads from both Illumina and 454 runs generated a consensus sequence 16,496 nt in length for the GLRaV-7 genome. The average coverage depth was 16, 891-fold per nucleotide (nt) with a minimum of 9 reads per nt at position 16,407and a maximum of 105,193 at position 8202. The virus genome sequence (GenBank accession # JN383343) was designated as GLRaV-7 Swi, after the original Swiss source of the PN-23 vinestock material. Other sequences only for the GLRaV-7 HSP70h gene previously recorded in the GenBank, ranging in length from 502 to 590 nt were found to be 91–97% identical to their homolog in the genome reported here. Consistent with a preliminary report on another GLRaV-7 isolate (Mikona et al., 2009) nine open reading frames were identified in the virus genome (Fig. 1). In this report, GLRaV-7 and other members of the phylogenetic cluster of viruses described in the next section are referred to as members of the “Velarivirus” genus (see Section 4).
Fig.1.
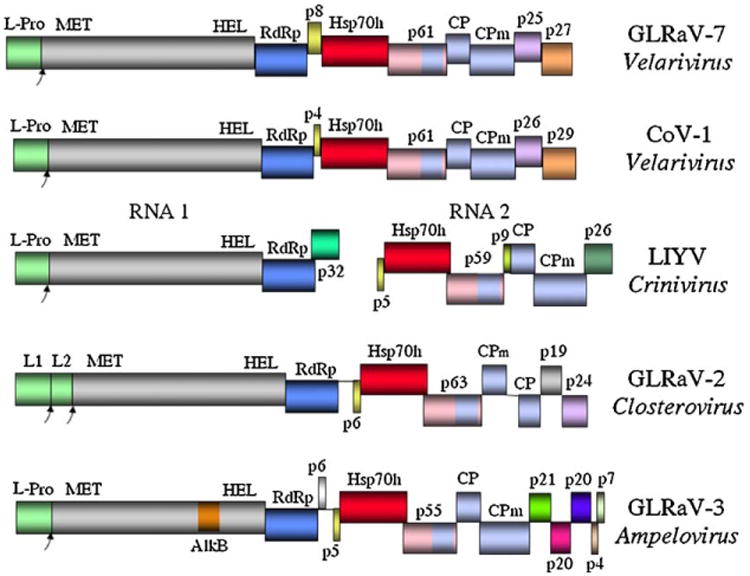
Genome organizations of GLRAV-7 in comparison to CoV-1, LIYV, GLRaV-2 and GLRaV-3 approximately to scale. The different segments represent open reading frames; their vertical heights represent the different frame registers. Shared colors represent conserved sequences. L-Pro, leader protease; MET, methyltransferase domain; HEL, RNA helicase domain; RdRp, RNA-dependent RNA polymerase; HSP70h, heat shock protein 70 homolog; CP, coat protein; CPm, minor coat protein.
3.5. Identification of an undescribed phylogenetic cluster including GLRaV-7
Systematic sequence comparisons with the GenBank database revealed that two other viruses infecting widely divergent hosts, Little cherry virus-1 (LChV-1; Jelkmann et al., 1997) and Cordy-line virus-1 (CoV-1; Melzer et al., 2011) were closely related to GLRaV-7. Using the Alignx Sequence Analysis tool (Vector NTI Advance™ 11.5, Invitrogen Inc., Carlsbad, CA), we found that 7 proteins encoded by these other two viruses consistently showed higher levels of identity with their homologs in GLRaV-7 than they did with other closteroviruses (Table 2; virus abbreviations are described in Table S1). These levels of identify reached 54% for the RNA-dependent RNA polymerase,∼41% for Hsp70h, and ∼30% for coat protein (CP). In contrast, identity levels of these three proteins were only 10–28% when compared with the GLRaV-3 (Table 2), which is the most closely related leafroll-associated virus to GLRaV-7, indicating the substantial divergence between these two viruses. GLRaV-4 group viruses show no CPm genes in their sequences (see Abou Ghanem-Sabanadzovic et al., 2011; Thompson et al., 2011) as is reflected in Table 2.
Table 2.
Similarities (percentage) between the amino acid sequences encoded by the genes in the GLRAV-7 genome, and amino acid sequences encoded by closteroviruses LChV-1,-2, GLRaV-2, -3, -Pr, BYV, BnYDV, CoV-1, LCV, SPaV, LIYV, and MV-1. MET, methyltransferase domain, HEL, helicase domain, RdRp, RNA-dependent RNA polymerase. hHSP70, heat shock 70-like protein homologs, CP, coat protein, CPm, minor coat protein.
| Virus | MTR | HEL | RdRp | hHSP70 | P60 | CP | CPm |
|---|---|---|---|---|---|---|---|
| LChV-1 | 25 | 39 | 54 | 43 | 25 | 31 | 18 |
| CoV-1 | 26 | 33 | 54 | 39 | 25 | 29 | 17 |
| BnYDV | 21 | 30 | 49 | 37 | 19 | 22 | 14 |
| LCV | 18 | 31 | 49 | 35 | 19 | 21 | 13 |
| SPaV | 20 | 40 | 49 | 38 | 17 | 20 | 12 |
| LIYV | 21 | 30 | 48 | 39 | 19 | 18 | 15 |
| GLRaV-3 | 14 | 22 | 28 | 25 | 10 | 10 | 8 |
| GLRaV-4 | 14 | 23 | 26 | 28 | 16 | 18 | – |
| GLRaV-5 | 14 | 22 | 26 | 27 | 15 | 17 | – |
| GLRaV-6 | 12 | 23 | 26 | 27 | 16 | 19 | – |
| GLRaV-Pr | 14 | 17 | 25 | 26 | 10 | 11 | – |
| LChV-2 | 11 | 19 | 25 | 25 | 12 | 14 | 9 |
| BYV | 13 | 21 | 27 | 26 | 13 | 12 | 13 |
| GLRaV-2 | 14 | 20 | 28 | 24 | 13 | 13 | 14 |
| MV-1 | 12 | 22 | 29 | 24 | 11 | 12 | 12 |
To investigate the phylogenetic affinities of GLRaV-7, multiple alignments of the amino acid sequences of viral proteins were generated and used to construct phylogenetic trees As shown in Fig.2a, GLRaV-7, LChV-1 and CoV-1 formed a distinct, strongly supported cluster within the Hsp70h phylogenetic tree that encompassed most of the recognized members of the family Closteroviridae. The analogous tree generated from CP sequences had similar topology, again with confident separation of a three-virus lineage (Fig. 2b). This three-virus cluster remained well separated in trees that were generated using the Minimum Evolution, Maximum Likelihood, or Maximum Parsimony methods (not shown).
Fig. 2.
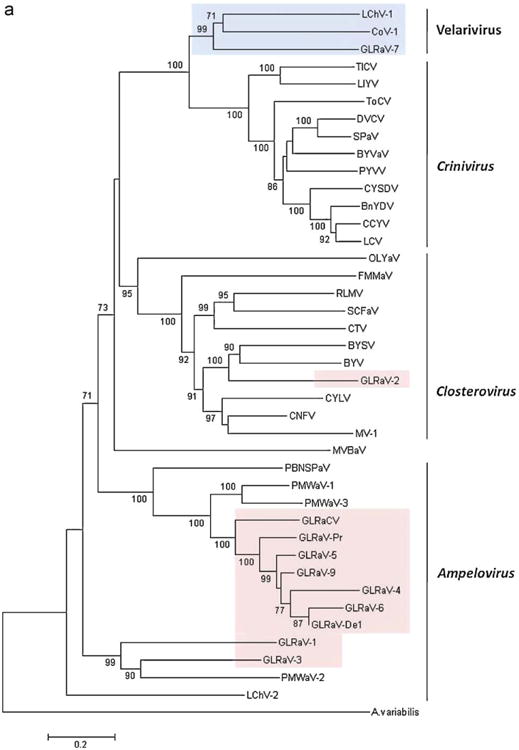
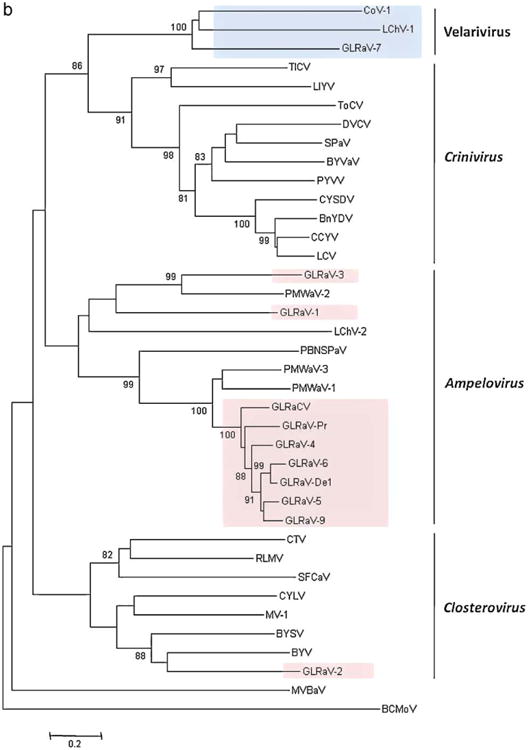
(a) Phylogenetic analysis of the heat shock protein 70 homolog sequences of GLRaV-7 and other species in Closteroviridae family. Bootstrap values are shown as percentages; percentages less than 70% are not show. Hsp70 of Anabaena variabilis (ABA20196) was used as an outgroup. The Velarivirus lineage is shaded blue, and all other leafroll viruses are shaded pink. (b) Phylogenetic analysis of the coat protein sequences of GLRaV-7 and other species in Closteroviridae family. Bootstrap values are shown as percentages; percentages less than 70% are not shown. The CP sequence of Bean calico mosaic virus (BCMoV) NC 003504 was used as an outgroup. The Velarivirus lineage is shaded blue, and all other leafroll viruses are shaded pink. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
The sister group to this cluster was found to be the Crinivirus genus, which is composed of viruses with bi-partite genomes and gene content distinct from that of GLRaV-7 or CoV-1 (Fig. 1). Criniviruses are transmitted by whiteflies (Ng and Falk, 2006), whereas no insect or other vectors have so far been identified for GLRaV-7, LChV-1, or CoV-1.
3.6. Comparative analysis of the members of the phylogenetic cluster
All identifiable protein domains encoded in the GLRaV-7 genome show higher similarity to the orthologous domains of the other two members in the identified phylogenetic cluster (Table 2) than to other closteroviruses. The only notable exception was the N-terminal portion of the GLRaV-7 polyprotein (∼400 residues) that showed only weak (E-value >0.05, Altschul et al., 1997) similarity to the corresponding region of the CoV-1 polyprotein and no similarity to the LChV-1 polyprotein. Instead, this part of the GLRaV-7 polyprotein was significantly similar to the respective sequences of the criniviruses (Fig. 3). Given the concordant topologies of the phylogenetic trees of the HSP70h and CP genes (Fig. 2a and b), in which criniviruses were found to be the sister group to the velariviruses, it seems likely that this ancestral leader segment degraded in the CoV-1-LChV-1 clade after its divergence from GLRaV-7.
Fig. 3.

Alignment of the leader regions of GLRaV-7 with those of criniviruses (positions in the polyprotein are indicated in parentheses). Asterisks denote amino acid residues identical in the aligned sequences; colons and dots show alignment columns occupied by similar residues depending on the degree of the similarity.
The monophyly of the three viruses in this cluster was further supported by characterization of the p25 protein. This protein is one of the poorly conserved predicted proteins encoded in the 3′-terminal portion of the GLRaV-7 genome; it appears to be homologous to the proteins encoded by the analogously positioned genes of LChV-1 and CoV-1 (Fig. 4). The p25 sequence showed no significant similarity to any sequence in a standard BLAST search of the non-redundant protein database at the NCBI. However, a search against protein sequences of positive-strand RNA viruses alone showed significant similarity with the p21 protein of LChV-1 (random expectation value below 0.001), and the second iteration of a PSI-BLAST search also retrieved the p26 sequence of CoV-1 (Fig. 4). The alignment of these three proteins revealed several conserved blocks with confidently predicted secondary structure, shown in Fig. 4 as spans of α helix (h) andβ strand (e) (the domains of that secondary structure are shown for GLRaV-7 in Fig. S1). The 3′-portions of the genomes of other closteroviruses encode proteins of similar size (e.g., Fig. 1) but sequence or structural similarity to velarivirus p25 could not be demonstrated despite extensive sequence comparison.
Fig. 4.

Alignment of the penultimate gene products of the three Velariviruses. Conserved blocks of predicted secondary structure are shown as spans of α helix (h) and β strand (e).
The 3′-most product of the GLRaV-7 genome, p27, showed no significant similarity to proteins of other closteroviruses (or any other proteins) although secondary structure prediction suggested that p27 is a globular protein with a distinct alpha-beta fold (Fig. S2). The presence of poorly conserved genes encoding ∼20–30kDa proteins in the 3′-region of the genome (Fig. 1) is a common feature of the closteroviruses (Dolja et al., 2006).
3.7. Asymptomatic character of the members of the proposed genus
GLRaV-7 was found to be asymptomatic in Pinot Noir and Cabernet Franc as reported here; GLRaV-7 has also been reported elsewhere as asymptomatic (Morales and Monis, 2007; Avgelis and Boscia, 2001). Both the LChV-1 and CoV-1 infections have been recorded as asymptomatic (Matic et al., 2009; Melzer et al., 2011). LChV-1 has been identified in symptomatic plants as a member of a mixed infection, but in no case has it been associated with symptomatic infections for which the presence of other symptomatic, co-infecting viruses has been conclusively ruled out.
4. Discussion
4.1. Simplified taxonomy of grapevine leafroll viruses
Recent phylogenetic analyses have simplified Closteroviridae traxonomy, especially as it relates to the grapevine leafroll viruses. The phylogenetic tree presented in Fig.2a shows that the grapevine leafroll viruses can now be divided into four categories. The Ampelovirus genus contains two of these clusters. One is the GLRaV-4 group as described by Abou Ghanem-Sabanadzovic et al. (2011). The second contains GLRaV-1 and -3; in this cluster, comparison of the HSP70 sequences (sources in Table S1) shows 43% similarity between them (c.f., Table 2).
GLRaV-2 stands alone in the Closterovirus genus. And a fourth division of the Closteroviridae is proposed in Fig. 2a, in which GLRaV-7 is grouped with other unassigned viruses in the family. That phylogenetic cluster is designated in this report as the Velarivirus genus.
4.2. The putative new genus “Velarivirus”
We describe in this work the common features that distinguish the cluster containing GLRaV-7, CoV-1, and LChV-1 from other generic groups in the Closteroviridae. This three virus group could constitute a new genus in that family.
We have brought this possibility to the attention of the ICTV Closteroviridae nomenclature study group. GLRaV-7 has been noted as being distinct from other closteroviruses that have been assigned to accepted genera within the family (G. Martelli, personal communication). For the purposes only of the discussion presented in this report, we refer to the potential new genus as the “Velariviruses” (from the latin velari, which means cryptic, or veiled). This provisional name derives from the asymptomatic nature of GLRaV-7 infection in grapevine as described here. Support for the putative new genus includes the following observations.
In Fig. 1 the genomic organization of the three proposed members of genus Velarivirus is shown to include a polyprotein encoding the papain-like leader protease, capping enzyme (in particular, the readily recognizable methyltransferase domain), and helicase; the RNA-dependent RNA polymerase (RdRp); an Hsp70h; a three-gene block encoding the major (CP) and two minor (p61 and CPm) capsid proteins; and two uncharacterized proteins, p25 and p27, at the 3′-terminus. This genomic organization was nearly identical among the viruses of the proposed genus, in contrast to substantial differences in comparisons with the genomic organizations of members of other genera in the Closteroviridae.
As seen in Table 2, all the viral protein sequences that were compared showed greater levels of similarity within the proposed genus Valerivirus than with other genera in the Closteroviridae.
As seen in Fig. 4, for the penultimate gene product (p26), there was detectable sequence similarity within the proposed Velarivirus genus; there was no detectable similarity of that gene product with members of other genera in the Closteroviridae.
As seen in Fig. 2a and b, comparisons based on two viral proteins formed a Velarivirus clade with 99–100% bootstrap support in phylogenetic analysis. Judging from these phylogenetic trees, the only alternative to establishing a new genus is to merge these viruses into the existing genus Crinivirus. However, the putatively named velariviruses possess a monopartite genome (a single long RNA) whereas criniviruses have a bipartite genome (two RNAs).
4.3. Other proteins encoded by members of the putative Velarivirus genus
The small open reading frame that overlaps the distal portion of the RdRp reading frame in GLRaV-7 encodes a 71 residue-long putative protein with prominent predicted transmembrane helices in the N-terminal and C-terminal regions. Although this small open reading frame did not show significant sequence similarity to any other proteins in current databases, similar small membrane proteins are encoded by other closteroviruses. In the case of Beet yellows virus, this small hydrophobic protein has been demonstrated to be expressed from a subgenomic messenger RNA and is targeted to the endoplasmic reticulum where it is involved in the critical function of virus cell-to-cell movement (Peremyslov and Dolja, 2002; Peremyslov et al., 2004).
The presence of genes encoding poorly conserved ∼20–30kDa proteins in the 3′-region of the genome is a common feature of the closteroviruses (Fig. 1) (Dolja et al., 2006). It seems likely that the homologous relationships between these proteins, e.g., as revealed here for the proposed Velariviruses in Fig. 4, extend further than sequence comparisons (even with the most sensitive available methods) can show. These proteins may be involved in virus counter-defense against specific host RNAi suppression (Reed et al., 2003), which could accelerate their divergence through the “arms race” in specific host-virus pairs (Lu et al., 2004).
4.4. Proposed renaming of GLRaV-7
We show here, as have others (Morales and Monis, 2007; Avgelis and Boscia, 2001) that leafroll symptoms are not associated with GLRaV-7 infection. In no case has GLRaV-7 been associated with symptomatic infection in which the presence of other co-infecting viruses has been conclusively ruled out. Together with previous reports (Choueiri et al., 1996; Turturo et al., 2000; Morales and Monis, 2007) the results presented here suggest the possibility of the existence of a broad yet unsuspected occurrence of asymptomatic GLRaV-7 in commercial wine, raisin, and table grape varieties.
We conclude that the current species designation of this virus as “leafroll-associated” is misleading, both in biological and phylogenomic terms. Therefore we propose that the name “GLRaV-7” should be considered for replacement by the ICTV with a new name that does not associate the virus with leafrolling in grapevine.
Supplementary Material
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.virusres.2011.10.018.
References
- Abou GhanemS-abanadzovic N, Sabanadzovic S, Uyemoto JK, Golino D, Rowhani A. A putative new Ampelovirus associated with grapevine leafroll disease. Arch Virol. 2010;155:1871–1876. doi: 10.1007/s00705-010-0773-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou GhanemS-abanadzovic N, Sabanadzovic S, Gugerli P, Rowhani A. Genome organization, serology and phylogeny of Grapevine leafroll-associated viruses 4 and 6: Taxonomic implications. Virus Res. 2011 Sep 8; doi: 10.1016/j.virusres.2011.09.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Alabi OJ, Al Rwahnih M, Karthikeyan G, Poojari S, Fuchs M, Rowhani A, Rayapati N. Grapevine leafroll-associated virus 1 occurs as genetically diverse populations. Phytopathology. 2011 Aug 10; doi: 10.1094/PHYTO-04-11-0114. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Alkowni R, Rowhani A, Daubert S, Golino D. Partial molecular characterization of a new ampelovirus associated with grapevine leafroll disease. J Plant Pathol. 2004;86:123–133. [Google Scholar]
- Al Rwahnih M, Daubert S, Golino D, Rowhani A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology. 2009;387:395–401. doi: 10.1016/j.virol.2009.02.028. [DOI] [PubMed] [Google Scholar]
- Al Rwahnih M, Osman F, Sudarshana M, Uyemoto J, Minafra A, Saldarelli P, Martelli G, Rowhani A. Detection of grapevine leafroll-associated virus 7 using real time qRT-PCR and conventional RT-PCR. J Virol Methods. 2011 doi: 10.1016/j.jviromet.2011.11.026. accepted. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgelis A, Boscia D. Grapevine leafroll-associated closterovirus 7 in Greece. Phytopathol Mediterr. 2001;40:289–292. [Google Scholar]
- Bertsch C, Beuve M, Dolja VV, Wirth M, Pelsy F, Herrbach E, Lemaire O. Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol Direct. 2009;4:1–11. doi: 10.1186/1745-6150-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson LF, Waterhouse AL, Ebeler SE, Walker MA, Lapsley JT. The present and future of the international wine industry. Nature. 2002;418:696–699. doi: 10.1038/nature01018. [DOI] [PubMed] [Google Scholar]
- Choueiri E, Boscia D, Digiaro M, Castellano MA, Martelli GP. Some properties of a hitherto undescribed filamentous virus of the grapevine. Vitis. 1996;35:91–93. [Google Scholar]
- Dolja VV, Kreuze JF, Valkonen JP. Comparative and functional genomics of closteroviruses. Virus Res. 2006;117:38–51. doi: 10.1016/j.virusres.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli CF, Rezaian AM. Nucleotide sequence and organization of ten open reading frames in the genome of grapevine leafroll-associated virus 1 and identification of three subgenomic RNAs. J Gen Virol. 2000;81:605–615. doi: 10.1099/0022-1317-81-3-605. [DOI] [PubMed] [Google Scholar]
- Jelkmann W, Fechtner B, Agranovsky AA. Complete genome structure and phylogenetic analysis of little cherry virus, a mealybug-transmissible closterovirus. J Gen Virol. 1997;78:2067–2071. doi: 10.1099/0022-1317-78-8-2067. [DOI] [PubMed] [Google Scholar]
- Karasev AV. Genetic diversity and evolution of closteroviruses. Ann Rev Phytopathol. 2000;38:293–324. doi: 10.1146/annurev.phyto.38.1.293. [DOI] [PubMed] [Google Scholar]
- Klaassen VA, Sim SY, Dangl GS, Osman M, Al Rwahnih M, Rowhani A, Golino DA. Vitis californica and Vitis californica ×Vitis vinifera hybrids are hosts for Grapevine leafroll-associated virus-2 and -3 and Grapevine virus A and B. Plant Dis. 2011;95:657–665. doi: 10.1094/PDIS-09-10-0621. [DOI] [PubMed] [Google Scholar]
- Ling KS, Zhu HY, Gonsalves D. Complete nucleotide sequence and genome organization of Grapevine leafroll-associated virus 3, type member of the genus Ampelovirus. J Gen Virol. 2004;85:2099–2102. doi: 10.1099/vir.0.80007-0. [DOI] [PubMed] [Google Scholar]
- Lu R, Folimonov AS, Shintaku M, Li WX, Falk BW, Dawson WO, Ding SW. Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. Proc Natl Acad Sci USA. 2004;101:15742–15747. doi: 10.1073/pnas.0404940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliogka VI, Dovas CI, Katis NI. Evolutionary relationships of virus species belonging to a distinct lineage within the Ampelovirus genus. Virus Res. 2008;135:125–135. doi: 10.1016/j.virusres.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Martelli GP, editor. Graft Transmissible Diseases of Grapevine: Handbook for Detection and Diagnosis. Food and Agriculture Organization of the United Nations; Rome: 1993. [Google Scholar]
- Martelli GP, Agranovsky AA, Bar-Joseph M, Boscia D, Candresse T, Coutts RH, Dolja VV, Falk BW, Gonsalves D, Jelkmann W, et al. The family Closteroviridae revised. Arch Virol. 2002;147:2039–2044. doi: 10.1007/s007050200048. [DOI] [PubMed] [Google Scholar]
- Matic S, Minafra A, Sánchez-Navarro JA, Pallás V, Myrta A, Martelli GP. ‘Kwanzan Stunting’ syndrome: detection and molecular characterization of an Italian isolate of Little cherry virus 1. Virus Res. 2009;143:61–67. doi: 10.1016/j.virusres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Melzer MJ, Sether DM, Borth WB, Mersino EF, Hu JS. An assemblage of closteroviruses infects Hawaiian ti (Cordyline fruticosa L.) Virus Genes. 2011;42:254–260. doi: 10.1007/s11262-010-0537-9. [DOI] [PubMed] [Google Scholar]
- Menzel W, Goetz R, Lesemann DE, Vetten HJ. Molecular characterization of a closterovirus from carrot. Arch Virol. 2009;154:1343–1347. doi: 10.1007/s00705-009-0428-3. [DOI] [PubMed] [Google Scholar]
- Mikona C, Turturo C, Navarro B, Menzel W, Minafra A, Rott ME, Martelli GP, Jelkmann W. Taxonomy, complete nucleotide sequence and genome organization of grapevine leafroll-associated virus–7. Proceedings of XVI Meet. Int. Counc. Study of Virus-like Diseases of the Grapevine (ICVG); Dijon, France. 2009. p. 275. [Google Scholar]
- Morales RZ, Monis J. First detection of Grapevine leafroll associated virus-7 in California vineyards. Plant Dis. 2007;91:465–469. doi: 10.1094/PDIS-91-4-0465B. [DOI] [PubMed] [Google Scholar]
- Ng JCK, Falk BW. Virus–vector interactions mediating nonpersistent and semipersistent transmission of plant viruses. Ann Rev Phytopathol. 2006;44:183–212. doi: 10.1146/annurev.phyto.44.070505.143325. [DOI] [PubMed] [Google Scholar]
- Peremyslov VV, Dolja VV. Identification of the subgenomic mRNAs that encode 6-kDa movement protein and Hsp70 homolog of beet yellows virus. Virology. 2002;295:299–306. doi: 10.1006/viro.2002.1396. [DOI] [PubMed] [Google Scholar]
- Peremyslov VV, Pan YW, Dolja VV. Movement protein of a closterovirus is a type III integral transmembrane protein localized to the endoplasmic retic-ulum. J Virol. 2004;78:3704–3709. doi: 10.1128/JVI.78.7.3704-3709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC, Kasschau KD, Prokhnevsky AI, Gopinath K, Pogue GP, Carrington JC, Dolja VV. Suppressor of RNA silencing encoded by beet yellows virus. Virology. 2003;306:203–209. doi: 10.1016/s0042-6822(02)00051-x. [DOI] [PubMed] [Google Scholar]
- Rowhani A, Uyemoto JK, Golino DA, Martelli GP. Pathogen testing and certification of Vitis and Prunus species. Ann Rev Phytopathol. 2005;43:261–278. doi: 10.1146/annurev.phyto.43.040204.135919. [DOI] [PubMed] [Google Scholar]
- Routh G, Zhang YP, Saldarelli P, Rowhani A. Use of degenerate primers for partial sequencing and RT-PCR-based assays of grapevine leafroll-associated viruses 4 and 5. Phytopathology. 1998;88:1238–1243. doi: 10.1094/PHYTO.1998.88.11.1238. [DOI] [PubMed] [Google Scholar]
- Saldarelli P, Rowhani A, Routh G, Minafra A, Digiaro M. Use of degenerate primersina RT-PCR assay for the identification and analysis of some filamentous viruses, with special reference to Clostero- and Vitiviruses of the Grapevine. Eur J Plant Pathol. 1998;104:945–950. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Bibsoul TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Fuchs M, Perry KL. Genomic analysis of Grapevine leafroll associated virus-5 and related viruses. Virus Res. 2011 Aug 30; doi: 10.1016/j.virusres.2011.08.006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Turturo C, Rott ME, Minafra A, Saldarelli P, Jelkmann W, Martelli GP. Partial molecular characterization and RT-PCR detection of grapevine leafroll associated virus 7. In the Proceedings of the 13 Meetings of the International Council for the Study of Viruses and Virus-Like Diseases of the Grapevine; March 12-17; Adelaide, Australia. 2000. pp. 17–18. [Google Scholar]
- Tzanetakis IE, Martin RR. Strawberry chlorotic fleck: Identification and characterization of a novel Closterovirus associated with the disease. Virus Res. 2007;124:88–94. doi: 10.1016/j.virusres.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Tzanetakis IE, Postman J, Martin RR. Characterization of a novel member of the family Closteroviridae from Mentha spp. Phytopathology. 2005;95:1043–1048. doi: 10.1094/PHYTO-95-1043. [DOI] [PubMed] [Google Scholar]
- Tzanetakis IE, Wintermantel WM, Poudel B, Zhou J. Diodia vein chlorosis virus is a group-1 crinivirus. Arch Virol. 2011 doi: 10.1007/s00705-011-1055-3. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- Wang J, Sharma AM, Duffy S, Almeida RP. Genetic diversity in the 3′ terminal 4.7-kb region of grapevine leafroll-associated virus 3. Phytopathology. 2011;101:445–450. doi: 10.1094/PHYTO-07-10-0173. [DOI] [PubMed] [Google Scholar]
- Zhu HY, Ling KS, Goszczynski DE, McFerson JR, Gonsalves D. Nucleotide sequence and genome organization of grapevine leafroll-associated virus-2 are similar to beet yellows virus, the closterovirus type member. J Gen Virol. 1998;79:1289–1298. doi: 10.1099/0022-1317-79-5-1289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.