

Summary
Abnormal folate one-carbon metabolism (FOCM) is implicated in neural tube defects (NTDs), severe malformations of the nervous system. MTHFR mediates unidirectional transfer of methyl groups from the folate cycle to the methionine cycle and, therefore, represents a key nexus in partitioning one-carbon units between FOCM functional outputs. Methionine cycle inhibitors prevent neural tube closure in mouse embryos. Similarly, the inability to use glycine as a one-carbon donor to the folate cycle causes NTDs in glycine decarboxylase (Gldc)-deficient embryos. However, analysis of Mthfr-null mouse embryos shows that neither S-adenosylmethionine abundance nor neural tube closure depend on one-carbon units derived from embryonic or maternal folate cycles. Mthfr deletion or methionine treatment prevents NTDs in Gldc-null embryos by retention of one-carbon units within the folate cycle. Overall, neural tube closure depends on the activity of both the methionine and folate cycles, but transfer of one-carbon units between the cycles is not necessary.
Keywords: one-carbon metabolism, folic acid, neural tube defects, spina bifida, glycine cleavage system, non-ketotic hyperglycinemia, eye, Mthfr, Gldc
Graphical Abstract
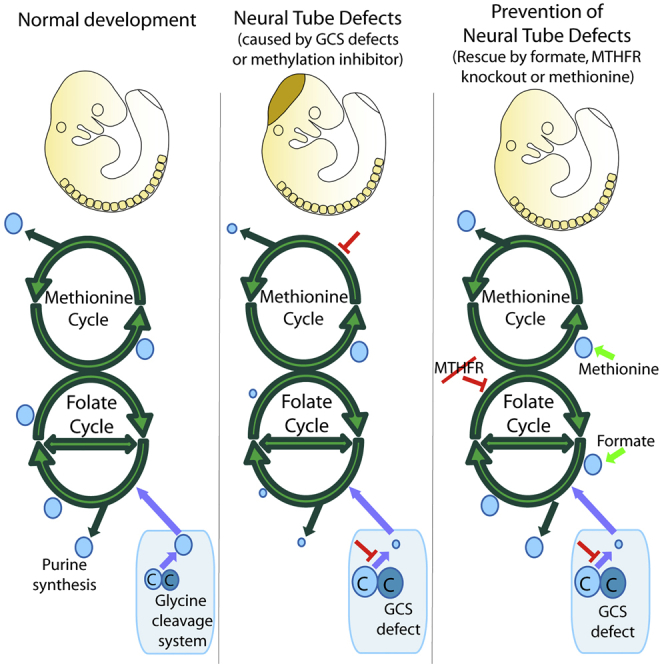
Highlights
-
•
Inhibition of methionine cycle activity prevents neural tube closure, causing NTDs
-
•
Loss of embryonic and maternal MTHFR activity does not prevent neural tube closure
-
•
Glycine is a 1C donor to the folate cycle via the glycine cleavage system in the embryo
-
•
Ablation of glycine cleavage causes NTDs, preventable by MTHFR inactivity or methionine
Leung at al. find that embryonic neural tube closure depends both on the supply of one-carbon units to the folate cycle from glycine cleavage and on the methionine cycle. In contrast, transfer of one-carbon units from the folate cycle to the methionine cycle by MTHFR is dispensable.
Introduction
Folate one-carbon metabolism (FOCM) comprises an interlinked network of reactions that transfer one-carbon (1C) units for numerous cellular functions (Figure 1A; Tibbetts and Appling, 2010, Locasale, 2013). Key outputs include provision of 1C units for biosynthesis of thymidylate and purines and generation of S-adenosylmethionine (SAM), the “universal” methyl donor for methylation of DNA, RNA, proteins, and lipids (Brosnan et al., 2015, Ducker and Rabinowitz, 2017). FOCM is also central to a larger metabolic network through links, for example, to polyamine synthesis and creatine synthesis and the transulfuration pathway. Abnormalities of FOCM are associated with a number of diseases, including cancers, fatty liver disease, cardiovascular disease, inborn errors of metabolism (such as non-ketotic hyperglycinemia), and age-related cognitive impairment. The association of folate status with birth defects, such as neural tube defects (NTDs), also implicates FOCM in playing a key role during development.
Figure 1.

Altered Abundance of Folates in Mthfr-Null Embryos
(A) Summary diagram of folate 1C metabolism (enzymes are indicated in purple text).
(B–E) LC-MS/MS profiling of folates in (B) post-natal brain, (C) post-natal liver, and embryos at E10.5 (D) and E12.5 (E) shows that the relative abundance of 5-methyl THF (CH3THF) is significantly reduced in Mthfr−/− (∗p < 0.0001 compared with Mthfr+/+). Conversely, the abundance of other folates was increased (∗p < 0.01, ∗∗p < 0.05; significant difference from wild-type, p < 0.05). Note that CH2-THF may be under-represented because of conversion to THF during analysis (maximal 20% of CH2-THF at pH 7 used). Number of samples, n = 3 per genotype for brain, liver, and E12.5. At E10.5, n = 4 Mthfr+/+, 7 Mthfr+/−, and 5 Mthfr−/−.
Flux through FOCM depends on both the availability of tetrahydrofolate (THF), which is the 1C “carrier” in the folate cycle, and on supply of 1C units, principally derived from serine (Davis et al., 2004, Ducker and Rabinowitz, 2017, Yang and Vousden, 2016). Mammals cannot synthesize folates de novo and depend on sources from the diet and microbiota. In mice, cellular uptake of folate in the embryo is essential for viability and for completion of neural tube closure. Embryos lacking Folr1, encoding folate receptor 1, require supplementation with folate (methyl-THF or formyl-THF) for survival to neurulation stages and display frequent NTDs (Spiegelstein et al., 2004). In contrast, mouse embryos are relatively resistant to maternal folate deficiency. Folate depletion, achieved through defined diets and antibiotic treatment, diminishes embryonic folate content (Heid et al., 1992, Burren et al., 2008). Folate availability limits growth and developmental progression but does not dissociate these parameters or cause NTDs in wild-type embryos (Burren et al., 2008). Nevertheless, maternal folate deficiency exacerbates NTDs in some genetically susceptible mouse strains (Burren et al., 2008, Burren et al., 2010, Beaudin et al., 2011).
In parallel with a requirement for availability of the THF backbone, as supplied by dietary folate or synthetic folic acid, mouse genetic mutants provide compelling evidence that an adequate supply of 1C units is essential for neural tube closure. Serine can be a 1C donor via the action of Shmt1 in the cytoplasm and nucleus (Beaudin et al., 2011, Herbig et al., 2002, MacFarlane et al., 2011). However, the majority of 1C units entering the folate and methionine cycles appear to derive from mitochondrial FOCM, with transfer of formate to the cytoplasm (Tibbetts and Appling, 2010). The inability to generate formate from 10-formyl THF in mouse embryos lacking the mitochondrial 10-formyl THF synthetase enzyme (Mthfd1L) results in growth retardation and frequent failure of neural tube closure (Momb et al., 2013). Similarly, loss of function of components of the mitochondrial glycine cleavage system, Amt or Gldc, also causes NTDs in mice (Narisawa et al., 2012, Pai et al., 2015). Functional mutations in the corresponding human genes AMT and GLDC have also been found in NTD patients (Narisawa et al., 2012, Shah et al., 2016). Prevention of NTDs by maternal formate supplementation in Mthfd1L and Gldc mouse mutants supports the hypothesis that impaired neural tube closure results from diminished supply of 1C into FOCM in these models (Momb et al., 2013, Pai et al., 2015). This concept is supported by alteration in the relative abundance of folates and normalization of the folate profile by formate in Gldc-deficient embryos (Pai et al., 2015). Loss of function of the glycine cleavage system also causes an elevated embryonic tissue glycine concentration (Pai et al., 2015). It is not yet known whether the ensuing abnormalities of the folate profile and NTDs in Gldc-null embryos result directly from lack of glycine-derived 1C. Alternatively, it could be hypothesized that elevated glycine acts through product inhibition to reverse the action of Shmt in using serine as the 1C donor, as observed in some cancer cell lines (Labuschagne et al., 2014).
Although the Amt, Gldc, and Mthfd1L loss-of-function mouse mutants emphasize an essential role for supply of 1C units from mitochondrial FOCM, the relative requirements for 1C consumption in the folate and methionine cycles (Figure 1A) during neural tube closure are not well defined. Inhibitors of the methionine cycle or knockout of DNA methyltransferase cause cranial NTDs in mouse embryos (Okano et al., 1999, Dunlevy et al., 2006, Burren et al., 2008) and, similarly, inhibit neural tube closure in chick and frog embryos (Afman et al., 2005, Toriyama et al., 2017). Moreover, among folate cycle functions, impaired thymidylate biosynthesis is associated with NTDs induced by folate deficiency in Pax3- or Shmt1-null mice (Fleming and Copp, 1998, Burren et al., 2008, Beaudin et al., 2011). Therefore, various functional outputs of FOCM may be required in neural tube closure, implying a necessity for regulated flux through multiple reaction pathways for partitioning of 1C units according to metabolic need.
The transfer of 1C units from the folate cycle to the methionine cycle is mediated by 5,10-methylene tetrahydrofolate reductase (MTHFR) in a unidirectional reaction, which therefore commits 1C units to the methionine cycle (Tibbetts and Appling, 2010). Computational modeling of hepatic FOCM predicts the outcome of altered flux through specific reaction(s) (Reed et al., 2006). However, the presence of several loops within the reaction network, the potential for differential cell and tissue context-dependent regulation (Leung et al., 2013, Ducker and Rabinowitz, 2017), as well as embryonic-maternal interactions highlight the complexity in embryonic FOCM. Hence, it is necessary to analyze the effect of FOCM on neural tube closure in the developing embryo. Here we investigated the requirement for partitioning of 1C units between mitochondrial FOCM, the folate cycle, and the methionine cycle using models with loss of transfer of 1C units from the folate cycle to the methionine cycle (Mthfr-null) or impaired supply of 1C units to the folate cycle from mitochondria (Gldc-deficient).
Results
Methyl Groups Derived from Embryonic or Maternal Folate Metabolism Are Not Required for Neural Tube Closure
Mthfr-null mice are viable and born in the expected Mendelian ratio but exhibit reduced survival and a range of phenotypes, including slower growth, liver steatosis, altered brain histology, and behavioral abnormalities (Chen et al., 2001, Schwahn et al., 2003, Jadavji et al., 2015, Lawrance et al., 2011). Tissues from post-natal Mthfr-null mice showed diminished abundance of 5-methyl THF as a proportion of total folate (Chen et al., 2001, Ghandour et al., 2004). We extended this analysis using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) methodology that allows quantification of the six major folates (Pai et al., 2015). Folates are quantified as individual polyglutamated forms, up to 7 glutamates (5 or 6 glutamates being the predominant form in mouse tissue) (Figure S1; Leung et al., 2013, Pai et al., 2015). The folate profiles confirmed that 5-methyl THF is virtually absent from the liver and brain of Mthfr-null mice at 3 weeks, with a corresponding increase in the relative abundance of other folates, principally THF and methylene-THF (CH2-THF; Figures 1B and 1C).
Among litters of embryos generated by intercrossing of Mthfr+/− mice, homozygous null embryos were present in the expected Mendelian ratio, and NTDs were not observed (n = 46 Mthfr−/− examined at embryonic day 9.5 [E9.5]–E12.5; Table S1), consistent with previous findings (Chen et al., 2001, De Castro et al., 2010). The major folate present in plasma is 5-methyl THF, and it is proposed that folate obtained from the maternal circulation may sustain Mthfr−/− embryos in utero, facilitating neural tube closure and allowing survival to birth (Schwahn et al., 2004). However, folate profiling at E10.5 and E12.5 showed that Mthfr−/− embryos contain minimal 5-methyl THF, comprising only approximately 1% or 2.5% of total folate in Mthfr−/− embryos at E10.5 and E12.5, respectively (Figures 1D and 1E; Figure S1). This is comparable with a 5-methyl THF relative abundance of 89% and 95% of total folate in Mthfr+/+ embryos at E10.5 and E12.5 (Figures 1D and 1E) and 80% and 93% of total folate at E10.5 and E12.5 in Mthfr+/−. In Mthfr−/− embryos, there was a corresponding increase in the relative abundance of other folates (Figures 1D and 1E; Figure S1).
At post-natal stages, 5-methyl THF generated by Mthfr contributes to remethylation of homocysteine, as shown by raised plasma homocysteine concentrations in heterozygous and null mice together with an increased abundance of SAH and lower SAM in most tissues analyzed (Chen et al., 2001; Figure 2). In contrast, SAM abundance was not significantly altered in Mthfr−/− embryos compared with Mthfr+/+ and Mthfr+/− littermates (Figure 2A). Nevertheless, Mthfr−/− embryos show a significantly elevated abundance of S-adenosylhomocysteine (SAH) (reflecting decreased remethylation of homocysteine) with a consequent reduction in the SAM/SAH ratio (Figures 2B and 2C). Although neural tube closure was not perturbed in Mthfr−/− embryos, these findings suggest that methylation reactions could potentially be compromised. For example, in adult tissues, elevated SAH was found to be a more consistent marker of DNA hypomethylation than SAM in cystathionine β-synthase heterozygous mice maintained on a methyl-deficient diet, although whether methylation changes were detectable varied with tissue (Caudill et al., 2001). However, we found no effect of Mthfr genotype on cytosine methylation of DNA in the embryo (Figure 2E).
Figure 2.

Methionine Cycle Intermediates Are Differentially Affected by Mthfr Genotype in Post-natal Tissue and Embryos
(A) The abundance of S-adenosylmethionine (SAM) was significantly lower in livers of Mthfr−/− mice than in wild-types (∗p < 0.05), and SAM showed a non-significant trend toward lower abundance in brains of Mthfr−/− (p = 0.07). SAM abundance was not altered in Mthfr−/− E12.5 embryos compared with littermates of other genotypes.
(B and C) In contrast, S-adensylhomocysteine concentration (B, SAH) was elevated, and the SAM/SAH ratio (C) was reduced in Mthfr−/− tissue and embryos compared with Mthfr+/+ (∗p < 0.01, significantly differs from Mthfr+/+). n = 3 samples per genotype for each tissue.
(D–F) The proportion of (D) 2-deoxycytosine, (E) 5-methyldeoxycytosine, and (F) 5-hydroxy-2-deoxycytosine did not differ with Mthfr genotype among embryos at E12.5 (individual samples and mean ± SEM are shown).
See also Table S2.
Although generation of 5-methyl THF in the embryo was not essential for neural tube closure or maintenance of SAM abundance, this did not rule out a requirement for folate cycle-derived methyl groups because null embryos developed in the context of an Mthfr+/− maternal environment. We hypothesized that maternal folate-derived methyl groups could plausibly contribute to the methionine cycle. Therefore, as a further step, we carried out a two-step breeding program to generate additional litters using Mthfr−/− dams. Mthfr−/− offspring of Mthfr−/− dams were indistinguishable from Mthfr+/− littermates (n = 10 Mthfr+/− and 7 Mthfr−/−). NTDs were not observed among these litters (Table S1), showing that embryonic and maternal Mthfr activity are both dispensable for neural tube closure. As observed for offspring of Mthfr+/− dams, there was no reduction in abundance of SAM in Mthfr−/− embryos from Mthfr−/− dams (Table S2), analyzed at E10.5, although the SAM/SAH ratio was similarly diminished because of elevated SAH (Table S2). Methionine itself is an essential amino acid for protein synthesis. Hence, although folate-dependent methionine synthesis is dispensable for neural tube closure, it seems likely that sufficient methionine for embryo survival is obtained via the maternal diet and/or the action of Bhmt.
Methionine Cycle Inhibitors Cause NTDs but Not via a Methyl Trap
Lack of methyl group transfer from the folate cycle to the methionine cycle does not cause NTDs, despite a significantly lower SAM/SAH ratio in Mthfr-null embryos. In contrast, we previously found that inhibitors of the methionine cycle (cycloleucine or ethionine) cause a high frequency of cranial NTDs in wild-type embryos in whole-embryo culture (Dunlevy et al., 2006). NTDs arise without generalized growth-retarding or toxic effects (Dunlevy et al., 2006). Cycloleucine is an inhibitor of methionine adenosyltransferase (MAT) activity that causes elevated SAH and a lower SAM/SAH ratio in treated embryos (Dunlevy et al., 2006). Ethionine is an ethyl analog of methionine that competes with methionine for MAT and is converted to S-adenosyl ethionine (SAE). Treated embryos have a significantly lower abundance of SAM, elevated SAH, and a lower SAM/SAH ratio (Dunlevy et al., 2006), whereas SAE also inhibits SAM-dependent methyltransferases directly (Alix, 1982). We cannot discount the possibility of non-specific effects, particularly of ethionine, but the induction of NTDs by two inhibitors that act via different mechanisms and without other embryotoxic effects suggests that methionine cycle function is required for neural tube closure. Two possible mechanisms could plausibly underlie NTDs induced by these inhibitors: direct suppression of methylation reactions or imposition of a “methyl trap.” The methyl trap hypothesis proposes that impaired methionine cycle flux leads to trapping of folates as 5-methyl THF because of the unidirectional nature of the Mthfr-mediated reaction. Inability to regenerate THF would suppress folate cycle functions, and this mechanism is postulated to explain some effects of vitamin B12 deficiency (Scott, 1999).
Mthfr-null embryos would be predicted to be resistant to a methyl trap because of their inability to generate 5-methyl THF. Arguing against a methyl trap mechanism underlying the effect of ethionine or cycloleucine, we found that NTDs were induced at similar frequencies in Mthfr+/+, Mthfr+/−, and Mthfr−/− embryos treated in whole-embryo culture from E8.5 (Figures 3A–3C). Moreover, analysis of embryonic folate profiles showed that ethionine treatment significantly decreased the relative abundance of 5-methyl THF in Mthfr+/+ embryos (Figure 3D). These findings suggest that SAE inhibits Mthfr activity, in common with the strong inhibitory effect of SAM, and show that ethionine does not cause a methyl trap in wild-type embryos. In Mthfr−/− embryos (which act as a methyl trap-resistant control), 5-methyl THF was virtually absent, irrespective of ethionine treatment (Figure 3E). Unlike ethionine, cycloleucine had relatively little effect on the folate profile of Mthfr+/+ or Mthfr−/− embryos (Figures 3D and 3E). Hence, these inhibitors are proposed to cause NTDs through suppression of the methionine cycle and not depletion of 1C carrying folates through a methyl trap.
Figure 3.

Methionine Cycle Inhibitors Cause NTDs without Imposition of a Methyl Trap
(A–C) Treatment in whole-embryo culture with ethionine (5 mM) or cycloleucine (15 mM) caused NTDs among embryos of all Mthfr genotypes (A). NTDs affected the cranial region (open midbrain-forebrain neural folds in cycloleucine-treated embryo; arrow in B), whereas closure was completed during the culture period in vehicle-treated controls (C). Scale bar represents 1 mm.
(D) In Mthfr+/+ embryos, ethionine treatment resulted in a significant increase in relative abundance of THF. Both ethionine and cycloleucine caused a decrease in relative abundance of 5-methyl THF compared with controls (∗p < 0.01, ANOVA).
(E) In cultured Mthfr−/−embryos, abundance of folate intermediates was not significantly affected by ethionine or cycloleucine, and, as in non-cultured embryos, the relative abundance of THF and 5-methyl THF differed from Mthfr+/+ embryos (compare with D, p < 0.001, two-way ANOVA).
In (D) and (E), n = 3–5 samples per genotype for each treatment
Neural Tube Closure Depends on Glycine Decarboxylase Function
Having analyzed Mthfr−/− embryos, in which partitioning of 1C units from the folate cycle to the methionine cycle is prevented, we analyzed embryos in which we hypothesize the supply of 1C units into the folate cycle is limited. Mitochondrial FOCM supplies 1C units to the cytoplasmic folate cycle as formate. This appears to be essential for neural tube closure because NTDs occur in embryos lacking expression of the mitochondrial FOCM components Amt, Mthfd1L, or Gldc (Narisawa et al., 2012, Momb et al., 2013, Pai et al., 2015) and may be rescued by formate supplementation. Gldc and Amt encode components of the glycine cleavage system (GCS), which mediates decarboxylation of glycine with transfer of a 1C unit to THF, generating 5,10-methylene THF (Figure 1A).
A hypomorphic gene-trap allele of Gldc (denoted GldcGT1) causes an approximately 90% reduction in mRNA abundance, elevated glycine concentration, and altered folate profile. NTDs arise in approximately 20% of homozygous GldcGT1/GT1 embryos (Pai et al., 2015; Table S3). To analyze the effect of a greater reduction in GCS activity, we generated an additional line of Gldc-deficient mice (GldcGT2) using an embryonic stem cell (ESC) line carrying a gene trap construct in intron 19 of Gldc that results in a truncated mRNA lacking exons 20–25. Gldc mRNA expression was undetectable in the homozygous mutant embryos by qRT-PCR. Neural tube closure was incomplete, leading to highly penetrant NTDs affecting 57% of homozygous GldcGT2/GT2 mutants analyzed at E9.5–E16.5 (Figure 4; Table S3). Maternal supplementation with formate during pregnancy prevented NTDs in Gldc-null embryos (Figure 4L), consistent with our findings in GldcGT1/GT1 hypomorphic embryos (Pai et al., 2015).
Figure 4.

Loss of Function of the GCS Causes Neural Tube Defects
(A–E) Cranial neural tube closure is complete in a Gldc+/+ embryo at E10.5 (A), whereas, in GldcGT2/GT2 embryos (B–E), the neural folds remain open (region between arrows) in the mid-hindbrain (B), fore-hindbrain (C), or throughout the entire mid-hindbrain and spinal region (craniorachischisis [Crn], D and E).
(F–H) At later stages (F–H, E15.5), failed closure in the brain, low spine, or entire brain and spine leads to the typical appearance of exencephaly (Ex, F and G), spina bifida (SB, G), and Crn (H), respectively (scale bars represent 1 mm).
(I–K) Among litters examined at E16.5–18.5, unilateral (J and J’) or bilateral (K and K’) eye defects were frequently observed among Gldc mutant fetuses but not in the wild-type (I and I’).
(L) Maternal formate supplementation prevents NTDs in GldcGT2/GT2 embryos (n = 10 untreated, 9 formate-treated; ∗p < 0.02, Fisher’s exact test).
(M) Frequency of NTDs among litters from GldcGT2/+ intercrosses (n = 38 litters analyzed).
Data for eye defects are included for a subset of litters analyzed at E14.5–16.5. See also Table S3.
Compound mutant embryos, GldcGT1/GT2, generated by intercrossing of GldcGT1/+ and GldcGT2/+ heterozygotes, also developed NTDs with similar penetrance (53%) as GldcGT2/GT2 homozygotes. NTDs also occurred at a low frequency among heterozygous GldcGT1/+ (0.7%) and GldcGT2/+ (6%) embryos (Table S3). As in GldcGT1 mutants, NTDs principally affected the cranial region (exencephaly) (Figures 4B, 4C, and 4F). However, in GldcGT2/GT2 and GldcGT1/GT2 mutants, we also observed occasional spina bifida (Figure 4G) or craniorachischisis (Figure 4H). Spina bifida results from failure to complete closure in the low spine, whereas craniorachischisis results from failure of initiation of closure at the hindbrain-cervical boundary, and the entire spinal neural tube remains open (Figures 4D and 4E). Hence, GCS function contributes to neural tube closure at all axial levels. In addition to NTDs, abnormalities of eye development occurred in 30% of GldcGT2/GT2 homozygous mutants examined at E14.5–E16.5 (Figure 4M). Eye defects tended to be unilateral and resembled anophthalmia or microphthalmia (Figures 4J and 4K).
Glycine Cleavage Provides 1C Units in Folate Metabolism and Contributes to De Novo Purine Biosynthesis
The lower abundance of 1C-carrying folates and rescue of NTDs by formate in Gldc-deficient embryos led us to hypothesize that NTDs caused by GCS disruption result from a diminished supply of glycine-derived 1C units to FOCM. On the other hand, whether there is a requirement for glycine as a 1C donor is questioned by metabolic labeling in cancer cell lines, which found that glycine cleavage does not contribute 1C units for nucleotide biosynthesis (Jain et al., 2012, Labuschagne et al., 2014, Fan et al., 2014). Instead, excess glycine led to SHMT-mediated conversion of glycine to serine and inhibition of proliferation, presumably because of depletion of 1C units that are required for nucleotide biosynthesis (Fan et al., 2014, Labuschagne et al., 2014).
Metabolic labeling in humans and in fetal lambs shows bidirectional interconversion of serine and glycine (Kalhan and Hanson, 2012, Lamers et al., 2007). In contrast to cancer cell lines, infusion of [13C2] glycine in adult humans indicated that glycine can contribute to serine synthesis via the “reverse” SHMT-mediated reaction, not only directly (as an intact molecule) but also as a 1C donor (via 5,10-methylene THF) following glycine cleavage (Kalhan and Hanson, 2012, Lamers et al., 2007, Lamers et al., 2009). Although this whole-body flux analysis confirmed that GCS activity provides 1C units for generation of 5,10-methylene THF, the majority of this is utilized in serine synthesis as opposed to other FOCM outputs (Lamers et al., 2009).
In embryos, including at neurulation stages, it has not yet been determined whether GCS activity contributes to production of 1C units for cytosolic functions of FOCM, including nucleotide biosynthesis. Given the known tissue heterogeneity in the regulation of FOCM (Brosnan et al., 2015, Ducker and Rabinowitz, 2017), we further investigated this question by labeling embryos with [1,2-13C] glycine or [1,2-12C] glycine in whole-embryo culture for a period of 24 hr from E9.5 (Figure 5A). Tissue extracts (n = 5 per genotype for each treatment) were analyzed by LC-MS.
Figure 5.

Glycine Is a 1C Donor in Neurulation-Stage Embryos
(A) Labeling with [1,2-13C] glycine was performed in whole-embryo culture with the yolk sac intact until E10.5 (typical stage of embryos after culture is shown; the arrow indicates cranial NTD in a GldcGT1/GT2 embryo).
(B) The ion abundance of labeled glycine was elevated in Gldc-null embryos compared with wild-types (n = 5 embryos per genotype).
(C–E) Fractional incorporation of labeled glycine was approximately 10% of total (C). Glycine was incorporated intact into serine (C) and purines (D and E), in which it contributes C4 and C5. In wild-type embryos, M+1 labeling of serine and purines (at C2 or C8) was also detected, implying GCS-mediated cleavage of glycine to generate 5,10-methylene THF and transfer of the 1C unit (indicated in red) via formate to formyl-THF in the cytoplasm (E). No M+1 or M+3 labeling of serine or purines was detected in Gldc-null embryos.
Wild-type and Gldc-deficient (GldcGT1/GT2) embryos exhibited uptake of labeled glycine (M+2, mass of unlabeled glycine + 2) from the culture medium (rat serum). As a proportion of total glycine, the maximum enrichment of labeled glycine was approximately 10% (Figure 5C). Gldc-deficient embryos contained a greater quantity of glycine, as shown by ion abundance (Figure 5B), as predicted by their lack of GCS activity (p < 0.001, t test). Some labeled glycine was converted to serine in both wild-type and Gldc-deficient embryos, showing that the Shmt1 and/or Shmt2 reaction is reversible in the embryo (Figure 5C). In addition to the M+2 isotopomer, a fraction of serine was the M+1 isotopomer in wild-type embryos but not in GldcGT1/GT2 mutants (Figure 5C). This labeling results from transfer of a glycine-derived 1C unit from 5,10-methylene THF and confirms the activity of the GCS. Arguing against a significant reversal of the Shmt1/2 reaction toward serine production, because of excess glycine in embryos lacking GCS activity, the ion abundance of labeled serine was not elevated in Gldc-deficient embryos (Figure 5B). Instead, the levels showed a trend toward reduction (p = 0.054), suggesting that demand for serine as a 1C donor may be increased. Similarly, in non-cultured embryos, the elevated concentration of tissue glycine observed in GldcGT1/GT1 embryos was not accompanied by an increase in total tissue serine content (Table S4).
M+1 labeling of serine in wild-type but not in Gldc-deficient embryos demonstrates that glycine cleavage contributes 1C units to FOCM, at least as far as 5,10-methylene THF in the mitochondria. It remained possible that all glycine-derived 1C units remain within mitochondrial FOCM, either in serine synthesis or through complete oxidation, in which 10-formyl THF is converted to CO2 and THF, via activity of mitochondrial 10-formyl THF dehydrogenase (ALDH1L2) (Krupenko et al., 2010). We therefore asked whether labeled glycine was utilized in de novo purine synthesis (Figure 5D). Intact glycine contributes C4, C5, and N7 of the purine ring in formation of the intermediate glycinamide ribonucleotide (GAR), catalyzed by Gart. In addition, C2 and C8 are contributed by 10-formyl THF (Figure 5E). Hence, purine nucleotide M+2 isotopomers can be generated from incorporation of intact [1,2-13C] glycine. We detected M+2 purines, including AMP, ADP, ATP, guanine, guanosine monophosphate (GMP), and guanosine diphosphate (GDP), in both Gldc+/+ and GldcGT1/GT2 embryos (Figure 5D). M+1 and M+3 isotopomers, which can only be generated by provision of glycine-derived 1C units via 5,10-methylene THF and formate, were detected in wild-type embryos, demonstrating contribution of the GCS to de novo purine synthesis. We assume that the abundance of M+4 isotopomers, if formed, was below the level of detection. Notably, no M+1 or M+3 purine nucleotides were present in GldcGT1/GT2 embryos. In summary, Gldc-deficient embryos can incorporate intact glycine into the purine ring but cannot contribute glycine-derived 1C units to purine synthesis.
Mthfr Activity or Methionine Treatment Modifies the Frequency of NTDs Caused by Abnormal Folate Metabolism
In humans, the rs1801133 SNP (C677T) in MTHFR is associated with increased risk of NTDs in some populations (Botto and Yang, 2000), and functional mutations in GLDC have been identified in NTD patients (Narisawa et al., 2012). We therefore generated compound heterozygous Mthfr+/−; Gldc+/GT2 mice to examine the effects of simultaneously reducing function in both genes. Two contrasting outcomes were considered. Because methionine cycle inhibitors cause NTDs, and the SAM/SAH ratio is lower in Mthfr-null embryos, it appeared possible that combined loss of Mthfr and GCS activity could have an additive, deleterious effect on neural tube closure. Alternatively, we reasoned that, if insufficiency of 1C units within the folate cycle is the cause of Gldc-related NTDs, then prevention of 1C transfer to the methionine cycle via Mthfr could have an ameliorating effect. The double heterozygous mice were viable and were intercrossed to generate experimental litters. As in previous crosses, Mthfr−/−; Gldc+/+ embryos did not develop NTDs, whereas Mthfr+/+; GldcGT2/GT2 embryos displayed a high frequency of cranial NTDs. Remarkably, we observed a protective effect of the Mthfr-null allele among GldcGT2/GT2 embryos, with a significant reduction in NTD frequency among compound null embryos (Figure 6A). Similarly, although infrequent NTDs were observed among GldcGT2/+ heterozygotes, they did not arise in Mthfr −/−; GldcGT2/+ embryos.
Figure 6.

Prevention of Gldc-Associated NTDs by Mthfr Loss of Function
(A) The frequency of NTDs among Gldc homozygous mutants is significantly affected by Mthfr genotype (different from Mthfr+/+, ∗p < 0.05 Fisher’s exact test).
(B) Among embryos carrying loss-of-function alleles of Gldc and Mthfr (n = 4–7 samples per genotype at E11.5), we observed significant variation between genotypes in the relative abundance of each folate (p < 0001, ANOVA; Holm-Sidak pairwise comparison: ∗∗ indicates difference to all other genotypes, ∗ bars do not differ from each other but significantly differ from all other genotypes; # indicates significant difference with Gldc genotype in Mthfr+/+ embryos).
(C) Abundance of SAH was significantly increased and the SAM/SAH ratio decreased in all Mthfr−/−embryos compared with other Mthfr genotypes, irrespective of Gldc genotype (∗p < 0.01, ANOVA).
LC-MS/MS-based folate profiling of the Gldc/Mthfr compound mutant embryos at E11.5 showed significant variation between genotypes in the relative abundance of each folate (Figure 6B). This was largely driven by the Mthfr genotype in that the Mthfr−/− embryos differed from Mthfr+/+ in abundance of every folate, irrespective of Gldc genotype (Figure 6B). Hence, the abundance of 5-methyl THF was significantly diminished in Mthfr-null embryos, and the relative abundance of the other folates, including formyl-THF and THF, was correspondingly increased in Mthfr−/−; GldcGT2/GT2 compared with Mthfr+/+; GldcGT2/GT2 embryos. Loss of function of Gldc caused an additional increase in dihydrofolate (DHF) and a decrease in methylene-THF in double-mutant Mthfr−/−; GldcGT2/GT2 embryos compared with Mthfr−/−; Gldc+/+ (Figure 6B; bars marked ∗∗ differ from all other genotypes). In the presence of wild-type Mthfr, loss of function of Gldc conferred an increase in abundance of DHF and THF and a reduction in 5-methyl THF, as observed in GldcGT1 embryos (Pai et al., 2015).
Quantification of embryonic tissue SAM showed no effect of Gldc or Mthfr genotype, whereas SAH abundance was significantly increased in the absence of Mthfr but not Gldc (Figure 6C). Hence, preventing transfer of 1C units from the folate cycle to the methionine cycle prevents NTDs in Gldc-deficient embryos.
We asked whether loss of Mthfr activity also modifies susceptibility to NTDs induced by pharmacological disruption of the folate cycle. Maternal treatment with methotrexate, an inhibitor of dihydrofolate reductase (Dhfr) is reported to induce NTDs (Zhao et al., 2013). However, using similar doses, we did not observe NTDs in embryos of any genotype among litters generated by intercrossing of Mthfr+/− mice (Table S5). As an alternative approach, we tested the effect of maternal treatment with 5-fluorouracil, a thymidylate synthase inhibitor. Single doses of 28 or 40 mg/kg at E8.5 resulted in embryonic death (resorption) or severe retardation of growth and development, so cranial neural tube closure could not be assessed. Among 22 litters (165 embryos) treated with a lower dose of 5-fluorouracil (12.5 or 20 mg/kg), NTDs were observed at low frequency (Table S5; Figure S2). Compared with Mthfr+/+ embryos (11.6% NTDs), we noted a trend toward a lower frequency of NTDs among littermates carrying one (Mthfr+/−; 5.0% NTDs) or two (Mthfr−/−; 2.9%) null alleles (Table S5).
Finally, because the Mthfr/Gldc mice (above) were not fully congenic, we further tested whether modulation of the methionine cycle affects neural tube closure in Gldc-deficient embryos even though there is no apparent deficit of methionine cycle metabolites. We predicted that provision of supplemental methionine may alter partitioning of 1C units from the folate cycle, as in Mthfr-null embryos. Litters were generated by intercrossing of GldcGT1/+ and GldcGT2/+ mice and treated by maternal supplementation with methionine (70 mg/kg, n = 20 control and 12 methionine litters). Notably, we observed a significantly lower frequency of cranial NTDs among methionine-treated GldcGT1/GT2 embryos compared with controls (Figure 7A). Methionine treatment appeared to normalize the folate profile of Gldc-deficient embryos. In control litters, GldcGT1/GT2 embryos exhibit a higher relative abundance of THF and a decreased relative abundance of 5-methyl THF at E10.5 compared with Gldc+/+ littermates. The magnitude of these abnormalities was diminished in methionine-treated embryos (Figure 7B). Notably, the relative abundance of formyl THF was extremely low in untreated Gldc-deficient embryos but was increased by methionine treatment (Figure 7B). These findings are consistent with the hypothesis that supplemental methionine “spares” folate-derived 1C units that are retained in the folate cycle and support functions that are essential for neural tube closure.
Figure 7.

Methionine Supplementation of Gldc-Deficient Embryos Is Associated with Normalization of the Folate Profile and Prevention of NTDs
(A) Compared with controls (n = 20 litters), maternal supplementation with methionine from E7.5 (n = 12 litters) resulted in a lower frequency of NTDs among homozygous mutant embryos (significant difference from controls; ∗p < 0.05, Fisher’s exact test).
(B) Folate profile of embryos collected at E10.5 following maternal methionine treatment (n = 3–6 embryos per group). Significant differences in relative abundance between genotypes and/or treatment were noted for THF, 5-methyl THF, and formyl THF (p < 0.001, two-way ANOVA; ∗, significant difference from other genotypes within treatment group; ∗∗, significant effect of methionine treatment within the same genotype).
Discussion
Folate status is associated with risk of NTDs in human clinical trials and population studies (Blom et al., 2006, Copp et al., 2013, Crider et al., 2011). The link between the folate cycle and methionine cycle in FOCM has led to the hypothesis of a mechanistic sequence from disruption of folate-dependent reactions to impaired methylation and consequent failure of neural tube closure. This model is widely cited but appears to be an over-simplification. Our findings suggest that regulation of the partitioning of 1C units between mitochondrial FOCM, the folate cycle, and flux through the methionine cycle is required for neural tube closure.
Although inhibitor studies need careful interpretation, for potential non-specific effects, they provide experimental evidence that methionine cycle function is essential for neural tube closure in mice (Dunlevy et al., 2006; this study), chicks (Afman et al., 2005) and Xenopus (Toriyama et al., 2017). Plausible mechanisms that may underlie NTDs include diminished methylation of genomic DNA (Okano et al., 1999), proteins (Moephuli et al., 1997, Toriyama et al., 2017), or other molecules. Nevertheless, analysis of Mthfr-null embryos shows that generation of 5-methyl THF within the neurulation stage embryo is not essential for maintenance of SAM abundance or for neural tube closure. Moreover, concomitant loss of both maternal and embryonic Mthfr activity does not cause NTDs. These observations suggest that, despite a significant reduction in the SAM/SAH ratio, the availability of SAM for methylation is adequate to meet demand during neurulation (despite post-natal abnormalities). Similarly, dietary folate deficiency causes a reduction in the SAM/SAH ratio without causing NTDs in wild-type embryos (Burren et al., 2008). These observations suggest that maternal methionine intake and/or use of choline and betaine as sources of 1C groups for methylation of homocysteine by betaine-homocysteine methyltransferase (Bhmt) is sufficient to meet the demand for methionine in the neurulating embryo (for production of SAM and for protein synthesis). In fact, both may play a role. We found that SAH is elevated in Mthfr-null embryos (suggesting a limit on homocysteine remethylation by Bhmt), whereas SAM is diminished in Mthfr-null tissues at 3 weeks of age (suggesting that dietary methionine is insufficient to meet the demand, at least post-natally). Interestingly, lowering of SAM levels in mouse blastocysts requires concomitant suppression of both the folate cycle and Bhmt expression (Zhang et al., 2015). The reduction in plasma betaine levels in Mthfr-null adult mice suggests increased used of the choline-betaine-dependent pathway for methylation reactions (Schwahn et al., 2003).
How do findings in mice relate to human NTDs? Although evaluation of the effect of maternal folate status on DNA methylation within the neurulation stage human embryo is practically difficult, associations have been observed in analysis of cord blood (Joubert et al., 2016). Notably however, the majority of reported studies show a negative association, arguing against a generalized methylation-promoting effect of folic acid. Whether specific genomic loci or other targets, such as proteins or RNA, are targeted remains to be determined. Human studies of association of blood choline levels with NTDs have been inconsistent, suggesting that, if an effect is present, then it is unlikely to be strong (Mills et al., 2014). Interestingly however, association with NTDs has been noted for polymorphisms in PEMT, encoding phosphatidylethanolamine N-methyltransferase, which mediates phosphatidylcholine biosynthesis (Zhang et al., 2006, Mills et al., 2014). Moreover, use of choline as a methyl donor in production of phosphatidylcholine appears to be favored in individuals carrying the rs1801133 polymorphism in MTHFR (Yan et al., 2011). It is possible that the potential association of choline status with NTDs needs to be analyzed in the context of genotype for both MTHFR and PEMT.
Although transfer of 1C units from the folate cycle to the methionine cycle in embryonic or maternal FOCM appears to be dispensable for neural tube closure in mice, we find that generation of 1C units in mitochondrial FOCM is essential. Hence, NTDs caused by loss of function of the GCS result from lack of 1C transfer to THF, with consequent suppression of 5,10-methylene THF synthesis and subsequent 1C-dependent reactions. This model is consistent with prevention of NTDs by formate, which occurs even though elevated glycine is still present in formate-treated embryos (Pai et al., 2015). The consequence of GCS disruption in the embryo therefore differs from effects of elevated glycine in some cancer cell lines, where elevated glycine reverses Shmt-mediated reactions and inhibits proliferation (Labuschagne et al., 2014) or even has toxic effects (Kim et al., 2015).
Isotope labeling demonstrated that glycine contributes to de novo purine synthesis both by direct incorporation into the purine ring and via donation of 1C groups via 10-formyl THF. The dependence on two 10-formyl THF units may confer particular sensitivity of purine biosynthesis to impaired glycine cleavage compared with thymidylate biosynthesis, which requires only one 1C group.
Having established that GCS-derived 1C units contribute to cytoplasmic FOCM in the embryo, we found no effect of Gldc deficiency on the abundance of SAM, SAH, or the SAM/SAH ratio, suggesting that Gldc-related NTDS are unlikely to result from impaired methionine cycle activity. This conclusion is consistent with the lack of NTDs in Mthfr−/− embryos even though SAH concentration is elevated, although SAM-mediated methylation is limiting for neural tube closure based on the evidence from use of methylation cycle inhibitors. On the other hand, the striking effect of Mthfr loss of function in preventing NTDs in Gldc-null embryos suggests that retention of 1C units within the folate cycle, at the expense of the methionine cycle, ameliorates the diminished supply from glycine cleavage. These findings suggest a crucial requirement for the folate cycle and/or mitochondrial FOCM in neural tube closure; key outputs likely include nucleotide biosynthesis as well as potentially other outputs of these cycles. The protective effect of Mthfr ablation is also consistent with the protective effect of methionine in Gldc-null embryos (this study) and in Amt−/− embryos (Narisawa et al., 2012). We hypothesize that addition of methionine spares 1C units from transfer to the methionine cycle. In both Mthfr-null and methionine-treated embryos, prevention of NTDs correlates with an increase in relative abundance of 10-formyl THF.
The protective effect of Mthfr loss of function in a mouse mutant with NTDs could be considered counterintuitive in the context that the rs1801133 (C677T) polymorphism is an established risk factor for NTD (Botto and Yang, 2000, Blom et al., 2006, Greene et al., 2009). However, this polymorphism is associated with lower plasma and red blood cell folate levels (Crider et al., 2011, Tsang et al., 2015). Our findings are consistent with the hypothesis that the NTD risk association of MTHFR is mediated through the effect on blood folate status (Stover et al., 2015) rather than an effect on enzymatic activity leading to diminished methylation.
Experimental Procedures
Mice
Mthfr-null mice were described previously (Chen et al., 2001). To generate Mthfr-null dams for experimental matings, early lethality was prevented by maternal supplementation with betaine until weaning (Schwahn et al., 2004). Gldc-deficient mice (denoted GldcGT1) were described previously (Pai et al., 2015). An additional line of Gldc-deficient mice (denoted GldcGT2) was generated using an embryonic stem cell line obtained from the North American Conditional Mouse Mutagenesis (NorCOMM) project. Animal studies were carried out under the regulations of the Animals (Scientific Procedures) Act 1986 of the United Kingdom Government and in accordance with the guidance issued by the Medical Research Council, United Kingdom in Responsibility in the Use of Animals for Medical Research (July 1993).
Litters were generated by timed matings in which mice (used from 7 weeks of age) were paired overnight, and the day of finding a copulation plug was designated E0.5. Whole-embryo culture was performed as described previously (Dunlevy et al., 2006, Pryor et al., 2012). Mice and embryos were genotyped by PCR of genomic DNA.
Quantification of FOCM Intermediates by Mass Spectrometry
Analysis of multiple folates was performed by ultra-performance LC (UPLC)-MS/MS as described previously (Pai et al., 2015). Folates were measured by multiple reaction monitoring (MRM) with optimized cone voltage and collision energy for precursor and product ions as described previously (Leung et al., 2013, Pai et al., 2015). SAM and SAH were quantified by LC-MS/MS as described previously (Burren et al., 2006) with minor modifications. Cytosine 5-methylation of DNA was analyzed by LC mass spectrometry using a method that is insensitive to co-occurring RNA methylation (Capuano et al., 2014). See the Supplemental Experimental Procedures for details.
Stable Isotope Tracing of [1,2-13C] Glycine
Embryo extracts were analyzed by LC-MS as described previously (Chen et al., 2012, Ismailoglu et al., 2014). We applied an in-house untargeted stable isotope tracing (USIT) workflow in which labeled metabolites were identified on the basis of differential abundance in embryos cultured in [1,2-13C] glycine compared with [1,2-12C] glycine-supplemented serum. Selected metabolites were identified on the basis of previously curated isotopologues.
Statistical Analysis
Quantitative data (e.g., metabolite abundance) was analyzed by t test (two groups) or ANOVA (three or more groups) with Holm-Sidak pairwise comparison for post hoc analysis. Prior to application of these methods, data were checked for normal distribution. Analysis of the frequency of Gldc mutant embryos that exhibited NTDs in different treatment groups or by Mthfr genotype was performed by Fisher’s exact test. Statistical analysis was performed using Sigmastat (v3.5, Systat Software).
Author Contributions
Conceptualization, N.D.E.G., K.-Y.L., and A.J.C.; Methodology, Q.C., S.S.G., K.-Y.L., and M.R.; Formal Analysis, N.D.E.G. and K.-Y.L.; Investigation, K.-Y.L., Y.J.P., Q.C., C.S., E.C., S.S., D.S., and N.D.E.G.; Writing – Original Draft, N.D.E.G; Writing – Review & Editing, N.D.E.G., K.-Y.L., and A.J.C.; Visualization, N.D.E.G., K.-Y.L., and Q.C.; Supervision, N.D.E.G., M.R., and S.S.G.
Acknowledgments
We thank Rima Rozen for supplying Mthfr mice and for helpful discussions, Evanthia Nikolopoulou for assistance with illustrations, Michael Muelleder and Christoph Messner for technical assistance, and the UCL Translational Mass Spectrometry Research group for ongoing collaboration. The project was funded by the Medical Research Council (N003713 and J003794 to N.D.E.G. and A.J.C.), NIH Research Grant PO1 HD067244 (to S.S.G.), Action Medical Research (GN2403 to N.D.E.G. and A.J.C.), and Great Ormond Street Hospital Children’s Charity (to N.D.E.G. and A.C.). M.R. is supported by the Francis Crick Institute, which receives its core funding from the Wellcome Trust, United Kingdom Medical Research Council, and Cancer Research UK (Programme FC001134). Research was supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children, the NHS Foundation Trust, and University College London.
Published: November 14, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.10.072.
Supplemental Information
References
- Afman L.A., Blom H.J., Drittij M.J., Brouns M.R., van Straaten H.W. Inhibition of transmethylation disturbs neurulation in chick embryos. Brain Res. Dev. Brain Res. 2005;158:59–65. doi: 10.1016/j.devbrainres.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Alix J.-H. Molecular aspects of the in vivo and in vitro effects of ethionine, an analog of methionine. Microbiol. Rev. 1982;46:281–295. doi: 10.1128/mr.46.3.281-295.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin A.E., Abarinov E.V., Noden D.M., Perry C.A., Chu S., Stabler S.P., Allen R.H., Stover P.J. Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. Am. J. Clin. Nutr. 2011;93:789–798. doi: 10.3945/ajcn.110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom H.J., Shaw G.M., den Heijer M., Finnell R.H. Neural tube defects and folate: case far from closed. Nat. Rev. Neurosci. 2006;7:724–731. doi: 10.1038/nrn1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto L.D., Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am. J. Epidemiol. 2000;151:862–877. doi: 10.1093/oxfordjournals.aje.a010290. [DOI] [PubMed] [Google Scholar]
- Brosnan M.E., MacMillan L., Stevens J.R., Brosnan J.T. Division of labour: how does folate metabolism partition between one-carbon metabolism and amino acid oxidation? Biochem. J. 2015;472:135–146. doi: 10.1042/BJ20150837. [DOI] [PubMed] [Google Scholar]
- Burren K.A., Mills K., Copp A.J., Greene N.D.E. Quantitative analysis of s-adenosylmethionine and s-adenosylhomocysteine in neurulation-stage mouse embryos by liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006;844:112–118. doi: 10.1016/j.jchromb.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burren K.A., Savery D., Massa V., Kok R.M., Scott J.M., Blom H.J., Copp A.J., Greene N.D.E. Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum. Mol. Genet. 2008;17:3675–3685. doi: 10.1093/hmg/ddn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burren K.A., Scott J.M., Copp A.J., Greene N.D. The genetic background of the curly tail strain confers susceptibility to folate-deficiency-induced exencephaly. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:76–83. doi: 10.1002/bdra.20632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano F., Mülleder M., Kok R., Blom H.J., Ralser M. Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal. Chem. 2014;86:3697–3702. doi: 10.1021/ac500447w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudill M.A., Wang J.C., Melnyk S., Pogribny I.P., Jernigan S., Collins M.D., Santos-Guzman J., Swendseid M.E., Cogger E.A., James S.J. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J. Nutr. 2001;131:2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- Chen Z., Karaplis A.C., Ackerman S.L., Pogribny I.P., Melnyk S., Lussier-Cacan S., Chen M.F., Pai A., John S.W., Smith R.S. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- Chen Q., Park H.C., Goligorsky M.S., Chander P., Fischer S.M., Gross S.S. Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PLoS ONE. 2012;7:e37149. doi: 10.1371/journal.pone.0037149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp A.J., Stanier P., Greene N.D. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol. 2013;12:799–810. doi: 10.1016/S1474-4422(13)70110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crider K.S., Bailey L.B., Berry R.J. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients. 2011;3:370–384. doi: 10.3390/nu3030370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S.R., Stacpoole P.W., Williamson J., Kick L.S., Quinlivan E.P., Coats B.S., Shane B., Bailey L.B., Gregory J.F., 3rd Tracer-derived total and folate-dependent homocysteine remethylation and synthesis rates in humans indicate that serine is the main one-carbon donor. Am. J. Physiol. Endocrinol. Metab. 2004;286:E272–E279. doi: 10.1152/ajpendo.00351.2003. [DOI] [PubMed] [Google Scholar]
- De Castro S.C., Leung K.Y., Savery D., Burren K., Rozen R., Copp A.J., Greene N.D.E. Neural tube defects induced by folate deficiency in mutant curly tail (Grhl3) embryos are associated with alteration in folate one-carbon metabolism but are unlikely to result from diminished methylation. Birth Defects Res. A Clin. Mol. Teratol. 2010;88:612–618. doi: 10.1002/bdra.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker G.S., Rabinowitz J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017;25:27–42. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlevy L.P.E., Burren K.A., Mills K., Chitty L.S., Copp A.J., Greene N.D.E. Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res. A Clin. Mol. Teratol. 2006;76:544–552. doi: 10.1002/bdra.20286. [DOI] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst J.J., Shlomi T., Thompson C.B., Rabinowitz J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A., Copp A.J. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280:2107–2109. doi: 10.1126/science.280.5372.2107. [DOI] [PubMed] [Google Scholar]
- Ghandour H., Chen Z., Selhub J., Rozen R. Mice deficient in methylenetetrahydrofolate reductase exhibit tissue-specific distribution of folates. J. Nutr. 2004;134:2975–2978. doi: 10.1093/jn/134.11.2975. [DOI] [PubMed] [Google Scholar]
- Greene N.D.E., Stanier P., Copp A.J. Genetics of human neural tube defects. Hum. Mol. Genet. 2009;18(R2):R113–R129. doi: 10.1093/hmg/ddp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid M.K., Bills N.D., Hinrichs S.H., Clifford A.J. Folate deficiency alone does not produce neural tube defects in mice. J. Nutr. 1992;122:888–894. doi: 10.1093/jn/122.4.888. [DOI] [PubMed] [Google Scholar]
- Herbig K., Chiang E.P., Lee L.R., Hills J., Shane B., Stover P.J. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J. Biol. Chem. 2002;277:38381–38389. doi: 10.1074/jbc.M205000200. [DOI] [PubMed] [Google Scholar]
- Ismailoglu I., Chen Q., Popowski M., Yang L., Gross S.S., Brivanlou A.H. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014;391:230–240. doi: 10.1016/j.ydbio.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadavji N.M., Deng L., Malysheva O., Caudill M.A., Rozen R. MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short-term memory and increased apoptosis in the hippocampus of wild-type offspring. Neuroscience. 2015;300:1–9. doi: 10.1016/j.neuroscience.2015.04.067. [DOI] [PubMed] [Google Scholar]
- Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A.L., Kafri R., Kirschner M.W., Clish C.B., Mootha V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert B.R., den Dekker H.T., Felix J.F., Bohlin J., Ligthart S., Beckett E., Tiemeier H., van Meurs J.B., Uitterlinden A.G., Hofman A. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat. Commun. 2016;7:10577. doi: 10.1038/ncomms10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhan S.C., Hanson R.W. Resurgence of serine: an often neglected but indispensable amino Acid. J. Biol. Chem. 2012;287:19786–19791. doi: 10.1074/jbc.R112.357194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Fiske B.P., Birsoy K., Freinkman E., Kami K., Possemato R.L., Chudnovsky Y., Pacold M.E., Chen W.W., Cantor J.R. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupenko N.I., Dubard M.E., Strickland K.C., Moxley K.M., Oleinik N.V., Krupenko S.A. ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 2010;285:23056–23063. doi: 10.1074/jbc.M110.128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne C.F., van den Broek N.J., Mackay G.M., Vousden K.H., Maddocks O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- Lamers Y., Williamson J., Gilbert L.R., Stacpoole P.W., Gregory J.F., 3rd Glycine turnover and decarboxylation rate quantified in healthy men and women using primed, constant infusions of [1,2-(13)C2]glycine and [(2)H3]leucine. J. Nutr. 2007;137:2647–2652. doi: 10.1093/jn/137.12.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers Y., Williamson J., Theriaque D.W., Shuster J.J., Gilbert L.R., Keeling C., Stacpoole P.W., Gregory J.F., 3rd Production of 1-carbon units from glycine is extensive in healthy men and women. J. Nutr. 2009;139:666–671. doi: 10.3945/jn.108.103580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrance A.K., Racine J., Deng L., Wang X., Lachapelle P., Rozen R. Complete deficiency of methylenetetrahydrofolate reductase in mice is associated with impaired retinal function and variable mortality, hematological profiles, and reproductive outcomes. J. Inherit. Metab. Dis. 2011;34:147–157. doi: 10.1007/s10545-010-9127-1. [DOI] [PubMed] [Google Scholar]
- Leung K.Y., De Castro S.C., Cabreiro F., Gustavsson P., Copp A.J., Greene N.D. Folate metabolite profiling of different cell types and embryos suggests variation in folate one-carbon metabolism, including developmental changes in human embryonic brain. Mol. Cell. Biochem. 2013;378:229–236. doi: 10.1007/s11010-013-1613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J.W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane A.J., Anderson D.D., Flodby P., Perry C.A., Allen R.H., Stabler S.P., Stover P.J. Nuclear localization of de novo thymidylate biosynthesis pathway is required to prevent uracil accumulation in DNA. J. Biol. Chem. 2011;286:44015–44022. doi: 10.1074/jbc.M111.307629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J.L., Fan R., Brody L.C., Liu A., Ueland P.M., Wang Y., Kirke P.N., Shane B., Molloy A.M. Maternal choline concentrations during pregnancy and choline-related genetic variants as risk factors for neural tube defects. Am. J. Clin. Nutr. 2014;100:1069–1074. doi: 10.3945/ajcn.113.079319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moephuli S.R., Klein N.W., Baldwin M.T., Krider H.M. Effects of methionine on the cytoplasmic distribution of actin and tubulin during neural tube closure in rat embryos. Proc. Natl. Acad. Sci. USA. 1997;94:543–548. doi: 10.1073/pnas.94.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momb J., Lewandowski J.P., Bryant J.D., Fitch R., Surman D.R., Vokes S.A., Appling D.R. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. USA. 2013;110:549–554. doi: 10.1073/pnas.1211199110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa A., Komatsuzaki S., Kikuchi A., Niihori T., Aoki Y., Fujiwara K., Tanemura M., Hata A., Suzuki Y., Relton C.L. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 2012;21:1496–1503. doi: 10.1093/hmg/ddr585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Pai Y.J., Leung K.Y., Savery D., Hutchin T., Prunty H., Heales S., Brosnan M.E., Brosnan J.T., Copp A.J., Greene N.D. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun. 2015;6:6388. doi: 10.1038/ncomms7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor S.E., Massa V., Savery D., Greene N.D.E., Copp A.J. Convergent extension analysis in mouse whole embryo culture. Methods Mol. Biol. 2012;839:133–146. doi: 10.1007/978-1-61779-510-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M.C., Nijhout H.F., Neuhouser M.L., Gregory J.F., 3rd, Shane B., James S.J., Boynton A., Ulrich C.M. A mathematical model gives insights into nutritional and genetic aspects of folate-mediated one-carbon metabolism. J. Nutr. 2006;136:2653–2661. doi: 10.1093/jn/136.10.2653. [DOI] [PubMed] [Google Scholar]
- Schwahn B.C., Chen Z., Laryea M.D., Wendel U., Lussier-Cacan S., Genest J., Jr., Mar M.H., Zeisel S.H., Castro C., Garrow T., Rozen R. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003;17:512–514. doi: 10.1096/fj.02-0456fje. [DOI] [PubMed] [Google Scholar]
- Schwahn B.C., Laryea M.D., Chen Z., Melnyk S., Pogribny I., Garrow T., James S.J., Rozen R. Betaine rescue of an animal model with methylenetetrahydrofolate reductase deficiency. Biochem. J. 2004;382:831–840. doi: 10.1042/BJ20030822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J.M. Folate and vitamin B12. Proc. Nutr. Soc. 1999;58:441–448. doi: 10.1017/s0029665199000580. [DOI] [PubMed] [Google Scholar]
- Shah R.H., Northrup H., Hixson J.E., Morrison A.C., Au K.S. Genetic association of the glycine cleavage system genes and myelomeningocele. Birth Defects Res. A Clin. Mol. Teratol. 2016;106:847–853. doi: 10.1002/bdra.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelstein O., Mitchell L.E., Merriweather M.Y., Wicker N.J., Zhang Q., Lammer E.J., Finnell R.H. Embryonic development of folate binding protein-1 (Folbp1) knockout mice: Effects of the chemical form, dose, and timing of maternal folate supplementation. Dev. Dyn. 2004;231:221–231. doi: 10.1002/dvdy.20107. [DOI] [PubMed] [Google Scholar]
- Stover P.J., MacFarlane A.J., Field M.S. Bringing clarity to the role of MTHFR variants in neural tube defect prevention. Am. J. Clin. Nutr. 2015;101:1111–1112. doi: 10.3945/ajcn.115.111088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts A.S., Appling D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- Toriyama M., Toriyama M., Wallingford J.B., Finnell R.H. Folate-dependent methylation of septins governs ciliogenesis during neural tube closure. FASEB J. 2017;31:3622–3635. doi: 10.1096/fj.201700092R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang B.L., Devine O.J., Cordero A.M., Marchetta C.M., Mulinare J., Mersereau P., Guo J., Qi Y.P., Berry R.J., Rosenthal J. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am. J. Clin. Nutr. 2015;101:1286–1294. doi: 10.3945/ajcn.114.099994. [DOI] [PubMed] [Google Scholar]
- Yan J., Wang W., Gregory J.F., 3rd, Malysheva O., Brenna J.T., Stabler S.P., Allen R.H., Caudill M.A. MTHFR C677T genotype influences the isotopic enrichment of one-carbon metabolites in folate-compromised men consuming d9-choline. Am. J. Clin. Nutr. 2011;93:348–355. doi: 10.3945/ajcn.110.005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Vousden K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- Zhang J., Zhu H., Yang W., Shaw G.M., Lammer E.J., Finnell R.H. Phosphatidylethanolamine N-methyltransferase (PEMT) gene polymorphisms and risk of spina bifida. Am. J. Med. Genet. A. 2006;140:785–789. doi: 10.1002/ajmg.a.31142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Denomme M.M., White C.R., Leung K.Y., Lee M.B., Greene N.D., Mann M.R., Trasler J.M., Baltz J.M. Both the folate cycle and betaine-homocysteine methyltransferase contribute methyl groups for DNA methylation in mouse blastocysts. FASEB J. 2015;29:1069–1079. doi: 10.1096/fj.14-261131. [DOI] [PubMed] [Google Scholar]
- Zhao J., Guan T., Wang J., Xiang Q., Wang M., Wang X., Guan Z., Xie Q., Niu B., Zhang T. Influence of the antifolate drug Methotrexate on the development of murine neural tube defects and genomic instability. J. Appl. Toxicol. 2013;33:915–923. doi: 10.1002/jat.2769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.