

Abstract
Background
Laodelphax striatellus Fallén (Hemiptera: Delphacidae) is one of the most destructive rice pests. L. striatellus is different from 2 other rice planthoppers with a released genome sequence, Sogatella furcifera and Nilaparvata lugens, in many biological characteristics, such as host range, dispersal capacity, and vectoring plant viruses. Deciphering the genome of L. striatellus will further the understanding of the genetic basis of the biological differences among the 3 rice planthoppers.
Findings
A total of 190 Gb of Illumina data and 32.4 Gb of Pacbio data were generated and used to assemble a high-quality L. striatellus genome sequence, which is 541 Mb in length and has a contig N50 of 118 Kb and a scaffold N50 of 1.08 Mb. Annotated repetitive elements account for 25.7% of the genome. A total of 17 736 protein-coding genes were annotated, capturing 97.6% and 98% of the BUSCO eukaryote and arthropoda genes, respectively. Compared with N. lugens and S. furcifera, L. striatellus has the smallest genome and the lowest gene number. Gene family expansion and transcriptomic analyses provided hints to the genomic basis of the differences in important traits such as host range, migratory habit, and plant virus transmission between L. striatellus and the other 2 planthoppers.
Conclusions
We report a high-quality genome assembly of L. striatellus, which is an important genomic resource not only for the study of the biology of L. striatellus and its interactions with plant hosts and plant viruses, but also for comparison with other planthoppers.
Keywords: comparative genomics, insects, genome sequencing, annotation, virus transmission
Background
The small brown planthopper, Laodelphax striatellus (Delphacidae, Hemiptera), is one of the most destructive pests in a variety of crops (Fig. 1). It is widespread in the Palearctic region, including countries such as China, Japan, Germany, Italy, Russia, Kazakhstan, Turkey, and United Kingdom [1]. L. striatellus is polyphagous and its hosts include rice, maize, oats, tall oatgrass, wheat, and barley. It injures plants by sap-sucking behavior using its piercing-sucking mouthpart, after which symptoms like stunting, chlorosis, and hopper burn may further develop in plants. Apart from feeding damage, L. striatellus transmits various plant viruses, such as rice stripe virus (RSV), rice black-streaked dwarf virus (RBSDV), barley yellow striate mosaic virus, maize rough dwarf virus, wheat rosette stunt virus, and wheat chlorotic streak virus [2]. Some of these viruses may cause serious damage to agricultural production, such as RSV and RBSDV. For example, rice stripe disease caused by RSV has broken out over the past several decades in many East Asian countries, including China, where rice field production was reduced by 30–50% and total loss of harvest was observed in some areas [3].
Figure 1:

Photograph of Laodelphax striatellus on a rice plant leaf. Scale bar, 1 mm.
L. striatellus is distinct from 2 other rice planthoppers, white-backed planthopper (Sogatella furcifera) and brown planthopper (Nilaparvata lugens), in several important traits such as host range, dispersal capacity, and plant viruses that they vector. N. lugens mostly feeds on rice plants, S. furcifera feeds on rice, wheat, and maize, and L. striatellus has an even broader host range. Both N. lugens and S. furcifera are known for migratory habits [4]. Whereas S. furcifera is the vector of Southern rice black streak dwarf virus (SRBSDV) [5] and N. lugens is the vector of rice ragged stunt virus (RRSV) and rice grassy stunt virus [6, 7], L. striatellus is the carrier of RSV, RBSDV, and several other viruses. Although the genome sequences of S. furcifera and N. lugens have been released recently [8, 9], no comparative genomic analyses were reported for the 2 planthoppers. Deciphering the genome of L. striatellus can help us understand the genetic basis underlying the differences in important traits between L. striatellus and the other 2 rice planthoppers.
Data Description
Sample and sequencing
The inbreeding line used for genome sequencing is an inbred laboratory strain that was derived from a field population collected in Hai’an, Jiangsu province, China. A single gravid female was selected, and her progenies were sib-mated for 22 generations to obtain the inbreeding line. Planthoppers were reared on 2–3-cm rice seedlings at 25°C and a photoperiod of 16:8 hours of light/dark. DNA was extracted by using Puregene Core Kit A (Qiagen Sciences, Maryland, USA) from the F22 specimens following the manufacturer's instruction. We built 5 libraries with insert size between 180 bp and 800 bp for paired-end sequencing and 9 libraries with insert size between 1.4 Kb and 24 Kb for mate-pair sequencing according to the standard protocols of the Illumina HiSeq 2500 sequencer (Table 1). We also constructed 33 Pacbio RSII libraries according to the standard Pacbio protocols (Table 1). In total, we generated 190 Gb of Illumina data (126 Gb of paired-end reads and 64 Gb of mated-pair reads) and 32.4 Gb of Pacbio data, representing 316× and 54× coverage of the genome, respectively.
Table 1:
Sequencing data used for genome assembly and annotation
| Category | Accession | Life stage | Sample type | Insert size, bp | Read length, bp | No. of reads |
|---|---|---|---|---|---|---|
| Survey | SRR5816389 | Adult | DNA | 230 | 2 × 125 | 127 772 669 |
| Assmebly | SRR5830088 | Adult | DNA | 180 | 2 × 100 | 123 459 791 |
| SRR5816388 | Adult | DNA | 250 | 2 × 125 | 137 013 558 | |
| SRR5816387 | Adult | DNA | 500 | 2 × 100 | 141 587 274 | |
| SRR5816386 | Adult | DNA | 500 | 2 × 125 | 30 520 480 | |
| SRR5816393 | Adult | DNA | 800 | 2 × 100 | 153 498 320 | |
| SRR5816392 | Adult | DNA | 1.4–1.6 K | 2 × 125 | 40 251 413 | |
| SRR5816391 | Adult | DNA | 2.6–2.8 K | 2 × 125 | 36 559 438 | |
| SRR5816390 | Adult | DNA | 5–5.6 K | 2 × 125 | 26 684 783 | |
| SRR5816385 | Adult | DNA | 5.6–6.5 K | 2 × 125 | 23 069 935 | |
| SRR5816384 | Adult | DNA | 9–11 K | 2 × 125 | 24 285 333 | |
| SRR5816377 | Adult | DNA | 11–13 K | 2 × 125 | 23 396 366 | |
| SRR5816376 | Adult | DNA | 13–15 K | 2 × 125 | 30 547 732 | |
| SRR5816379 | Adult | DNA | 15–18 K | 2 × 125 | 25 926 919 | |
| SRR5816378 | Adult | DNA | 18–24 K | 2 × 125 | 26 325 395 | |
| SRR5817574 | Adult | DNA | - | 8559 | 99 701 | |
| SRR5817559 | Adult | DNA | - | 8947 | 77 038 | |
| SRR5817582 | Adult | DNA | - | 8474 | 104 288 | |
| SRR5817569 | Adult | DNA | - | 8518 | 114 320 | |
| SRR5817560 | Adult | DNA | - | 9202 | 80 599 | |
| SRR5817562 | Adult | DNA | - | 9211 | 100 089 | |
| SRR5817573 | Adult | DNA | - | 8610 | 102 997 | |
| SRR5817558 | Adult | DNA | - | 9007 | 86 083 | |
| SRR5817581 | Adult | DNA | - | 8452 | 89 374 | |
| SRR5817570 | Adult | DNA | - | 8419 | 101 715 | |
| SRR5817550 | Adult | DNA | - | 9192 | 82 657 | |
| SRR5817576 | Adult | DNA | - | 8597 | 105 080 | |
| SRR5817553 | Adult | DNA | - | 8586 | 77 467 | |
| SRR5817557 | Adult | DNA | - | 8821 | 75 712 | |
| SRR5817567 | Adult | DNA | - | 8363 | 106 634 | |
| SRR5817575 | Adult | DNA | - | 8620 | 105 795 | |
| SRR5817552 | Adult | DNA | - | 8985 | 66 096 | |
| SRR5817556 | Adult | DNA | - | 8573 | 83 500 | |
| SRR5817568 | Adult | DNA | - | 8357 | 104 295 | |
| SRR5817578 | Adult | DNA | - | 8528 | 108 299 | |
| SRR5817565 | Adult | DNA | - | 8728 | 69 694 | |
| SRR5817555 | Adult | DNA | - | 8480 | 86 385 | |
| SRR5817571 | Adult | DNA | - | 8437 | 106 314 | |
| SRR5817577 | Adult | DNA | - | 8686 | 106 337 | |
| SRR5817566 | Adult | DNA | - | 8890 | 52 889 | |
| SRR5817554 | Adult | DNA | - | 8648 | 85 970 | |
| SRR5817572 | Adult | DNA | - | 8437 | 101 258 | |
| SRR5817580 | Adult | DNA | - | 8490 | 104 459 | |
| SRR5817563 | Adult | DNA | - | 8954 | 91 218 | |
| SRR5817561 | Adult | DNA | - | 8724 | 84 033 | |
| SRR5817579 | Adult | DNA | - | 8776 | 107 138 | |
| SRR5817564 | Adult | DNA | - | 9054 | 68 294 | |
| SRR5817551 | Adult | DNA | - | 8508 | 88 776 | |
| Annotation | SRR5816381 | Larva | RNA | 250–300 | 2 × 150 | 23 733 333 |
| SRR5816380 | Adult | RNA | 250–300 | 2 × 150 | 24 933 333 | |
| SRR5816383 | Egg | RNA | 250–300 | 2 × 150 | 24 633 333 | |
| SRR5816382 | Fat body | RNA | 250–300 | 2 × 150 | 31 300 000 | |
| SRR5816375 | Brain | RNA | 250–300 | 2 × 150 | 40 333 333 | |
| SRR5816374 | Gonad | RNA | 250–300 | 2 × 150 | 33 300 000 | |
| SRR5816394 | Tentacle | RNA | 250–300 | 2 × 150 | 24 966 666 |
Survey library in the Category column was used to estimate the genome size of Laodelphax striatellus. Libraries of insert size >1 Kb were mate-paired. For gene annotation, data from 2 previously sequenced tissues were used under accession SRR1619428 for salivary gland and SRR1617617 for alimentary canal.
For transcriptome sequencing, total RNA was isolated from 4 tissues (antenna, brain, fatty body, and gonad) and whole bodies of 3 developmental stages (egg, nymph, adult) of L. striatellus using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Nanodrop (Thermo Scientific, Wilmington, DE, USA) was used to determine RNA quantity, and gel electrophoresis was used to examine RNA quality. cDNA libraries were constructed according to the manufacturer's instructions and sequenced on an Illumina HiSeq 2500 sequencer.
Estimation of genome size and determination of chromosome number
We estimated the genome size of L. striatellus using 2 independent approaches: flow cytometry [10] and k-mer analyses [11]. The flow cytometry analysis was carried out according to a published procedure [10]. Briefly, a female adult was ground in PBS-T buffer. The mixture was filtered by a 40-μm cell filter, incubated with 2 μg/mL RNase A at 37°C for 15 minutes, and then stained with 5 μg/mL propidium iodide at 25°C for 30 minutes. The fluorescence signal was detected by a FACSCallbur Analyzer (Becton-Dickinson, San Jose, CA, USA). Heads of Drosophila melanogaster and cytoblasts of Gallus gallus were treated with the same procedure as genome size references. The genome sizes of D. melanogaster and G. gallus are known to be 0.18 pg and 1.25 pg, respectively [12]. As shown in Fig. S1, the genome size of L. striatellus was estimated to be 0.60 pg (587 Mb) by the flow cytometry method. In k-mer analysis, 31.94 Gb of clean reads were utilized to generate a k-mer (k = 17) depth distribution curve (Fig. S1D), based on which the genome size was estimated to be 550 Mb. Accordingly, the haploid genome size of L. striatellus was estimated to be 550–587 Mb.
The chromosome number was determined by cytological analysis of testes cells. The testes of newly emerged males were dissected in insect Ringer solution, fixed in Carnoy's fixative for 15 minutes. The testes were washed with 0.01 mol/L PBS solution, stained at 0.5 μg/mL Hoechst 33 258, and sealed with Antifade Mounting Medium (Beyotime, Jiangsu, China). Cells in meiosis phase were selected for chromosome counting under a confocal microscope Zeiss LSM710 (Carl Zeiss, Oberkochen, Germany). In most cases, 15 haploid chromosomes were observed (30 for diploid chromosomes) (Fig. S2), although sometimes only 14 were visible. Thus the number of chromosomes in L. striatellus was determined to be 2n = 30.
Genome assembly and assessment
We assembled the genome with both Illumina sequencing and Pacbio sequencing data. Illumina data were used to build contigs and scaffolds as follows. First, all reads with ≥10% unidentified nucleotides, or with >10 nt aligned to the adapter sequences, or being putative PCR duplicates were removed to obtain clean reads. Mate-pair reads from libraries with insert sizes >2 kb were classified as paired-end, unpaired, negative, and mate-pair reads, and only the negative and mate-pair reads were retained for the assembly. Second, we employed SOAPdenovo v. 3.0 (SOAPdenovo, RRID:SCR_010752) [13,14] with the parameters “pregraph -K 33 -p 30 -d 30; contig –k 33 –M 3” to build de Bruijn graph and assemble sequencing reads into contigs. Third, all mate-pair reads were mapped to the contigs, and mate-pair information was added in a stepwise manner to connect contigs into scaffolds. GapCloser v. 1.12 (GapCloser, RRID:SCR_015026) [13] was used to fill the gaps between scaffolds with a local assembly strategy. Afterwards, PBJelly v. 15.8.24 (PBJelly, RRID:SCR_012091) [15] was used to fill the gaps between scaffolds using the 32.4 Gb (∼54×) of Pacbio data. Briefly, all the gaps (length > 25 bp) on the assembly were identified first, and the Pacbio reads were mapped to the assembly using PBJelly. The BLASR alignments were parsed to identify gap-supporting reads by comparing aligned and unaligned base positions within each read [16]. Overlap-layout-consensus engine ALLORA within PBSuite (v. 15.8.24, Pacific Biosciences Menlo Park) [17] was used to assemble the reads for each gap to generate consensus gap-filling sequences. As the final step, the consensus gap-filling sequences were spliced into the corresponding gap position in the draft assembly, replacing all Ns if the gap was closed and leaving the appropriate number of Ns if the gap was only reduced.
With the above assembly procedure, we obtained a final assembly of 541 Mb, having 38 193 scaffolds with a contig N50 length of 118 Kb and a scaffold N50 length of 1.1 Mb. The length of the assembly accounts for 91.7% and 98.4% of the estimated genome size by flow cytometry and k-mer analysis, respectively. The longest contig and scaffold were 2.0 Mb and 10.4 Mb, respectively (Table 2). The Pacbio sequencing data greatly improved the length of contigs compared with the published genomes of N. lugens (contig N50, 24.2 Kb [8]) and S. furcifera (contig N50, 70.7 Kb [9]), which were assembled with Illumina data only (Table 2). We aligned clean reads onto the genome assembly using BWA (BWA, RRID:SCR_010910) [18] and calculated the fraction of bases at a given sequencing depth. The results showed a very small fraction of low-coverage bases, suggesting high coverage and accuracy of the genome assembly (Fig. S3).
Table 2:
Statistics comparison of genome assembly and annotation among 3 planthoppers
| Laodelphax striatellus | Nilaparvata lugens a | Sogatella furcifera b | ||||
|---|---|---|---|---|---|---|
| Category | Contig | Scaffold | Contig | Scaffold | Contig | Scaffold |
| Total size, Mb | 530.2 | 541.0 | 993.8 | 1140.8 | 673.9 | 720.7 |
| Total number | 48 574 | 38 193 | 80 046 | 46 558 | 50 020 | 20 450 |
| Maximum length, Kb | 1990 | 10 350 | 230 | 2254 | 800 | 12 789 |
| N50 length, Kb | 118 | 1085 | 24 | 357 | 71 | 1185 |
| GC content, % | 34.5 | 34.6 | 31.6 | |||
| TE proportion, % | 23.0 | 38.9 | 39.7 | |||
| BUSCO evaluation, % | 92 | 81 | 92 | |||
| Gene number | 17 736 | 27 571 | 21 254 | |||
| Average gene length, bp | 14 342 | 11 216 | 12 597 | |||
| Average CDS length, bp | 1289 | 1135 | 1526 | |||
| Average exon per gene | 6 | 4 | 6 | |||
| Average exon length, bp | 213 | 264 | 240 | |||
| Average intron length, bp | 2587 | 3062 | 2064 | |||
Validation and quality control
The completeness and accuracy of the genome assembly were assessed by 4 independent approaches. First, the overall base composition and the percentage of Ns were calculated. As shown in Table S1, the assembled genome had a low percentage (1.99%) of Ns and an expected base composition, which is similar to that of the other 2 planthoppers. The overall GC content of L. striatellus was 34.54%, similar to that of N. lugens [8] and slightly higher than that of S. furcifera [9]. Second, we remapped Illumina paired-end reads to the assembly using BWA [18], and we found that 93.2% of reads could be mapped back, covering 96.83% of the assembled genome, including 95.08% of the genome with ≥×20 coverage (Table S2). Third, we performed de novo transcriptome assembly using Trinity v. 2.0.2 (Trinity, RRID:SCR_013048) for RNA-seq data from multiple developmental stages and tissues (Table 1). We also included 2 published RNA sequencing datasets from salivary glands and alimentary canal [19] in the transcriptome assembly. We mapped the assembled transcripts to the genome assembly using TopHat (TopHat, RRID:SCR_013035) with default parameters and found that 90.31% of the transcripts with >90% transcript coverage were aligned to 1 scaffold (Table S3), indicating that most expressed genes were correctly assembled in the genome. When the RNA reads from the 9 transcriptome datasets were directly mapped to the genome, 78% to 94% could be correctly mapped to the genome with appropriate splicing, indicating that the genome assembly had a good representative of gene regions (Table S4). Finally, the benchmarking universal single-copy orthologs v. 1 (BUSCO, RRID:SCR_015008) dataset representing 2675 genes for arthropoda was used for genome assessment [20]. Our assembled genome captured 92% (2470/2675) of the BUSCO genes, suggesting that a gene repertoire was nearly complete (Table S5). Taken together, these results suggest that our assembled genome was highly accurate and nearly covered the whole genome.
Annotation of repetitive elements
Two independent methods, namely homology-based and de novo prediction, were applied for repetitive element annotation. For the homology-based method, the assembled genome was compared with Repbase, issued on 13 January 2014 [21], using RepeatMasker v. 4.0.5 (RepeatMasker, RRID:SCR_012954) and RepeatProteinMasker (v. 1.36) with default settings [22]. For the de novo prediction, we built a de novo repeat library with LTR_FINDER v. 1.0.5 (LTR_Finder, RRID:SCR_015247) [23], Piler (v. 1.06) [24], RepeatScout v. 1.0.5 (RepeatScout, RRID:SCR_014653) [25], and RepeatModeler v. 1.0.8 (RepeatModeler, RRID:SCR_015027). Tandem Repeat Finder (v. 4.07b) [26] was used to search tandem repeats. Furthermore, RepeatProteinMask [22] was used to identify putative transposable element (TE)–related proteins. After merging all the repetitive elements identified by abovementioned tools, we identified a total of 139.1 Mb of repetitive sequences, accounting for 25.7% of the genome (Table S6). The percentage of repetitive elements in the L. striatellus genome was much lower than those of N. lugens (48.6%) [8] and S. furcifera (44.3%) [9]. Of all the repetitive sequences, 10.59% were the class I transposable elements (retrotransposon), including 5.01% long interspersed nuclear elements, 1.32% long terminal repeats, and 4.26% short interspersed nuclear elements. Class II elements (DNA transposons) represented only 4.92% of the genome (Table 3). L. striatellus had the lowest TE fraction and the smallest genome size compared with N. lugens and S. furcifera (Table 3).
Table 3:
Comparison of transposable element contents of the 3 planthoppers
| Laodelphax striatellus | Nilaparvata lugens | Sogatella furcifera | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| De novo + Repbase | TE proteins | Combined TEs | Combined TEs | Combined TEs | ||||||
| Class | Length, bp | % of genome | Length, bp | % of genome | Length, bp | % of genome | Length, bp | % of genome | Length, bp | % of genome |
| DNA | 24 818 676 | 4.59 | 2 550 902 | 0.47 | 26 592 872 | 4.92 | 162 024 958 | 14.20 | 126 002 323 | 17.33 |
| LINE | 24 160 245 | 4.47 | 4 889 094 | 0.90 | 27 124 925 | 5.01 | 182 652 892 | 16.00 | 69 257 982 | 9.52 |
| LTR | 7 122 249 | 1.32 | 0 | 0.00 | 7 122 249 | 1.32 | 168 492 299 | 14.80 | 31 286 552 | 4.30 |
| SINE | 22 739 683 | 4.20 | 743 909 | 0.14 | 23 044 510 | 4.26 | 8 272 412 | 0.70 | 10 730 722 | 1.48 |
| Other | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 41 262 | 0.00 | 23 167 338 | 3.18 |
| Unknown | 27 609 625 | 5.10 | 0 | 0.00 | 27 609 625 | 5.10 | 21 890 733 | 1.90 | 28 395 639 | 3.90 |
| Total | 119 645 576 | 22.12 | 8 177 428 | 1.51 | 124 360 921 | 22.99 | 443 765 874 | 38.90 | 288 840 556 | 39.73 |
De novo + Repbase refers to TE integrated between de novo and Repbase prediction. TE proteins refers to TE identified by RepeatProteinMask. Combined TEs refers to the 2 TE combined results above. Other means TE that can be classified but doesn’t belong given classes. Unknown means TE that can’t be classified.
DNA: DNA transposon; LINE: long interspersed nuclear element; LTR: long terminal repeat; SINE: short interspersed nuclear element.
Annotation of protein-coding genes
The protein-coding genes were annotated with evidence from the homology-base method, ab initio prediction, and RNA-seq data. For the homology-based method, the annotated gene sets from 8 species, N. lugens, Acyrthosiphon pisum, Pediculus humanus, Nasonia vitripennis, D. melanogaster, Bombyx mori, Rhodnius prolixus, and Daphnia pulex (Table S7), were aligned to the L. striatellus genome using TBLASTN (TBLASTN, RRID:SCR_011822) [27] with an E-value cutoff of 1E−5. GeneWise v. 2.2.0 (GeneWise, RRID:SCR_015054) [28] was used to define gene models. For ab initio prediction, we utilized Augustus v. 3.1 (Augustus: Gene Prediction, RRID:SCR_008417) [29], GlimmerHMM v. 3.0.4 (GlimmerHMM, RRID:SCR_002654 [30], SNAP (v. 2013–11-29) [31], GeneID (v. 1.4) [32, 33], and GENSCAN v. 1.0 (GENSCAN, RRID:SCR_012902) [34] to predict potential protein-coding genes from the repeat-masked genome. Furthermore, we identified gene structures with the assistance of 9 transcriptomes assembled by Tophat-Cufflinks (v. 2.2.1) [35] and Trinity-PASA (v. 2.0.2) [36], respectively. Then we integrated all predicted gene structures above with EvidenceModeler (v. 1.1.1) [37] to obtain a nonredundant set of 17 736 protein-coding genes with an average gene length of around 16.17 Kb (Table S8–S9, Fig. S4). We constructed the orthologous gene families using annotated genes from 22 closely related species (Table S7) and found that L. striatellus had 4210 species-specific genes, fewer than those of N. lugens (10 163) and S. furcifera (7743) (Fig. 2). This may be attributed to the smaller genome size and lower gene number in L. striatellus.
Figure 2:

Gene cluster analysis among 22 arthropod species. 1:1:1 and N: N: N represent universal orthologs with single-copy or multiple-copy numbers, respectively. Insect, Diptera, Hemiptera, Hymenoptera, Lepidoptera, and Coleoptera stand for taxon-specific orthologs, respectively. Other indicates orthlogs that do not belong to any abovementioned ortholog categories. SD indicates species-specifically duplicated genes. ND indicates genes that cannot be classified into any other categories. The location of Laodelphax striatellus is indicated by an arrow.
We used 3 methods to evaluate the gene models that we obtained. First, we examined the 2 Kb upstream and downstream regions of annotated genes and found that the majority (16 525, 93.17%) of genes did not contain any ambiguous bases (Ns) in the 2 Kb up- and downstream regions, indicating that these gene models are not located near an assembly gap and thus the gene models are unlikely to be a fragment. Second, we compared our annotated genes with the corresponding orthlogous genes in D. melanogaster. We performed BLASTX (BLASTX, RRID:SCR_001653) [27] searches against the D. melanogaster gene set using the de novo assembled transcripts in L. striatellus. A total of 8484 assembled transcripts that had identity >60% with a D. melanogaster gene and covered >90% of the coding region were regarded as full-length transcripts. Among them, 3728 transcripts (excluding redundant protein isoforms) containing a complete ORF were searched against the annotated genes, and 3093 (82.97%) of them had a near perfect match to an annotated gene, indicating that most annotated genes were complete. Third, we compared our annotated genes to the 2 sets of BUSCO (v. 2) genes (1066 arthropoda genes and 303 eukaryote genes) [20] and found that our predicted genes were considered complete BUSCO genes in 97.6% and 98.0% of the eukaryote genes and arthropoda genes, respectively (Fig. S5), suggesting that a nearly complete repertoire of protein-coding gene set was determined.
To estimate the level of heterozygosity in the gene model, we aligned 23× reads to the genome assembly with BWA [18]. After removing duplicates, heterozygous SNPs were identified using BCFtools [38]. The heterozygous SNPs in the coding regions of each gene were used to compute read coverage and heterozygosity. Only a single heterozygosity peak of around 0.3 was detected (Fig. S6A). We ranked the heterozygosity rate of all the gene set and took the top 20% as high heterozygosity (the remainder was designated low heterozygosity). Coverage histograms of high and low heterozygosity showed similar ranges of coverage distribution (Fig. S6B). Therefore, the heterozygosity did not influence the gene annotation.
In order to obtain putative functional assignments to the annotated genes, we compared the annotated protein sequences of L. striatellus to proteins in the Kyoto Encyclopedia of Genes and Genomes (KEGG, RRID:SCR_012773) [39], NR [40], and Swiss-Prot [41] databases using BLASTP (BLASTP, RRID:SCR_001010) [27] with an E-value cutoff of 1E−5. Domains and motifs were scanned in Interpro [42] database by InterProScan (InterProScan, RRID:SCR_005829) [43]. There were 78.7%, 66.3%, 63.6%, and 69.5% of annotated proteins showing significant sequence similarity with the proteins in NR, Swiss-prot, KEGG, and InterPro (InterPro, RRID:SCR_006695), respectively. Among the 12 322 genes with an InterPro hit, 11 159 (90.6%) had Pfam (Pfam, RRID:SCR_004726) annotations and 8935 (72.5%) had gene ontology (GO, RRID:SCR_002811) associations. After removing redundancy, 14 182 of 17 736 genes (80.0%) were assigned to known databases (Fig. 3). Among the 3554 unannotated genes, 1391 (7.8%) were L. striatellus–specific genes.
Figure 3:
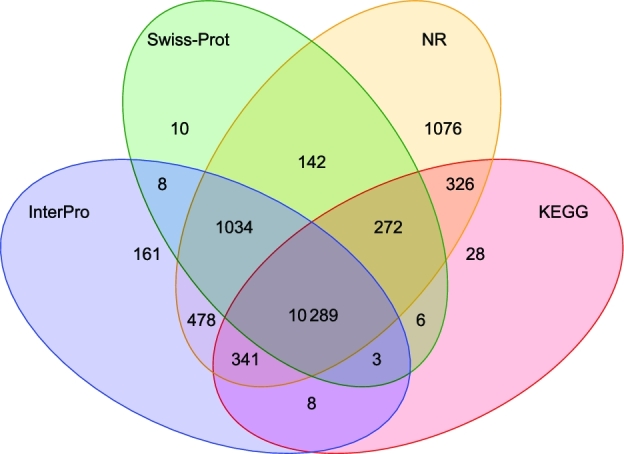
Venn diagram of functional annotation by 4 databases. NR: nonredundant protein databases.
Gene orthology prediction
Twenty-one sequenced insects (Zootermopsis nevadensis, Tribolium castaneum, Anoplophora glabripennis, Anopheles gambiae, D. melanogaster, A. pisum, Diuraphis noxia, Cimex lectularius, L. striatellus, R. prolixus, N. lugens, S. furcifera, Diaphorina citri, Oncopeltus fasciatus, Apis mellifera, N. vitripennis, B. mori, B. tabaci, Danaus plexippus, Locusta migratoria, and P. humanus) and 1 noninsect arthropoda sequenced species (D. pulex) were used to infer gene orthology and reconstruct the phylogenetic tree. The annotated coding sequences were downloaded from the websites listed in Table S7. The homologous gene families were identified using TreeFam [44, 45] and ascribed in different categories (Fig. 2). The gene families were identified following these steps: (i) BLASTP [27] was used to compare all protein sequences for the 22 species with an E-value cutoff of 1E−7; (ii) the blast alignments were concatenated by Solar (v. 0.9.6) [45], followed by homology identification among protein sequences; and (iii) gene families were identified using hcluster_sg (v. 0.5.0) [45]. RAxML (v. 8.0.19) [46] was used to reconstruct the phylogenetic tree based on the concatenated single-copy protein sequences under the PROTGAMMAAUTO model with 100 bootstrap replicates. R8s (v. 1.7.1) [47] and MCMCtree (PAML package, v. 4.7; PAML, RRID:SCR_014932) [48] were used to estimate the divergence times among species. The parameters used in MCMCtree were “–rootage 510 -clock 3 -alpha 0.977999 -model 7.” To examine gene family expansion and contraction in the 3 planthoppers, we chose 1 additional hemipteran species, R. prolixus, as outgroup to infer expanded/contracted gene families using CAFE (v. 3.1) [49]. A conditional P-value was calculated for each gene family, and the gene families with P-values <0.05 were considered as significantly expanded or contracted. The phylogenetic analysis revealed that L. striatellus clustered together with the other 2 planthoppers and had a closer relationship to S. furcifera than N. lugens (Fig. 4). The divergence times of nonplanthopper insect species were generally consistent with those estimated in the previous study [8]. The results of molecular dating analysis indicated that the ancestor of L. striatellus and S. furcifera split with N. lugens about 87.5 million years ago and that L. striatellus diverged from S. furcifera approximately 31 million years ago (Fig. S7).
Figure 4:

Phylogenetic analysis of 22 arthropod species. The phylogenetic tree was constructed based on amino acid sequences of 277 single-copy orthologs among 22 arthropod species (Anopheles gambiae, Anoplophora glabripennis, Apis mellifera, Acyrthosiphon pisum, Bombyx mori, Bemisia tabaci, Cimex lectularius, Diaphorina citri, Drosophila melanogaster, Diuraphis noxia, Danaus plexippus, Daphnia pulex, Locusta migratoria, Laodelphax striatellus, Nilaparvata lugens, Nasonia vitripennis, Oncopeltus fasciatus, Pediculus humanus, Rhodnius prolixus, Sogatella furcifera, Tribolium castaneum, Zootermopsis nevadensis) using the maximum likelihood algorithm. The tree was rooted with D. pulex.
Compared with N. lugens and S. furcifera, L. striatellus had fewer expanded gene families and more contracted gene families (Fig. S8). This might partially explain why L. striatellus has the lowest gene number among the 3 planthopper species. Since the divergence of L. striatellus and S. furcifera, L. striatellus and S. furcifera have had 95 and 547 expanded gene families, respectively (Fig. S8). The significantly expanded gene families in L. striatellus included some specific members of multigene families, such as odorant receptor, cytochrome P450, and serine protease (especially trypsin) (Table S10). The specific members of chemosensory protein, odorant binding protein, carboxylesterase, and ATP-binding cassette transporter families were also increased in L. striatellus although their P-values were higher than 0.05 (Table S10). Expansion of these gene families may have contributed to the widest host plant range of L. striatellus among the 3 planthoppers. The specific members of gene families associated with energy metabolism were significantly expanded in S. furcifera, such as acyl-CoA synthetase, fatty acyl-CoA reductase, acyl-CoA-binding protein, and acyl-coenzyme A thioesterase. The specific members of glyceraldehyde-3-phosphate dehydrogenase, D-beta-hydroxybutyrate dehydrogenase, ADP/ATP translocase, acyl-CoA transporter, and ATP synthase families also increased, although with P-values higher than 0.05 (Table S10). N. lugens had 433 expanded gene families (Fig. S8). A bunch of specific members from energy metabolism–related gene families, including Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, ATP-citrate synthase, malonyl-CoA decarboxylase, NADH dehydrogenase (ubiquinone) 1α subcomplex subunit 7 and subunit 8, acyl-CoA synthetase, ATP synthase, and enoyl-CoA delta isomerase increased in N. lugens although their P-values were higher than 0.05 (Table S10). Expansion in the energy metabolism–related gene families is in accordance with the migratory habits of S. furcifera and N. lugens.
Olfaction and detoxification system
It is essential for herbivorous insects to recognize and locate their host plants utilizing their sense of gustation and olfaction. Chemicals from the environment are received and recognized by chemoreceptor genes, including odorant receptors (ORs), gustatory receptors (GRs), and ionotropic receptors (IRs) in gustatory and olfactory organs. Detoxification gene families also play an essential role in defense against natural xenobiotics from host plants or synthetic xenobiotics including insecticides. To identify chemoreception and detoxification-related genes in L. striatellus, we retrieved corresponding gene sequences of other insect species from previous studies and used them as queries. These genes were searched against the L. striatellus gene set using BLASTP [27] with an E-value cutoff of 1E−5. In addition, we scanned the gene sets of 3 planthoppers for domain information using InterProScan and extracted genes with domains corresponding to each family. Finally, we integrated results from both BLASTP and InterProScan to obtain the final set of protein families.
There were 106 ORs, 38 IRs, and 12 GRs identified in L. striatellus (Table S11). The numbers of ORs and GRs in L. striatellus were more than twice as many as those in N. lugens and S. furcifera, representing a significant expansion in these 2 families. This is consistent with the fact that L. striatellus is the most polyphagous among the 3 planthoppers because polyphagous insects tend to have more OR genes than monophagous [8]. Moreover, we identified 2 protein families important for odor recognition and pheromone perception, namely odorant binding proteins (OBPs) and chemosensory proteins (CSPs). There were 16 OBPs and 31 CSPs in L. striatellus, the most among the 3 planthoppers (Table S11). The relatively higher number of odor-related genes in L. striatellus might be closely related to its polyphagous habit.
We manually annotated families of detoxification-related genes, including 26 UDP-glycosyltransferases, 29 glutathione-S-transferases, 54 carboxyl/cholinesterase, 73 ATP-binding cassette transporters, and 76 cytochrome P450s in L. striatellus (Table S12). The total number of detoxification-related genes in L. striatellus was smaller than that in N. lugens, but larger than that in S. furcifera.
Immune-related genes
We identified immune gene repertoires of the 3 planthoppers using a homology-based method. Immune genes from D. melanogaster, A. gambiae, Aedes aegypti, and Culex quinquefasciatus were downloaded from ImmunoDB [50]. Gene sets from the 3 planthoppers were used as queries and searched against the immune genes of the 4 insects, respectively, using BLASTX with an E-value cutoff of 1E−5. The best hits were selected for further domain architecture analysis using InterProScan and then were confirmed manually. The number of immune-related genes in L. striatellus was 330, which was more than that in N. lugens (289) and S. furcifera (280) (Table S13). The redundant copies of immune genes in L. striatellus mainly included autophagy genes, 1,3-beta-D glucan binding protein genes, clip-domain serine protease genes, and genes of small RNA regulatory pathway members. However, the numbers of C-type lectin genes and Toll-like receptor genes were lower in L. striatellus compared with the other 2 planthoppers.
Transcriptomic responses of 3 planthoppers to their borne plant viruses
L. striatellus, S. furcifera, and N. lugens transmit different rice viruses. To explore the molecular response to respective plant viruses, we analyzed and compared the transcriptomic responses of L. striatellus to RSV, S. furcifera to SRBSDV, and N. lugens to RRSV. The 3 viruses are transmitted in a persistent-propagative way. For L. striatellus, RSV was incubated in the fourth-instar nymphs for 5 days, as described previously [51]. Three replicates of infected or noninfected insects were used to construct paired-end RNA-seq libraries for sequencing on an Illumina Hiseq 2500 sequencer. The transcriptomic data of S. furcifera infected with SRBSDV were retrieved from a previous study [52]. The third-instar nymphs of N. lugens were infected by RRSV for 7 days before being collected for RNA extraction using the SV Total RNA Isolation System (Promega, Madison, WI, USA). The gene expression libraries for RRSV-infected and noninfected samples were constructed and sequenced on an Illumina HiSeq 2000 sequencer. RNA-seq reads were mapped to the corresponding genome using TopHat2 (v. 2.1.1) [53]. For L. striatellus and S. furcifera, HTSeq [54] was used to count the number of reads mapped to each gene model, and the edgeR package was used to identify differentially expressed genes (DEGs) with a fold change cutoff of 2 and FDR cutoff of 0.01. For N. lugens, generalized fold change for ranking differentially expressed genes from RNA-seq data was used to detect DEGs without biological replicates. The gene annotation files were downloaded from the corresponding websites (Table S14). We referred to genes with higher expressions in the viruliferous group as upregulated genes and lower as downregulated. The results showed that 460 (185 up and 275 down), 162 (48 up and 114 down), and 1070 (515 up and 555 down) genes were differentially expressed in L. striatellus, N. lugens, and S. furcifera, respectively, when bearing their respective plant viruses.
The DEGs in the 3 planthoppers were compared in GO terms, and the common GO terms were retrieved (Table S15). The upregulated genes in the 3 planthoppers were involved in the biological processes of regulation of transcription (GO:0 006355) and protein phosphorylation (GO:0 006468). The downregulated genes in the 3 planthoppers took part in the biological processes of carbohydrate metabolic process (GO:0 005975), chitin catabolic process (GO:0 006032), and proteolysis (GO:0 006508).
Two zinc finger proteins of L. striatellus, 1 zinc finger protein of N. lugens, and 6 zinc finger proteins of S. furcifera were commonly upregulated while genes of chitinases, cytochrome P450 CYP4s, and trypsins were commonly downregulated in the 3 planthoppers (Table S16) in response to their respective plant viruses. We also identified homologous genes that were commonly regulated in the 3 planthoppers by aligning N. lugens DEGs with those of L. striatellus and S. furcifera using BLASTP with a cutoff of 1E−3, a sequence identity higher than 60%, and a coverage higher than 50%. Three groups of homologous genes, including 1 group of commonly upregulated genes and 2 groups of commonly downregulated genes, were retrieved from the 3 planthoppers (Table S17). The protein lengths of these homologous genes ranged from 120 to 472 amino acids. We used these proteins as queries to search the NR database and found no homologous genes in other species with a cutoff of 1E−7, indicating that these genes are likely planthopper-specific genes.
Differences in immune response to virus infection in the 3 planthoppers were also observed. The RNAi pathway genes, RISC-loading complex TARBP2 and argonaute-3, were upregulated in S. furcifera and N. lugens, respectively, but genes in the RNAi pathway did not respond to virus infection in L. striatellus. The antimicrobial peptide defensin was upregulated in L. striatellus and N. lugens but was downregulated in S. furcifera. The expression of the Down syndrome cell adhesion molecule gene increased in L. striatellus [55] and decreased in S. furcifera, but did not show significant change in N. lugens in response to their respective plant viruses.
In summary, we reported a high-quality of genome of L. striatellus, a notorious rice pest insect. L. striatellus has the smallest genome and the lowest number of protein-coding genes compared with the other 2 rice planthoppers, S. furcifera and N. lugens. Comparative genomic analyses identified expansions and contractions in olfactory genes, detoxification genes, immune genes, and energy metabolism genes among the 3 rice planthoppers, which may have contributed to their differences in important traits such as host range, migratory habit, and plant virus transmission. Despite having the smallest genome, L. striatellus has the widest host plant range among the 3 planthoppers. This situation is different from that of the genome evolution in Aphididae, where the soybean aphid, Aphis glycines, which is an extreme specialist, has the smallest genome compared with another 3 aphid species with published genome sequences [56]. With the addition of the L. striatellus genome, the genome data of the 3 rice planthoppers will aid studies in various areas of planthoppers and promote control strategies in the future.
Availability of supporting data
Genome sequencing and transcriptome data used for genome assembly and gene annotation are deposited in the SRA under bioproject number PRJNA393384. Further supporting data, including annotations, gene expression data, alignments, and BUSCO results, are available via the GigaScience repository, GigaDB (GigaDB, RRID:SCR_004002) [57].
Abbreviations
BUSCO: benchmarking universal single-copy ortholog; CSP: chemosensory protein; DEG: differentially expressed gene; GO: gene ontology; GR: gustatory receptor; IR: ionotropic receptor; KEGG: Kyoto Encyclopedia of Genes and Genomes; OBP: odorant binding protein; OR: odorant receptor; RBSDV: rice black-streaked dwarf virus; RRSV: rice ragged stunt virus; RSV: rice stripe virus; SRBSDV: Southern rice black streak dwarf virus; TE: transposable element.
Additional file
Table S1. Base composition of the Laodelphax striatellus genome assembly.
Table S2. Summary of reads mapping to the genome assembly of Laodelphax striatellus.
Table S3. Transcript-based evaluation of the genome assembly of Laodelphax striatellus.
Table S4. Statistics of 9 transcriptomic reads mapped to different genomic regions.
Table S5. Genome completeness assessment using benchmarking universal single copy orthologs in 5 insects.
Table S6. Repetitive elements predicted by different programs.
Table S7. Sources of genome data of 22 arthropod species.
Table S8. Gene models predicted by different methods.
Table S9. Statistical comparison of gene sets of Laodelphax striatellus and 9 other arthropod species.
Table S10. Expanded gene families in the 3 planthoppers.
Table S11. Chemoreception-related genes in the 3 planthoppers.
Table S12. Detoxification-related genes in the 3 planthoppers.
Table S13. Immune genes in the 3 planthoppers.
Table S14. Sources of gene annotation files for the 3 planthoppers.
Table S15. Shared gene ontology terms for differentially expressed genes in the 3 planthoppers responding to plant viruses.
Table S16. Commonly regulated genes with similar functions in the 3 planthoppers responding to plant viruses.
Table S17. Homologous genes in the 3 planthoppers responding to plant viruses.
Figure S1. Laodelphax striatellus genome size estimation by flow cytometry and k-mer analyses. (A), (B), and (C) Fluorescence peaks for Drosophila melanogaster, Gallus gallus, and L. striatellus, respectively. The genome sizes of D. melanogaster and G. gallus were 0.18 pg and 1.25 pg, respectively. The genome size of L. striatellus was calculated to be 0.60 pg. (D) The depth distribution of k-mers (k = 17).
Figure S2. Laodelphax striatellus chromosomes dyed with Hoechst 33 258. (A) Haploid chromosomes. (B) Diploid chromosomes.
Figure S3. Sequencing depth distribution. The x-axis shows sequencing depth, and the y-axis shows fraction of bases with certain sequencing depth.
Figure S4. Summary of gene structures of Laodelphax striatellus and 8 other species used for gene annotation.
Figure S5. BUSCO assessment of the Laodelphax striatellus gene set. The completeness of the gene set was assessed with 2 BUSCO v. 2 datasets (arthropoda and eukaryote). The recovered matches are classified as “complete” if their lengths are within the expectation of the BUSCO profile match lengths. If these are found only once, they are classified as “complete single,” and other “complete” matches are classified as “complete duplicated.” The matches that are only partially recovered are classified as “fragmented,” and BUSCO groups for which there are no matches that pass the tests of orthology are classified as “missing.” For each species, the right bar shows the arthropoda results and the left bar shows the eukaryote results. Aga: Anopheles gambiae; Agl: Anoplophora glabripennis; Ame: Apis mellifera; Api: Acyrthosiphon pisum; Bmo: Bombyx mori; Bta: Bemisia tabaci; Cle: Cimex lectularius; Dci: Diaphorina citri; Dme: Drosophila melanogaster; Dno: Diuraphis noxia; Dpl: Danaus plexippus; Dpu: Daphnia pulex; Lmi: Locusta migratoria; Lst: Laodelphax striatellus; Nlu: Nilaparvata lugens; Nvi: Nasonia vitripennis; Ofa: Oncopeltus fasciatus; Phu: Pediculus humanus; Rpr: Rhodnius prolixus; Sfu: Sogatella furcifera; Tca: Tribolium castaneum; Zne: Zootermopsis nevadensis.
Figure S6. Determination of genomic heterozygosity. (A) Density distribution of heterozygous rates. (B) Frequency distribution of read coverage of both high and low heterozygosity. All heterozygosity rates were ranked, and the top 20% were chosen as high heterozygosity (high_het in the legend) and the remainder as low heterozygosity (low_het in the legend).
Figure S7. Divergence times estimation of 22 arthropod species. The number on each node stands for the divergence time from the present (million years ago [Mya]), with 95% confidence interval values noted in brackets. Four calibration times were used in the estimation: D. pulex–D. melanogaster divergence (445∼530 Mya), N. vitripennis–D. melanogaster divergence (279∼306 Mya), A. gambiae–D. melanogaster divergence (235∼269 Mya), and A. mellifera–N. vitripennis divergence (175∼215 Mya). The location of L. striatellus was indicated by an arrow.
Figure S8. Gene family expansion and contraction in the 3 planthoppers. R. prolixus was used as an outgroup to construct the phylogenetic tree and infer expanded/contracted gene families by CAFÉ. A conditional P-value was calculated for each gene family, and families with P-values <0.05 were considered significantly expanded (green) or contracted (red).
Competing interests
The authors declare that there are no financial and nonfinancial competing interests in this study.
Funding
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB11040200), Major State Basic Research Development Program of China (973 Program; No. 2014CB13840402), and Natural Science Foundation of China (No. 31371934).
Author contributions
J.Z. and F.J. collected the samples, prepared the DNA and RNA, analyzed the data, and drafted the paper. X.W. and P.Y. coordinated the project. Y.B. sequenced the transcriptomes. W.Z., W.W., H.L., Q.W., N.C., J.L., X.C., L.L., and J.Y. analyzed the data. L.K. and F.C. designed the research and wrote and revised the paper.
Supplementary Material
23 Aug 2017 Reviewed
26 Sep 2017 Reviewed
19 Sep 2017 Reviewed
Acknowledgements
We thank Prof. Thomas Sicheritz-Pontén from Technical University of Denmark and Prof. Renyi Liu from Shanghai Center for Plant Stress Biology and Center of Excellence for Molecular Plant Sciences, Chinese Academy of Sciences, for comments and language suggestions.
References
- 1. Dmitriev DA. 3I Interactive Keys and Taxonomic Databases.2003. http://dmitriev.speciesfile.org/index.asp. Accessed 1 May 2017.
- 2. Genus Laodelphax Fennah, 1963. College of Agriculture and Natural Resources, University of Delaware; http://ag.udel.edu/research/delphacid/species/Laodelphax.htm. Accessed 1 May 2017. [Google Scholar]
- 3. Sun DZ, Jiang L. Research on the inheritance and breeding of rice stripe resistance. Chin Agricult Sci Bull 2006;12:073. [Google Scholar]
- 4. Huang H, Xue J, Zhuo J et al. Comparative analysis of the transcriptional responses to low and high temperatures in three rice planthopper species. Mol Ecol 2017;26(10):2726–37. [DOI] [PubMed] [Google Scholar]
- 5. Zhou G, Wen J, Cai D et al. Southern rice black-streaked dwarf virus: a new proposed Fijivirus species in the family Reoviridae. Chin Sci Bull 2008;53(23):3677–85. [Google Scholar]
- 6. Jia D, Guo N, Chen H et al. Assembly of the viroplasm by viral non-structural protein Pns10 is essential for persistent infection of rice ragged stunt virus in its insect vector. J Gen Virolo 2012;93(Pt_10):2299–309. [DOI] [PubMed] [Google Scholar]
- 7. Zheng L, Mao Q, Xie L et al. Infection route of rice grassy stunt virus, a tenuivirus, in the body of its brown planthopper vector, Nilaparvata lugens (Hemiptera: Delphacidae) after ingestion of virus. Virus Res 2014;188:170–3. [DOI] [PubMed] [Google Scholar]
- 8. Xue J, Zhou X, Zhang C et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol 2014;15(12):521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang L, Tang N, Gao X et al. Genome sequence of a rice pest, the white-backed planthopper (Sogatella furcifera). Gigascience 2017;6(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hare EE, Johnston JS. Genome size determination using flow cytometry of propidium iodide-stained nuclei. Mol Methods Evol Genet 2011:3–12. [DOI] [PubMed] [Google Scholar]
- 11. Li R, Fan W, Tian G et al. The sequence and de novo assembly of the giant panda genome. Nature 2010;463(7279):311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bennett MD. Comparisons with caenorhabditis (100 Mb) and drosophila (175 Mb) using flow cytometry show genome size in arabidopsis to be 157 Mb and thus 25 % larger than the arabidopsis genome initiative estimate of 125 Mb. Ann Botany 2003;91(5):547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang J, Zhang CM, Zhao X et al. The Jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet 2016;12(12):e1006433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang S, Zhang J, Jiao W et al. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat Ecol Evol 2017;1(5):0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. English AC, Richards S, Han Y et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 2012;7(11):e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 2012;13(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelley RK, Wang G, Venook AP. Biomarker use in colorectal cancer therapy. J Natl Compr Canc Netw 2011;9(11):1293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao W, Lu L, Yang P et al. Organ-specific transcriptome response of the small brown planthopper toward rice stripe virus. Insect Biochem Mol Biol 2016;70:60–72. [DOI] [PubMed] [Google Scholar]
- 20. Simão FA, Waterhouse RM, Ioannidis P et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 21. Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 2015;6(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. TarailoGraovac M, Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 2009:4.10.1–14. [DOI] [PubMed] [Google Scholar]
- 23. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007;35(Web Server):W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics 2005;21(Suppl 1):i152–8. [DOI] [PubMed] [Google Scholar]
- 25. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics 2005;21(Suppl 1):i351–8. [DOI] [PubMed] [Google Scholar]
- 26. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27(2):573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altschul SF, Gish W, Miller W et al. Basic local alignment search tool. J Mol Biol 1990;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- 28. Birney E. GeneWise and Genomewise. Genome Res 2004;14(5):988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keller O, Kollmar M, Stanke M et al. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011;27(6):757–63. [DOI] [PubMed] [Google Scholar]
- 30. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004;20(16):2878–9. [DOI] [PubMed] [Google Scholar]
- 31. Bedell JA, Korf I, Gish W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 2000;16(11):1040–1. [DOI] [PubMed] [Google Scholar]
- 32. Blanco E, Abril JF. Computational gene annotation in new genome assemblies using GeneID. Bioinformatics DNA Seq Anal 2009:243–61. [DOI] [PubMed] [Google Scholar]
- 33. Blanco E, Parra G, Guigó R. Using Geneid to identify genes. Curr Protoc Bioinformatics 2007:4.3.1–28. [DOI] [PubMed] [Google Scholar]
- 34. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997;268(1):78–94. [DOI] [PubMed] [Google Scholar]
- 35. Trapnell C, Roberts A, Goff L et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7(3):562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haas BJ, Delcher AL, Mount SM et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 2003;31(19):5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haas BJ, Salzberg SL, Zhu W et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 2008;9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H, Handsaker B, Wysoker A et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2006;35(suppl_1):D61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.UniProt Consortium.UniProt: a hub for protein information. Nucleic Acids Res 2014:43(Database issue):D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hunter S, Apweiler R, Attwood TK et al. InterPro: the integrative protein signature database. Nucleic Acids Res 2008;37(suppl_1):D211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001;17(9): 847–8. [DOI] [PubMed] [Google Scholar]
- 44. Li H, Coghlan A, Ruan J et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res 2006;34(90001):D572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruan J, Li H, Chen Z et al. TreeFam: 2008 update. Nucleic Acids Res 2008;36(Database issue):D735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30(9):1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003;19(2):301–2. [DOI] [PubMed] [Google Scholar]
- 48. Van De Wiel MA, Leday GGR, Pardo L et al. Bayesian analysis of RNA sequencing data by estimating multiple shrinkage priors. Biostatistics 2013;14(1):113–28. [DOI] [PubMed] [Google Scholar]
- 49. De Bie T, Cristianini N, Demuth JP et al. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 2006;22(10):1269–71. [DOI] [PubMed] [Google Scholar]
- 50. Immuno DB. EM Zdobnov Group. 2008. http://cegg.unige.ch/Insecta/immunodb. Accessed 29 May 2016.
- 51. Zhao W, Yang P, Kang L et al. Different pathogenicities of rice stripe virus from the insect vector and from viruliferous plants. New Phytol 2016;210(1):196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Tang N, Gao XL et al. Understanding the immune system architecture and transcriptome responses to Southern rice black-streaked dwarf virus in Sogatella furcifera. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim D, Pertea G, Trapnell C et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang F, Li Q, Chen X et al. Roles of the L aodelphax striatellus down syndrome cell adhesion molecule in rice stripe virus infection of its insect vector. Insect Mol Biol 2016;25(4):413–21. [DOI] [PubMed] [Google Scholar]
- 56. Wenger JA, Cassone BJ, Legeai F et al. Whole genome sequence of the soybean aphid, Aphis glycines. Insect Biochem Mol Biol 2017. [DOI] [PubMed] [Google Scholar]
- 57. Zhu JJ, Jiang F, Wang XH et al. Supporting data for “Genome sequence of the small brown planthopper, Laodelphax striatellus.” GigaScience Database 2017. http://dx.doi.org/10.5524/100361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
23 Aug 2017 Reviewed
26 Sep 2017 Reviewed
19 Sep 2017 Reviewed