

Abstract
The Protein Data Bank (PDB) is the global archive for structural information on macromolecules, and a popular resource for researchers, teachers, and students, amassing more than one million unique users each year. Crystallographic structure models in the PDB (more than 100,000 entries) are optimized against the crystal diffraction data and geometrical restraints. This process of crystallographic refinement typically ignored hydrogen bond (H‐bond) distances as a source of information. However, H‐bond restraints can improve structures at low resolution where diffraction data are limited. To improve low‐resolution structure refinement, we present methods for deriving H‐bond information either globally from well‐refined high‐resolution structures from the PDB‐REDO databank, or specifically from on‐the‐fly constructed sets of homologous high‐resolution structures. Refinement incorporating HOmology DErived Restraints (HODER), improves geometrical quality and the fit to the diffraction data for many low‐resolution structures. To make these improvements readily available to the general public, we applied our new algorithms to all crystallographic structures in the PDB: using massively parallel computing, we constructed a new instance of the PDB‐REDO databank (https://pdb-redo.eu). This resource is useful for researchers to gain insight on individual structures, on specific protein families (as we demonstrate with examples), and on general features of protein structure using data mining approaches on a uniformly treated dataset.
Keywords: hydrogen bonds, homology, restraints, X‐ray crystallography, refinement, PDB, PDB‐REDO, databank, high‐throughput computing
Introduction
Crystallographic structure models are optimized against the crystallographic diffraction data and a priori known geometrical targets, the geometrical restraints. In any crystallographic refinement procedure, low‐resolution diffraction data means that fewer observations of diffracted X‐ray intensities are available, and as resolution declines the crystallographic refinement problem becomes increasingly underdetermined.1 Restraint dictionaries2, 3 describing “ideal” refinement targets for bond lengths, angles, planar groups, and other well‐defined stereochemical features, at low resolution become gradually insufficient to yield high‐quality structure models. Additional, external restraints4 can be defined and, for example, hydrogen bond restraints5, 6 (H‐bonds), and Ramachandran torsion angle restraints5, 7 have been used to enhance protein secondary structure quality, particularly at lower resolution.
Macromolecular crystals diffract X‐rays to higher or lower resolution in an unpredictable manner: even very similar proteins or the same protein bound to different ligands (e.g., drug candidates), can yield crystallographic data at very different resolutions. This allows refinement methods to harvest information from a high‐resolution “reference” model and use it to refine low‐resolution models.5, 6, 8, 9, 10, 11 Available implementations of this principle focus on harvesting restraints from a single external reference structure model of high quality, and transferring that information to the low‐resolution structure under refinement. Thus the crystallographer is faced with the often difficult and inevitably subjective decision of selecting the “best” model from a group of protein structure models as a reference.12 Recently, this process was partly automated in the LORESTR pipeline,13 which uses a series of different refinement protocols and reference restraints from ProSMART,6, 8 to ultimately return the best result using restraints from the optimal reference model.
A set of reference models consisting of many available homologous higher resolution structures would take conformational flexibility implicitly into account and may therefore help obtaining a better measure for the variation of certain distances, while idiosyncrasies of a single reference model will not cause bad restraint targets. However, heterogeneity in the reference data (e.g., multiple conformational states of a protein) will often be present in the structure ensemble. Therefore, flexibility toward local dissimilarities between the homologs is required. Such flexibility can be achieved by focusing on real interactions such as hydrogen bonds instead of distances or angles that do not represent chemical bonds or interactions. H‐bond networks are well conserved between homologous proteins,14 and if a specific H‐bond is not, inspection of the molecular geometry reveals this immediately. In addition, H‐bonds are omnipresent in proteins: more than 90% of all main‐chain donors and acceptors are involved in at least one H‐bond and side‐chain donors and acceptors make more than one H‐bond on average.15 Main‐chain H‐bonds form the secondary structure elements,14 and have been restrained in low‐resolution refinement before.5, 6 H‐bonds that involve side‐chains describe the tertiary and quaternary structure of a protein and are therefore more informative about the specific molecular details of a protein.
We are developing the PDB‐REDO procedure that rerefines and rebuilds macromolecular structures before16 and after17 they are submitted to the PDB. Here, methods are presented that improve the PDB‐REDO pipeline and low‐resolution refinement in general. We have developed a system that employs H‐bond restraints to improve the geometry of low‐resolution structure models. First, we optimize targets for H‐bond restraints based on global high‐resolution structure data from the PDB‐REDO databank, and show that these restraints improve protein structure models. Then, we describe how restraint targets can be redefined based on homologous structure data and how both global and homology‐based H‐bond restraints are implemented in the PDB‐REDO pipeline. Subsequently we apply our HOmology DErived Restraints (HODER) to the entire PDB data bank, using a highly parallel computational architecture that allowed 60 CPU years of computation to be performed in about a week, allowing a new resource (https://pdb-redo.eu /) to be made publically available. Finally, we present examples of the information that can be derived from this novel resource, and how this can help scientists gain a better understanding of protein structure.
Results
Derivation, application, and validation of general H‐bond restraints
We based the detection of H‐bonds on the geometrical criteria defined by McDonald and Thornton,15 which were slightly loosened to obtain a complete H‐bond set (Supporting Information, Fig. S1). This set is then subjected to numerous filters to finally arrive at a concise set of high‐quality H‐bonds that will be restrained. For example, we check each main‐chain H‐bond against secondary structure information derived from DSSP, the de facto standard for secondary structure assignment,18 and donors are not allowed to donate more H‐bonds than the number of hydrogen atoms that are bound to them. Not all H‐bonds are equal: the distance between the donor and acceptor atom differs between different secondary structure elements and different types of side‐chain H‐bonds. Therefore, we derived specific targets for each H‐bond type from high‐quality structural data from PDB‐REDO17 models with a resolution ≤1.8 Å and an R free ≤ 0.20, 10,173 entries in total. H‐bonds were detected in all these entries and separated per category. Main‐chain H‐bonds were separated in six secondary structure categories (α‐helix, π‐helix, 310‐helix, antiparallel β‐strand, parallel β‐strand, and others) based on the assignments in DSSP. Side‐chain H‐bonds were divided into categories where all H‐bonds have the same donor and acceptor type. Hence, for example, one category contains all Lys‐Nζ to Gln‐Oɛ H‐bonds. The full procedure is detailed in the Supporting Information.
We detected approximately two million main‐chain H‐bonds and two million side‐chain H‐bonds, which were used to derive a target for each H‐bond type. The observed H‐bond‐length distributions were modeled with a two‐sided normal distribution to obtain ideal target values (see Supporting Information). Main‐chain targets vary between 2.86 Å and 2.98 Å for different secondary structure elements (Supporting Information, Table S1) and side‐chain H‐bonds between 2.60 Å and 3.36 Å for different types (Supporting Information, Table S2). Notably, H‐bond restraints previously incorporated into ProSMART/Refmac58 and Phenix5 use a single distance target for all hydrogen bonds they restrain (2.8 Å for Refmac5 and 2.9Å for Phenix); the secondary structure and atom pair‐dependent mining of hydrogen bonds here, brings a more accurate target function into play.
Defining the weight of H‐bond restraints against other restraints during crystallographic refinement is key. This weight was optimized based on the premise that high‐resolution structures accurately reflect hydrogen bonding in proteins. Hence, the distribution of H‐bond distances was evaluated for the same set of high‐quality PDB‐REDO models used to derive the targets and also for PDB‐REDO entries with a resolution ≥2.5 Å. The restraint weight was optimized selecting a value that transformed the H‐bond length distribution of the low‐resolution set to become most similar to that of the high‐resolution set after refinement (Supporting Information, Fig. S2).
The effect of the H‐bond restraints was initially evaluated by running refinements with and without restraints: the effect of H‐bond restraints was greater at lower resolution, while at resolution better than 2.5 Å, the effect of H‐bond restraints was negligible. We thus constructed a test set containing 155 low‐resolution entries (for details, see Supporting Information) and proceeded by validating the effect of our method in refinement.
H‐bond restraints on the basis of general targets improve the refinement of the test set of low‐resolution structure models in the majority of cases (Supporting Information, Fig. S3 and Table S3). Mainly the geometry of the protein, measured by packing and Ramachandran angle quality, is improved, while marginal average effects are observed for R work and R free. As expected, main‐chain H‐bond restraints had more impact than side‐chain restraints (Supporting Information, Table S3). To further test the effect of our new H‐bond detection algorithms we repeated calculations with H‐bond restraints generated by ProSMART and Phenix: for all model quality criteria our method performs comparably or better than previous methods (Supporting Information, Table S4).
Analyzing the general H‐bond restraints in more detail showed specific shortcomings: at places the restraints were too tight, distorting the backbone; in some categories specific H‐bonds could be relatively weak and should be restrained at greater target length; variation in H‐bond length was larger in variable regions such as loops and side‐chains; and there are small systematic differences within groups that were assigned a single target (e.g., a systematic difference in H‐bond lengths between the middle of a long α‐helix and its C‐terminus19). Because the variability inherent to H‐bond lengths cannot be captured in any sensible general division, we set out to define a target on the basis of homologous structure models, expecting a much more accurate measure of the molecular context of the H‐bond than the general data‐mining described in this section.
Homology‐based H‐bond restraints
To generate homology‐based H‐bond restraints, we first need to extract the protein sequence from the working PDB file. The program pdb2fasta (see Supporting Information for details) was developed to extract the sequence for modeled and unmodeled parts of the structure, and maps 73 common types of noncanonical (mostly post‐translationally modified) amino acids to their parent amino acid. The program has been tried and tested for PDB‐wide stability, gives information on unmodeled parts of the sequence, and may therefore be used also for purposes outside the scope of this work.
The sequence file produced by pdb2fasta is then passed to BLAST,20 which is run against the PDB‐REDO databank. BLAST results are passed to our new program HODER (HOmology DErived Restraints), to first identify suitable homologs from a databank of structural data. Briefly, in default settings, we consider hits with ≥70% sequence identity and a resolution higher than the query (see Supporting Information for details). Importantly, users can also add their own PDB files to HODER, to be used as extra homologs: this functionality is important if one is e.g. working on a series of ligand soaks.
After the residues of the working structure are mapped onto their homologous residues, HODER attempts to derive the H‐bond distance restraints. For every H‐bond in the working structure, the equivalent H‐bond distance is computed in all homologs, wherever possible. Then, these distances are clustered using an optimized 1D k‐means algorithm,21 the optimal number of clusters is determined by the Bayesian information criterion,22 within some constraints, and corresponding target distances for each cluster are computed, wherever possible (for details on all the above criteria see Supporting Information).
We then repeated the same calculations for H‐bond restraints based on general targets for restraints based on homology: as the latter differ from general H‐bond restraints only in how their target is derived, the same restraint weight was used. In our test set (see above) 87 ± 16% of the H‐bond restraints in each structure were based on homology; for H‐bonds where no homology‐based target could be defined, we apply the general target values described above. Altogether, homology‐based H‐bond restraints do not deliver a uniform global improvement in performance compared to our general restraints, but neither did they show obvious drawbacks. Importantly, however, when the structures in the test set were recalculated using the implementation in PDB‐REDO, which we shall discuss now, the homology‐based restraints work better than general restraints. Hence, homology‐based H‐bond restraints do work better in more extensive model optimization protocols.
Homology‐based H‐bond restraints in PDB‐REDO
The H‐bond restraint procedures have been incorporated into the PDB‐REDO pipeline (see Supporting Information for details). About one quarter of the crystallographic structures in the PDB (24,506 out of 101,347 PDB‐REDO databank entries) with a resolution equal to or worse than 2.5 Å, could benefit from H‐bonds restrains. Homology‐based H‐bond restraints can be generated for 17,824 of these entries (73%), and 82% of all generated restraints for this set were homology‐based (the remaining 18% were defined using the fallback general targets).
In general, the PDB‐REDO pipeline already improves both the geometry and the fit to the data of published structure models.17 With the application of H‐bond restraints in PDB‐REDO, these improvements are enhanced in the same test set as used for the refinements above (Fig. 1 and Supporting Information, Fig. S4 and Table S5). Importantly, and in contrast to refinements discussed in the previous section, homology‐based restraints work decidedly better than general restraints in the PDB‐REDO pipeline (see Supporting Information for details).
Figure 1.
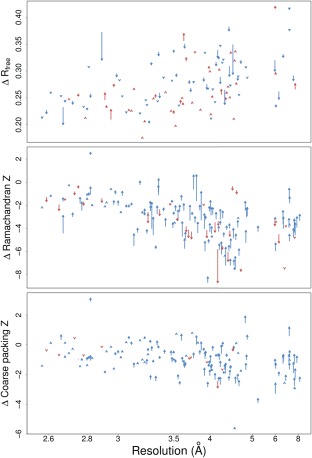
Comparison of PDB‐REDO runs with and without homology‐based H‐bond restraints for all entries in the test set of 155 entries. Each arrow represents the scores from two rerefinements on a single PDB entry. Arrow tails indicate scores from refinement without restraints; arrowheads indicate scores from refinement with restraints. Blue and red arrows indicate improvement and deterioration of the score, respectively. The shown scores are the R free (top) calculated by Refmac5,23 and the Ramachandran Z score (middle) and first generation packing Z score (bottom) from WHAT_CHECK.24 Arrows at the same resolution have been shifted up to 0.05 Å to reduce clutter. Packing Z score and Ramachandran Z score are not shown if they were not computed by WHAT_CHECK; R free is not shown if a new R free set was chosen by PDB‐REDO.17
When models are subjected to the PDB‐REDO pipeline using homology‐based H‐bond restraints, they are influenced by their homologous PDB‐REDO entries. In turn, the PDB‐REDO models subjected to homology‐based restraints may also become the basis of the restraints for other homologous structure models of even lower resolution, which could cause a feedback loop and structure families converging to a consensus structure over multiple rounds of optimization. Then, the true differences between the different structures could be lost. We assessed this risk by subjecting all entries in six protein families (hemoglobin, BRCA1, MutS/MutL, OmpF porin, F1‐ATPase, and alcohol dehydrogenase) to PDB‐REDO refinement five times. Differences between structure models do not decrease when multiple cycles of PDB‐REDO with H‐bond restraints are applied (Supporting Information, Table S6), suggesting that weight optimization and the tolerance to external restraint outliers in Refmac5 prevent bias toward other, possibly incorrect conformations (see Supporting Information for details and additional observations).
Massively parallel computing for a novel PDB‐REDO databank with homology information
The observations that global and homology‐based H‐bond restraints improve low‐resolution structure models after a single PDB‐REDO refinement in a test set of 155 entries encouraged us to update all entries in the PDB‐REDO databank17 with the most recent version of the (fully automated) PDB‐REDO software that includes the refinement strategies based on H‐bond restraints.
Self‐contained Docker (www.docker.com) and Singularity25 images with all PDB‐REDO core and third‐party components (more than fifty independent pieces of software) were created to facilitate massive deployment on any (High Performance Computing or HPC) host (see Supporting Information). Running the complete PDB‐REDO pipeline with 101,570 entries finally required about 60 CPU years (half a million hours) and all computations were finished within about a week using ∼3072 cores on the Gordon HPC cluster, and the large memory nodes on the Comet HPC cluster, at the San Diego Supercomputer Centre. These runs were assembled in a new PDB‐REDO databank.
This new databank is a resource of consistent and high‐quality protein structure models. The introduction of homology‐based restraints has improved the quality of low‐resolution structure models in a consistent manner, as all low‐resolution structure models in the databank were allowed to refine using information derived from high‐resolution homologs (Fig. 2). Importantly, this is not only a new resource in its own right, but it can also serve for better homology restraint generation for future structure refinement. It should be noted that the improvement over the entire resolution range is also the result of other improvements to the PDB‐REDO pipeline and the external programs therein, including better treatment of twinning, general improvements to TLS, NCS, and ADP refinement,17 validation, and correction of structural zinc sites,26 better handling of carbohydrates,27 improved selection of resolution cut‐off and the generation of anomalous difference maps when possible. All these developments are consistently and uniformly applied in all entries, in addition to the applicable homology‐derived restraints. These results in an “internally‐consistent” PDB‐REDO databank that is constructed with a single software version (in contrast to the previous instance of the databank (Supporting Information, Fig. S9).
Figure 2.
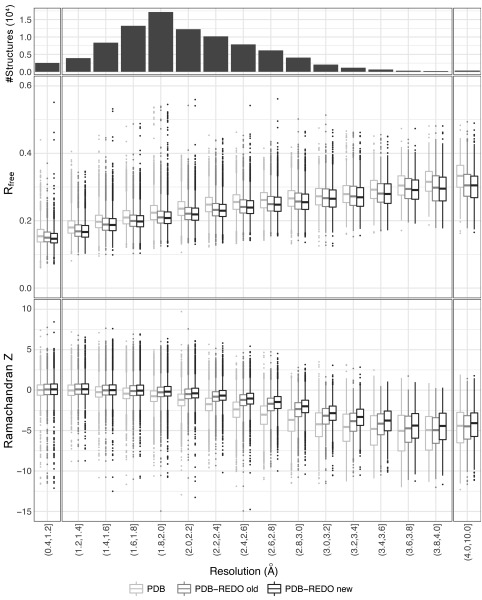
R free and Ramachandran Z score as a function of crystallographic resolution for entries present in PDB, in the PDB‐REDO databank prior to the introduction of homology‐derived H‐bond restraints (PDB‐REDO version 6.23), and in the PDB‐REDO databank calculated with version 7.00. Outliers are shown when they are located beyond 1.5 times the interquartile range. R free for PDB entries was determined by PDB‐REDO for consistency.
Interestingly, the information source for homology‐derived restraints can be analyzed in detail for every structure. In Figure 3 and interactive figures in Supporting Information HTML, we show a directed graph to represent information transfer from any higher‐resolution homolog to any lower resolution homolog. About half of the PDB‐REDO structures (nodes) in the network are connected (edges) to other structures, as they donate or receive H‐bond restraints by satisfying the criteria for homolog use (Supporting Information). The connected graphs in the network typically correspond to protein families. The clusters have widely varying topologies and may be highly connected [Fig. 3(A)] or may consist of a few structures that receive restraints from structures that only donate [Fig. 3(B)]. Interestingly, we found one single very large cluster, consisting of many smaller clusters connected to each other mainly by antibodies and lysozyme (Supporting Information HTML). We show, using community structure detection,28 that the modules in this graph also correspond to clusters of homogenous function [Fig. 3(C,D) and Supporting Information HTML]. Visualizing and analyzing these clusters is an important tool for detecting genuine structural differences within specific family members.
Figure 3.
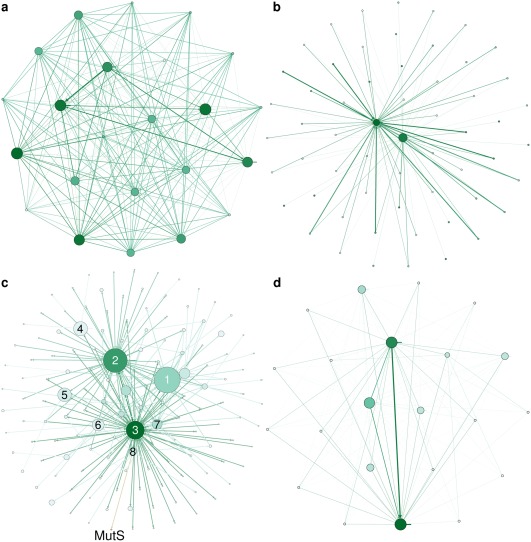
Network representations of H‐bond information transfer between homologs. The nodes represent structures in the PDB‐REDO databank. Node size and color correspond to the number of incoming edges and used resolution (darker is lower), respectively. The edge weight corresponds to the number of homologous chains. (A) Breast cancer 1 (BRCA1). (B) Alcohol dehydrogenase (ADH). (C) Modules detected in the largest network. Node size reflects module size. The three most frequent terms in PDB TITLE records (stripped from English articles, punctuation, etc.) of the structure members in the labeled modules are (1) lysozyme, carbonic, anhydrase; (2) Fab, antibody, fragment; (3) antibody, Fab, HIV; (4) trypsin, inhibitor, thrombin; (5) HLA, peptide, class; (6) hsp90, bound, inhibitor; (7) ubiquitin, nucleosome, histone; (8) binding, maltose, bound. The MutS community (orange; MutS, mismatch, coli) is linked to community 8. (D) The MutS community.
The family of maltose transporters is an example where examining the cluster can aid analysis: the 3fh629 structure in this family is receiving information from every other node/structure in this family [Fig. 4(B)]. Examining the structure in more detail indeed shows that the introduction of homology restraints has led to local improvements, i.e., better definition of the secondary structure content (despite the fact that the original model was already refined with secondary structure restraints29). The secondary structure for this protein became now more similar to the family [Fig. 4(A,C,D)] in an unsupervised, automated manner, and would thus not mislead a potentially interested researcher to believe that this model is genuinely different to other homologues in secondary structure content.
Figure 4.
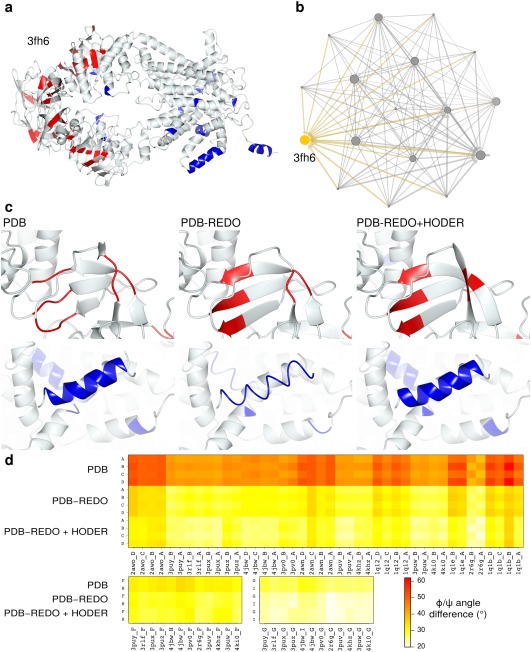
(A) The 4.5 Å structure model of E. coli maltose transporter (PDB entry 3fh629) after PDB‐REDO with HODER. All colored residues (strand in red, helix in blue) in the full structure are residues that changed in secondary structure between PDB, PDB‐REDO, and PDB‐REDO with restraints from HODER. Secondary structure elements are defined by CCP4mg30 which defines secondary structure based on DSSP algorithms.18 The secondary structure content is highest after using homology‐based H‐bond restraints, which coincides with improvements of quality scores compared to the PDB structure: R free (0.338 vs 0.3770), Ramachandran Z score (−6.7 vs −7.8), first‐generation packing Z score (−2.2 vs −2.8) and Molprobity overall percentile (60.0 vs 6.0). The all‐atom rmsd is 0.9Å and the biggest coordinate shift is 5.6Å. (B) The network neighborhood of homologous PDB entries that were used to define the restraints. The target entry, 3fh6, is shown in yellow. The node size corresponds to the number of incoming edges and edge thickness represents the number of homologous chains used. Small nodes are the high‐resolution homologs that only donate information. (C, top) Details of a β‐strand region are shown for PDB, PDB‐REDO and PDB‐REDO with HODER‐generated restraints. The regularity of the strand is improved by PDB‐REDO compared to the PDB and still further improved when restraints are used. (C, bottom) Details of an α‐helical region in the same structure models. At such a low resolution, PDB‐REDO requires the restraints from HODER to retain helical regularity. (D) The average absolute difference of φ/ψ torsion angles between 3fh6 chains and homologous chains for each homologous chain in the PDB, in PDB‐REDO and in the new version of PDB‐REDO with restraints from HODER. The chains A, B, C, and D are homologous mixed α/β domains and there are two pairs of homologous α‐helical domains: chains F and H and G and I, respectively. These three groups of homologous chains are shown separately. Especially the mixed α/β domains become much more similar to their homologous counterparts. All chains become still more similar to homologs when restraints are applied. Some homologs are clearly more similar in conformation to 3fh6 than others. All average angle differences fall in the range between 20° and 62° presented in the legend.
Apart from making changes of local interest to specific structures, the new PDB‐REDO databank can also provide a more reliable resource for data mining: for example, properties such as the Molprobity31 percentile and the ΔG of folding (calculated by FoldX32) are improved and become more uniform for protein families in PDB‐REDO (Fig. 5). Such uniform distributions can be much better learning sets for deriving empirical information by data mining protein structures, and can help improve modeling and analysis initiatives.
Figure 5.
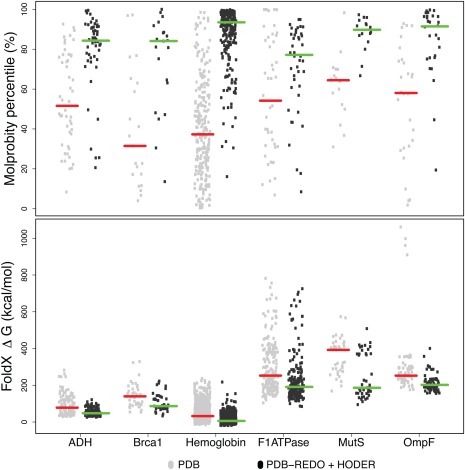
The MolProbity31 score percentiles (top) and energy of folding (ΔG) from FoldX32 (bottom) for each chain in the six investigated protein families. Data are shown for PDB and PDB‐REDO with restraints from HODER. For the Molprobity percentiles, a single data point is shown per entry; for ΔG, a score is shown per chain. The red and green horizontal bars indicate the median values.
Discussion
We describe general and homology‐based H‐bond restraints targets, obtained by new algorithms mining the PDB‐REDO databank, and show that these improve geometrical quality and the fit to X‐ray data for low‐resolution crystallographic structure models. This improvement often goes beyond the reach of current methods. In standalone refinements, homology‐based H‐bond restraints perform equally well to restraints based on general data mining. Within the PDB‐REDO pipeline, however, homology‐based restraints perform better than general restraints.
A difficulty with many methods based on reference structures is that their performance is dependent on the similarity of the reference structure to the target structure model. For example, in the LORESTR pipeline,13 different reference models are tested. In that approach, separate restraints are generated for each reference model and the refinement will adhere to those restraints that are closest to the current model. In such an implementation, including more homologs leads to a higher likelihood of generating distance targets close to the current distance, effectively keeping the model from changing sufficiently to get optimal results. This likely explains the authors' observation that more homologs did not improve refinement. In our approach, all available homologous structures are used to generate one or a few targets per interaction, and therefore more homologs only lead to a better definition of the restraint targets and their expected deviations. Using our approach also mitigates user dilemmas on model choice: a comprehensive search of likely models or combinations is computationally very expensive, but using a subset of homologs makes model selection semi‐arbitrary. The methods presented here have the advantage of using all homologous structure models, making them more computationally efficient and more robust to differences in homologs than methods based on a single reference structure model. Additionally, the width of the H‐bond‐length distribution is represented in the restraints, allowing regions with more structural variation to be less tightly restrained and vice versa. This information is not available if only one reference model is used.
We expect that the multihomolog methods presented here will not work as well if short‐range atom pairs are restrained that do not represent chemical bonds or interactions. Unlike such pairs, H‐bonds can be validated based on well‐established geometric criteria. Therefore the selection of restraints is more reliable, albeit the number of restraints is smaller. With this in mind, the restraints defined here for H‐bonds could be extended to other intramolecular interactions in a protein, such as π–π‐, cation–π‐, and anion–π‐interactions. Unlike H‐bonds, more than two atoms are involved in such interactions, hence more than a simple distance restraint is necessary to improve their geometry. The framework for restraining plane stacking interactions (as a proxy for π–π‐interactions) is available in Refmac5 and is used by the program LibG for nucleic acid restraints.33 Detailed studies into the geometry34 and thermodynamics35 of these interactions can aid in inferring which geometric parameters are best restrained.
The application of H‐bond restraints PDB‐wide in a massively parallel manner using HPC resources has generated a new resource for the biology community: a PDB‐REDO databank that incorporates homology information and is uniformly rerefined and rebuilt with a single software version. By eliminating software idiosyncrasies from the generation of the final structure model and by using homology information, structure similarity within a protein family can be analyzed optimally. That is, in PDB‐REDO, differences between models are more likely to be true differences instead of refinement‐related inconsistencies and the models are therefore more informative Moreover, using a higher‐quality structure data resource may prevent incorrect conclusions from dubious data. For example, a recent study36 detected a number of “novel zinc coordination geometries,” most, if not all, of which were simply errors in the input PDB data.37
Importantly, H‐bond restraints are aimed at improving the geometry of protein structure models and are therefore not solely applicable to models solved by X‐ray crystallography, but also to models obtained from cryo‐EM. Models solved by cryo‐EM still have a relatively low resolution compared to X‐ray crystallography and often have homologous domains of higher resolution present in the PDB. H‐bond restraints could also be applied to NMR and homology models but only once there is independent evidence that H‐bonding partners are actually close; in these cases H‐bond restraints should best be introduced at a final polishing stage of model optimization.
The new PDB‐REDO databank is a valuable novel resource for two audiences. Structure‐minded biologists can use the improved models to identify true features of particular structures in the context of a protein family. Bioinformaticians gain a resource in which the number of errors and inconsistencies from structural models is reduced such that applications like homology modeling or automated feature analysis of protein structures are more reliable.
Materials and Methods
Detailed materials and methods are given as Supporting Information.
Supporting Information
The Supporting Information consists of detailed methods and some additional results (SupplementalText.docx), supporting results in the form of tables and figures (SupplementalFigures.docx).
Supporting information
Supporting Information 1
Supporting Information 2
Acknowledgments
This work is supported from the Netherlands Organization for Scientific Research (NWO) Vidi grant 723.013.003 to RPJ, and the Horizon 2020 programs West‐Life (e‐Infrastructure Virtual Research Environment project No. 675858) and iNEXT (project No. 653706). Computational resources and other support were granted by Johnson and Johnson. The methods discussed in this article have been incorporated into the PDB‐REDO pipeline that is available as a webserver16 at https://pdb-redo.eu or as downloadable software. The new PDB‐REDO databank is also available at https://pdb-redo.eu and rsync://rsync.pdb‐redo.eu/pdb‐redo. The source code of HODER, pdb2fasta, and detectHbonds and the containerized versions of the entire PDB‐REDO pipeline are available on request.
Significance The Protein Data Bank is the oldest public source of biological data and a popular reference site: more than 150,000 files are downloaded each day. PDB‐REDO challenged the notion of PDB as a historical archive, by proactively updating crystallographic PDB structure models based on the original data. Here, we describe new PDB‐REDO algorithms that utilize hydrogen‐bonding patterns to take into account evolutionary relationships between PDB entries. These allow improving quality indicators of structural models, particularly at the low‐resolution regime. Using high performance computing and cloud‐computing deployment tools, we “redid” the entire PDB (more than 100,000 structure models). The algorithms and the novel PDB‐REDO resource that we describe and analyze are available to the entire community (https://pdb-redo.eu).
References
- 1. Kleywegt GJ, Jones TA (2002) Homo crystallographicus ‐ quo vadis? Structure 10:465–472. [DOI] [PubMed] [Google Scholar]
- 2. Engh RA, Huber R (1991) Accurate bond and angle parameters for X‐ray protein structure refinement. Acta Cryst 47:392–400. [Google Scholar]
- 3. Parkinson G, Vojtechovsky J, Clowney L, Brünger AT, Berman HM (1996) New parameters for the refinement of nucleic acid‐containing structures. Acta Cryst 52:57–64. [DOI] [PubMed] [Google Scholar]
- 4. Mooij WTM, Cohen SX, Joosten K, Murshudov GN, Perrakis A (2009) “Conditional restraints”: restraining the free atoms in ARP/wARP. Structure 17:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Headd JJ, Echols N, Afonine PV, Grosse‐Kunstleve RW, Chen VB, Moriarty NW, Richardson DC, Richardson JS, Adams PD (2012) Use of knowledge‐based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Cryst D68:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicholls RA, Fischer M, McNicholas S, Murshudov GN (2014) Conformation‐independent structural comparison of macromolecules with ProSMART. Acta Cryst D 70:2487–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haddadian EJ, Gong H, Jha AK, Yang X, Debartolo J, Hinshaw JR, Rice PA, Sosnick TR, Freed KF (2011) Automated real‐space refinement of protein structures using a realistic backbone move set. Biophys J 101:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nicholls RA, Long F, Murshudov GN (2012) Low‐resolution refinement tools in REFMAC5. Acta Cryst 68:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schröder GF, Levitt M, Brunger AT (2010) Super‐resolution biomolecular crystallography with low‐resolution data. Nature 464:1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2012) Exploiting structure similarity in refinement: automated NCS and target‐structure restraints in BUSTER. Acta Cryst 68:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang C, Wang Q, Ma J (2015) Deformable complex network for refining low‐resolution X‐ray structures. Acta Cryst 71:2150–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Beusekom B, Perrakis A, Joosten RP (2016) Data mining of macromolecular structures. Methods Mol Biol 1415:107–138. [DOI] [PubMed] [Google Scholar]
- 13. Kovalevskiy O, Nicholls RA, Murshudov GN (2016) Automated refinement of low‐resolution macromolecular structures using prior information. Acta Cryst 72:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker EN, Hubbard RE (1984) Hydrogen bonding in globular proteins. Prog Biophys Mol Biol 44:97–179. [DOI] [PubMed] [Google Scholar]
- 15. McDonald IK, Thornton JM (1994) Satisfying hydrogen bonding potential in proteins. J Mol Biol 238:777–793. [DOI] [PubMed] [Google Scholar]
- 16. Joosten RP, Long F, Murshudov GN, Perrakis A (2014) The PDB_REDO server for macromolecular structure model optimization. IUCR J 1:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joosten RP, Joosten K, Murshudov GN, Perrakis A (2012) PDB_REDO: constructive validation, more than just looking for errors. Acta Cryst 68:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 19. Grishaev A, Bax A (2004) An empirical backbone‐backbone hydrogen‐bonding potential in proteins and its applications to NMR structure refinement and validation. J Am Chem Soc 126:7281–7292. [DOI] [PubMed] [Google Scholar]
- 20. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H, Song M (2011) Ckmeans.1d.dp: optimal k‐means clustering in one dimension by dynamic programming. R J 3:29–33. [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464. [Google Scholar]
- 23. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hooft RWW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272–272. [DOI] [PubMed] [Google Scholar]
- 25. Kurtzer (2016) Singularity 2.1.2 ‐ Linux application and environment containers for science. Available from: https://zenodo.org/record/60736
- 26. Touw WG, van Beusekom B, Evers JMG, Vriend G, Joosten RP (2016) Validation and correction of Zn‐CysxHisy complexes. Acta Cryst 72:1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joosten RP, Lütteke T (2017) Carbohydrate 3D structure validation. Curr Opin Struct Biol 44:9–17. [DOI] [PubMed] [Google Scholar]
- 28. Clauset A, Newman MEJ, Moore C (2004) Finding community structure in very large networks. Phys Rev E 70:066111. [DOI] [PubMed] [Google Scholar]
- 29. Khare D, Oldham ML, Orelle C, Davidson AL, Chen J (2009) Alternating access in maltose transporter mediated by rigid‐body rotations. Mol Cell 33:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McNicholas S, Potterton E, Wilson KS, Noble MEM (2011) Presenting your structures: theCCP4mgmolecular‐graphics software. Acta Cryst 67:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen VB, Arendall WB, 3rd , Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst 66:12–21. : [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L (2005) The FoldX web server: an online force field. Nucleic Acids Res 33:W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G (2015) Tools for macromolecular model building and refinement into electron cryo‐microscopy reconstructions. Acta Cryst D 71:136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakrabarti P, Bhattacharyya R (2007) Geometry of nonbonded interactions involving planar groups in proteins. Prog Biophys Mol Biol 95:83–137. [DOI] [PubMed] [Google Scholar]
- 35. Marsili S, Simone M, Riccardo C, Vincenzo S, Piero P (2008) Thermodynamics of stacking interactions in proteins. Phys Chem Chem Phys 10:2673. [DOI] [PubMed] [Google Scholar]
- 36. Yao S, Flight RM, Rouchka EC, Moseley HNB (2015) A less‐biased analysis of metalloproteins reveals novel zinc coordination geometries. Proteins 83:1470–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raczynska JE, Wlodawer A, Jaskolski M (2016) Prior knowledge or freedom of interpretation? A critical look at a recently published classification of “novel” Zn binding sites. Proteins 84:770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Supporting Information 2