

Abstract
IMP3 (insulin-like growth factor-2 mRNA binding protein 3) is an oncofetal protein whose expression is prognostic for poor outcome in several cancers. Although IMP3 is expressed preferentially in triple- negative breast cancer (TNBC), its function is poorly understood. We observed that IMP3 expression is significantly higher in tumor initiating than non-tumor initiating breast cancer cells and we demonstrate that IMP3 contributes to self-renewal and tumor initiation, properties associated with cancer stem cells (CSCs). The mechanism by which IMP3 contributes to this phenotype involves its ability to induce the stem cell factor SOX2. IMP3 does not interact with SOX2 mRNA significantly or regulate SOX2 expression directly. We discovered that IMP3 binds avidly to SNAI2 (SLUG) mRNA and regulates its expression by binding to the 5′ UTR. This finding is significant because SLUG has been implicated in breast CSCs and TNBC. Moreover, we show that SOX2 is a transcriptional target of SLUG. These data establish a novel mechanism of breast tumor initiation involving IMP3 and they provide a rationale for its association with aggressive disease and poor outcome.
Keywords: IMP3, SLUG, SOX2, self-renewal and tumor initiation
Introduction
Many cancers harbor a small population of cells, often referred to as cancer stem cells (CSCs), which exhibit the ability to self-renew and initiate new tumors1, 47. These characteristics have important implications for our understanding of tumor recurrence and dormancy47. Despite the evidence supporting the existence of CSCs, much less is known about the origin of these cells and the mechanisms that regulate their function. The concept that solid tumors contain stem-like cells was pioneered in breast cancer and subsequent studies have used breast cancer as a model to study the genesis of CSCs1, 34.Triple-negative breast cancers (TNBCs) are of particular interest in this discussion because these aggressive cancers harbor a relatively high-frequency of CSCs compared to other breast cancer subtypes34. For these reasons, TNBCs are useful for elucidating mechanisms that contribute to the formation and function of CSCs. In this context, we are interested in IMP3, which is a member of a family of IGF2 mRNA binding proteins that function in RNA stabilization, trafficking and localization32, because it is expressed preferentially in TNBC48. These observations are consistent with the fact that IMP3 expression correlates with the aggressive behavior of many cancers and that it has been exploited for the prognostic assessment of specific cancers23. What is not known is whether there is a causal link between IMP3 and aggressive behavior and, if so, the mechanism by which this RNA binding protein contributes to such behavior.
In this study, we pursued the potential contribution of IMP3 to breast CSCs and TNBC and sought to investigate the mechanisms involved.
Results
IMP3 expression is elevated in breast CSCs and contributes to self-renewal and tumor initiation
Analysis of a published gene expression profile29 revealed that IMP3 expression is significantly higher in the tumor initiating CD44+CD24−ESA+ population1 isolated from human breast tumor tissues compared to the bulk population of tumor cells (Fig. 1A). There was no significant difference in the expression of IMP1 and IMP2 (two other members of the IGF2 mRNA binding protein family) between these populations (Fig. 1A). Based on this observation and the report that IMP3 is preferentially expressed in TNBCs48, we assessed the contribution of IMP3 to the genesis and function of the CD44+CD24−ESA+ population in TNBC. Depletion of IMP3 in TNBC cells (SUM1315 and MDA435) and in cells isolated from a human breast tumor decreased the frequency of CD44+CD24−ESA+ cells (Figs. 1B, S1B). The schematic for FACS analysis of the CD44+CD24−ESA+ population is presented in Fig. S1A.
Figure 1.
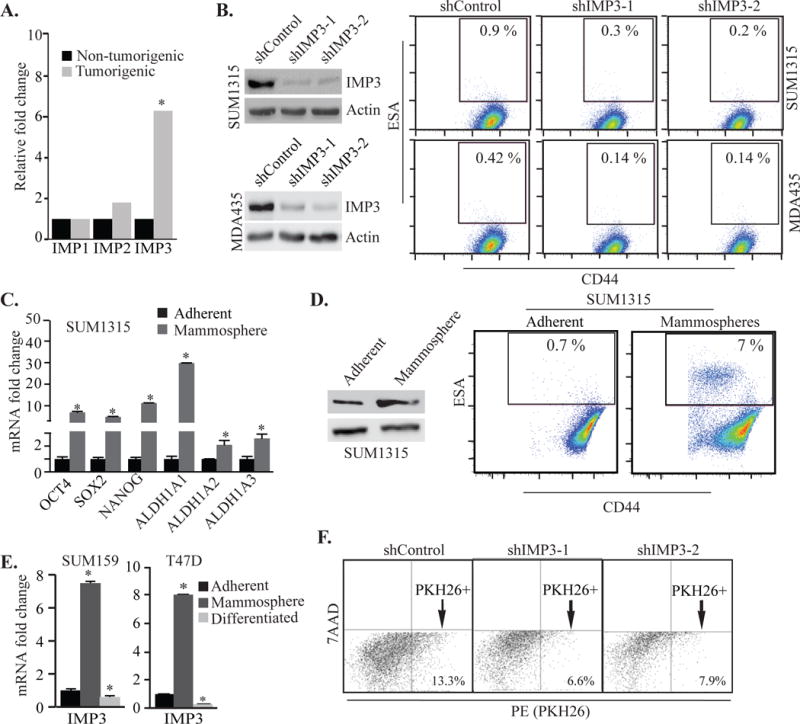
IMP3 expression is elevated in breast CSCs. (A) Expression of IMP1, IMP2 and IMP3 was analyzed in the CD44+CD24−ESA+ and bulk populations of breast tumor cells using a published database (GEO accession no. GSE6883). Gene expression was analyzed in GEO2R. (B) IMP3 expression was depleted using shRNAs (shIMP3-1 and shIMP3-2) in SUM1315 and MDA435 cells and analyzed for CD44+CD24−ESA+ population by FACS. Immunoblots show IMP3 protein expression. shGFP infected cells were used as control (shControl). FACS profiles represent CD44+ESA+ population which were preselected for CD24−. See Fig. S1A. (C) Total RNA was extracted from SUM1315 cells grown as either adherent cultures or mammospheres and expression of the genes indicated in the figures was quantified by qPCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as reference gene. (D) Flow cytometric analysis of CD44+CD24−ESA+ population in SUM1315 cells grown as adherent culture or mammospheres. Immonoblot shows IMP3 protein expression in SUM1315 cells grown as mammospheres. (E) Total RNA was isolated from SUM159 and T47D cells grown as either adherent cultures, mammospheres or differentiated mammospheres induced by collagen-1, and assayed for IMP3 expression by qPCR. (F) Control or IMP3-depleted SUM1315 cells were labelled with PKH26 dye and quantified for PKH26+ cells by FACS after 3 weeks of culture. 7-AAD was used to discriminate dead cells. p value (*) < 0.05.
Given that mammosphere culture can increase the frequency of breast CSCs18, we characterized mammosphere-derived cells for their expression of stem cell markers and frequency of CD44+CD24−ESA+ cells. For this purpose, we used SUM1315 cells and patient-derived xenografts (PDX) of TNBC46, which express IMP3 (Fig. S1C). As shown in Figs. 1C, D & S1D, expression of stem cell genes (SOX2, OCT4, NANOG and ALDH1A) along with IMP3, as well as the frequency of CD44+CD24−ESA+ cells, are significantly elevated in mammospheres generated from these cells compared to adherent cells. Importantly, collagen-1 induced differentiation of mammosphere-derived PDX cells resulted in a decrease of IMP3 expression and in the frequency of CD44+CD24−ESA+ cells (Fig. S1D). This observation is strengthened by our analysis of SUM159 and T47D cells, which do not express IMP3 when grown as adherent cultures. Interestingly, IMP3 expression is induced significantly in these cells when grown as mammospheres, and collagen-1 induced differentiation of these mammospheres13 resulted in a dramatic loss of IMP3 expression (Fig. 1E). Moreover, IMP3 loss also decreased the ability of TNBC cells to retain the lipophilic dye PKH26, a measure of the quiescent nature of CSCs34 (Fig. 1F).
Elevated expression of IMP3 in mammospheres and its reduction upon collagen induced differentiation suggests that IMP3 may regulate the mammosphere forming ability of TNBC cells. Indeed, depletion of IMP3 expression in SUM1315 cells and cells isolated from a human breast tumor decreased their ability to form mammospheres (Figs. 2A, S1E). The ability of IMP3-depleted cells to form mammospheres was restored upon transfection of IMP3-depleted SUM1315 cells with an IMP3 expressing construct that is resistant to shIMP3-1 (IMP3-mut), demonstrating the specificity of IMP3 (Fig. 2B). Moreover, IMP3 depletion specifically in the CD44+CD24−ESA+ population isolated from SUM1315 cells also decreased mammosphere formation (Fig. 2C). Given that self-renewal is a defining feature of stem cells, we assessed the role of IMP3 in self-renewal by quantifying the ability of mammospheres to be passaged serially. As shown in Fig. 2D, depletion of IMP3 significantly reduced the number of mammospheres with increasing passage. The ability of IMP3 to regulate self-renewal and the frequency of CD44+CD24−ESA+ cells suggested that it contributes to tumor initiation. Indeed, transplantation of IMP3-depleted SUM1315 cells in the mammary fat pads of non-obese diabetic (NOD)-Cg-PrkdcscidIL2rgtm1Wjl (NSG) mice significantly increased tumor-free survival compared to control cells (Fig. 2E, left panel). Tumor initiating ability was restored by transfecting IMP3-depleted cells with the IMP3-expressing construct that is resistant to the shRNA (IMP3-mut) (right panel).
Figure 2.
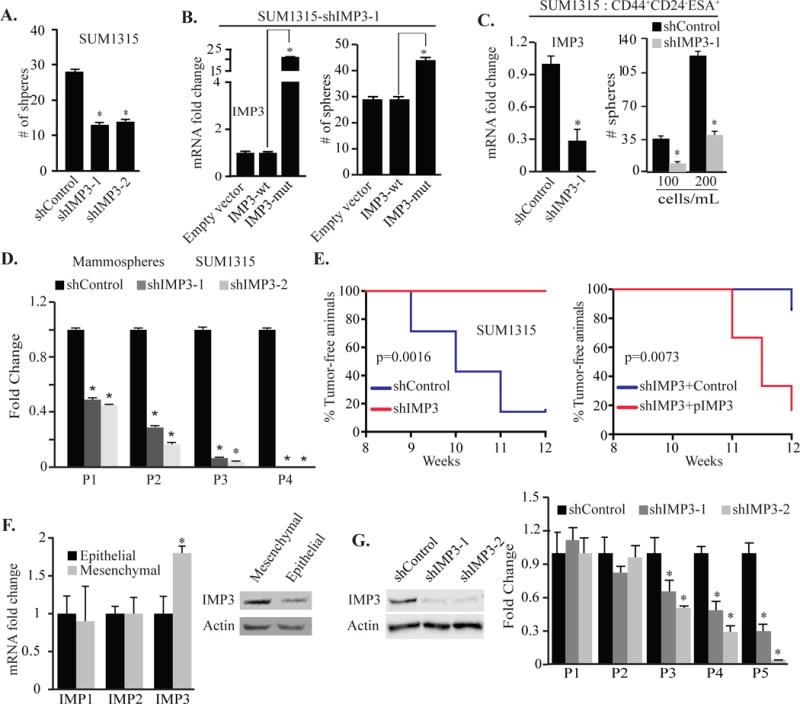
IMP3 regulates self-renewal and tumor initiation. (A) Control and IMP3-depleted SUM1315 cells were grown as mammospheres for 7 days and quantified. (B) IMP3-depleted SUM1315 cells (SUM1315-shIMP3-1) were transfected with constructs expressing either wild type (IMP3-wt) or a mutated IMP3 (shIMP3-1 resistant) and evaluated for mammosphere formation. (C) IMP3 expression was depleted in CD44+CD24−ESA+ population isolated from SUM1315 cells, grown as mammospheres for 7 days and quantified. (D) SUM1315 cells were grown as mammospheres (P1), quantified and serially passaged (P2, P3 & P4) every 7 days. (E) Control, IMP3-depleted SUM1315 cells (left panel, 7 mice per group) or IMP3-depleted cells expressing either mutant IMP3 (pIMP3) or empty vector (control) (right panel, 6 mice per group) were transplanted into the mammary fat pads of NSG mice. Formation of palpable tumors was used to evaluate tumor initiation. The curve comparison was done using Log-rank test. (F) Total RNA and protein extracts were prepared from the epithelial or mesenchymal population of CD44+CD24− cells isolated from SRC-transformed MCF10A cells and the expression of IMP1, IMP2 and IMP3 was quantified by qPCR (left) and immunoblotting (right). (G) IMP3 expression was depleted in mesenchymal cells using shRNAs (immunoblot, left), grown as mammospheres, serially passaged every 7 days and quantified (right). p value (*) < 0.05.
To validate our findings, we employed another model for studying breast CSCs. Inducible Src transformation of MCF10A cells increases the frequency of CD44+CD24−/low cells21. Recently, we reported that this CD44+CD24−/low population is comprised of distinct epithelial and mesenchymal populations, which differ markedly in their tumorigenic properties17. Specifically, the mesenchymal population is highly tumorigenic and enriched for breast CSCs compared to the epithelial cells. We observed that the expression of IMP3, but not IMP1 and IMP2, is significantly higher in the mesenchymal population of CD44+CD24−/low cells compared to the epithelial population and that depletion of IMP3 in the mesenchymal cells reduced their self-renewal ability (Fig. 2F & G). Taken together, our data demonstrate that IMP3 promotes self-renewal and tumor initiation.
IMP3 regulation of SOX2 underlies its contribution to mammosphere formation
To investigate the mechanism by which IMP3 contributes to mammosphere formation and tumor initiation, we focused initially on SOX2 for several reasons. Its expression is enhanced in mammospheres (Fig. 1C) and it has been implicated in the genesis of TNBC37. Moreover, SOX2 expression is associated with basal-like breast cancers12, the majority of which are triple-negative and harbor a high frequency of tumorigenic cells20. These observations suggest a causal link between IMP3 and SOX2. Indeed, we found that SOX2 expression is elevated in CD44+CD24−ESA+ cells and in the mesenchymal population isolated from SRC-transformed MCF10A cells (Fig. 3A). Moreover, SOX2 expression increases in mammosphere culture and decreases upon differentiation of these cells (Fig. 3B). The observed similarities between IMP3 and SOX2 prompted us to investigate whether IMP3 can regulate SOX2 expression. Depletion of IMP3 in TNBC cell lines using shRNAs resulted in a decrease in SOX2 mRNA and protein expression (Fig. 3C). Similar results were observed when IMP3 was depleted using smart-pool siRNAs (Fig. S2A).
Figure 3.
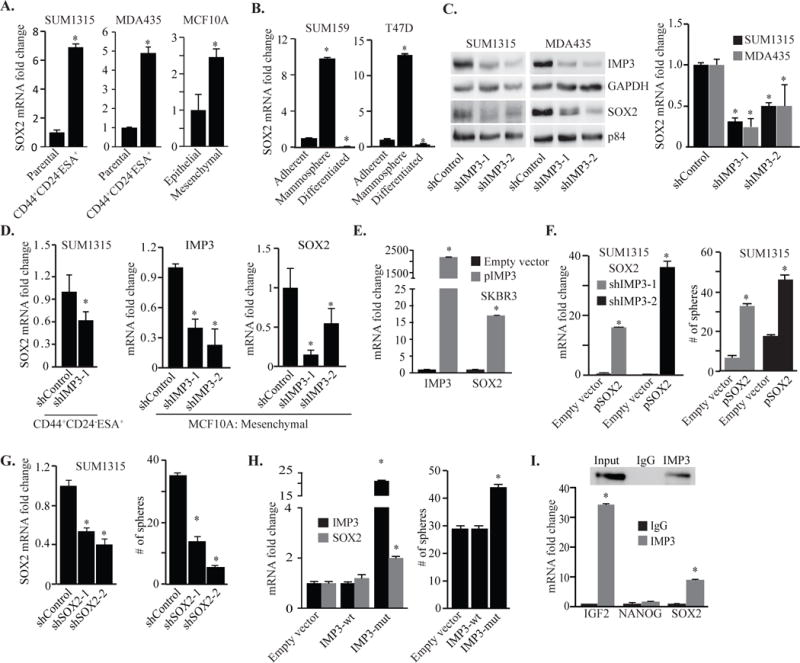
IMP3 regulation of SOX2 underlies its contribution to mammosphere formation. (A) SOX2 mRNA was quantified by qPCR using total RNA isolated from CD44+CD24−ESA+ population sorted from SUM1315 (left), MDA435 (middle) and the mesenchymal population of SRC-transformed MCF10A cells (right). (B) Total RNA was isolated from SUM159 and T47Dcells grown as either adherent cultures, mammospheres or differentiated mammospheres induced by collagen-1, and assayed for SOX2 expression by qPCR. (C) Nuclear and cytoplasmic extracts were prepared from IMP3-depleted or control SUM1315 and MDA435 cells, and SOX2 expression was assessed by immunoblotting and qPCR. (D) IMP3 expression was depleted in the CD44+CD24−ESA+ population sorted from SUM1315 cells and the mesenchymal population of SRC-transformed MCF10A cells, and SOX2 mRNA was quantified by qPCR. (E) IMP3 and SOX2 expression was quantified by qPCR in SKBR3 cells transiently transfected with an empty vector or IMP3 expression construct (pIMP3). (F) SOX2 expression was rescued in IMP3-depleted SUM1315 cells (shIMP3-1 & shIMP3-2) using a lentivirus- based expression construct (pSOX2, left). These IMP3-depleted and SOX2 rescued cells were grown as mammospheres for 7 days and quantified (right). (G) SOX2 expression was depleted in SUM1315 cells using shRNAs (shSOX2-1 & shSOX2-2, left) and these cells were evaluated for mammosphere formation in comparison to control cells (right). (H) SOX2 expression and mammosphere formation were evaluated using IMP3-depleted and IMP3-depleted cells that had been transfected with wild-type IMP3 (IMP3-wt) or a mutant construct that is resistant to shIMP3-1 (IMP3-mut). (I) RIP was performed on extracts prepared from SUM1315 cells using an IMP3-specific antibody or isotype matched IgG. SOX2, NANOG and IGF2 mRNAs were quantified by qPCR. IGF2 mRNA was used as positive control and NANOG was used as negative control. Immunoblot shows the specificity of IMP3 antibody. p value (*) < 0.05.
To extend our results on IMP3 and SOX2 to populations enriched for CSCs, we depleted IMP3 in mammosphere cultures of SUM1315 cells (Fig. S2B), the CD44+CD24−ESA+ population isolated from SUM1315 cells and the mesenchymal population of CD44+CD24-/low cells isolated from SRC- transformed MCF10A cells (Fig. 3D). In all cases, diminishing IMP3 decreased SOX2 expression. The same effect was seen in tumor cells isolated from a human breast tumor tissue (Fig. S2C). Also, exogenous expression of IMP3 in luminal SKBR3 cells increased SOX2 expression (Fig. 3E). Furthermore, exogenous expression of SOX2 in IMP3-depleted SUM1315 cells rescued mammosphere formation (Fig. 3F), indicating that the ability of IMP3 to promote mammosphere formation is SOX2-dependent. Moreover, depletion of SOX2 in SUM1315 cells significantly reduced mammosphere formation compared to control cells (Fig. 3G). Importantly, transfection of IMP3-depleted SUM1315 cells with the shRNA-resistant IMP3 construct increased SOX2 expression and mammosphere formation (Fig. 3H), providing specificity for IMP3 regulation of SOX2. Interestingly, BMI1, which has been implicated in self-renewal33, is not regulated by IMP3 (Fig. S2D). Since IMP3 was originally identified as IGF2 binding protein and that IGF2 promotes tumor formation45, we explored the possible contribution of IGF2 to SOX2 expression. Depletion of IGF2 did not alter SOX2 mRNA expression even though IMP3 regulates the expression of IGF2 (Fig. S2E). Collectively, our data demonstrate that IMP3 contributes to mammosphere formation by regulating SOX2.
In pursuit of the mechanism by which IMP3 regulates SOX2, we performed an RNA- immunoprecipitation (RIP) assay to determine whether IMP3 can bind to SOX2 mRNA directly. As shown in Fig. 3I, we detected modest binding of IMP3 to SOX2 mRNA compared to IGF2 mRNA. Given that IMP3 usually binds to 3′ or 5′ UTR of its target mRNAs, we generated reporter constructs in which luciferase mRNA was fused with either the 3′UTR or 5′UTR of SOX2. Transfection of these constructs into control (empty vector) or IMP3-expressing (pIMP3) SUM159 cells resulted in no significant changes in luciferase activity, diminishing the possibility of a direct interaction between IMP3 protein and SOX2 mRNA (Fig. S2F).
SLUG phenocopies the functions of IMP3
SLUG (SNAI2) is a zinc-finger transcription factor, expressed preferentially in poorly differentiated breast cancers and it has been implicated in the function of breast CSCs and the initiation of basal-like breast cancer26, 36, 43. Based on these observations, we hypothesized that SLUG and IMP3 function in concert. We observed elevated expression of SLUG in CD44+CD24−ESA+ cells isolated from human breast tumors compared to the bulk population (Fig. 4A) in the same dataset that we used to analyze IMP329. SLUG expression is also elevated in CD44+CD24−ESA+ cells isolated from TNBC cell lines (Fig. 4B). Moreover, SLUG expression is enhanced by mammosphere culture and repressed by collagen-I-induced differentiation of these cultures (Fig. 4C). These similarities between IMP3 and SLUG coupled with the previous report that SLUG can induce SOX2 expression in hepatocellular carcinoma cells prompted us to investigate whether SLUG can also regulate SOX2 expression in TNBC50. In fact, we found that depletion of SLUG in TNBC and human breast tumor cells decreased SOX2 expression and mammosphere formation (Figs. 4D, S2G). SLUG depletion in SUM1315, MDA435 cells and in cells isolated from a human breast tumor, also reduced the frequency of CD44+CD24−ESA+ cells (Figs. 4E, S2G). Moreover, exogenous expression of SLUG in luminal SKBR3 cells increased SOX2 expression (Fig. 4F). We observed similar results with the CD44+CD24−ESA+ population isolated from SUM1315 cells and mesenchymal cells isolated from SRC-transformed MCF10A cells (Fig. 4G).
Figure 4.
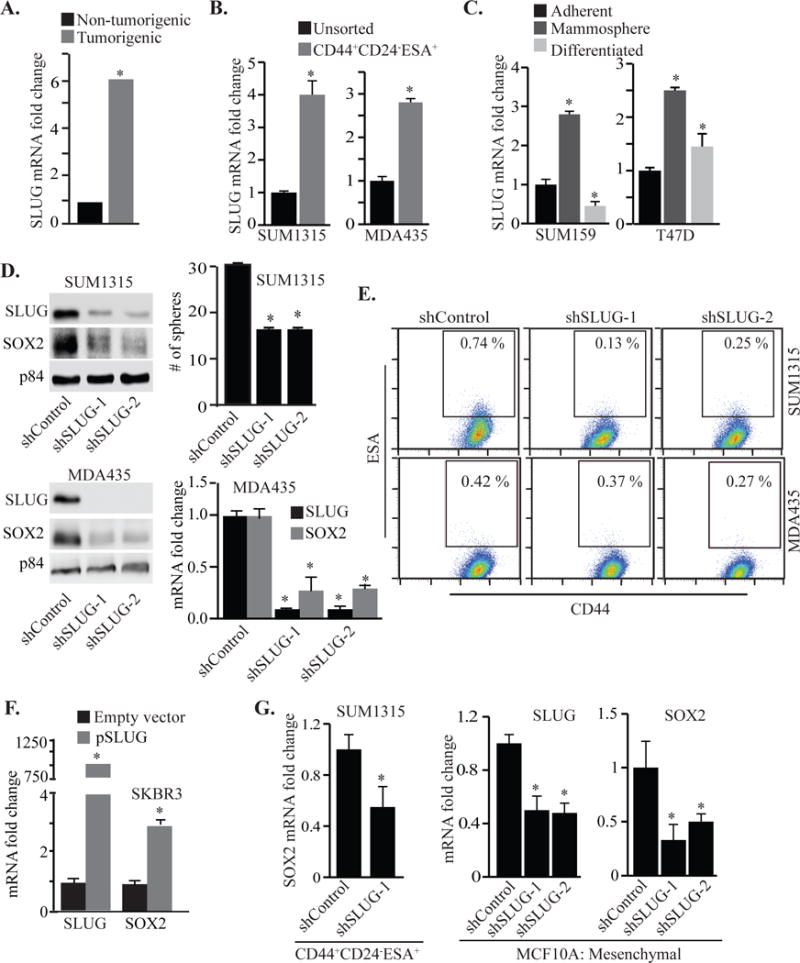
SLUG phenocopies the functions of IMP3. (A) SLUG expression was analyzed in the CD44+CD24−ESA+ and bulk populations of breast tumor cells using a published database (GEO accession no. GSE6883). (B) SLUG mRNA was quantified by qPCR using total RNA prepared from CD44+CD24−ESA+ population sorted from SUM1315 (left) and MDA435 cells (right). (C) Total RNA was isolated from SUM159 and T47D cells grown as either adherent cultures, mammospheres or differentiated mammospheres induced by collagen-1, and assayed for SLUG expression by qPCR. (D) SLUG expression was depleted in SUM1315 and MDA435 cells using shRNAs (shSLUG-1 & shSLUG-2) and these cells were maintained in mammosphere culture for 7 days. SOX2 expression was assessed in these SLUG-depleted cells by immunoblotting and qPCR. (E) FACS analysis of the CD44+CD24−ESA+ population in shControl and SLUG-depleted SUM1315 and MDA435 cells. (F) SLUG and SOX2 expression was quantified by qPCR in SKBR3 cells transiently transfected with a SLUG expression construct (pSLUG). (G) SLUG expression was depleted in CD44+CD24−ESA+ population sorted from SUM1315 cells (left) and the mesenchymal population of SRC-transformed MCF10A cells (right) using shRNAs and these SLUG- depleted cells were used for quantifying SOX2 mRNA by qPCR. p value (*) < 0.05.
The parallels between IMP3 and SLUG expression and function prompted us to investigate the possibility that IMP3 regulates SLUG expression and that SLUG mediates the contribution of IMP3 to tumor initiation. In fact, IMP3 was shown recently to regulate SLUG expression by binding to its mRNA and SLUG rescue in IMP3-depleted TNBC cells restored their migration and invasive ability. These data implicate SLUG as an important functional target of IMP344. We confirmed this finding and demonstrated that IMP3 binds to the 5′UTR of SLUG mRNA (Fig. S3A–C & Fig. 5A). Moreover, we found that IMP3 can regulate SLUG expression in CD44+CD24−ESA+ cells (Fig. 5B). IMP2, however, does not appear to impact SLUG expression (Fig. 5C) and no correlation between IMP2 and SLUG was detected in a gene expression dataset (Fig. D)8, 9, 15. SLUG depletion did not affect IMP3 expression, indicating that SLUG functions downstream of IMP3 (Fig. S3D & E). Collectively, our data demonstrate that IMP3 can regulate SLUG expression and that SLUG functions downstream of IMP3.
Figure 5.
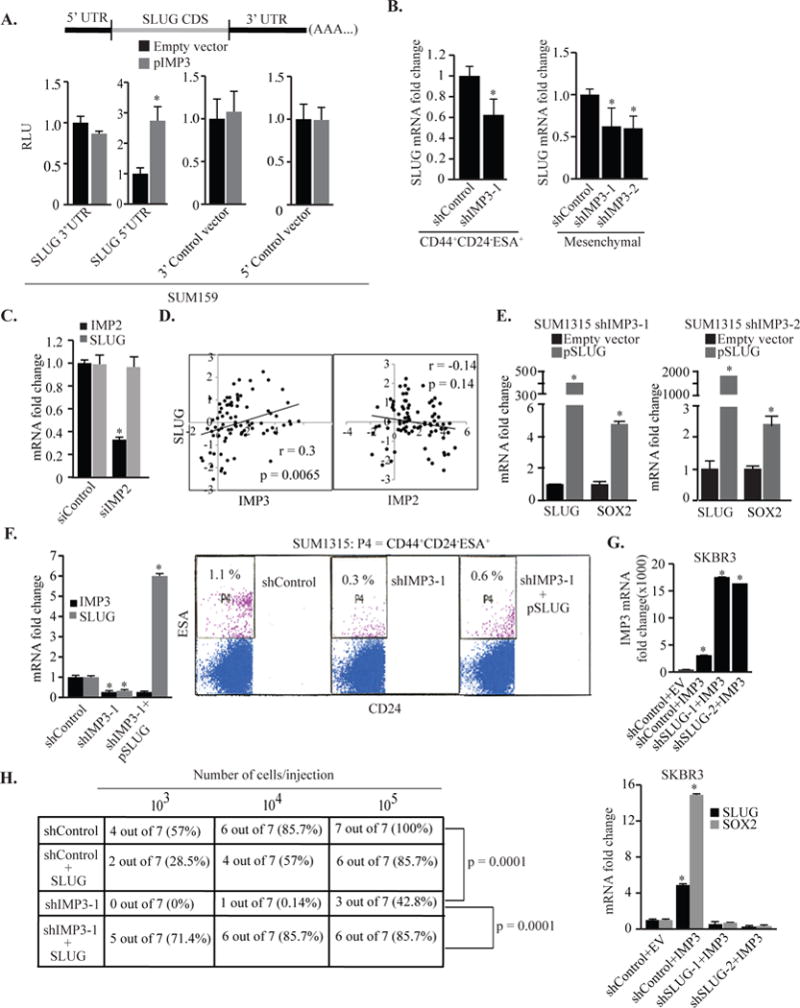
IMP3 regulation of SOX2 and tumor initiation is mediated by SLUG. (A) Parental (transfected with an empty vector) or IMP3-expressing SUM159 cells were transiently transfected with renilla luciferase reporter constructs carrying the 3′ & 5′ UTRs of SLUG mRNA or corresponding empty vector and luciferase expression was measured 24 h post-transfection. A construct expressing firefly luciferase (PGL3 control vector) was used as transfection control. The relative luminescence unit (RLU) was determined as renilla luciferase normalized to firefly luciferase activity. (B) SLUG mRNA was quantified by qPCR using total RNA prepared from IMP3-depleted CD44+CD24− ESA+ population sorted from SUM1315 cells (left) and the mesenchymal CD44+CD24− population of Src-transformed MCF10A cells (right). (C) Expression of IMP2 and SLUG was measured by qPCR using total RNA extracted from control or IMP2-depleted SUM1315 cells. IMP2 was depleted using smart-pool siRNA (20 nM) and transfection was carried out using Dharmafect-4 for 72 h. An off-target siRNA pool (20 nM) was used as control. (D) Expression of IMP3, IMP2 and SLUG was correlated using a published gene expression database comprising 81 BLBCs (cBioportal). Correlation coefficient (r) was calculated using Pearson’s correlation. (E) IMP3-depleted SUM1315 cells (shIMP3-1 & shIMP3-2) were transiently transfected with either an empty vector or a SLUG expression construct (pSLUG). SLUG and SOX2 mRNA expression was quantified by qPCR. (F) IMP3-depleted SUM1315 cells (shIMP3-1) were transiently transfected with either an empty vector or a SLUG expression construct and these cells were used to quantify CD44+CD24−ESA+ population by FACS (right). IMP3 and SLUG expression was measured by qPCR (left). (G) SKBR3 cells pre-transfected with either control shRNA (shControl) or SLUG specific shRNAs (shSLUG-1 & shSLUG-2) were transfected with an empty vector or IMP3-expressing construct. RNA isolated from these cells was used to quantify IMP3, SLUG and SOX2 expression by qPCR. (H) Control, IMP3-depleted or IMP3-depleted SUM1315 cells, stably infected with a SLUG-expressing lentivirus construct, were mixed with Matrigel (1:1, v/v) and injected into the mammary fat pads of NSG mice. Number of cells injected is indicated in the figure. Each group comprised 7 mice. p value (*) < 0.05.
The above findings prompted us to interrogate whether SLUG mediates IMP3 regulation of SOX2 and tumor initiation. Expression of SLUG in IMP3-depleted SUM1315 cells, which express low levels of SLUG, rescued SOX2 expression (Fig. 5E) and increased the CD44+CD24−ESA+ population (Fig. 5F). Moreover, IMP3 expression in SKBR3 cells that had been pre-transfected with SLUG-specific shRNA, failed to induce SOX2 expression (Fig. 5G). These findings demonstrate that SLUG mediates the IMP3 regulation of SOX2 expression. This conclusion was substantiated in vivo by showing that expression of SLUG in IMP3-depleted SUM1315 cells increased their tumor initiating ability (Fig. 5H). The role of the IMP3/SLUG axis in breast CSCs is also supported by our analysis of a published gene expression profile of doxorubicin-resistant subline derived from MCF7 cells, a luminal breast cancer cell line. These drug-resistant cells are highly enriched for the tumor initiating CD44+CD24−/low population and are much more tumorigenic than parental MCF7 cells7. Interestingly, these drug-resistant cells were found to be slow-cycling and express much higher levels of both IMP3 and SLUG compared to parental MCF7 cells (Fig. S3F). Finally, we observed that the expression of IMP3, SLUG and SOX2 correlates in a breast cancer database30 (Fig. 6A).
Figure 6.
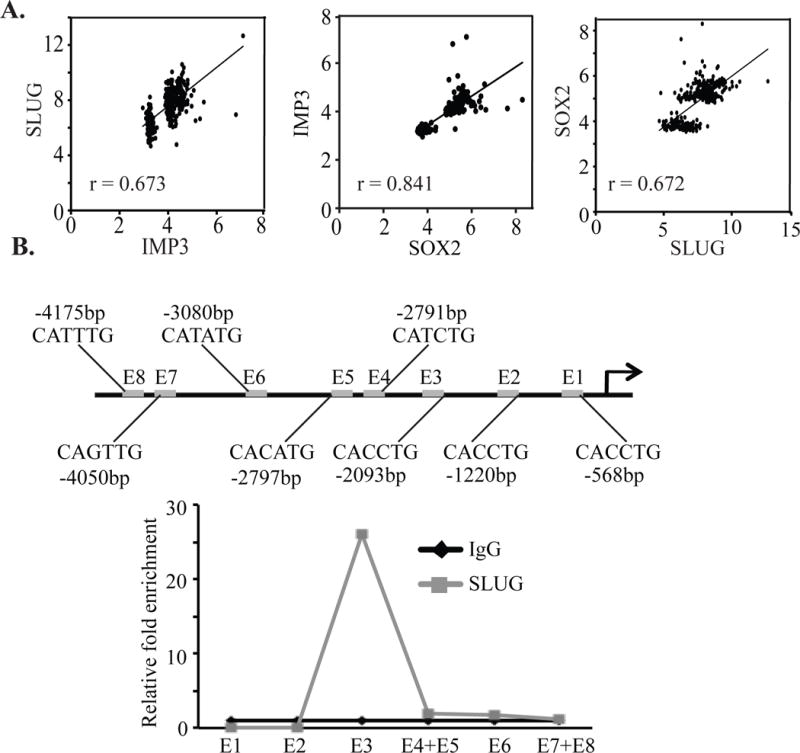
SLUG binds to the SOX2 promoter. (A) IMP3, SLUG and SOX2 mRNA expression was correlated in a published database comprising 327 samples (GEO accession no. GSE6532). Statistical significance was determined using Pearson’s correlation. (B) ChIP was performed using genomic DNA isolated from SUM1315 cells using a SLUG-specific antibody, and the SLUG-bound SOX2 fragments were quantified by qPCR. Isotype matched IgG was used as control for this experiment.
Although most studies on SLUG in breast cancer have focused on its ability to repress transcription5, 19, our data suggest that it may contribute to the activation of SOX2 transcription. We detected several E-boxes (CAnnTG)10 within the proximal region of the SOX2 promoter (Fig. 6B) and chromatin immunoprecipitation analysis detected a significant enrichment of SLUG in the region containing E-box-3 (E3) at −2093 bp. In contrast, we did not detect any SLUG binding in the E-boxes proximal or distal to E3 (Fig. 6B).
Discussion
In this study, we implicate IMP3 in the function of breast CSCs in TNBC, and demonstrate that this function is mediated by IMP3 regulation of SLUG. This key conclusion provides a rationale for the observation that IMP3 is expressed preferentially in TNBC48 because these tumors exhibit a high frequency of CSCs compared to other sub-types of breast cancer34. Our findings may also explain why IMP3 expression is prognostic for aggressive disease and metastatic potential based on the evidence that CSCs contribute to these clinical parameters3, 23.
Our findings reveal a post-transcriptional mechanism of SLUG regulation mediated by IMP3. This mode of regulation may function in concert with mechanisms that regulate SLUG transcription to ensure that SLUG expression is tightly controlled in breast cancer, especially given its ability to promote the genesis of CSCs, basal differentiation and the genesis of basal-like breast cancer35, 36. However, the mechanisms that regulate SLUG expression in breast cancer had not been determined. Clearly, IMP3-mediated regulation of SLUG mRNA provides one such mechanism. Although we demonstrate that IMP3 contributes to the genesis and function of breast CSCs by regulating SLUG expression, it likely contributes to these processes in other ways as well. In this direction, we note the recent finding that IMP3-ribonucleoproteins (RNPs) function as ‘cytoplasmic safe houses’ that prevent miRNA-mediated mRNA decay of specific oncogenes24.
A critical result is that SOX2 is a transcriptional target of SLUG establishing a causal relationship between two prominent factors in the biology of CSCs. Although SOX2 is not expressed in normal mammary stem cells, recent studies have highlighted its importance in breast cancer and CSCs6, 27, 41, 49. The fact that IMP3 regulates SOX2 by a SLUG-dependent mechanism enhances the significance of this mRNA binding protein in CSCs and the initiation of TNBC. Moreover, both IMP3 and SOX2 are expressed in embryonic stem cells, and there is a considerable similarity in the gene expression signatures between TNBCs and embryonic stem cells3. In addition to its critical role in embryonic stem cells, SOX2 has also been shown to regulate self-renewal and tumor initiation in osteosarcomas and squamous cell carcinomas2, 6. Moreover, the epidermal growth factor receptor (EGFR) can regulate self-renewal and CSC function by regulating SOX238, 42. Interestingly, we reported that EGFR regulates IMP3 expression in TNBC cells39.
Our findings suggest that IMP3 could have a significant role in resistance to chemotherapy and that it could be an attractive candidate for targeted therapy itself. Chemoresistance is an important feature of CSCs28, which results from the elevated expression of several ATP-binding cassette (ABC) transporters including breast cancer resistance protein (BCRP/ABCG2) that is also regulated by IMP340. In hepatocellular carcinoma, for example, IMP3 has been implicated in the abrogation of TGFβ signaling and consequent chemoresistance11. In contrast, both IMP340 and TGFβ signaling4 have been implicated in the chemoresistance of TNBC cells, suggesting that IMP3 does not inhibit TGFβ signaling in TNBC. Moreover, the observation that IMP3 expression is elevated in slow-cycling drug-resistant cells that display CSC properties is significant because chemotherapeutic drugs typically target rapidly proliferating cells and residual disease is enriched in slow-cycling cells with a higher frequency of CSCs that may be responsible for metastasis and relapse28, 29.
Targeting IMP3 is a potentially feasible and effective approach for the clinical management of TNBC because it is not expressed in normal breast and its mechanism of action is known (binding to specific RNA sequences). Also, as mentioned above, IMP3 contribute to residual disease following chemotherapy. The feasibility of targeting IMP3 is supported by the ability of IMP3-derived peptides to elicit a positive CTL response in patients with advanced esophageal cancer25. Interestingly, IMP2 has been implicated in the function of glioblastoma CSCs by a mechanism that involves regulation of key mRNAs involved in oxidative phosphorylation22. Although, both proteins are preferentially expressed in TNBCs, IMP2 did not appear to be important for breast CSCs function, partly because of its inability to regulate SLUG expression. Nonetheless, an emerging paradigm is that IMPs are critical for the function of CSCs and potential therapeutic targets11, 22.
Materials and Methods
Cells and reagents
The human breast cancer cell lines SUM1315 and SUM159 were obtained from Dr. Stephen Ethier (Kramanos Institute, MI, USA). MCF10A, MDA435, MDA468, SKBR3, BT549, T47D and HEK293T cell lines were obtained from American Type Culture Collection (ATCC). SUM1315 cells were maintained in F-12 medium supplemented with 5% fetal bovine serum (FBS), insulin (5μg/mL), epidermal growth factor (10ng/mL) and 1% penicillin-streptomycin. SUM159 cells were maintained in F-12 medium supplemented with 5 % FBS, insulin (5μg/mL), hydrocortisone (1 μg/mL) and 1% penicillin-streptomycin. The MDA468, MDA435, BT549, T47D and SKBR3 cell lines were maintained in RPMI supplemented with 10% FBS and 1% penicillin-streptomycin. HEK293T cells were cultured in DMEM (high glucose) supplemented with 10% FBS, non-essential amino acids (1×, Gibco), HEPES (pH 7.4, 1 mM, Gibco), sodium pyruvate (1mM, Gibco) and 1% penicillin-streptomycin. ER-SRC- transformed MCF10A cells was provided by Dr. Kevin Struhl (Harvard Medical School) and maintained as described17.
shRNAs specific for GFP (RHS4459), IMP3 (TRCN0000074675, TRCN0000074677), SLUG (TRCN0000015389, TRCN0000015390), SOX2 (TRCN0000003252, TRCN0000003250) and IGF2 (TRCN0000062430, TRCN0000062432) were obtained from Open Biosystems (Rockford, IL, USA). Smartpool siRNA specific for IMP3 and IMP2 were purchased from Thermo Scientific (Lafayette, CO, USA). Antibody for IMP3 was purchased from DAKO (CA, USA). SLUG, SOX2 and BMI1 antibodies were purchased from Cell Signaling technology (Danvers, MA, USA). GAPDH and p84 antibodies were obtained from GenTex (CA, USA). The luciferase reporter constructs PsiCheck-2 and pLightSwitch_5UTR were obtained from Dr. Sean Ryder (UMass Medical School, Worcester, MA, USA) and SwitchGear Genomics (CA, USA), respectively. Lipofectamine-2000 and Fugene-6 were procured from Invitrogen (CA, USA) and Promega Corporation (WI, USA), respectively. Collagen-1 was purchased from BD Biosciences (San Jose, CA, USA).
Antibodies used for FACS are: CD44-PE, CD24-APC (BD Biosciences); mouse-ESA (AbCam) and anti-mouse-488 (AbCam).
IMP3, SLUG and SOX2-depleted cell lines were generated by infecting them with pLKO.1 based lentiviruses expressing the corresponding shRNAs and subsequent selection in puromycin (2μg/mL). Stable cell lines were maintained regularly in puromycin (1μg/mL). IMP3 and SLUG expression constructs were generated by inserting their cDNAs into a lentivirus vector (pCDH-CMV- MCS-EF1-copGFP) into EcoRI/NotI and EcoRI/BamHI sites respectively. Primers used are listed in Supplementary Table 1.
Human breast tumor tissue
Human breast tumor tissue was obtained in compliance with the Institutional Review Board of the University of Massachusetts Medical School. Tumor cells were isolated as described previously14. Briefly, the tissue was minced and digested for 6h with a mixture of collagenase-A (Roche, Indianapolis, IN, USA) and hyaluronidase (MP Biomedicals, Solon, OH, USA). The digested cells were spun down, washed twice with serum-free DMEM/F12 medium and resuspended cells were plated in complete medium (DMEM/F12, 10% FBS, 1% penicillin-streptomycin and 5μg/mL insulin) for 2h to deplete mammary fibroblasts. The collected organoids were dissociated into a single cell suspension by trypsinization and filtered through 40 μm filter (BD Biosciences) to remove residual clustered cells and plated for subsequent experiments.
Patient derived xenograft (PDX)
Breast tumor tissue (triple-negative) samples were obtained from breast cancer surgery patients, through Brigham and Women’s Hospital and Massachusetts General Hospital. These breast tumors samples were cut into small fragments, which were then surgically implanted into the fourth mammary fat pad of NSG mice. Tumor xenografts were expanded into multiple recipient NSG mice for several passages. Tumors were harvested and analyzed as described in the text.
Flow cytometry
To quantify CD44+CD24−ESA+ population, cells were harvested by trypsinization, washed with phosphate buffered saline (PBS) and incubated with the desired antibodies as follows: ESA (1:200, 30′ at 4°C); anti-mouse 488 (1:500, 30′ at 4°C); CD44-PE (1:1000) and CD24-APC (1:100) for 30′ at 4°C. Cells were washed twice with PBS following antibody incubation. For sorting CD44+CD24−ESA+ population, cells were incubated with the same antibodies, re-suspended in serum free DMEM-F12 (1:1) with 1mM EDTA and collected in same medium with 10% FBS. All analyses were performed using BD LSRII flow cytometer and data analysis were performed using FlowJo software.
Mammosphere, self-renewal and tumor initiation experiment
Mammospheres were cultured as described previously16. For self-renewal assays, mammospheres were collected by centrifugation, washed with PBS, trypsinized for 5 minutes and single cell suspension was prepared. This single cell suspension was counted and seeded for subsequent mammosphere formation. Mammospheres were counted by taking bright field images of 15-20 different areas per well. Differentiation was induced by culturing the mammosphere derived cells on collagen-1 coated dish with serum containing medium13. Trypsin was inactivated by soybean-trypsin inhibitor (1mg/mL, Sigma).
For tumor initiation experiment, cells were mixed with matrigel (1:1,v/v) and 50 μL of the cell-matrigel mix was injected into the mammary fat pads of NSG mice (Jackson Laboratory, Bar Harbor, ME) as described previously16. Tumor onset was determined by palpation and visual observation.
PKH26 labeling
For PKH26 staining, control and IMP3-depleted SUM1315 cells were stained with PKH26 Red Fluorescent Cell Linker Kit (Sigma, MO, USA). Labeled cells were maintained for three weeks and PKH26+ cells were quantified using FACS. 7-Aminoactinomycin-D (7-AAD) was used to discriminate dead cells. The same procedure was followed to sort PKH26+ population from MDA435 cells.
Biochemical assays
For immunoblotting, cell extracts were prepared using RIPA buffer containing EDTA and EGTA (Boston Bioproducts, MA, USA). Cytoplasmic and nuclear extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Lafayette, CO, USA). A protease and phosphatase inhibitor cocktail was added separately (Roche Applied Biosciences, IN, USA). Protein extracts were blotted with the appropriate primary Abs and then incubated with either mouse or rabbit IgG horseradish peroxidase-conjugated secondary antibody. An ECL kit (Thermo Scientific, IL, USA) was used to develop the blots. qPCR was performed by isolating total RNA from adherent cells or cells grown as mammospheres using Trizol reagent (Invitrogen) following the manufacturer’s protocol. cDNA was synthesized using superscript-II reverse transcriptase (Invitrogen). mRNAs were quantified by real time PCR analysis (ABI Prism, Applied Biosystems) using Power Syber Green PCR master mix (Applied Biosystems). Quantification was performed using ΔΔCt method and GAPDH was used as reference gene. Primers used for qPCR are listed in Supplementary Table 2.
Transfections and reporter assays
Transient transfection of IMP3 and SLUG expression constructs was performed using Fugene-6 as the transfection agent, and total mRNA and protein extracts were prepared 48 h post-transfection. siRNAs specific to IMP3 and IMP2 (25 nM) was transfected using Dharmafect-4 (Thermo Scientific) as the transfection agent and protein extracts were prepared 72 h post-transfection. For siRNA experiments, a pool of non-targeting siRNA (Thermo Scientific) was used as control (25 mM).
To generate SOX2 and SLUG 3′/5′ UTR reporter constructs, human SOX2 or SLUG 3′ UTR and 5′ UTR were amplified using cDNA prepared from SUM1315 cells and cloned into the psiCheck-2 and pLightSwitch-5′UTR vectors, respectively. The 3′UTR was cloned into XhoI/NotI sites and the 5′ UTR was cloned into BglII/NcoI sites. Same restriction sites were used for both SLUG and SOX2 UTRs. PCR amplified UTR fragments were confirmed by restriction mapping and cloned reporter constructs were confirmed by sequence analysis. Primers used are listed in Supplementary Table 3. Parental or IMP3 expressing SUM159 cells were transfected using Fugene-6 with either the control or UTR- constructs. Firefly reporter construct was used as transfection control for both 3′ and 5′ UTR constructs which express renilla luciferase. The Relative Light Units (RLU) value was calculated as the ratio of renilla luciferase to firefly luciferase activity (normalized luciferase activity). The protocol used for transfection and measurement of luciferase activity has been described previously31. Luciferase activity was measured in a Beckman Coultier, DTX 880 luminometer.
Binding assays
The interaction between IMP3 protein and associated mRNAs was determined using RNA-immunoprecipitation assay (RIP) followed by qPCR analysis. The assay was performed using RiboCluster Profiler RIP-Assay kit (MBL International, MA, USA). RIP grade antibody for IMP3 was purchased from the same company. Isotype matched IgG was used as control for immunoprecipitation. The chromatin immunoprecipitation (ChIP) assay was performed using ChIP-IT Express kit (Active motif, CA, USA). Chromatin was prepared by sonication using MISONIX Sonicator-3000 (power level 2, 15 second pulse X 4). DNA fragments were amplified by qPCR. Primers are listed in Supplementary Table 4.
Statistical analysis
The data are shown as the ± S. E. p values (*) were determined using student’s t test and p ≤ 0.05 was considered as significant. For in vivo serial dilution experiment, p values were calculated using Extended Mantel-Haenszel Stratified Test of Association. For correlation studies, statistical significance was calculated by Pearson’s correlation.
Supplementary Material
Acknowledgments
We thank Dr. Chung-Cheng Hsieh (University of Massachusetts Medical School) for statistical analysis of our in vivo data.
Financial Support: This work was supported by the Megan Lally Memorial Fund and NIH Grants CA168464, CA034196 and AI46629.
Footnotes
Conflicts of Interest: DLG and LDS are consultants for Viacord, Inc. The other authors declare that they have no conflict of interest.
References
- 1.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH, et al. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012;31:2270–2282. doi: 10.1038/onc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature genetics. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. Journal of cell science. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- 6.Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–250. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- 7.Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. Journal of the National Cancer Institute. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhary J, Skinner MK. Basic helix-loop-helix proteins can act at the E-box within the serum response element of the c-fos promoter to influence hormone-induced promoter activation in Sertoli cells. Molecular endocrinology. 1999;13:774–786. doi: 10.1210/mend.13.5.0271. [DOI] [PubMed] [Google Scholar]
- 11.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-beta in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. The Journal of biological chemistry. 2008;283:17969–17978. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 13.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & development. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. GLI1 regulates a novel neuropilin-2/alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO molecular medicine. 2013;5:488–508. doi: 10.1002/emmm.201202078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goel HL, Gritsko T, Pursell B, Chang C, Shultz LD, Greiner DL, et al. Regulated splicing of the alpha6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell reports. 2014;7:747–761. doi: 10.1016/j.celrep.2014.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, et al. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast cancer research : BCR. 2008;10:R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer research. 2002;62:1613–1618. [PubMed] [Google Scholar]
- 20.Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, et al. CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Human pathology. 2012;43:364–373. doi: 10.1016/j.humpath.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes & development. 2012;26:1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Z, Chu PG, Woda BA, Rock KL, Liu Q, Hsieh CC, et al. Analysis of RNA-binding protein IMP3 to predict metastasis and prognosis of renal-cell carcinoma: a retrospective study. The Lancet Oncology. 2006;7:556–564. doi: 10.1016/S1470-2045(06)70732-X. [DOI] [PubMed] [Google Scholar]
- 24.Jonson L, Christiansen J, Hansen TV, Vikesa J, Yamamoto Y, Nielsen FC. IMP3 RNP safe houses prevent miRNA-directed HMGA2 mRNA decay in cancer and development. Cell reports. 2014;7:539–551. doi: 10.1016/j.celrep.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Kono K, Iinuma H, Akutsu Y, Tanaka H, Hayashi N, Uchikado Y, et al. Multicenter, phase II clinical trial of cancer vaccination for advanced esophageal cancer with three peptides derived from novel cancer-testis antigens. J Transl Med. 2012;10:141. doi: 10.1186/1479-5876-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–1365. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 29.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England journal of medicine. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 30.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 31.Mak P, Leung YK, Tang WY, Harwood C, Ho SM. Apigenin suppresses cancer cell growth through ERbeta. Neoplasia. 2006;8:896–904. doi: 10.1593/neo.06538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Wewer UM, Nielsen FC. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Molecular and cellular biology. 1999;19:1262–1270. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, et al. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell stem cell. 2010;6:279–286. doi: 10.1016/j.stem.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Phillips S, Prat A, Sedic M, Proia T, Wronski A, Mazumdar S, et al. Cell-State Transitions Regulated by SLUG Are Critical for Tissue Regeneration and Tumor Initiation. Stem cell reports. 2014;2:633–647. doi: 10.1016/j.stemcr.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell stem cell. 2011;8:149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, Hernandez L, et al. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2007;20:474–481. doi: 10.1038/modpathol.3800760. [DOI] [PubMed] [Google Scholar]
- 38.Rybak AP, Tang D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cellular signalling. 2013;25:2734–2742. doi: 10.1016/j.cellsig.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 39.Samanta S, Sharma VM, Khan A, Mercurio AM. Regulation of IMP3 by EGFR signaling and repression by ERbeta: implications for triple-negative breast cancer. Oncogene. 2012;31:4689–4697. doi: 10.1038/onc.2011.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samanta S, Pursell B, Mercurio AM. IMP3 protein promotes chemoresistance in breast cancer cells by regulating breast cancer resistance protein (ABCG2) expression. The Journal of biological chemistry. 2013;288:12569–12573. doi: 10.1074/jbc.C112.442319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegle JM, Basin A, Sastre-Perona A, Yonekubo Y, Brown J, Sennett R, et al. SOX2 is a cancer-specific regulator of tumour initiating potential in cutaneous squamous cell carcinoma. Nature communications. 2014;5:4511. doi: 10.1038/ncomms5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, et al. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Molecular cancer. 2012;11:73. doi: 10.1186/1476-4598-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C, et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. The Journal of pathology. 2008;214:25–37. doi: 10.1002/path.2254. [DOI] [PubMed] [Google Scholar]
- 44.Su P, Hu J, Zhang H, Li W, Jia M, Zhang X, et al. IMP3 expression is associated with epithelial-mesenchymal transition in breast cancer. International journal of clinical and experimental pathology. 2014;7:3008–3017. [PMC free article] [PubMed] [Google Scholar]
- 45.Tada Y, Yamaguchi Y, Kinjo T, Song X, Akagi T, Takamura H, et al. The stem cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-independent growth in cancer cells. Oncogene. 2015;34:752–760. doi: 10.1038/onc.2013.599. [DOI] [PubMed] [Google Scholar]
- 46.Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer cell. 2013;24:347–364. doi: 10.1016/j.ccr.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell stem cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Walter O, Prasad M, Lu S, Quinlan RM, Edmiston KL, Khan A. IMP3 is a novel biomarker for triple negative invasive mammary carcinoma associated with a more aggressive phenotype. Human pathology. 2009;40:1528–1533. doi: 10.1016/j.humpath.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2014 doi: 10.1038/onc.2014.257. [DOI] [PubMed] [Google Scholar]
- 50.Zhao X, Sun B, Sun D, Liu T, Che N, Gu Q, et al. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncology reports. 2015;33:149–156. doi: 10.3892/or.2014.3562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.