

Abstract
Although hereditary kidney cancer syndrome accounts for approximately five percent of all kidney cancers, the mechanistic insight into tumor development in these rare conditions has provided the foundation for the development of molecular targeting agents currently used for sporadic kidney cancer. In the late 1980s, the comprehensive study for hereditary kidney cancer syndrome was launched in the National Cancer Institute, USA and the first kidney cancer‐associated gene, VHL, was identified through kindred analysis of von Hippel‐Lindau (VHL) syndrome in 1993. Subsequent molecular studies on VHL function have elucidated that the VHL protein is a component of E3 ubiquitin ligase complex for hypoxia‐inducible factor (HIF), which provided the basis for the development of tyrosine kinase inhibitors targeting the HIF‐VEGF/PDGF pathway. Recent whole‐exome sequencing analysis of sporadic kidney cancer exhibited the recurrent mutations in chromatin remodeling genes and the later study has revealed that several chromatin remodeling genes are altered in kidney cancer kindred at the germline level. To date, more than 10 hereditary kidney cancer syndromes together with each responsible gene have been characterized and most of the causative genes for these genetic disorders are associated with either metabolism or epigenome regulation. In this review article, we describe the molecular mechanisms of how an alteration of each kidney cancer‐associated gene leads to renal tumorigenesis as well as denote therapeutic targets elicited by studies on hereditary kidney cancer.
Keywords: Birt‐Hogg‐Dubé syndrome, cancer metabolism, epigenome regulation, hereditary leiomyomatosis renal cell cancer, von Hippel‐Lindau syndrome
Abbreviations
- AML
angiomyolipoma
- AMPK
5′‐AMP‐activated protein kinase
- BHD syndrome
Birt‐Hogg‐Dubé syndrome
- CA‐IX
carbonic anhydrase IX
- CIMP
CpG island methylator phenotype
- DENN
differentially expressed in neoplastic vs normal cells
- FH
fumarate hydratase
- FLCN
folliculin
- FNIP1 and FNIP2
folliculin‐interacting proteins 1 and 2
- GAP
GTPase‐activating protein
- GEF
guanine nucleotide exchange factor
- HGF
hepatocyte growth factor
- HIF
hypoxia inducible factor
- HLRCC
hereditary leiomyomatosis and renal cell cancer
- HMOX1
haem oxygenase 1
- HOCT
hybrid oncocytic/chromophobe tumor
- HPRCC
hereditary papillary renal cell carcinoma
- KEAP1
kelch‐like ECH‐associated protein 1
- LAM
lymphangioleiomyomatosis
- MEF
mouse embryonic fibroblast
- mTOR
mammalian target of rapamycin
- PDGF
platelet‐derived growth factor
- PHD
prolyl‐hydroxylase
- RCC
renal cell carcinoma
- SDHB, SDHC and SDHD
succinate dehydrogenase B,C and D
- SEGA
subependymal giant cell astrocytoma
- TCEB1
transcription elongation factor B polypeptide 1
- TCGA
the Cancer Genome Atlas
- TGF‐α
transforming growth factor alpha
- TKI
tyrosine kinase inhibitor
- TSC
tuberous sclerosis
- VEGF
vascular endothelial growth factor
- VHL syndrome
von Hippel‐Lindau syndrome
1. VON HIPPEL‐LINDAU (VHL) SYNDROME
von Hippel‐Lindau (VHL) syndrome is a rare hereditary neoplastic syndrome, which predisposes patients to develop retinal angioma, hemangioblastoma of the central nervous system, pheochromocytoma, pancreatic cystadenoma and neuroendocrine tumor, and clear cell renal cell carcinoma (RCC) (Figure 1). The gene responsible for the disease, located at chromosome 3p25.3, was identified as VHL tumor suppressor by positional cloning method in 1993.1, 2 Subsequent molecular studies have shown that VHL is a component of the E3 ubiquitin ligase complex which specifically recognizes HIF protein for degradation through the ubiquitin proteasome pathway; therefore, VHL alteration leads to the accumulation of HIF as well as increased transcription of its downstream genes, VEGF, PDGF and TGF‐α, which promote tumor progression.3 In 2013, independent research groups of The Cancer Genome Atlas (TCGA) project and in the University of Tokyo conducted whole‐exosome sequencing of sporadic clear cell RCC using next‐generation sequencing technology and elucidated that nearly 90% of sporadic clear cell RCC harbors alterations in VHL itself or in TCEB1, a component of the VHL complex.4, 5 These findings have provided robust evidence for using antiangiogenic agents or tyrosine kinase inhibitors (TKIs), including bevacizumab, sorafenib, sunitinib, axitinib and pazopanib, which target the VHL‐HIF‐VEGF/PDGF pathway as standardized therapeutics for sporadic RCC. However, in addition to VEGF/PDGF, HIF transcriptionally regulates a variety of genes, including cyclin D1, glut1 and CA‐IX etc. Thus, this partial inhibition of HIF downstream genes may limit the efficacy of TKI for RCC treatment.6 In this notion, HIF2α antagonist has been developed and its efficacy is under investigation.7
Figure 1.
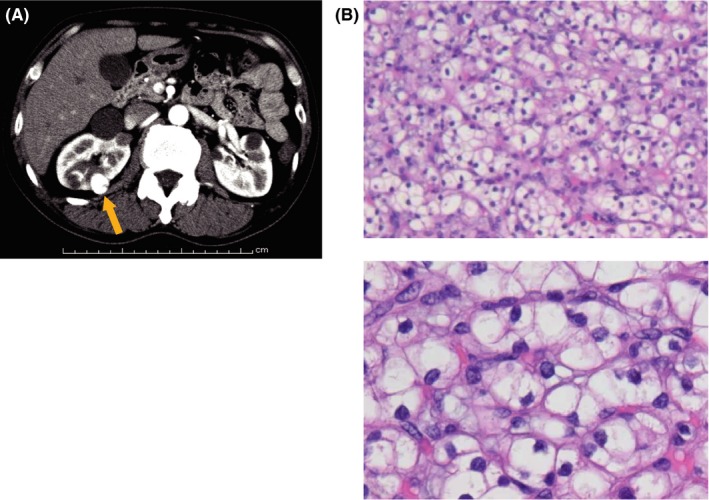
von Hippel‐Lindau (VHL) syndrome‐associated kidney cancer. A, Computed tomography with contrast material of VHL patient shows hypervascular tumor in the right kidney (orange arrow) and multiple cysts in both kidneys. Partial nephrectomy was done to the right kidney. B, Four out of 5 tumors and 1 out of 4 cyst walls exhibited the histology of clear cell renal cell carcinoma. Upper panel shows low magnification and lower panel shows high magnification of H&E staining
2. BIRT‐HOGG‐DUBÉ’ (BHD) SYNDROME
Birt‐Hogg‐Dubé (BHD) syndrome is a rare genetic disorder that causes development of lung cysts, fibrofolliculomas, and renal tumors with various histological subtypes, including chromophobe RCC, hybrid oncocytic/chromophobe tumor (HOCT), clear cell RCC, papillary RCC, and oncocytoma8, 9, 10, 11, 12 (Figure 2). In 2002, the responsible gene, FLCN was identified and the majority of germline FLCN mutations were either nonsense mutations or frameshift mutations with a few exceptions of missense mutations, including H255Y and K508R.13, 14, 15 Folliculin (FLCN) binds to its two interacting partners, folliculin‐interacting protein 1 and 2 (FNIP1 and FNIP2), and senses energy through the interaction between FNIPs and 5′AMP‐activated protein kinase (AMPK), an important energy‐sensing molecule.16, 17, 18, 19 Disruption of FLCN‐FNIPs interaction drives upregulated mTORC1‐dependent protein synthesis, upregulated PGC1α‐dependent mitochondrial oxidative metabolism and aberrant kidney cell proliferation.20, 21, 22, 23, 24 Crystallography of FLCN protein exhibited that FLCN has a DENN domain in its C‐terminus, suggesting FLCN may act as a modifier for Rab small GTPase family as well as a regulator for membranous trafficking.25, 26 In addition, FLCN shows either GAP activity towards RagC/D GTPases or GEF activity towards RagA/B GTPases, which consequently regulates mTORC1 localization on lysosomes, implying that FLCN may regulate multiple small GTPases.27, 28 These findings highlight that FLCN plays important roles in metabolism, and disruption of metabolism under FLCN deficiency may drive aberrant kidney cell proliferation. Kidney‐specific Flcn knockout mouse develops hyperproliferative polycystic kidney. However, this mouse model dies at 3 weeks of age as a result of renal failure before developing kidney cancer.23 Therefore, it is suggested that an additional mutation may be necessary for developing kidney cancer in cooperation with FLCN deficiency.
Figure 2.
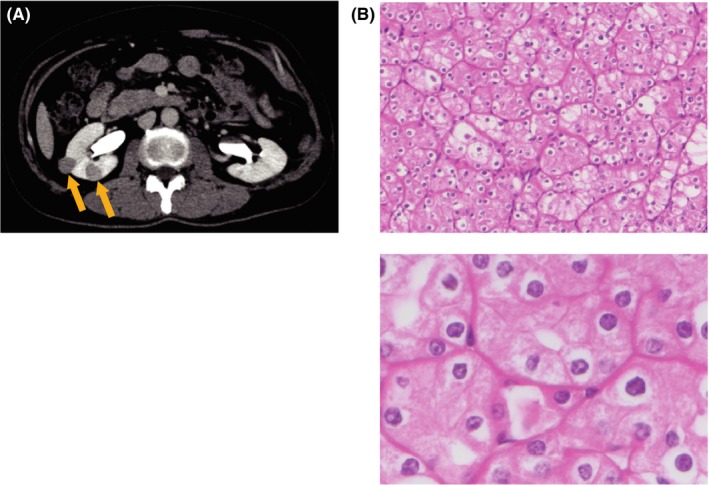
Birt‐Hogg‐Dubé (BHD) syndrome‐associated kidney cancer. A, Computed tomography with contrast material of BHD patient shows weakly stained tumors in the right kidney (orange arrows). Partial nephrectomy was done to the right kidney. B, H&E stain shows the most predominant forms of BHD‐associated kidney cancer, hybrid oncocytic/chromophobe tumors (HOCT). Low magnification (upper panel) and high magnification (lower panel). Figures are from Hasumi et al8
3. HEREDITARY LEIOMYOMATOSIS AND RENAL CELL CANCER (HLRCC)
Hereditary leiomyomatosis and renal cell cancer (HLRCC) predisposes patients to develop leiomyomatosis of skin and uterus with high frequency as well as type 2 papillary RCC in 10%‐16% of the affected patients, which presents a very aggressive behavior and metastasizes even from a small‐sized tumor, leading to very poor prognosis.29 In 2002, FH was identified as a causative gene for HLRCC.30 Alteration of FH drives the metabolic shift towards glycolysis as well as the accumulation of fumarate, an oncometabolite which inhibits α‐ketoglutarate‐dependent enzymes, including PHD and DNA demethylases, leading to HIF accumulation or genome‐wide methylated status called CpG island methylator phenotype (CIMP).31, 32, 33, 34 In FH‐deficient cells, KEAP1, E3 ubiquitin ligase for Nrf2 antioxidant transcription factor, is inactivated by its succinylated residues, leading to Nrf2 accumulation and resistance of FH‐deficient cells to reactive oxygen species.35 In fact, an inhibitor for HMOX1, a downstream target of Nrf2, suppressed cell proliferation of Fh‐deficient mouse embryonic fibroblasts (MEFs).36
4. HEREDITARY PARAGANGLIOMA‐PHEOCHROMOCYTOMA SYNDROME
Germline mutations in SDHB, SDHC, and SDHD, genes responsible for hereditary paraganglioma‐pheochromocytoma syndrome, cause the development of kidney cancer .37 Alteration of SDH leads to the metabolic shift towards glycolysis as well as to the accumulation of succinate, which drives tumor progression in the same way as does the accumulation of fumarate in FH‐deficient kidney cells.38, 39
5. HEREDITARY PAPILLARY RENAL CELL CARCINOMA (HPRCC)
Hereditary papillary renal cell carcinoma (HPRCC) is a very rare type of hereditary kidney cancer syndrome compared to VHL syndrome, BHD syndrome and HLRCC, and predisposes patients to develop bilateral type 1 papillary RCC. In 1997, activating mutation of MET was identified as a responsible genetic alteration. c‐MET, encoded by the MET gene is a tyrosine kinase receptor for HGF and the constitutive active form of c‐MET drives kidney cell proliferation.40, 41, 42 Whole‐exosome sequencing of sporadic kidney cancer showed alterations in the c‐MET/HGF pathway in 12% of clear cell RCC and in 10% of papillary RCC, indicating that targeting the c‐MET/HGF pathway is rational for the treatment of these histological types of kidney cancer and, in fact, the efficacy of cabozantinib which targets both c‐MET and VEGFR has been reported.43, 44
6. COWDEN SYNDROME
Cowden syndrome predisposes patients to develop intestinal hamartomatous polyps, benign skin tumors and macrocephaly. Patients are also at risk of malignancies in breast, thyroid, uterus and prostate, and 4%‐16% of patients develop kidney cancer with various types of histology, including papillary, chromophobe, and clear cell RCC.45 Alteration of PTEN, a causative gene for Cowden syndrome, drives activation of the PI3K‐AKT‐mTOR pathway.
7. TUBEROUS SCLEROSIS (TSC)
Tuberous sclerosis (TSC), a hamartoma syndrome with a triad of facial angiofibromas, seizure and developmental delay, predisposes patients to develop subepedymal giant cell astrocytoma (SEGA), angiomyolipoma (AML) in kidney, lymphangioleiomyomatosis (LAM) in lung and kidney cancer in 3% of affected patients. TSC1 and TSC2 have been identified as causative genes for TSC.46 TSC2 is a GTPase activating protein for Rheb GTPase whereas TSC1 regulates stability of TSC2 protein; either TSC1 or TSC2 mutation increases GTP‐bound Rheb GTPase, leading to constitutive activation of mTORC1 complex.47 Targeted next‐generation sequencing analysis of TSC‐associated kidney cancer demonstrated a relatively small number of somatic mutations in addition to TSC1/2 mutations, suggesting that mutations in TSC1/2 themselves may be strong driver mutations.46
8. CHROMOSOME 3P TRANSLOCATION‐ASSOCIATED KIDNEY CANCER SYNDROME
While sporadic clear cell RCC frequently harbors a large chromosomal deletion at chromosome 3p, hereditary kidney cancer with germline chromosomal 3p translocation has been reported.48 Chromosomal rearrangement involving chromosome 3p leads to loss of multiple kidney cancer‐associated genes including VHL, BAP1, PBRM1 and SETD2. Single inactivation of either Vhl, Bap1 or Pbrm1 does not cause development of kidney cancer, whereas double inactivation of Vhl/Bap1 or Vhl/Pbrm1 does cause development of kidney cancer, indicating that a large chromosomal deletion involving this locus is a critical event triggering renal tumorigenesis.49, 50
9. BAP1 CANCER SUSCEPTIBILITY SYNDROME
One of the biggest findings in whole‐exome sequencing for sporadic kidney cancer using next‐generation sequencing technology are the recurrent alterations in chromatin remodeling genes in clear cell and papillary RCC. Among these alterations, BAP1 mutations were found in 15% of sporadic RCC.4, 5 BAP1 mutation is a critical driver for renal tumorigenesis as double inactivation of murine Vhl and Bap1 develops malignant lesions in mouse kidney.50 Interestingly, a later study on hereditary kidney cancer reported that a germline BAP1 mutation was found in kidney cancer kindred.51 BAP1 is a tumor suppressor for multiple organs and germline BAP1 mutation drives malignant mesothelioma and malignant melanoma in uvea and skin. BAP1 deubiquitinates histone H2A at K119 and chromatin immunoprecipitation and DNA sequencing (ChIP‐seq) for BAP1 protein showed that significant BAP1 peaks locate near the transcription start sites of 5731 genes which may include the targets for BAP1‐deficient kidney cancer.52
10. OTHER HEREDITARY KIDNEY CANCERS
Germline PBRM1 mutation has been reported in a kindred of kidney cancer.53 PBRM1 remodels chromatin structure as well as regulates other tumor suppressors through its bromodomain interaction with acetylated lysine in histone H3 at K14 or in tumor suppressor proteins.54 PBRM1 mutation is an important driver mutation for kidney cancer development as its alteration was found in 40% of sporadic RCC, and double inactivation of murine Vhl and Pbrm1 causes development of kidney cancer in mouse.4, 5, 49 Additionally, germline CDKN2B mutation was found in kidney cancer kindred.55 Thus, a subset of genes found to be altered in sporadic kidney cancer by next‐generation sequencing analysis may be candidates for causative genes of hereditary kidney cancer. In addition, kindred with multiple germline mutations in cancer‐associated genes have been reported: neurofibromatosis type I with BHD syndrome, Li‐Fraumeni syndrome with BHD syndrome and Lynch syndrome with BHD syndrome. In these kindred, symptoms that are not observed in each syndrome were observed when the two syndromes occurred together, suggesting that we have to treat these patients with precautions.56
11. CONCLUSION
Although hereditary kidney cancer accounts for approximately five percent of all kidney cancers, mechanistic insight into tumorigenesis of these rare genetic disorders has provided the basis for the development of novel therapeutics for sporadic kidney cancer. Recent genome‐wide analysis on sporadic kidney cancer using next‐generation sequencing technology has further identified novel kidney cancer‐associated genes and later studies showed that some of these genes are altered in kidney cancer kindred at the germline level. Thus, to sort out driver mutations of kidney cancer, it is important to integrate data of genome‐wide analysis on sporadic kidney cancer with germline genomic data of patients with hereditary kidney cancer. Notably, most of the kidney cancer‐associated genes have roles in either metabolism or chromatin remodeling, suggesting that disruption of metabolism, dysregulation of chromatin remodeling, or loss of crosstalk between metabolism and the epigenome may drive renal tumorigenesis (Figure 3). In conclusion, understanding the metabolic and epigenetic abnormalities underlying deficiencies of kidney cancer‐associated genes may lead to the development of novel diagnostic biomarkers, diagnostic imaging modalities and novel therapeutics for kidney cancer.
Figure 3.
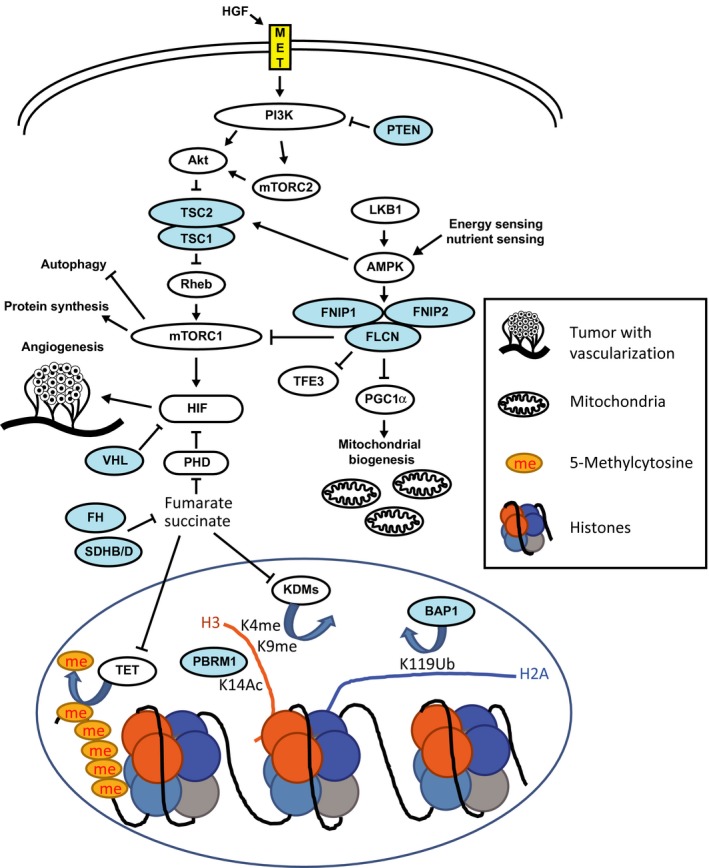
Hereditary kidney cancer‐associated genes. Blue shows tumor suppressor and yellow shows oncogene. PHD, prolyl hydroxylase; KDMs, lysine demethylases; TET, Ten‐eleven translocation methylcytosine dioxygenase
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
We acknowledge Dr Mitsuko Furuya and Dr Yoji Nagashima for the critical assessment on the histology of kidney cancer.
Hasumi H, Yao M. Hereditary kidney cancer syndromes: Genetic disorders driven by alterations in metabolism and epigenome regulation. Cancer Sci. 2018;109:581–586. https://doi.org/10.1111/cas.13503
Funding information
The Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT)/Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant/Award Number: ‘n/a’), JSPS KAKENHI Grant (Grant/Award Number: ‘16K11020’).
REFERENCES
- 1. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel‐Lindau disease tumor suppressor gene. Science. 1993;260:1317‐1320. [DOI] [PubMed] [Google Scholar]
- 2. Yao M, Latif F, Orcutt ML, et al. von Hippel‐Lindau disease: identification of deletion mutations by pulsed‐field gel electrophoresis. Hum Genet. 1993;92:605‐614. [DOI] [PubMed] [Google Scholar]
- 3. Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel‐Lindau protein. Cancer Cell. 2002;1:237‐246. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear‐cell renal cell carcinoma. Nat Genet. 2013;45:860‐867. [DOI] [PubMed] [Google Scholar]
- 6. Manalo DJ, Rowan A, Lavoie T, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF‐1. Blood. 2005;105:659‐669. [DOI] [PubMed] [Google Scholar]
- 7. Ricketts CJ, Crooks DR, Linehan WM. Targeting HIF2alpha in clear‐cell renal cell carcinoma. Cancer Cell. 2016;30:515‐517. [DOI] [PubMed] [Google Scholar]
- 8. Hasumi H, Baba M, Hasumi Y, Furuya M, Yao M. Birt‐Hogg‐Dube syndrome: clinical and molecular aspects of recently identified kidney cancer syndrome. Int J Urol. 2016;23:204‐210. [DOI] [PubMed] [Google Scholar]
- 9. Schmidt LS, Linehan WM. FLCN: the causative gene for Birt‐Hogg‐Dube syndrome. Gene. 2018;640:28‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kato I, Iribe Y, Nagashima Y, et al. Fluorescent and chromogenic in situ hybridization of CEN17q as a potent useful diagnostic marker for Birt‐Hogg‐Dube syndrome‐associated chromophobe renal cell carcinomas. Hum Pathol. 2016;52:74‐82. [DOI] [PubMed] [Google Scholar]
- 11. Furuya M, Hasumi H, Baba M, et al. Establishment and characterization of BHD‐F59RSVT, an immortalized cell line derived from a renal cell carcinoma in a patient with Birt‐Hogg‐Dube syndrome. Lab Invest. 2017;97:343‐351. [DOI] [PubMed] [Google Scholar]
- 12. Furuya M, Yao M, Tanaka R, et al. Genetic, Epidemiologic and Clinicopathologic Studies of Japanese Asian Patients with Birt‐Hogg‐Dube Syndrome. Clin Genet. 2016;90:403‐412. [DOI] [PubMed] [Google Scholar]
- 13. Hasumi Y, Baba M, Ajima R, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA. 2009;106:18722‐18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt‐Hogg‐Dube syndrome. Cancer Cell. 2002;2:157‐164. [DOI] [PubMed] [Google Scholar]
- 15. Hasumi H, Hasumi Y, Baba M, et al. H255Y and K508R missense mutations in tumour suppressor folliculin (FLCN) promote kidney cell proliferation. Hum Mol Genet. 2017;26:354‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baba M, Hong SB, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. 2006;103:15552‐15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hasumi H, Baba M, Hong SB, et al. Identification and characterization of a novel folliculin‐interacting protein FNIP2. Gene. 2008;415:60‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baba M, Keller JR, Sun HW, et al. The folliculin‐FNIP1 pathway deleted in human Birt‐Hogg‐Dube syndrome is required for murine B‐cell development. Blood. 2012;120:1254‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takagi Y, Kobayashi T, Shiono M, et al. Interaction of folliculin (Birt‐Hogg‐Dube gene product) with a novel Fnip1‐like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339‐5347. [DOI] [PubMed] [Google Scholar]
- 20. Baba M, Toyama H, Sun L, et al. Loss of folliculin disrupts hematopoietic stem cell quiescence and homeostasis resulting in bone marrow failure. Stem Cells. 2016;34:1068‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hasumi Y, Baba M, Hasumi H, et al. Folliculin (Flcn) inactivation leads to murine cardiac hypertrophy through mTORC1 deregulation. Hum Mol Genet. 2014;23:5706‐5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasumi H, Baba M, Hasumi Y, et al. Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J Natl Cancer Inst. 2012;104:1750‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baba M, Furihata M, Hong SB, et al. Kidney‐targeted Birt‐Hogg‐Dube gene inactivation in a mouse model: Erk1/2 and Akt‐mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hasumi H, Baba M, Hasumi Y, et al. Folliculin‐interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc Natl Acad Sci USA. 2015;112:E1624‐E1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laviolette LA, Mermoud J, Calvo IA, et al. Negative regulation of EGFR signalling by the human folliculin tumour suppressor protein. Nat Commun. 2017;8:15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nookala RK, Langemeyer L, Pacitto A, et al. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsun ZY, Bar‐Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petit CS, Roczniak‐Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid‐dependent activation of Rag GTPases. J Cell Biol. 2013;202:1107‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vocke CD, Ricketts CJ, Merino MJ, et al. Comprehensive genomic and phenotypic characterization of germline FH deletion in hereditary leiomyomatosis and renal cell carcinoma. Genes Chromosom Cancer. 2017;56:484‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406‐410. [DOI] [PubMed] [Google Scholar]
- 31. Tong WH, Sourbier C, Kovtunovych G, et al. The glycolytic shift in fumarate‐hydratase‐deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143‐153. [DOI] [PubMed] [Google Scholar]
- 33. Xiao M, Yang H, Xu W, et al. Inhibition of alpha‐KG‐dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research Network , Linehan WM, Spellman PT, et al. Comprehensive molecular characterization of papillary renal‐cell carcinoma. N Engl J Med. 2016;374:135‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adam J, Hatipoglu E, O'Flaherty L, et al. Renal cyst formation in Fh1‐deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frezza C, Zheng L, Folger O, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225‐228. [DOI] [PubMed] [Google Scholar]
- 37. Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype‐phenotype‐proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41‐51. [DOI] [PubMed] [Google Scholar]
- 38. Saxena N, Maio N, Crooks DR, et al. SDHB‐deficient cancers: the role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016;108 https://doi.org/10.1093/jnci/djv287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739‐752. [DOI] [PubMed] [Google Scholar]
- 40. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto‐oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68‐73. [DOI] [PubMed] [Google Scholar]
- 41. Haas NB, Nathanson KL. Hereditary kidney cancer syndromes. Adv Chronic Kidney Dis. 2014;21:81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakaigawa N, Yao M, Baba M, et al. Inactivation of von Hippel‐Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006;66:3699‐3705. [DOI] [PubMed] [Google Scholar]
- 43. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal‐cell carcinoma. N Engl J Med. 2015;373:1814‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xie Z, Lee YH, Boeke M, et al. MET inhibition in clear cell renal cell carcinoma. J Cancer. 2016;7:1205‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shuch B, Ricketts CJ, Vocke CD, et al. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. 2013;190:1990‐1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tyburczy ME, Jozwiak S, Malinowska IA, et al. A shower of second hit events as the cause of multifocal renal cell carcinoma in tuberous sclerosis complex. Hum Mol Genet. 2015;24:1836‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577‐590. [DOI] [PubMed] [Google Scholar]
- 48. Foster RE, Abdulrahman M, Morris MR, et al. Characterization of a 3;6 translocation associated with renal cell carcinoma. Genes Chromosom Cancer. 2007;46:311‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nargund AM, Pham CG, Dong Y, et al. The SWI/SNF protein PBRM1 restrains VHL‐loss‐driven clear cell renal cell carcinoma. Cell Rep. 2017;18:2893‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang SS, Gu YF, Wolff N, et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci USA. 2014;111:16538‐16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farley MN, Schmidt LS, Mester JL, et al. A novel germline mutation in BAP1 predisposes to familial clear‐cell renal cell carcinoma. Mol Cancer Res. 2013;11:1061‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dey A, Seshasayee D, Noubade R, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337:1541‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Benusiglio PR, Couve S, Gilbert‐Dussardier B, et al. A germline mutation in PBRM1 predisposes to renal cell carcinoma. J Med Genet. 2015;52:426‐430. [DOI] [PubMed] [Google Scholar]
- 54. Liao L, Testa JR, Yang H. The roles of chromatin‐remodelers and epigenetic modifiers in kidney cancer. Cancer Genet. 2015;208:206‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jafri M, Wake NC, Ascher DB, et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015;5:723‐729. [DOI] [PubMed] [Google Scholar]
- 56. Whitworth J, Skytte AB, Sunde L, et al. Multilocus inherited Neoplasia Alleles syndrome: a case series and review. JAMA Oncol. 2016;2:373‐379. [DOI] [PubMed] [Google Scholar]